
Role of Non-Coding Regulatory Elements in the Control of GR-Dependent Gene Expression


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
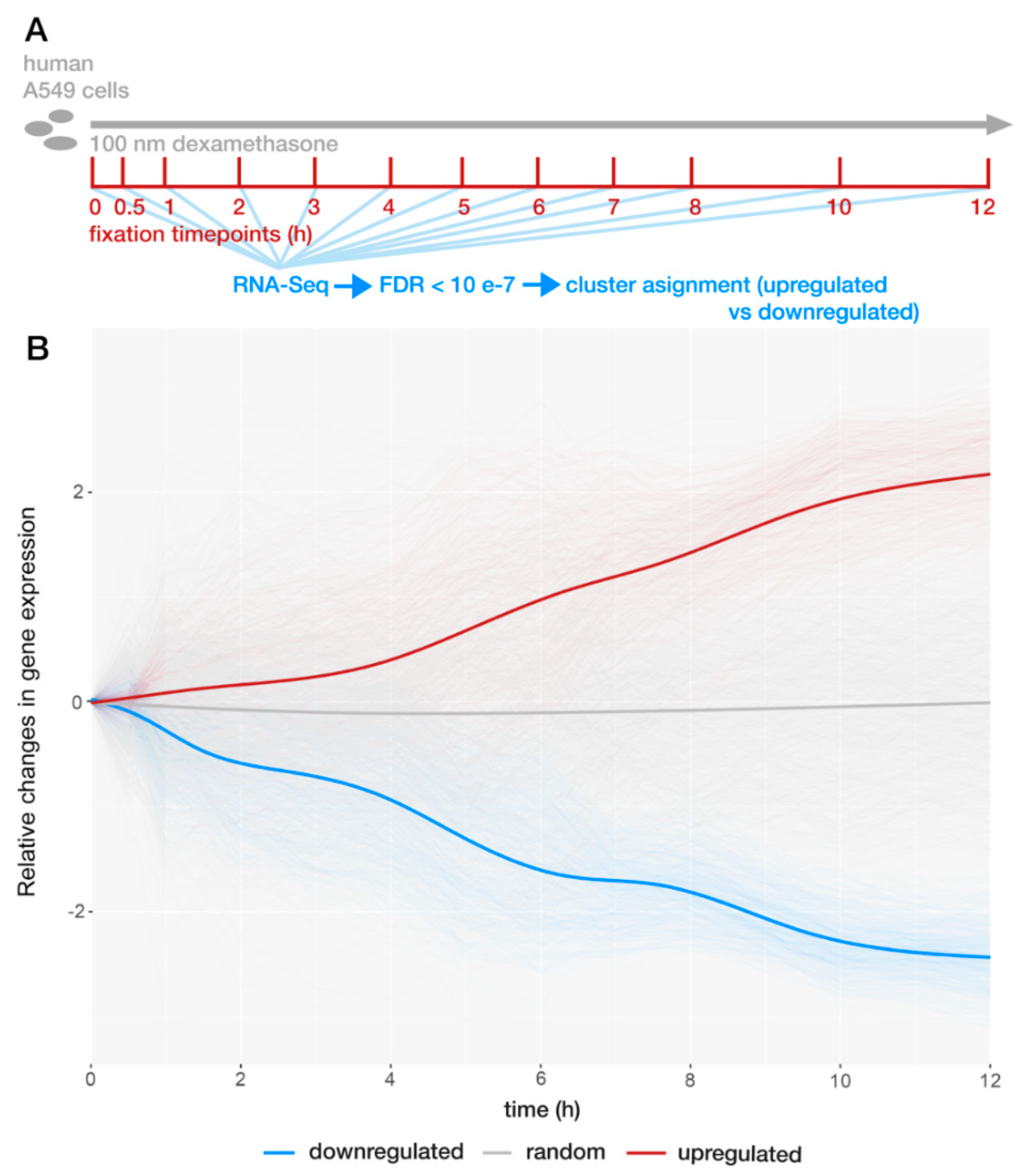
2.1. GR Regulates Expression Bidirectionally
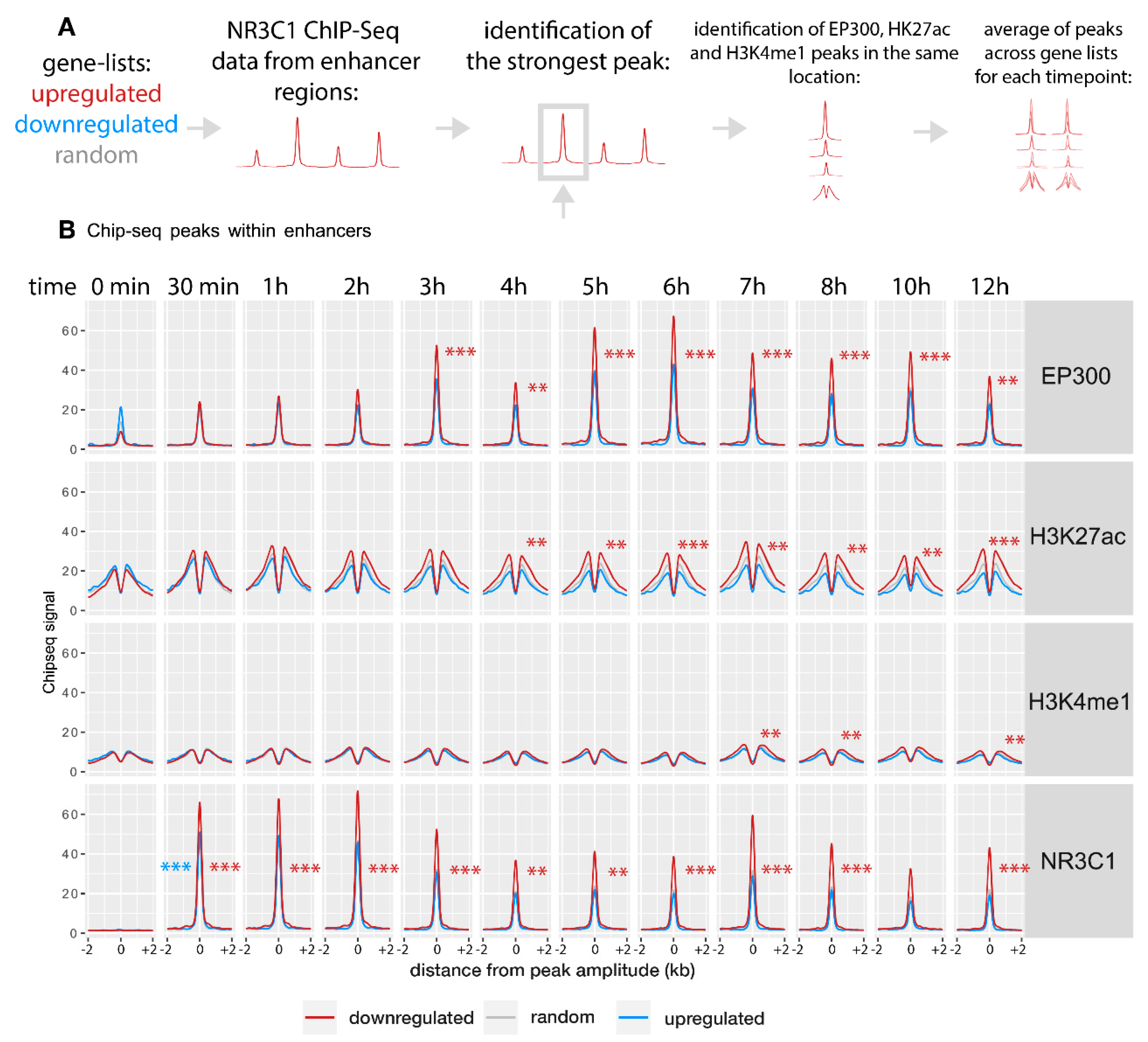
2.2. Gene Activation Is Associated with Enhancer Remodeling
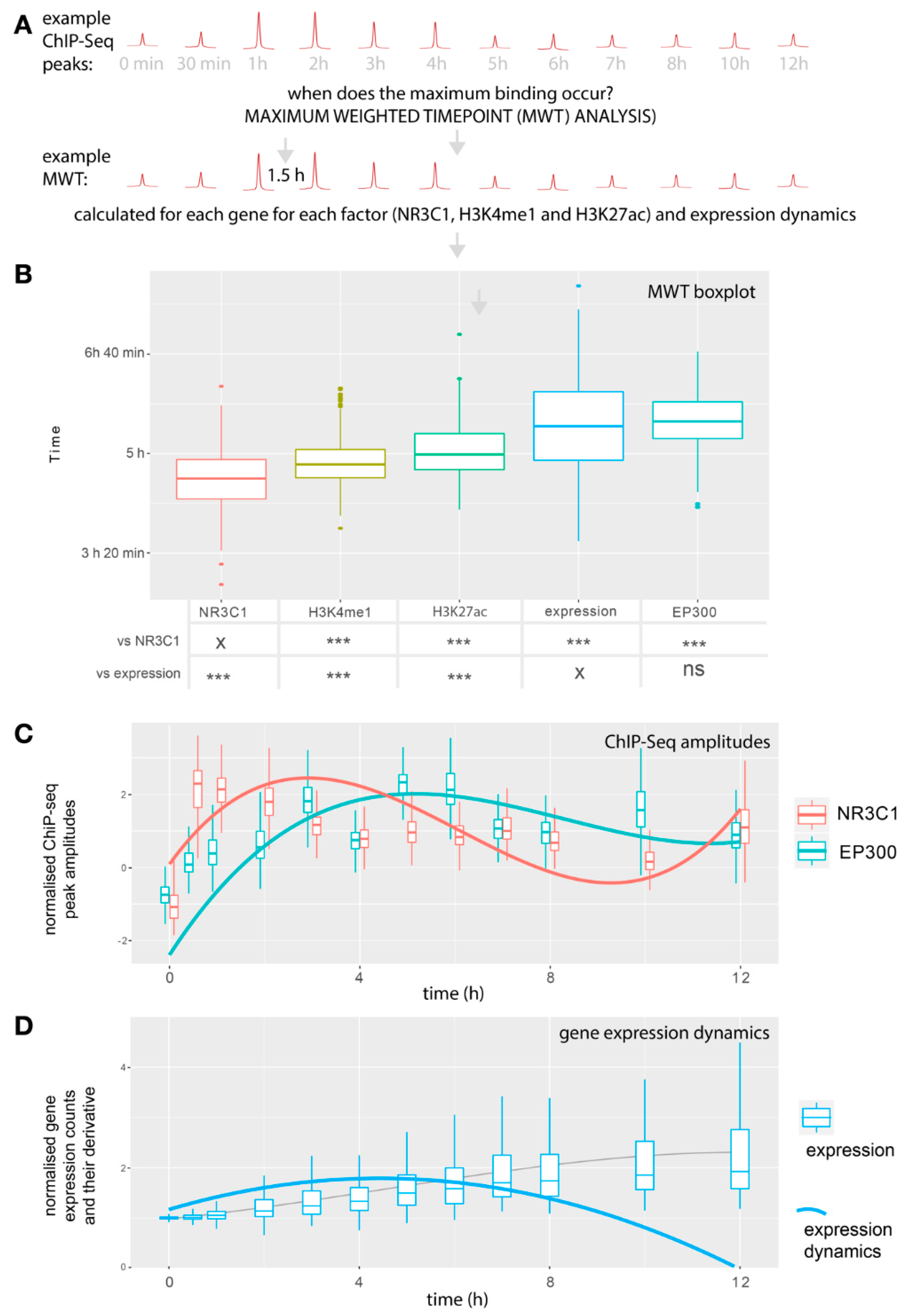
2.3. The Largest EP300 Recruitment Coincides with the Most Dynamic Gene Expression Changes
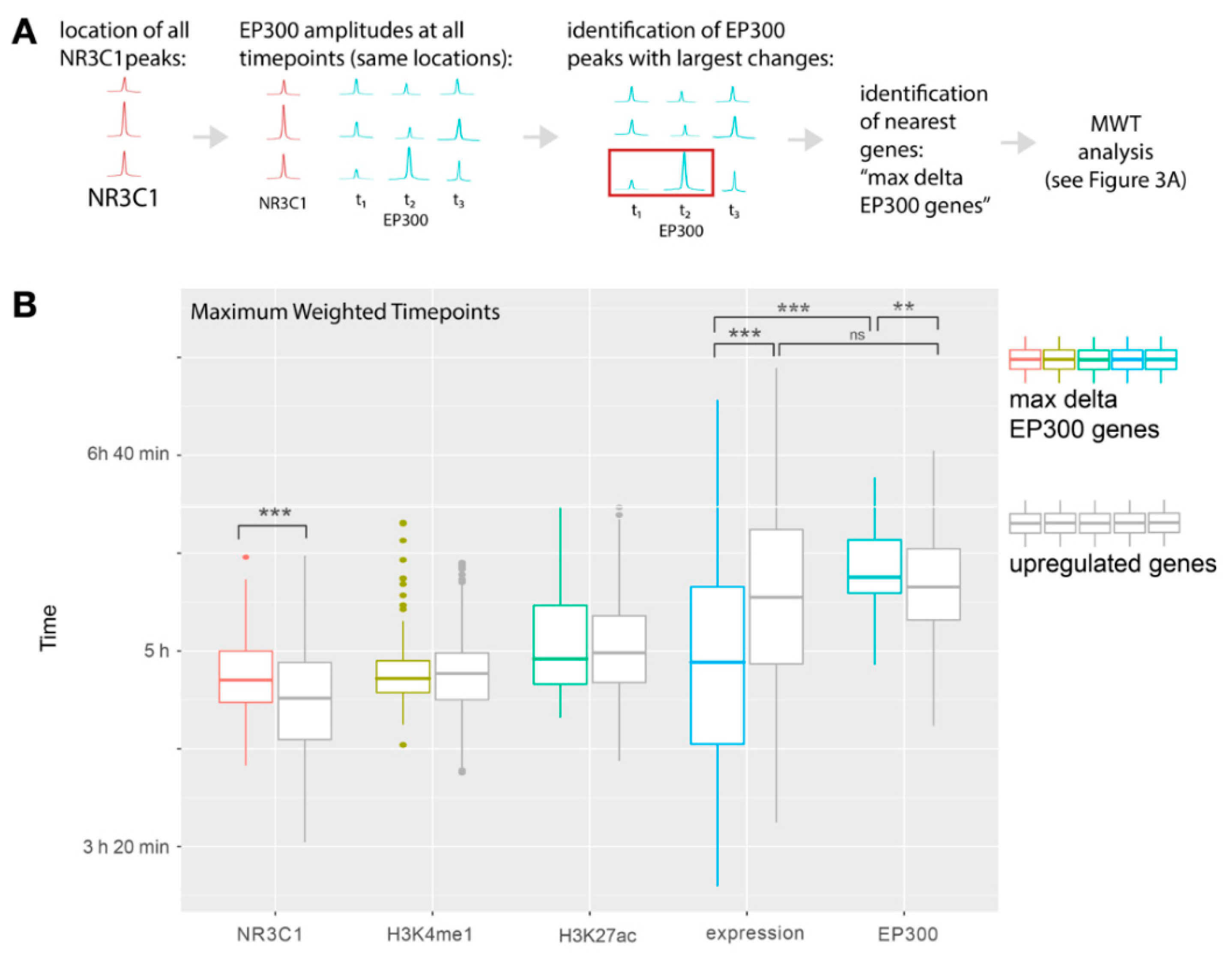
2.4. Peaks with the Highest Changes in EP300 Recruitment Occur near Upregulated Genes and EP300 Is Recruited to Them after the Largest Expression Dynamics Are Reached
3. Discussion
4. Methods
4.1. Datasets
4.2. RNA-Seq Analysis
4.3. ChIP-Seq analysis
5. Conclussions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Reddy, T.E.; Pauli, F.; Sprouse, R.O.; Neff, N.F.; Newberry, K.M.; Garabedian, M.J.; Myers, R.M. Genomic determination of the glucocorticoid response reveals unexpected mechanisms of gene regulation. Genome Res. 2009, 19, 2163–2171. [Google Scholar] [CrossRef] [Green Version]
- Piechota, M.; Korostynski, M.; Golda, S.; Ficek, J.; Jantas, D.; Barbara, Z.; Przewlocki, R. Transcriptional signatures of steroid hormones in the striatal neurons and astrocytes. BMC Neurosci. 2017, 18, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Slezak, M.; Korostynski, M.; Gieryk, A.; Golda, S.; Dzbek, J.; Piechota, M.; Wlazlo, E.; Bilecki, W.; Przewlocki, R. Astrocytes are a neural target of morphine action via glucocorticoid receptor-dependent signaling. Glia 2013, 61, 623–635. [Google Scholar] [CrossRef]
