
Viral Infection Modulates Mitochondrial Function

{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Physiological Morphology of Mitochondria
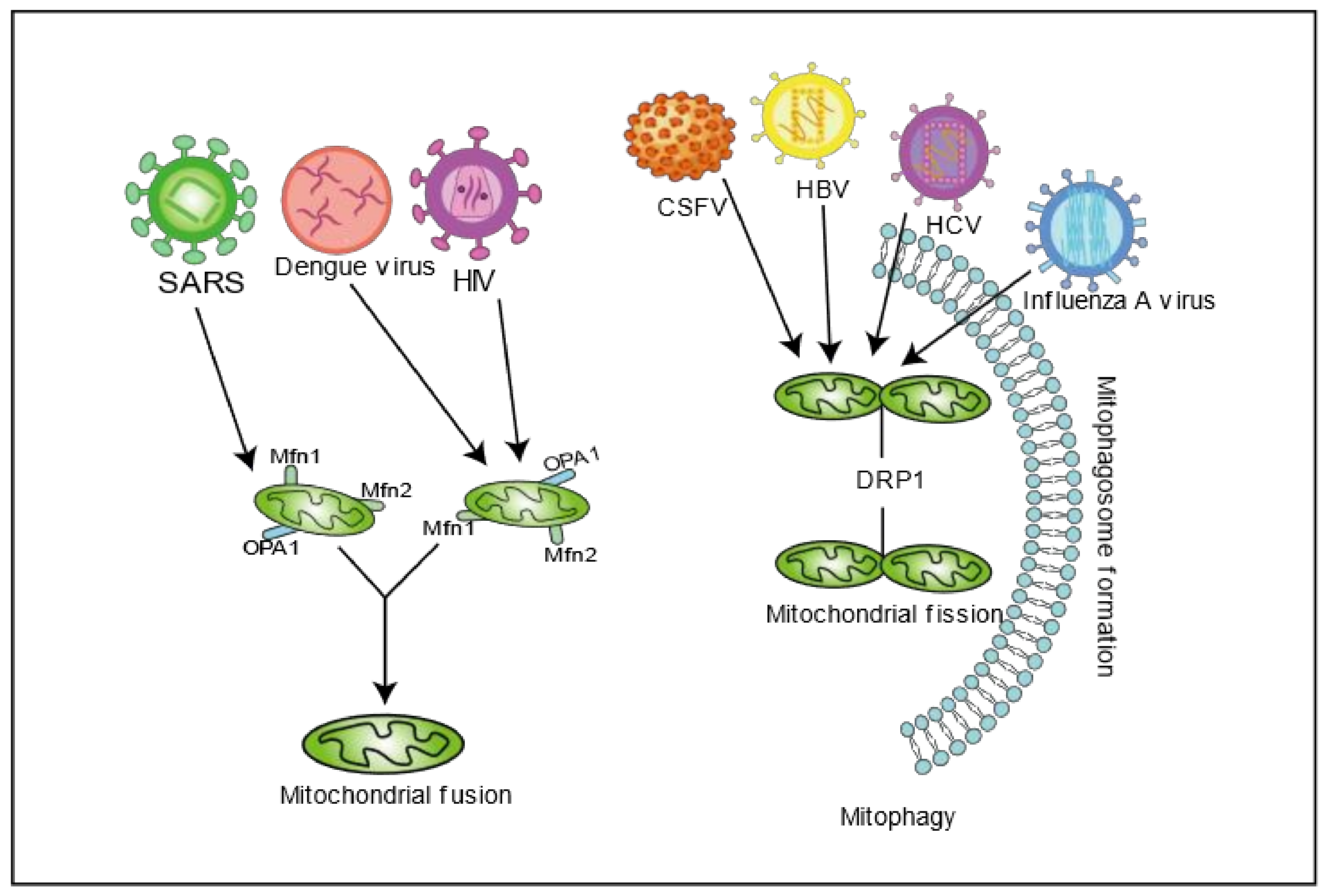
2. Viral Infection Disrupts Mitochondrial Dynamics
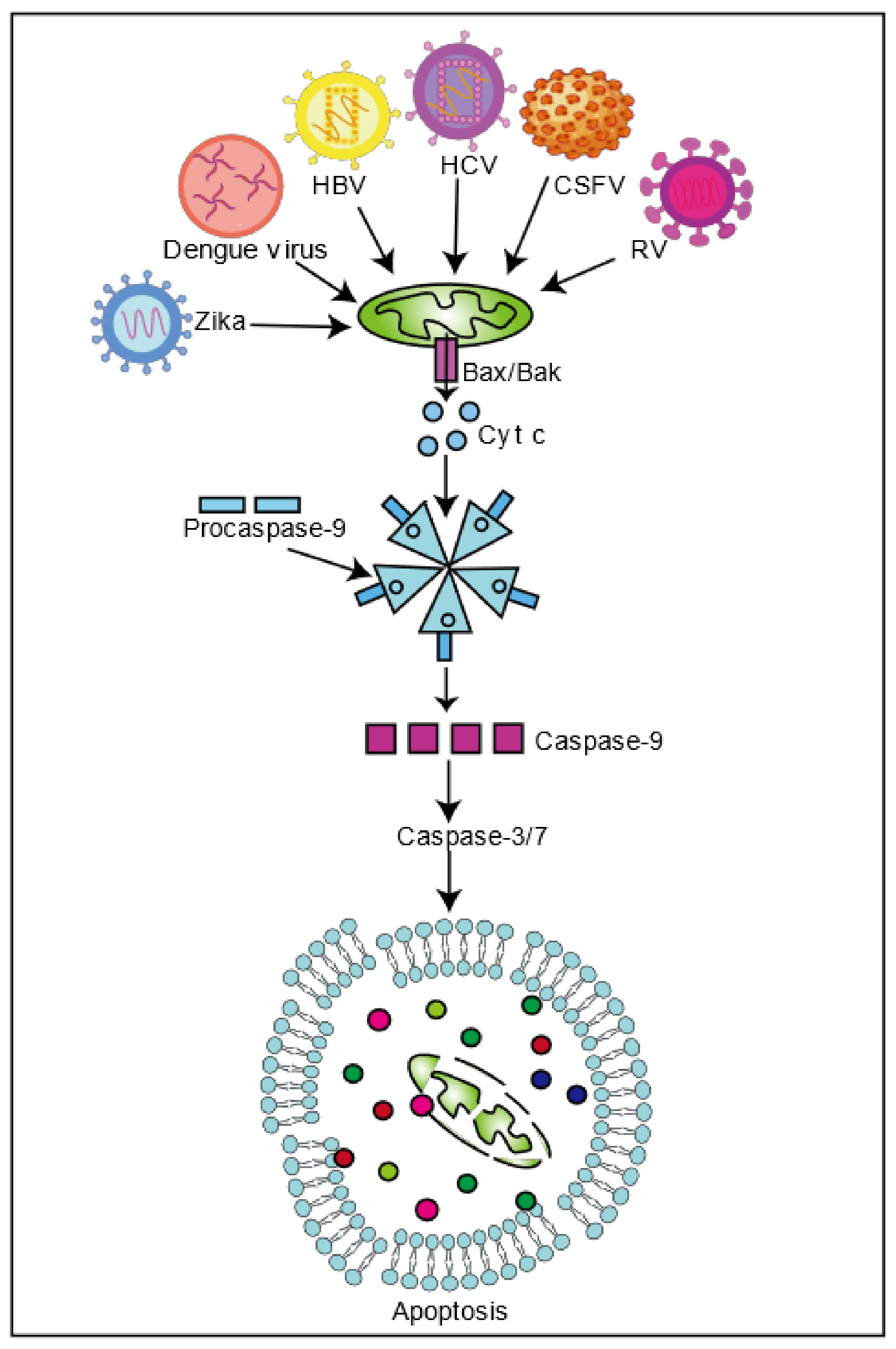
3. Viral Infection Regulates Mitochondria-Induced Cell Death
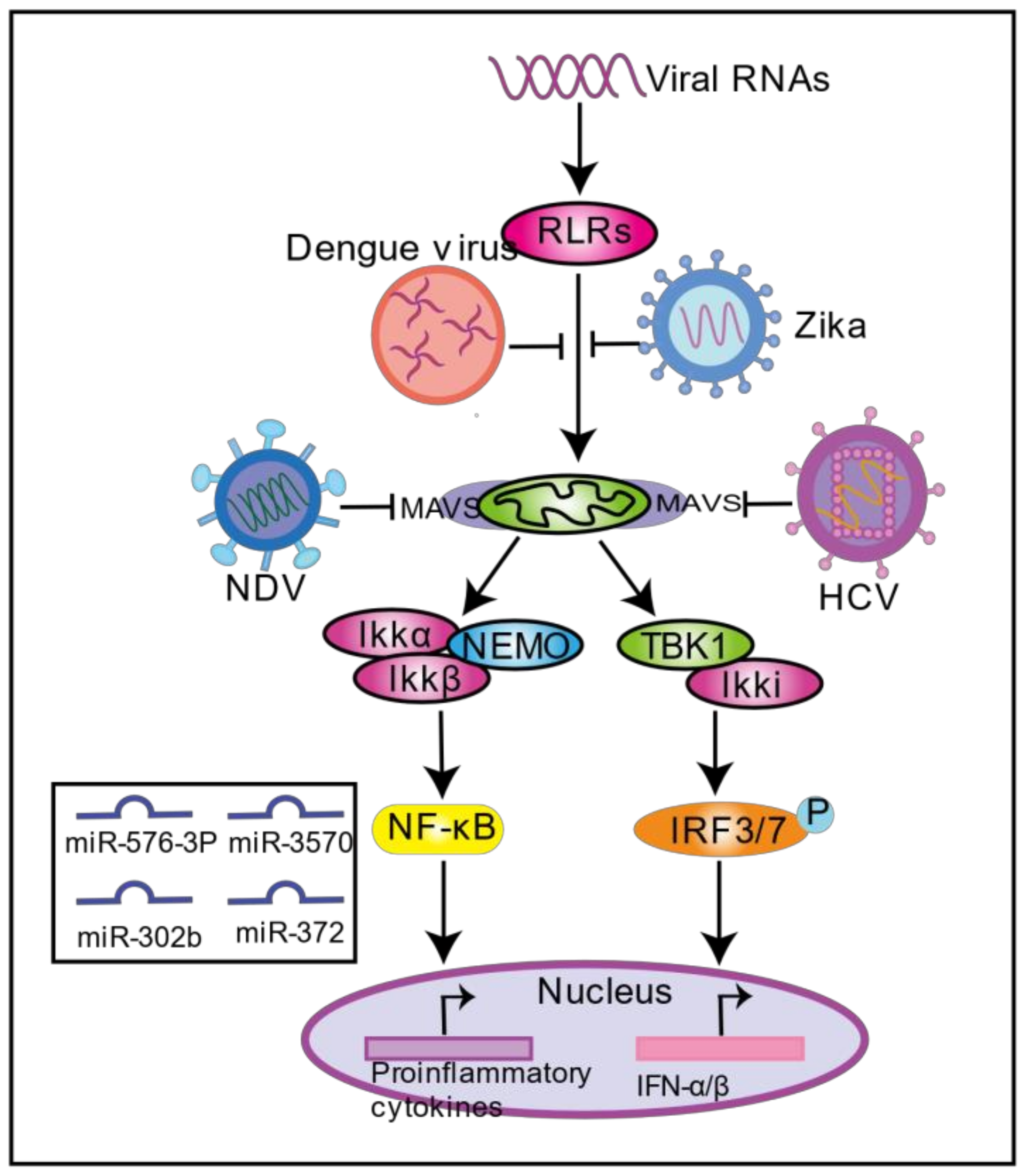
4. Viral Infection Regulates Mitochondria-Induced Innate Immunity
5. Viral Infection Regulates Mitochondrial Metabolism
6. Concluding Remarks
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kiriyama, Y.; Nochi, H. Intra- and Intercellular Quality Control Mechanisms of Mitochondria. Cells 2017, 7, 1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hales, K.G.; Fuller, M.T. Developmentally regulated mitochondrial fusion mediated by a conserved, novel, predicted GTPase. Cell 1997, 90, 121–129. [Google Scholar] [CrossRef] [Green Version]
- Chen, H.; Detmer, S.A.; Ewald, A.J.; Griffin, E.E.; Fraser, S.E.; Chan, D.C. Mitofusins Mfn1 and Mfn2 coordinately regulate mitochondrial fusion and are essential for embryonic development. J. Cell Biol. 2003, 160, 189–200. [Google Scholar] [CrossRef]
- Chen, H.; Chomyn, A.; Chan, D.C. Disruption of fusion results in mitochondrial heterogeneity and dysfunction. J. Biol. Chem. 2005, 280, 26185–26192. [Google Scholar] [CrossRef] [Green Version]
- Cipolat, S.; Martins de Brito, O.; Dal Zilio, B.; Scorrano, L. OPA1 requires mitofusin 1 to promote mitochondrial fusion. Proc. Natl. Acad. Sci. USA 2004, 101, 15927–15932. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koshiba, T.; Detmer, S.A.; Kaiser, J.T.; Chen, H.; McCaffery, J.M.; Chan, D.C. Structural basis of mitochondrial tethering by mitofusin complexes. Science 2004, 305, 858–862. [Google Scholar] [CrossRef] [Green Version]
- Cao, Y.L.; Meng, S.; Chen, Y.; Feng, J.X.; Gu, D.D.; Yu, B.; Li, Y.J.; Yang, J.Y.; Liao, S.; Chan, D.C.; et al. Mfn1 structures reveal nucleotide-triggered dimerization critical for mitochondrial fusion. Nature 2017, 542, 372–376. [Google Scholar] [CrossRef]
- Otera, H.; Mihara, K. Molecular mechanisms and physiologic functions of mitochondrial dynamics. J. Biochem. 2011, 149, 241–251. [Google Scholar] [CrossRef] [Green Version]
- Rojo, M.; Legros, F.; Chateau, D.; Lombès, A. Membrane topology and mitochondrial targeting of mitofusins, ubiquitous mammalian homologs of the transmembrane. J. Cell Sci. GTPase Fzo. 2002, 115, 1663–1674. [Google Scholar]
- Anand, R.; Wai, T.; Baker, M.J.; Kladt, N.; Schauss, A.C.; Rugarli, E.; Langer, T. The i-AAA protease YME1L and OMA1 cleave OPA1 to balance mitochondrial fusion and fission. J. Cell Biol. 2014, 204, 919–929. [Google Scholar] [CrossRef] [PubMed]
- Tondera, D.; Grandemange, S.; Jourdain, A.; Karbowski, M.; Mattenberger, Y.; Herzig, S.; Da Cruz, S.; Clerc, P.; Raschke, I.; Merkwirth, C.; et al. SLP-2 is required for stress-induced mitochondrial hyperfusion. EMBO J. 2009, 28, 1589–1600. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ingerman, E.; Perkins, E.M.; Marino, M.; Mears, J.A.; McCaffery, J.M.; Hinshaw, J.E.; Nunnari, J. Dnm1 forms spirals that are structurally tailored to fit mitochondria. J. Cell Biol. 2005, 170, 1021–1027. [Google Scholar] [CrossRef] [Green Version]
- Ji, W.K.; Hatch, A.L.; Merrill, R.A.; Strack, S.; Higgs, H.N. Actin filaments target the oligomeric maturation of the dynamin GTPase Drp1 to mitochondrial fission sites. Elife 2015, 4, e11553. [Google Scholar] [CrossRef] [PubMed]
- Taguchi, N.; Ishihara, N.; Jofuku, A.; Oka, T.; Mihara, K. Mitotic phosphorylation of dynamin-related GTPase Drp1 participates in mitochondrial fission. J. Biol. Chem. 2007, 282, 11521–11529. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saito, T.; Nah, J.; Oka, S.I.; Mukai, R.; Monden, Y.; Maejima, Y.; Ikeda, Y.; Sciarretta, S.; Liu, T.; Li, H.; et al. An alternative mitophagy pathway mediated by Rab9 protects the heart against ischemia. J. Clin. Investig. 2019, 129, 802–819. [Google Scholar] [CrossRef] [Green Version]
