
Liquid Biomarkers for Improved Diagnosis and Classification of CNS Tumors

 , ,
, , 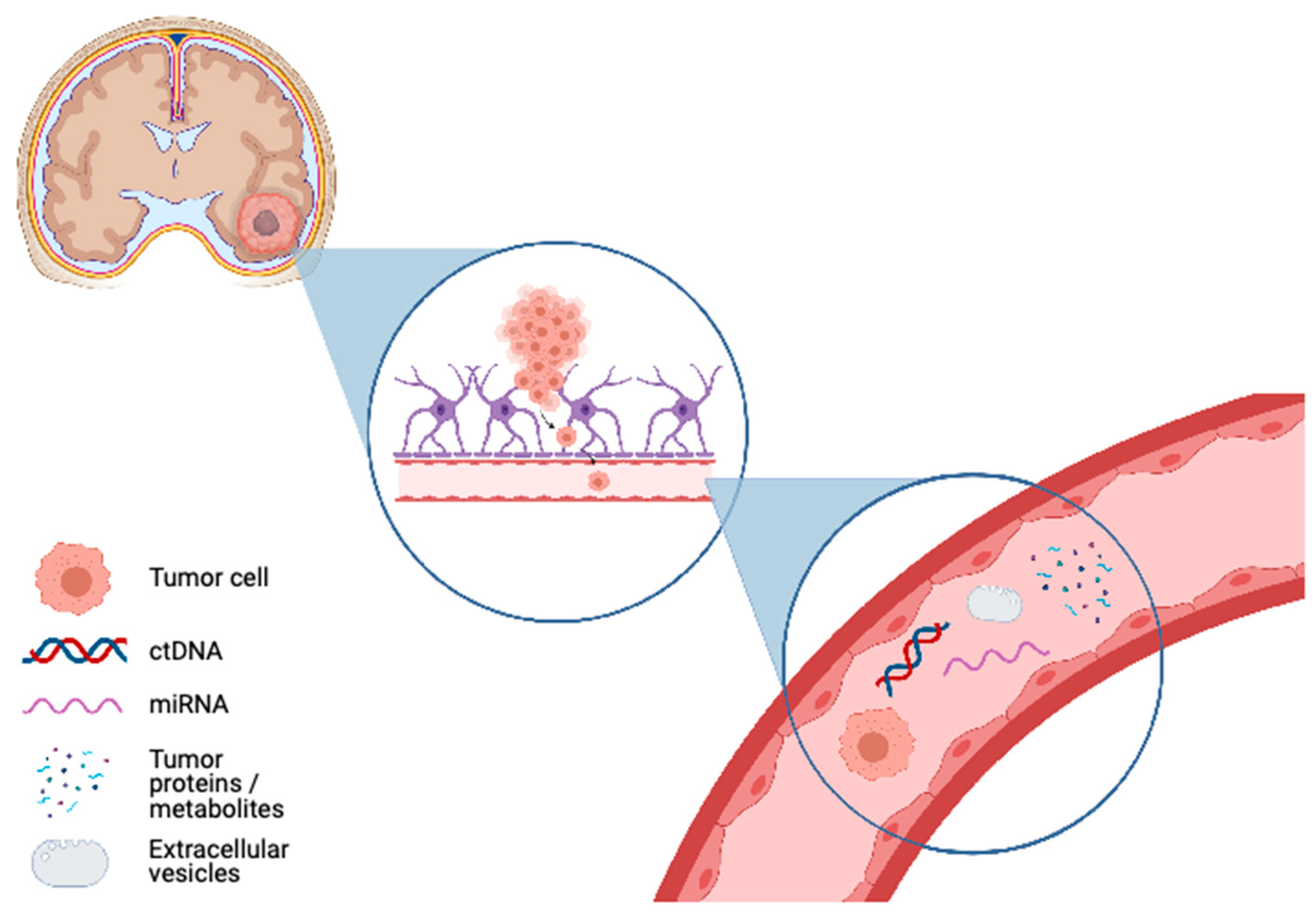{kind=link}
Abstract
:1. Background
2. Circulating Tumor Cells
3. Circulating Tumor DNA
4. Circulating Tumor MicoRNA
5. Circulating Proteome
6. Extracellular Vesicle
7. Future Directions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Mandel, P.; Metais, P. Les acides nucléiques du plasma sanguin chez l’homme [Nuclear Acids in Human Blood Plasma]. Comptes Rendus Seances Soc. Biol. Fil. 1948, 142, 241–243. [Google Scholar]
- Koffler, D.; Agnello, V.; Winchester, R.; Kunkel, H.G. The Occurrence of Single-Stranded DNA in the Serum of Patients with Systemic Lupus Erythematosus and Other Diseases. J. Clin. Investig. 1973, 52, 198–204. [Google Scholar] [CrossRef] [Green Version]
- Di Meo, A.; Bartlett, J.; Cheng, Y.; Pasic, M.D.; Yousef, G.M. Liquid biopsy: A step forward towards precision medicine in urologic malignancies. Mol. Cancer 2017, 16, 1–14. [Google Scholar] [CrossRef]
- Alix-Panabières, C.; Pantel, K. Clinical Applications of Circulating Tumor Cells and Circulating Tumor DNA as Liquid Biopsy. Cancer Discov. 2016, 6, 479–491. [Google Scholar] [CrossRef] [Green Version]
- Sun, K.; Jiang, P.; Chan, K.C.A.; Wong, J.; Cheng, Y.K.Y.; Liang, R.H.S.; Chan, W.-K.; Ma, E.S.K.; Chan, S.L.; Cheng, S.H.; et al. Plasma DNA tissue mapping by genome-wide methylation sequencing for noninvasive prenatal, cancer, and transplantation assessments. Proc. Natl. Acad. Sci. USA 2015, 112, E5503–E5512. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heitzer, E.; Ulz, P.; Geigl, J.B. Circulating Tumor DNA as a Liquid Biopsy for Cancer. Clin. Chem. 2015, 61, 112–123. [Google Scholar] [CrossRef] [PubMed]
- Yu, W.; Hurley, J.; Roberts, D.; Chakrabortty, S.; Enderle, D.; Noerholm, M.; Breakefield, X.; Skog, J. Exosome-based liquid biopsies in cancer: Opportunities and challenges. Ann. Oncol. 2021, 32, 466–477. [Google Scholar] [CrossRef]
- Duffy, M.J.; McDermott, E.W.; Crown, J. Blood-based biomarkers in breast cancer: From proteins to circulating tumor cells to circulating tumor DNA. Tumor Biol. 2018, 40. [Google Scholar] [CrossRef] [Green Version]
- Sato, Y.; Matoba, R.; Kato, K. Recent Advances in Liquid Biopsy in Precision Oncology Research. Biol. Pharm. Bull. 2019, 42, 337–342. [Google Scholar] [CrossRef] [Green Version]
- McGirt, M.J.; Chaichana, K.L.; Gathinji, M.; Attenello, F.J.; Than, K.; Olivi, A.; Weingart, J.D.; Brem, H.; redo Quiñones-Hinojosa, A. Independent association of extent of resection with survival in patients with malignant brain astrocytoma: Clinical article. J. Neurosurg. 2009, 110, 156–162. [Google Scholar] [CrossRef] [Green Version]
- Yong, R.L.; Lonser, R.R. Safety of Closed Brain Biopsy: Population-Based Studies Weigh in. World Neurosurg. 2013, 79, 53–54. [Google Scholar] [CrossRef] [Green Version]