- D’Ippolito, A.M.; McDowell, I.C.; Barrera, A.; Hong, L.K.; Leichter, S.M.; Bartelt, L.C.; Vockley, C.M.; Majoros, W.H.; Safi, A.; Song, L.; et al. Pre-established Chromatin Interactions Mediate the Genomic Response to Glucocorticoids. Cell Syst. 2018, 7, 146–160.e7. [Google Scholar] [CrossRef] [Green Version]
- John, S.; Sabo, P.J.; Thurman, R.E.; Sung, M.-H.; Biddie, S.C.; Johnson, T.A.; Hager, G.L.; Stamatoyannopoulos, J.A. Chromatin accessibility pre-determines glucocorticoid receptor binding patterns. Nat. Genet. 2011, 43, 264–268. [Google Scholar] [CrossRef]
- McDowell, I.C.; Barrera, A.; D’Ippolito, A.M.; Vockley, C.M.; Hong, L.K.; Leichter, S.M.; Bartelt, L.C.; Majoros, W.H.; Song, L.; Safi, A.; et al. Glucocorticoid receptor recruits to enhancers and drives activation by motif-directed binding. Genome Res. 2018, 28, 1272–1284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuznetsova, T.; Wang, S.-Y.; Rao, N.A.; Mandoli, A.; Martens, J.H.A.; Rother, N.; Aartse, A.; Groh, L.; Janssen-Megens, E.M.; Li, G.; et al. Glucocorticoid receptor and nuclear factor kappa-b affect three-dimensional chromatin organization. Genome Biol. 2015, 16, 264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shlyueva, D.; Stampfel, G.; Stark, A. Transcriptional enhancers: From properties to genome-wide predictions. Nat. Rev. Genet. 2014, 15, 272–286. [Google Scholar] [CrossRef] [PubMed]
- Creyghton, M.P.; Cheng, A.W.; Welstead, G.G.; Kooistra, T.G.; Carey, B.W.; Steine, E.J.; Hanna, J.; Lodato, M.A.; Frampton, G.M.; Sharp, P.A.; et al. Histone H3K27ac separates active from poised enhancers and predicts developmental state. Proc. Natl. Acad. Sci. USA 2010, 107, 21931–21936. [Google Scholar] [CrossRef] [Green Version]