- Chang, C.R.; Blackstone, C. Cyclic AMP-dependent protein kinase phosphorylation of Drp1 regulates its GTPase activity and mitochondrial morphology. J. Biol. Chem. 2007, 282, 21583–21587. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Richter, V.; Singh, A.P.; Kvansakul, M.; Ryan, M.T.; Osellame, L.D. Splitting up the powerhouse: Structural insights into the mechanism of mitochondrial fission. Cell Mol. Life Sci. 2015, 72, 3695–3707. [Google Scholar] [CrossRef]
- Yoon, Y.; Krueger, E.W.; Oswald, B.J.; McNiven, M.A. The mitochondrial protein hFis1 regulates mitochondrial fission in mammalian cells through an interaction with the dynamin-like protein DLP1. Mol. Cell Biol. 2003, 23, 5409–5420. [Google Scholar] [CrossRef] [Green Version]
- Otera, H.; Wang, C.; Cleland, M.M.; Setoguchi, K.; Yokota, S.; Youle, R.J.; Mihara, K. Mff is an essential factor for mitochondrial recruitment of Drp1 during mitochondrial fission in mammalian cells. J. Cell Biol. 2010, 191, 1141–1158. [Google Scholar] [CrossRef] [Green Version]
- Yu, R.; Jin, S.B.; Lendahl, U.; Nistér, M.; Zhao, J. Human Fis1 regulates mitochondrial dynamics through inhibition of the fusion machinery. EMBO J. 2019, 38, e99748. [Google Scholar] [CrossRef]
- Palmer, C.S.; Osellame, L.D.; Laine, D.; Koutsopoulos, O.S.; Frazier, A.E.; Ryan, M.T. MiD49 and MiD51, new components of the mitochondrial fission machinery. EMBO Rep. 2011, 12, 565–573. [Google Scholar] [CrossRef]
- Zhao, J.; Liu, T.; Jin, S.; Wang, X.; Qu, M.; Uhlén, P.; Tomilin, N.; Shupliakov, O.; Lendahl, U.; Nistér, M. Human MIEF1 recruits Drp1 to mitochondrial outer membranes and promotes mitochondrial fusion rather than fission. EMBO J. 2011, 30, 2762–2778. [Google Scholar] [CrossRef]
- Barbaro, G.; Di Lorenzo, G.; Asti, A.; Ribersani, M.; Belloni, G.; Grisorio, B.; Filice, G.; Barbarini, G. Hepatocellular mitochondrial alterations in patients with chronic hepatitis C: Ultrastructural and biochemical findings. Am. J. Gastroenterol. 1999, 94, 2198–2205. [Google Scholar] [CrossRef]
- Schwer, B.; Ren, S.; Pietschmann, T.; Kartenbeck, J.; Kaehlcke, K.; Bartenschlager, R.; Yen, T.S.; Ott, M. Targeting of hepatitis C virus core protein to mitochondria through a novel C-terminal localization motif. J. Virol. 2004, 78, 7958–7968. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Korenaga, M.; Wang, T.; Li, Y.; Showalter, L.A.; Chan, T.; Sun, J.; Weinman, S.A. Hepatitis C virus core protein inhibits mitochondrial electron transport and increases reactive oxygen species (ROS) production. J. Biol. Chem. 2005, 280, 37481–37488. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Machida, K.; Cheng, K.T.; Lai, C.K.; Jeng, K.S.; Sung, V.M.; Lai, M.M. Hepatitis C virus triggers mitochondrial permeability transition with production of reactive oxygen species, leading to DNA damage and STAT3 activation. J. Virol. 2006, 80, 7199–7207. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, T.; Campbell, R.V.; Yi, M.K.; Lemon, S.M.; Weinman, S.A. Role of Hepatitis C virus core protein in viral-induced mitochondrial dysfunction. J. Viral. Hepat. 2010, 17, 784–793. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.J.; Syed, G.H.; Khan, M.; Chiu, W.W.; Sohail, M.A.; Gish, R.G.; Siddiqui, A. Hepatitis C virus triggers mitochondrial fission and attenuates apoptosis to promote viral persistence. Proc. Natl. Acad. Sci. USA 2014, 111, 6413–6418. [Google Scholar] [CrossRef] [Green Version]
- Siu, G.K.; Zhou, F.; Yu, M.K.; Zhang, L.; Wang, T.; Liang, Y.; Chen, Y.; Chan, H.C.; Yu, S. Hepatitis C virus NS5A protein cooperates with phosphatidylinositol 4-kinase IIIα to induce mitochondrial fragmentation. Sci. Rep. 2016, 6, 23464. [Google Scholar] [CrossRef] [Green Version]
- Hara, Y.; Yanatori, I.; Ikeda, M.; Kiyokage, E.; Nishina, S.; Tomiyama, Y.; Toida, K.; Kishi, F.; Kato, N.; Imamura, M.; et al. Hepatitis C virus core protein suppresses mitophagy by interacting with Parkin in the context of mitochondrial depolarization. Am. J. Pathol. 2014, 184, 3026–3039. [Google Scholar] [CrossRef]