- Johnson, D.R.; O’Neill, B.P.; Decker, P.A.; Kosel, M.L.; Lanzino, G.; Hammack, J.E. Mortality and Discharge to Home After Closed Brain Biopsy: Analysis of 3523 Cases from the State of California, 2003–2009. World Neurosurg. 2013, 79, 110–115. [Google Scholar] [CrossRef] [PubMed]
- Whittle, I.R. The dilemma of low grade glioma. J. Neurol. Neurosurg. Psychiatry 2004, 75, ii31–ii36. [Google Scholar] [CrossRef] [Green Version]
- Yan, P.F.; Yan, L.; Zhang, Z.; Salim, A.; Wang, L.; Hu, T.T.; Zhao, H.Y. Accuracy of conventional MRI for preoperative diagnosis of intracranial tumors: A single center report of 762 cases. Int. J. Surg. 2016, 36, 109–117. [Google Scholar] [CrossRef]
- Daneman, R.; Prat, A. The blood–brain barrier. Cold Spring Harb. Perspect. Biol. 2015, 7, a020412. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, L.; Cheng, G.; Ye, D.; Nazeri, A.; Yue, Y.; Liu, W.; Wang, X.; Dunn, G.P.; Petti, A.A.; Leuthardt, E.C.; et al. Focused Ultrasound-enabled Brain Tumor Liquid Biopsy. Sci. Rep. 2018, 8, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Whitin, J.C.; Jang, T.; Merchant, M.; Yu, T.T.-S.; Lau, K.; Recht, B.; Cohen, H.J.; Recht, L. Alterations in Cerebrospinal Fluid Proteins in a Presymptomatic Primary Glioma Model. PLoS ONE 2012, 7, e49724. [Google Scholar] [CrossRef] [Green Version]
- Khwaja, F.W.; Reed, M.S.; Olson, J.J.; Schmotzer, B.J.; Gillespie, G.Y.; Guha, A.; Groves, M.D.; Kesari, S.; Pohl, J.; Van Meir, E.G. Proteomic Identification of Biomarkers in the Cerebrospinal Fluid (CSF) of Astrocytoma Patients. J. Proteome Res. 2007, 6, 559–570. [Google Scholar] [CrossRef] [Green Version]
- Shen, F.; Zhang, Y.; Yao, Y.; Hua, W.; Zhang, H.-S.; Wu, J.-S.; Zhong, P.; Zhou, L.-F. Proteomic analysis of cerebrospinal fluid: Toward the identification of biomarkers for gliomas. Neurosurg. Rev. 2014, 37, 367–380. [Google Scholar] [CrossRef]
- Castro-Giner, F.; Aceto, N. Tracking cancer progression: From circulating tumor cells to metastasis. Genome Med. 2020, 12, 31. [Google Scholar] [CrossRef] [Green Version]
- Masuda, T.; Hayashi, N.; Iguchi, T.; Ito, S.; Eguchi, H.; Mimori, K. Clinical and biological significance of circulating tumor cells in cancer. Mol. Oncol. 2016, 10, 408–417. [Google Scholar] [CrossRef]
- Ross, A.A.; Cooper, B.W.; Lazarus, H.M.; Mackay, W.; Moss, T.J.; Ciobanu, N.; Tallman, M.S.; Kennedy, M.J.; Davidson, N.E.; Sweet, D.; et al. Detection and viability of tumor cells in peripheral blood stem cell collections from breast cancer patients using immunocytochemical and clonogenic assay techniques. Blood 1993, 82, 2605–2610. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.-P.; Eisenberger, M.A.; Carducci, M.A.; Partin, A.W.; Scher, H.I.; Ts’O, P.O.P. Identification and characterization of circulating prostate carcinoma cells. Cancer 2000, 88, 2787–2795. [Google Scholar] [CrossRef]