- Visel, A.; Blow, M.J.; Li, Z.; Zhang, T.; Akiyama, J.A.; Holt, A.; Plajzer-Frick, I.; Shoukry, M.; Wright, C.; Chen, F.; et al. ChIP-seq accurately predicts tissue-specific activity of enhancers. Nat. Cell Biol. 2009, 457, 854–858. [Google Scholar] [CrossRef] [Green Version]
- Heintzman, N.D.; Stuart, R.K.; Hon, G.; Fu, Y.; Ching, C.W.; Hawkins, R.D.; Barrera, L.O.; Van Calcar, S.; Qu, C.; Ching, K.A.; et al. Distinct and predictive chromatin signatures of transcriptional promoters and enhancers in the human genome. Nat. Genet. 2007, 39, 311–318. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Fish, A.E.; Capra, J.A. Prediction of gene regulatory enhancers across species reveals evolutionarily conserved sequence properties. PLoS Comput. Biol. 2018, 14, e1006484. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clément, Y.; Torbey, P.; Gilardi-Hebenstreit, P.; Crollius, H.R. Enhancer–gene maps in the human and zebrafish genomes using evolutionary linkage conservation. Nucleic Acids Res. 2020, 48, 2357–2371. [Google Scholar] [CrossRef] [PubMed]
- Jin, F.; Li, Y.; Dixon, J.R.; Selvaraj, S.; Ye, Z.; Lee, A.Y.; Yen, C.-A.; Schmitt, A.D.; Espinoza, C.A.; Ren, B. A high-resolution map of the three-dimensional chromatin interactome in human cells. Nature 2013, 503, 290–294. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kvon, E.Z.; Zhu, Y.; Kelman, G.; Novak, C.S.; Plajzer-Frick, I.; Kato, M.; Garvin, T.H.; Pham, Q.; Harrington, A.N.; Hunter, R.D.; et al. Comprehensive in vivo interrogation reveals phenotypic impact of human enhancer variants. Cell 2020, 180, 1262–1271. [Google Scholar] [CrossRef]