- Gou, H.; Zhao, M.; Xu, H.; Yuan, J.; He, W.; Zhu, M.; Ding, H.; Yi, L.; Chen, J. CSFV induced mitochondrial fission and mitophagy to inhibit apoptosis. Oncotarget 2017, 8, 39382–39400. [Google Scholar] [CrossRef] [Green Version]
- Fan, S.; Wu, K.; Zhao, M.; Yuan, J.; Ma, S.; Zhu, E.; Chen, Y.; Ding, H.; Yi, L.; Chen, J. LDHB inhibition induces mitophagy and facilitates the progression of CSFV infection. Autophagy 2020, 16, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Xie, B.; Zhao, M.; Song, D.; Wu, K.; Yi, L.; Li, W.; Li, X.; Wang, K.; Chen, J. Induction of autophagy and suppression of type I IFN secretion by CSFV. Autophagy 2020, 16, 1–23. [Google Scholar] [CrossRef]
- Barbier, V.; Lang, D.; Valois, S.; Rothman, A.L.; Medin, C.L. Dengue virus induces mitochondrial elongation through impairment of Drp1-triggered mitochondrial fission. Virology 2017, 500, 149–160. [Google Scholar] [CrossRef]
- Yu, C.Y.; Liang, J.J.; Li, J.K.; Lee, Y.L.; Chang, B.L.; Su, C.I.; Huang, W.J.; Lai, M.M.; Lin, Y.L. Dengue Virus Impairs Mitochondrial Fusion by Cleaving Mitofusins. PLoS Pathog. 2015, 11, e1005350. [Google Scholar] [CrossRef] [Green Version]
- Yoo, Y.S.; Park, Y.J.; Lee, H.S.; Oanh, N.T.K.; Cho, M.Y.; Heo, J.; Lee, E.S.; Cho, H.; Park, Y.Y.; Cho, H. Mitochondria ubiquitin ligase, MARCH5 resolves hepatitis B virus X protein aggregates in the liver pathogenesis. Cell Death Dis. 2019, 10, 938. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.J.; Khan, M.; Quan, J.; Till, A.; Subramani, S.; Siddiqui, A. Hepatitis B virus disrupts mitochondrial dynamics: Induces fission and mitophagy to attenuate apoptosis. PLoS Pathog. 2013, 9, e1003722. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, W.; Calvo, P.A.; Malide, D.; Gibbs, J.; Schubert, U.; Bacik, I.; Basta, S.; O’Neill, R.; Schickli, J.; Palese, P.; et al. A novel influenza A virus mitochondrial protein that induces cell death. Nat. Med. 2001, 7, 1306–1312. [Google Scholar] [CrossRef]
- Gibbs, J.S.; Malide, D.; Hornung, F.; Bennink, J.R.; Yewdell, J.W. The influenza A virus PB1-F2 protein targets the inner mitochondrial membrane via a predicted basic amphipathic helix that disrupts mitochondrial function. J. Virol. 2003, 77, 7214–7224. [Google Scholar] [CrossRef] [Green Version]
- Yamada, H.; Chounan, R.; Higashi, Y.; Kurihara, N.; Kido, H. Mitochondrial targeting sequence of the influenza A virus PB1-F2 protein and its function in mitochondria. FEBS Lett. 2004, 578, 331–336. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoshizumi, T.; Ichinohe, T.; Sasaki, O.; Otera, H.; Kawabata, S.; Mihara, K.; Koshiba, T. Influenza A virus protein PB1-F2 translocates into mitochondria via Tom40 channels and impairs innate immunity. Nat. Commun. 2014, 5, 4713. [Google Scholar] [CrossRef]
- Wang, R.; Zhu, Y.; Ren, C.; Yang, S.; Tian, S.; Chen, H.; Jin, M.; Zhou, H. Influenza A virus protein PB1-F2 impairs innate immunity by inducing mitophagy. Autophagy 2021, 17, 496–511. [Google Scholar] [CrossRef]
- Pila-Castellanos, I.; Molino, D.; McKellar, J.; Lines, L.; Da Graca, J.; Tauziet, M.; Chanteloup, L.; Mikaelian, I.; Meyniel-Schicklin, L.; Codogno, P.; et al. Mitochondrial morphodynamics alteration induced by influenza virus infection as a new antiviral strategy. PLoS Pathog. 2021, 17, e1009340. [Google Scholar]
- Cornillez-Ty, C.T.; Liao, L.; Yates, J.R., 3rd; Kuhn, P.; Buchmeier, M.J. Severe acute respiratory syndrome coronavirus nonstructural protein 2 interacts with a host protein complex involved in mitochondrial biogenesis and intracellular signaling. J. Virol. 2009, 83, 10314–10318. [Google Scholar] [CrossRef] [Green Version]
- Merkwirth, C.; Langer, T. Prohibitin function within mitochondria: Essential roles for cell proliferation and cristae morphogenesis. Biochim. Biophys. Acta 2009, 1793, 27–32. [Google Scholar] [CrossRef] [Green Version]