- Ladanyi, A.; Molnár, B.; Tanko, L.M.; Sréter, L.; Tulassay, Z. Detection of circulating tumor cell clusters in the peripheral blood of colorectal cancer patients. Gastroenterology 2000, 118, A278. [Google Scholar] [CrossRef]
- Adamczyk, L.A.; Williams, H.R.; Frankow, A.; Ellis, H.P.; Haynes, H.R.; Perks, C.; Holly, J.M.P.; Kurian, K.M. Current Understanding of Circulating Tumor Cells—Potential Value in Malignancies of the Central Nervous System. Front. Neurol. 2015, 6, 174. [Google Scholar] [CrossRef] [Green Version]
- Bankó, P.; Lee, S.Y.; Nagygyörgy, V.; Zrínyi, M.; Chae, C.H.; Cho, D.H.; Telekes, A. Technologies for circulating tumor cell separation from whole blood. J. Hematol. Oncol. 2019, 12, 1–20. [Google Scholar] [CrossRef] [Green Version]
- Potdar, P.D.; Lotey, N.K. Role of circulating tumor cells in future diagnosis and therapy of cancer. J. Cancer Metastasis Treat. 2015, 1, 44. [Google Scholar] [CrossRef] [Green Version]
- Hamilton, J.D.; Rapp, M.; Schneiderhan, T.M.; Sabel, M.; Hayman, A.; Scherer, A.; Kröpil, P.; Budach, W.; Kretschmar, U.; Gerber, P.A.; et al. Glioblastoma Multiforme Metastasis Outside the CNS: Three Case Reports and Possible Mechanisms of Escape. J. Clin. Oncol. 2014, 32, e80–e84. [Google Scholar] [CrossRef]
- Krol, I.; Castro-Giner, F.; Maurer, M.; Gkountela, S.; Szczerba, B.M.; Scherrer, R.; Coleman, N.; Carreira, S.; Bachmann, F.; Anderson, S.; et al. Detection of circulating tumour cell clusters in human glioblastoma. Br. J. Cancer 2018, 119, 487–491. [Google Scholar] [CrossRef] [Green Version]
- Bang-Christensen, S.R.; Pedersen, R.S.; Pereira, M.A.; Clausen, T.M.; Løppke, C.; Sand, N.T.; Ahrens, T.D.; Jørgensen, A.M.; Lim, Y.C.; Goksøyr, L.; et al. Capture and Detection of Circulating Glioma Cells Using the Recombinant VAR2CSA Malaria Protein. Cells 2019, 8, 998. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chistiakov, D.A.; Chekhonin, V.P. Circulating tumor cells and their advances to promote cancer metastasis and relapse, with focus on glioblastoma multiforme. Exp. Mol. Pathol. 2018, 105, 166–174. [Google Scholar] [CrossRef]
- MacArthur, K.M.; Kao, G.D.; Chandrasekaran, S.; Alonso-Basanta, M.; Chapman, C.; Lustig, R.A.; Wileyto, E.P.; Hahn, S.M.; Dorsey, J.F. Detection of Brain Tumor Cells in the Peripheral Blood by a Telomerase Promoter-Based Assay. Cancer Res. 2014, 74, 2152–2159. [Google Scholar] [CrossRef] [Green Version]
- Müller, C.; Holtschmidt, J.; Auer, M.; Heitzer, E.; Lamszus, K.; Schulte, A. Cancer: Hematogenous dissemination of glioblastoma multiforme. Sci. Transl. Med. 2014, 6, ra101–ra247. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, J.P.; Nahed, B.V.; Madden, M.W.; Oliveira, S.M.; Springer, S.; Bhere, D.; Chi, A.S.; Wakimoto, H.; Rothenberg, S.M.; Sequist, L.V.; et al. Brain Tumor Cells in Circulation Are Enriched for Mesenchymal Gene Expression. Cancer Discov. 2014, 4, 1299–1309. [Google Scholar] [CrossRef] [Green Version]