- Zhang, G.; Shi, J.; Zhu, S.; Lan, Y.; Xu, L.; Yuan, H.; Liao, G.; Liu, X.; Zhang, Y.; Xiao, Y.; et al. DiseaseEnhancer: A resource of human disease-associated enhancer catalog. Nucleic Acids Res. 2018, 46, D78–D84. [Google Scholar] [CrossRef] [Green Version]
- Ramamoorthy, S.; Cidlowski, J.A. Corticosteroids. Rheum. Dis. Clin. N. Am. 2016, 42, 15–31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ratman, D.; Berghe, W.V.; Dejager, L.; Libert, C.; Tavernier, J.; Beck, I.M.; De Bosscher, K. How glucocorticoid receptors modulate the activity of other transcription factors: A scope beyond tethering. Mol. Cell. Endocrinol. 2013, 380, 41–54. [Google Scholar] [CrossRef]
- Sacta, M.A.; Chinenov, Y.; Rogatsky, I. Glucocorticoid Signaling: An Update from a Genomic Perspective. Annu. Rev. Physiol. 2016, 78, 155–180. [Google Scholar] [CrossRef] [PubMed]
- Weikum, E.R.; Knuesel, M.T.; Ortlund, E.R.W.E.A.; Yamamoto, M.T.K.K.R. Glucocorticoid receptor control of transcription: Precision and plasticity via allostery. Nat. Rev. Mol. Cell Biol. 2017, 18, 159–174. [Google Scholar] [CrossRef]
- ENCODE Project Consortium. An integrated encyclopedia of DNA elements in the human genome. Nature 2012, 489, 57–74. [Google Scholar] [CrossRef] [PubMed]
- Chen, E.Y.; Tan, C.M.; Kou, Y.; Duan, Q.; Wang, Z.; Meirelles, G.V.; Clark, N.R.; Ma’Ayan, A. Enrichr: Interactive and collaborative HTML5 gene list enrichment analysis tool. BMC Bioinform. 2013, 14, 128. [Google Scholar] [CrossRef] [Green Version]