- Shi, C.S.; Qi, H.Y.; Boularan, C.; Huang, N.N.; Abu-Asab, M.; Shelhamer, J.H.; Kehrl, J.H. SARS-coronavirus open reading frame-9b suppresses innate immunity by targeting mitochondria and the MAVS/TRAF3/TRAF6 signalosome. J. Immunol. 2014, 193, 3080–3089. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, M.; Bansal, V.; Feschotte, C. A Single-Cell RNA Expression Map of Human Coronavirus Entry Factors. Cell Rep. 2020, 32, 108175. [Google Scholar] [CrossRef] [PubMed]
- Gordon, D.E.; Jang, G.M.; Bouhaddou, M.; Xu, J.; Obernier, K.; White, K.M.; O’Meara, M.J.; Rezelj, V.V.; Guo, J.Z.; Swaney, D.L.; et al. A SARS-CoV-2 protein interaction map reveals targets for drug repurposing. Nature 2020, 583, 459–468. [Google Scholar] [CrossRef] [PubMed]
- Acehan, D.; Jiang, X.; Morgan, D.G.; Heuser, J.E.; Wang, X.; Akey, C.W. Three-dimensional structure of the apoptosome: Implications for assembly, procaspase-9 binding, and activation. Mol. Cell 2002, 9, 423–432. [Google Scholar] [CrossRef]
- Nomura-Takigawa, Y.; Nagano-Fujii, M.; Deng, L.; Kitazawa, S.; Ishido, S.; Sada, K.; Hotta, H. Non-structural protein 4A of Hepatitis C virus accumulates on mitochondria and renders the cells prone to undergoing mitochondria-mediated apoptosis. J. Gen. Virol. 2006, 87, 1935–1945. [Google Scholar] [CrossRef] [PubMed]
- Chiou, H.L.; Hsieh, Y.S.; Hsieh, M.R.; Chen, T.Y. HCV E2 may induce apoptosis of Huh-7 cells via a mitochondrial-related caspase pathway. Biochem. Biophys. Res. Commun. 2006, 345, 453–458. [Google Scholar] [CrossRef]
- Lee, S.K.; Park, S.O.; Joe, C.O.; Kim, Y.S. Interaction of HCV core protein with 14-3-3epsilon protein releases Bax to activate apoptosis. Biochem. Biophys. Res. Commun. 2007, 352, 756–762. [Google Scholar] [CrossRef] [PubMed]
- Zhao, P.; Han, T.; Guo, J.J.; Zhu, S.L.; Wang, J.; Ao, F.; Jing, M.Z.; She, Y.L.; Wu, Z.H.; Ye, L.B. HCV NS4B induces apoptosis through the mitochondrial death pathway. Virus Res. 2012, 169, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Javed, F.; Manzoor, S. HCV non-structural NS4A protein of genotype 3a induces mitochondria mediated death by activating Bax and the caspase cascade. Microb. Pathog. 2018, 124, 346–355. [Google Scholar] [CrossRef] [PubMed]
- Takada, S.; Shirakata, Y.; Kaneniwa, N.; Koike, K. Association of hepatitis B virus X protein with mitochondria causes mitochondrial aggregation at the nuclear periphery, leading to cell death. Oncogene 1999, 18, 6965–6973. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nasirudeen, A.M.; Wang, L.; Liu, D.X. Induction of p53-dependent and mitochondria-mediated cell death pathway by dengue virus infection of human and animal cells. Microbes Infect. 2008, 10, 1124–1132. [Google Scholar] [CrossRef]
- Gao, W.Y.; Li, D.; Cai, D.E.; Huang, X.Y.; Zheng, B.Y.; Huang, Y.H.; Chen, Z.X.; Wang, X.Z. Hepatitis B virus X protein sensitizes HL-7702 cells to oxidative stress-induced apoptosis through modulation of the mitochondrial permeability transition pore. Oncol. Rep. 2017, 37, 48–56. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, H.J.; Kim, S.Y.; Kim, J.; Lee, H.; Choi, M.; Kim, J.K.; Ahn, J.K. Hepatitis B virus X protein induces apoptosis by enhancing translocation of Bax to mitochondria. IUBMB Life 2008, 60, 473–480. [Google Scholar] [CrossRef]
- Padhan, K.; Minakshi, R.; Towheed, M.A.B.; Jameel, S. Severe acute respiratory syndrome coronavirus 3a protein activates the mitochondrial death pathway through p38 MAP kinase activation. J. Gen. Virol. 2008, 89, 1960–1969. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Wei, L.; Jiang, D.; Wang, J.; Cong, X.; Fei, R. SARS-CoV nucleocapsid protein induced apoptosis of COS-1 mediated by the mitochondrial pathway. Artif. Cells Blood Substit. Immobil. Biotechnol. 2007, 35, 237–253. [Google Scholar] [CrossRef] [Green Version]
- Chan, C.M.; Ma, C.W.; Chan, W.Y.; Chan, H.Y. The SARS-Coronavirus Membrane protein induces apoptosis through modulating the Akt survival pathway. Arch Biochem. Biophys. 2007, 459, 197–207. [Google Scholar]