- Gao, F.; Cui, Y.; Jiang, H.; Sui, D.; Wang, Y.; Jiang, Z.; Zhao, J.; Lin, S. Circulating tumor cell is a common property of brain glioma and promotes the monitoring system. Oncotarget 2016, 7, 71330–71340. [Google Scholar] [CrossRef]
- Liu, T.; Xu, H.; Huang, M.; Ma, W.; Saxena, D.; Lustig, R.A.; Alonso-Basanta, M.; Zhang, Z.; Rourke, D.M.O.; Zhang, L.; et al. Circulating glioma cells exhibit stem cell-like properties. Cancer Res. 2018. [Google Scholar] [CrossRef] [Green Version]
- Shankar, G.M.; Balaj, L.; Stott, S.L.; Nahed, B.; Carter, B.S. Liquid biopsy for brain tumors. Expert Rev. Mol. Diagn. 2017, 17, 943–947. [Google Scholar] [CrossRef] [PubMed]
- Stucklin, A.S.G.; Ramaswamy, V.; Daniels, C.; Taylor, M.D. Review of molecular classification and treatment implications of pediatric brain tumors. Curr. Opin. Pediatr. 2018, 30, 3–9. [Google Scholar] [CrossRef]
- Heller, G.; McCormack, R.; Kheoh, T.; Molina, A.; Smith, M.R.; Dreicer, R.; Saad, F.; De Wit, R.; Aftab, D.T.; Hirmand, M.; et al. Circulating Tumor Cell Number as a Response Measure of Prolonged Survival for Metastatic Castration-Resistant Prostate Cancer: A Comparison With Prostate-Specific Antigen Across Five Randomized Phase III Clinical Trials. J. Clin. Oncol. 2018, 36, 572–580. [Google Scholar] [CrossRef]
- Zhao, Y.; Jiang, F.; Wang, Q.; Wang, B.; Han, Y.; Yang, J.; Wang, J.; Wang, K.; Ao, J.; Guo, X.; et al. Cytoplasm protein GFAP magnetic beads construction and application as cell separation target for brain tumors. J. NanobioTechnol. 2020, 18, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Achrol, A.S.; Rennert, R.C.; Anders, C.; Soffietti, R.; Ahluwalia, M.S.; Nayak, L.; Peters, S.; Arvold, N.D.; Harsh, G.R.; Steeg, P.S.; et al. Brain metastases. Nat. Rev. Dis. Prim. 2019, 5, 5. [Google Scholar] [CrossRef]
- Klotz, R.; Yu, M. Insights into brain metastasis: Recent advances in circulating tumor cell research. Cancer Rep. 2020, e1239. [Google Scholar] [CrossRef] [PubMed]
- Baccelli, I.; Schneeweiss, A.; Riethdorf, S.; Stenzinger, A.; Schillert, A.; Vogel, V.; Klein, C.; Saini, M.; Bäuerle, T.; Wallwiener, M.; et al. Identification of a population of blood circulating tumor cells from breast cancer patients that initiates metastasis in a xenograft assay. Nat. Biotechnol. 2013, 31, 539–544. [Google Scholar] [CrossRef]
- Hodgkinson, C.L.; Morrow, C.J.; Li, Y.; Metcalf, R.L.; Rothwell, D.G.; Trapani, F.; Polanski, R.; Burt, D.J.; Simpson, K.L.; Morris, K.; et al. Tumorigenicity and genetic profiling of circulating tumor cells in small-cell lung cancer. Nat. Med. 2014, 20, 897–903. [Google Scholar] [CrossRef] [PubMed]