- Farmer, P.; Bonnefoi, H.; Becette, V.; Tubiana-Hulin, M.; Fumoleau, P.; Larsimont, D.; MacGrogan, G.; Bergh, J.; Cameron, D.A.; Goldstein, D.R.; et al. Identification of molecular apocrine breast tumours by microarray analysis. Oncogene 2005, 24, 4660–4671. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hendriksen, P.J.; Dits, N.F.; Kokame, K.; Veldhoven, A.; Van Weerden, W.M.; Bangma, C.H.; Trapman, J.; Jenster, G. Evolution of the Androgen Receptor Pathway during Progression of Prostate Cancer. Cancer Res. 2006, 66, 5012–5020. [Google Scholar] [CrossRef] [Green Version]
- Mikkonen, L.; Pihlajamaa, P.; Sahu, B.; Zhang, F.-P.; Jänne, O.A. Androgen receptor and androgen-dependent gene expression in lung. Mol. Cell. Endocrinol. 2010, 317, 14–24. [Google Scholar] [CrossRef] [Green Version]
- Wirapati, P.; Sotiriou, C.; Kunkel, S.; Farmer, P.; Pradervand, S.; Haibe-Kains, B.; Desmedt, C.; Ignatiadis, M.; Sengstag, T.; Schütz, F.; et al. Meta-analysis of gene expression profiles in breast cancer: Toward a unified understanding of breast cancer subtyping and prognosis signatures. Breast Cancer Res. 2008, 10, 1–11. [Google Scholar] [CrossRef]
- Liu, R.; States, D.J. Consensus Promoter Identification in the Human Genome Utilizing Expressed Gene Markers and Gene Modeling. Genome Res. 2002, 12, 462–469. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vandevyver, S.; Dejager, L.; Tuckermann, J.; Libert, C. New Insights into the Anti-inflammatory Mechanisms of Glucocorticoids: An Emerging Role for Glucocorticoid-Receptor-Mediated Transactivation. Endocrinology 2013, 154, 993–1007. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raisner, R.; Kharbanda, S.; Jin, L.; Jeng, E.; Chan, E.; Merchant, M.; Haverty, P.M.; Bainer, R.; Cheung, T.; Arnott, D.; et al. Enhancer Activity Requires CBP/P300 Bromodomain-Dependent Histone H3K27 Acetylation. Cell Rep. 2018, 24, 1722–1729. [Google Scholar] [CrossRef] [Green Version]