- Guerrero, R.; Guerrero, C.; Acosta, O. Induction of Cell Death in the Human Acute Lymphoblastic Leukemia Cell Line Reh by Infection with Rotavirus Isolate Wt1-5. Biomedicines 2020, 8, 242. [Google Scholar] [CrossRef] [PubMed]
- Martin-Latil, S.; Mousson, L.; Autret, A.; Colbère-Garapin, F.; Blondel, B. Bax is activated during rotavirus-induced apoptosis through the mitochondrial pathway. J. Virol. 2007, 81, 4457–4464. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhowmick, R.; Halder, U.C.; Chattopadhyay, S.; Nayak, M.K.; Chawla-Sarkar, M. Rotavirus-encoded nonstructural protein 1 modulates cellular apoptotic machinery by targeting tumor suppressor protein p53. J. Virol. 2013, 87, 6840–6850. [Google Scholar] [CrossRef] [Green Version]
- Mukherjee, A.; Patra, U.; Bhowmick, R.; Chawla-Sarkar, M. Rotaviral nonstructural protein 4 triggers dynamin-related protein 1-dependent mitochondrial fragmentation during infection. Cell Microbiol. 2018, 20, e12831. [Google Scholar] [CrossRef] [PubMed]
- Zamarin, D.; García-Sastre, A.; Xiao, X.; Wang, R.; Palese, P. Influenza virus PB1-F2 protein induces cell death through mitochondrial ANT3 and VDAC1. PLoS Pathog. 2005, 1, e4. [Google Scholar] [CrossRef]
- Yang, S.; Gorshkov, K.; Lee, E.M.; Xu, M.; Cheng, Y.S.; Sun, N.; Soheilian, F.; de Val, N.; Ming, G.; Song, H.; et al. Zika Virus-Induced Neuronal Apoptosis via Increased Mitochondrial Fragmentation. Front. Microbiol. 2020, 11, 598203. [Google Scholar] [CrossRef]
- Neumann, S.; El Maadidi, S.; Faletti, L.; Haun, F.; Labib, S.; Schejtman, A.; Maurer, U.; Borner, C. How do viruses control mitochondria-mediated apoptosis? Virus Res. 2015, 209, 45–55. [Google Scholar] [CrossRef] [PubMed]
- Maiuri, M.C.; Zalckvar, E.; Kimchi, A.; Kroemer, G. Self-eating and self-killing: Crosstalk between autophagy and apoptosis. Nat. Rev. Mol. Cell Biol. 2007, 8, 741–752. [Google Scholar] [CrossRef]
- Li, S.; Wang, J.; Zhou, A.; Khan, F.A.; Hu, L.; Zhang, S. Porcine reproductive and respiratory syndrome virus triggers mitochondrial fission and mitophagy to attenuate apoptosis. Oncotarget 2016, 7, 56002–56012. [Google Scholar]
- Meng, G.; Xia, M.; Wang, D.; Chen, A.; Wang, Y.; Wang, H.; Yu, D.; Wei, J. Mitophagy promotes replication of oncolytic Newcastle disease virus by blocking intrinsic apoptosis in lung cancer cells. Oncotarget 2014, 5, 6365–6374. [Google Scholar]
- Freundt, E.C.; Yu, L.; Park, E.; Lenardo, M.J.; Xu, X.N. Molecular determinants for subcellular localization of the severe acute respiratory syndrome coronavirus open reading frame 3b protein. J. Virol. 2009, 83, 6631–6640. [Google Scholar] [CrossRef] [Green Version]
- Bojkova, D.; Klann, K.; Koch, B.; Krause, D.; Ciesek, S.; Cinatl, J.; Münch, C. Proteomics of SARS-CoV-2-infected host cells reveals therapy targets. Nature 2020, 583, 469–472. [Google Scholar] [CrossRef] [PubMed]
- Meylan, E.; Curran, J.; Hofmann, K.; Moradpour, D.; Binder, M.; Bartenschlager, R.; Tschopp, J. Cardif is an adaptor protein in the RIG-I antiviral pathway and is targeted by hepatitis C virus. Nature 2005, 437, 1167–1172. [Google Scholar] [CrossRef] [PubMed]
- Li, X.D.; Sun, L.; Seth, R.B.; Pineda, G.; Chen, Z.J. Hepatitis C virus protease NS3/4A cleaves mitochondrial antiviral signaling protein off the mitochondria to evade innate immunity. Proc. Natl. Acad. Sci. USA 2005, 102, 17717–17722. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feng, H.; Sander, A.L.; Moreira-Soto, A.; Yamane, D.; Drexler, J.F.; Lemon, S.M. Hepatovirus 3ABC proteases and evolution of mitochondrial antiviral signaling protein (MAVS). J. Hepatol. 2019, 71, 25–34. [Google Scholar] [CrossRef] [PubMed]
- Qian, S.; Fan, W.; Liu, T.; Wu, M.; Zhang, H.; Cui, X.; Zhou, Y.; Hu, J.; Wei, S.; Chen, H.; et al. Seneca Valley Virus Suppresses Host Type I Interferon Production by Targeting Adaptor Proteins MAVS, TRIF, and TANK for Cleavage. J. Virol. 2017, 91, e00823-17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wei, C.; Ni, C.; Song, T.; Liu, Y.; Yang, X.; Zheng, Z.; Jia, Y.; Yuan, Y.; Guan, K.; Xu, Y.; et al. The hepatitis B virus X protein disrupts innate immunity by downregulating mitochondrial antiviral signaling protein. J. Immunol. 2010, 185, 1158–1168. [Google Scholar] [CrossRef]