- De Almeida Nagata, D.E.; Chiang, E.Y.; Jhunjhunwala, S.; Caplazi, P.; Arumugam, V.; Modrusan, Z.; Chan, E.; Merchant, M.; Jin, L.; Arnott, D.; et al. Regulation of Tumor-Associated Myeloid Cell Activity by CBP/EP300 Bromodomain Modulation of H3K27 Acetylation. Cell Rep. 2019, 27, 269–281.e4. [Google Scholar] [CrossRef] [Green Version]
- Fergelot, P.; Van Belzen, M.; Van Gils, J.; Afenjar, A.; Armour, C.M.; Arveiler, B.; Beets, L.; Burglen, L.; Busa, T.; Collet, M.; et al. Phenotype and genotype in 52 patients with Rubinstein-Taybi syndrome caused byEP300mutations. Am. J. Med Genet. Part A 2016, 170, 3069–3082. [Google Scholar] [CrossRef]
- Zimmermann, N.; Acosta, A.M.B.F.; Kohlhase, J.; Bartsch, O. Confirmation of EP300 gene mutations as a rare cause of Rubinstein–Taybi syndrome. Eur. J. Hum. Genet. 2007, 15, 837–842. [Google Scholar] [CrossRef] [Green Version]
- Becskei, A.; Séraphin, B.; Serrano, L. Positive feedback in eukaryotic gene networks: Cell differentiation by graded to binary response conversion. EMBO J. 2001, 20, 2528–2535. [Google Scholar] [CrossRef] [Green Version]
- Zhang, B.; Day, D.S.; Ho, J.W.; Song, L.; Cao, J.; Christodoulou, D.; Seidman, J.G.; Crawford, G.E.; Park, P.J.; Pu, W.T. A dynamic H3K27ac signature identifies VEGFA-stimulated endothelial enhancers and requires EP300 activity. Genome Res. 2013, 23, 917–927. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, C.; Lee, J.-E.; Lai, B.; Macfarlan, T.S.; Xu, S.; Zhuang, L.; Liu, C.; Peng, W.; Ge, K. Enhancer priming by H3K4 methyltransferase MLL4 controls cell fate transition. Proc. Natl. Acad. Sci. USA 2016, 113, 11871–11876. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sérandour, A.A.; Avner, S.; Percevault, F.; DeMay, F.; Bizot, M.; Lucchetti-Miganeh, C.; Barloy-Hubler, F.; Brown, M.; Lupien, M.; Métivier, R.; et al. Epigenetic switch involved in activation of pioneer factor FOXA1-dependent enhancers. Genome Res. 2011, 21, 555–565. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharifi-Zarchi, A.; Gerovska, D.; Adachi, K.; Totonchi, M.; Pezeshk, H.; Taft, R.J.; Schöler, H.R.; Chitsaz, H.; Sadeghi, M.; Baharvand, H.; et al. DNA methylation regulates discrimination of enhancers from promoters through a H3K4me1-H3K4me3 seesaw mechanism. BMC Genom. 2017, 18, 1–21. [Google Scholar] [CrossRef] [PubMed]
- Dorighi, K.M.; Swigut, T.; Henriques, T.; Bhanu, N.V.; Scruggs, B.S.; Nady, N.; Still, C.D., II; Garcia, B.A.; Adelman, K.; Wysocka, J. Mll3 and Mll4 facilitate enhancer RNA synthesis and transcription from promoters independently of H3K4 monomethylation. Mol. Cell 2017, 66, 568–576.e4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Local, A.; Huang, H.; Albuquerque, C.P.; Singh, N.; Lee, A.Y.; Wang, W.; Wang, C.; Hsia, J.E.; Shiau, A.K.; Ge, K.; et al. Identification of H3K4me1-associated proteins at mammalian enhancers. Nat. Genet. 2018, 50, 73–82. [Google Scholar] [CrossRef] [PubMed]
- Rickels, R.; Herz, H.-M.; Sze, C.C.; Cao, K.; Morgan, M.A.; Collings, C.K.; Gause, M.; Takahashi, Y.; Wang, L.; Rendleman, E.J. Histone H3K4 monomethylation catalyzed by Trr and mammalian COMPASS-like proteins at enhancers is dispensable for development and viability. Nat. Genet. 2017, 49, 1647. [Google Scholar] [CrossRef] [Green Version]