- Sun, Y.; Zheng, H.; Yu, S.; Ding, Y.; Wu, W.; Mao, X.; Liao, Y.; Meng, C.; Ur Rehman, Z.; Tan, L.; et al. Newcastle Disease Virus V Protein Degrades Mitochondrial Antiviral Signaling Protein to Inhibit Host Type I Interferon Production via E3 Ubiquitin Ligase RNF5. J. Virol. 2019, 93, e00322-19. [Google Scholar] [CrossRef] [Green Version]
- Ding, S.; Zhu, S.; Ren, L.; Feng, N.; Song, Y.; Ge, X.; Li, B.; Flavell, R.A.; Greenberg, H.B. Rotavirus VP3 targets MAVS for degradation to inhibit type III interferon expression in intestinal epithelial cells. Elife 2018, 7, e39494. [Google Scholar] [CrossRef] [PubMed]
- Riedl, W.; Acharya, D.; Lee, J.H.; Liu, G.; Serman, T.; Chiang, C.; Chan, Y.K.; Diamond, M.S.; Gack, M.U. Zika Virus NS3 Mimics a Cellular 14-3-3-Binding Motif to Antagonize RIG-I- and MDA5-Mediated Innate Immunity. Cell Host Microbe. 2019, 26, 493–503.e6. [Google Scholar] [CrossRef] [PubMed]
- He, Z.; Zhu, X.; Wen, W.; Yuan, J.; Hu, Y.; Chen, J.; An, S.; Dong, X.; Lin, C.; Yu, J.; et al. Dengue Virus Subverts Host Innate Immunity by Targeting Adaptor Protein MAVS. J. Virol. 2016, 90, 7219–7230. [Google Scholar] [CrossRef] [Green Version]
- Yarbrough, M.L.; Zhang, K.; Sakthivel, R.; Forst, C.V.; Posner, B.A.; Barber, G.N.; White, M.A.; Fontoura, B.M. Primate-specific miR-576-3p sets host defense signalling threshold. Nat. Commun. 2014, 5, 4963. [Google Scholar] [CrossRef]
- Xu, T.; Chu, Q.; Cui, J.; Bi, D. Inducible MicroRNA-3570 Feedback Inhibits the RIG-I-Dependent Innate Immune Response to Rhabdovirus in Teleost Fish by Targeting MAVS/IPS-1. J. Virol. 2018, 92, e01594-17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yasukawa, K.; Kinoshita, D.; Yaku, K.; Nakagawa, T.; Koshiba, T. The microRNAs miR-302b and miR-372 regulate mitochondrial metabolism via the SLC25A12 transporter, which controls MAVS-mediated antiviral innate immunity. J. Biol. Chem. 2020, 295, 444–457. [Google Scholar] [CrossRef] [PubMed]
- Zhou, L.; He, R.; Fang, P.; Li, M.; Yu, H.; Wang, Q.; Yu, Y.; Wang, F.; Zhang, Y.; Chen, A.; et al. Hepatitis B virus rigs the cellular metabolome to avoid innate immune recognition. Nat. Commun. 2021, 12, 98. [Google Scholar] [CrossRef]
- Zhang, W.; Wang, G.; Xu, Z.G.; Tu, H.; Hu, F.; Dai, J.; Chang, Y.; Chen, Y.; Lu, Y.; Zeng, H.; et al. Lactate Is a Natural Suppressor of RLR Signaling by Targeting MAVS. Cell 2019, 178, 176–189.e15. [Google Scholar] [CrossRef] [PubMed]
- Bandiera, S.; Billie Bian, C.; Hoshida, Y.; Baumert, T.F.; Zeisel, M.B. Chronic hepatitis C virus infection and pathogenesis of hepatocellular carcinoma. Curr. Opin. Virol. 2016, 20, 99–105. [Google Scholar] [CrossRef] [Green Version]
- Irshad, M.; Gupta, P.; Irshad, K. Molecular basis of hepatocellular carcinoma induced by hepatitis C virus infection. World J. Hepatol. 2017, 9, 1305–1314. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gerresheim, G.K.; Roeb, E.; Michel, A.M.; Niepmann, M. Hepatitis C Virus Downregulates Core Subunits of Oxidative Phosphorylation, Reminiscent of the Warburg Effect in Cancer Cells. Cells 2019, 8, 1410. [Google Scholar] [CrossRef] [Green Version]
- Gerresheim, G.K.; Bathke, J.; Michel, A.M.; Andreev, D.E.; Shalamova, L.A.; Rossbach, O.; Hu, P.; Glebe, D.; Fricke, M.; Marz, M.; et al. Cellular Gene Expression during Hepatitis C Virus Replication as Revealed by Ribosome Profiling. Int. J. Mol. Sci. 2019, 20, 1321. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wolski, D.; Foote, P.K.; Chen, D.Y.; Lewis-Ximenez, L.L.; Fauvelle, C.; Aneja, J.; Walker, A.; Tonnerre, P.; Torres-Cornejo, A.; Kvistad, D.; et al. Early Transcriptional Divergence Marks Virus-Specific Primary Human CD8+ T Cells in Chronic versus Acute Infection. Immunity 2017, 47, 648–663.e8. [Google Scholar] [CrossRef] [Green Version]