- Provençal, N.; Arloth, J.; Cattaneo, A.; Anacker, C.; Cattane, N.; Wiechmann, T.; Röh, S.; Ködel, M.; Klengel, T.; Czamara, D.; et al. Glucocorticoid exposure during hippocampal neurogenesis primes future stress response by inducing changes in DNA methylation. Proc. Natl. Acad. Sci. USA 2020, 117, 23280–23285. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sorrells, S.F.; Caso, J.R.; Munhoz, C.D.; Sapolsky, R.M. The Stressed CNS: When Glucocorticoids Aggravate Inflammation. Neuron 2009, 64, 33–39. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chapman, A.B.; Knight, D.M.; Ringold, G.M. Glucocorticoid regulation of adipocyte differentiation: Hormonal triggering of the developmental program and induction of a differentiation-dependent gene. J. Cell Biol. 1985, 101, 1227–1235. [Google Scholar] [CrossRef] [PubMed]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Borczyk, M.; Zieba, M.; Korostyński, M.; Piechota, M. Role of Non-Coding Regulatory Elements in the Control of GR-Dependent Gene Expression. Int. J. Mol. Sci. 2021, 22, 4258. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22084258
Borczyk M, Zieba M, Korostyński M, Piechota M. Role of Non-Coding Regulatory Elements in the Control of GR-Dependent Gene Expression. International Journal of Molecular Sciences. 2021; 22(8):4258. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22084258
Chicago/Turabian StyleBorczyk, Malgorzata, Mateusz Zieba, Michał Korostyński, and Marcin Piechota. 2021. "Role of Non-Coding Regulatory Elements in the Control of GR-Dependent Gene Expression" International Journal of Molecular Sciences 22, no. 8: 4258. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22084258