- Wolski, D.; Lauer, G.M. Hepatitis C Virus as a Unique Human Model Disease to Define Differences in the Transcriptional Landscape of T Cells in Acute versus Chronic Infection. Viruses 2019, 11, 683. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Piccoli, C.; Scrima, R.; Quarato, G.; D’Aprile, A.; Ripoli, M.; Lecce, L.; Boffoli, D.; Moradpour, D.; Capitanio, N. Hepatitis C virus protein expression causes calcium-mediated mitochondrial bioenergetic dysfunction and nitro-oxidative stress. Hepatology 2007, 46, 58–65. [Google Scholar] [CrossRef]
- Ripoli, M.; D’Aprile, A.; Quarato, G.; Sarasin-Filipowicz, M.; Gouttenoire, J.; Scrima, R.; Cela, O.; Boffoli, D.; Heim, M.H.; Moradpour, D.; et al. Hepatitis C virus-linked mitochondrial dysfunction promotes hypoxia-inducible factor 1 alpha-mediated glycolytic adaptation. J. Virol. 2010, 84, 647–660. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jung, G.S.; Jeon, J.H.; Choi, Y.K.; Jang, S.Y.; Park, S.Y.; Kim, S.W.; Byun, J.K.; Kim, M.K.; Lee, S.; Shin, E.C.; et al. Pyruvate dehydrogenase kinase regulates hepatitis C virus replication. Sci. Rep. 2016, 6, 30846. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Diamond, D.L.; Syder, A.J.; Jacobs, J.M.; Sorensen, C.M.; Walters, K.A.; Proll, S.C.; McDermott, J.E.; Gritsenko, M.A.; Zhang, Q.; Zhao, R.; et al. Temporal proteome and lipidome profiles reveal hepatitis C virus-associated reprogramming of hepatocellular metabolism and bioenergetics. PLoS Pathog. 2010, 6, e1000719. [Google Scholar] [CrossRef] [Green Version]
- Ramière, C.; Rodriguez, J.; Enache, L.S.; Lotteau, V.; André, P.; Diaz, O. Activity of hexokinase is increased by its interaction with hepatitis C virus protein NS5A. J. Virol. 2014, 88, 3246–3254. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fontaine, K.A.; Sanchez, E.L.; Camarda, R.; Lagunoff, M. Dengue virus induces and requires glycolysis for optimal replication. J. Virol. 2015, 89, 2358–2366. [Google Scholar] [CrossRef] [Green Version]
- Allonso, D.; Andrade, I.S.; Conde, J.N.; Coelho, D.R.; Rocha, D.C.; da Silva, M.L.; Ventura, G.T.; Silva, E.M.; Mohana-Borges, R. Dengue Virus NS1 Protein Modulates Cellular Energy Metabolism by Increasing Glyceraldehyde-3-Phosphate Dehydrogenase Activity. J. Virol. 2015, 89, 11871–11883. [Google Scholar] [CrossRef] [Green Version]
- Silva, E.M.; Conde, J.N.; Allonso, D.; Ventura, G.T.; Coelho, D.R.; Carneiro, P.H.; Silva, M.L.; Paes, M.V.; Rabelo, K.; Weissmuller, G.; et al. Dengue virus nonstructural 3 protein interacts directly with human glyceraldehyde-3-phosphate dehydrogenase (GAPDH) and reduces its glycolytic activity. Sci. Rep. 2019, 9, 2651. [Google Scholar] [CrossRef]
- Fernandes-Siqueira, L.O.; Zeidler, J.D.; Sousa, B.G.; Ferreira, T.; Da Poian, A.T. Anaplerotic Role of Glucose in the Oxidation of Endogenous Fatty Acids during Dengue Virus Infection. MSphere 2018, 3, e00458-17. [Google Scholar] [CrossRef] [Green Version]
- Rasmussen, A.L.; Diamond, D.L.; McDermott, J.E.; Gao, X.; Metz, T.O.; Matzke, M.M.; Carter, V.S.; Belisle, S.E.; Korth, M.J.; Waters, K.M.; et al. Systems virology identifies a mitochondrial fatty acid oxidation enzyme, dodecenoyl coenzyme A delta isomerase, required for hepatitis C virus replication and likely pathogenesis. J. Virol. 2011, 85, 11646–11654. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Douglas, D.N.; Pu, C.H.; Lewis, J.T.; Bhat, R.; Anwar-Mohamed, A.; Logan, M.; Lund, G.; Addison, W.R.; Lehner, R.; Kneteman, N.M. Oxidative Stress Attenuates Lipid Synthesis and Increases Mitochondrial Fatty Acid Oxidation in Hepatoma Cells Infected with Hepatitis C Virus. J. Biol. Chem. 2016, 291, 1974–1990. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thaker, S.K.; Chapa, T.; Garcia, G., Jr.; Gong, D.; Schmid, E.W.; Arumugaswami, V.; Sun, R.; Christofk, H.R. Differential Metabolic Reprogramming by Zika Virus Promotes Cell Death in Human versus Mosquito Cells. Cell Metab. 2019, 29, 1206–1216.e4. [Google Scholar] [CrossRef]
- Beys-da-Silva, W.O.; Rosa, R.L.; Santi, L.; Berger, M.; Park, S.K.; Campos, A.R.; Terraciano, P.; Varela, A.P.M.; Teixeira, T.F.; Roehe, P.M.; et al. Zika Virus Infection of Human Mesenchymal Stem Cells Promotes Differential Expression of Proteins Linked to Several Neurological Diseases. Mol. Neurobiol. 2019, 56, 4708–4717. [Google Scholar] [CrossRef] [PubMed]
- Tiwari, S.K.; Dang, J.; Qin, Y.; Lichinchi, G.; Bansal, V.; Rana, T.M. Zika virus infection reprograms global transcription of host cells to allow sustained infection. Emerg. Microbes Infect. 2017, 6, e24. [Google Scholar] [CrossRef] [Green Version]
- Rothan, H.A.; Fang, S.; Mahesh, M.; Byrareddy, S.N. Zika Virus and the Metabolism of Neuronal Cells. Mol. Neurobiol. 2019, 56, 2551–2557. [Google Scholar] [CrossRef]
- Frauwirth, K.A.; Thompson, C.B. Regulation of T lymphocyte metabolism. J. Immunol. 2004, 172, 4661–4665. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hollenbaugh, J.A.; Munger, J.; Kim, B. Metabolite profiles of human immunodeficiency virus infected CD4+ T cells and macrophages using LC-MS/MS analysis. Virology 2011, 415, 153–159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palmer, C.S.; Ostrowski, M.; Gouillou, M.; Tsai, L.; Yu, D.; Zhou, J.; Henstridge, D.C.; Maisa, A.; Hearps, A.C.; Lewin, S.R.; et al. Increased glucose metabolic activity is associated with CD4+ T-cell activation and depletion during chronic HIV infection. AIDS 2014, 28, 297–309. [Google Scholar] [CrossRef] [PubMed]
- Kavanagh Williamson, M.; Coombes, N.; Juszczak, F.; Athanasopoulos, M.; Khan, M.B.; Eykyn, T.R.; Srenathan, U.; Taams, L.S.; Dias Zeidler, J.; Da Poian, A.T.; et al. Upregulation of Glucose Uptake and Hexokinase Activity of Primary Human CD4+ T Cells in Response to Infection with HIV-1. Viruses 2018, 10, 114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hegedus, A.; Kavanagh Williamson, M.; Huthoff, H. HIV-1 pathogenicity and virion production are dependent on the metabolic phenotype of activated CD4+ T cells. Retrovirology 2014, 11, 98. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hegedus, A.; Kavanagh Williamson, M.; Khan, M.B.; Dias Zeidler, J.; Da Poian, A.T.; El-Bacha, T.; Struys, E.A.; Huthoff, H. Evidence for Altered Glutamine Metabolism in Human Immunodeficiency Virus Type 1 Infected Primary Human CD4+ T Cells. AIDS Res. Hum. Retrovir. 2017, 33, 1236–1247. [Google Scholar] [CrossRef] [Green Version]
- Valle-Casuso, J.C.; Angin, M.; Volant, S.; Passaes, C.; Monceaux, V.; Mikhailova, A.; Bourdic, K.; Avettand-Fenoel, V.; Boufassa, F.; Sitbon, M.; et al. Cellular Metabolism Is a Major Determinant of HIV-1 Reservoir Seeding in CD4+ T Cells and Offers an Opportunity to Tackle Infection. Cell Metab. 2019, 29, 611–626.e5. [Google Scholar] [CrossRef] [Green Version]
- Kishimoto, N.; Yamamoto, K.; Abe, T.; Yasuoka, N.; Takamune, N.; Misumi, S. Glucose-dependent aerobic glycolysis contributes to recruiting viral components into HIV-1 particles to maintain infectivity. Biochem. Biophys. Res. Commun. 2021, 549, 187–193. [Google Scholar] [CrossRef] [PubMed]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, X.; Wu, K.; Zeng, S.; Zhao, F.; Fan, J.; Li, Z.; Yi, L.; Ding, H.; Zhao, M.; Fan, S.; et al. Viral Infection Modulates Mitochondrial Function. Int. J. Mol. Sci. 2021, 22, 4260. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22084260
Li X, Wu K, Zeng S, Zhao F, Fan J, Li Z, Yi L, Ding H, Zhao M, Fan S, et al. Viral Infection Modulates Mitochondrial Function. International Journal of Molecular Sciences. 2021; 22(8):4260. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22084260
Chicago/Turabian StyleLi, Xiaowen, Keke Wu, Sen Zeng, Feifan Zhao, Jindai Fan, Zhaoyao Li, Lin Yi, Hongxing Ding, Mingqiu Zhao, Shuangqi Fan, and et al. 2021. "Viral Infection Modulates Mitochondrial Function" International Journal of Molecular Sciences 22, no. 8: 4260. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22084260