- Drapkin, B.J.; George, J.; Christensen, C.L.; Mino-Kenudson, M.; Dries, R.; Sundaresan, T.; Phat, S.; Myers, D.T.; Zhong, J.; Igo, P.; et al. Genomic and Functional Fidelity of Small Cell Lung Cancer Patient-Derived Xenografts. Cancer Discov. 2018, 8, 600–615. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Ridgway, L.D.; Wetzel, M.D.; Ngo, J.; Yin, W.; Kumar, D.; Goodman, J.C.; Groves, M.D.; Marchetti, D. The Identification and Characterization of Breast Cancer CTCs Competent for Brain Metastasis. Sci. Transl. Med. 2013, 5, 180ra48. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boral, D.; Vishnoi, M.; Liu, H.N.; Yin, W.; Sprouse, M.L.; Scamardo, A.; Hong, D.S.; Tan, T.Z.; Thiery, J.P.; Chang, J.C.; et al. Molecular characterization of breast cancer CTCs associated with brain metastasis. Nat. Commun. 2017, 8, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Ruan, H.; Zhou, Y.; Shen, J.; Zhai, Y.; Xu, Y.; Pi, L. Circulating tumor cell characterization of lung cancer brain metastasis in the cerebrospinal fluid through single-cell transcriptome analysis. medRxiv 2020, 10, e246. [Google Scholar]
- Bronkhorst, A.J.; Ungerer, V.; Holdenrieder, S. The emerging role of cell-free DNA as a molecular marker for cancer management. Biomol. Detect. Quantif. 2019, 17, 100087. [Google Scholar] [CrossRef]
- Corcoran, R.B.; Chabner, B.A. Application of Cell-free DNA Analysis to Cancer Treatment. N. Engl. J. Med. 2018, 379, 1754–1765. [Google Scholar] [CrossRef] [Green Version]
- Newman, A.M.; Bratman, S.V.; To, J.; Wynne, J.F.; Eclov, N.C.W.; Modlin, L.A.; Liu, C.L.; Neal, J.W.; Wakelee, H.A.; Merritt, R.E.; et al. An ultrasensitive method for quantitating circulating tumor DNA with broad patient coverage. Nat. Med. 2014, 20, 548–554. [Google Scholar] [CrossRef] [PubMed]
- Jones, J.; Nguyen, H.; Drummond, K.; Morokoff, A. Circulating Biomarkers for Glioma: A Review. Neurosurg. 2021, 88, E221–E230. [Google Scholar] [CrossRef]
- Wang, J.; Zhao, Y.Y.; Li, J.F.; Guo, C.C.; Chen, F.R.; Su, H.K. IDH1 mutation detection by droplet digital PCR in glioma. Oncotarget 2015, 6, 39651. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hindson, B.J.; Ness, K.D.; Masquelier, D.A.; Belgrader, P.; Heredia, N.J.; Makarewicz, A.J. High-throughput droplet digital PCR system for absolute quantitation of DNA copy number. Anal. Chem. 2011, 83, 8604–8610. [Google Scholar] [CrossRef]
- Reid, A.L.; Freeman, J.B.; Millward, M.; Ziman, M.; Gray, E.S. Detection of BRAF-V600E and V600K in melanoma circulating tumour cells by droplet digital PCR. Clin. Biochem. 2015, 48, 999–1002. [Google Scholar] [CrossRef] [PubMed]
- Zhu, G.; Ye, X.; Dong, Z.; Lu, Y.C.; Sun, Y.; Liu, Y.; McCormack, R.; Gu, Y.; Liu, X. Highly Sensitive Droplet Digital PCR Method for Detection of EGFR-Activating Mutations in Plasma Cell–Free DNA from Patients with Advanced Non–Small Cell Lung Cancer. J. Mol. Diagn. 2015, 17, 265–272. [Google Scholar] [CrossRef]
- Raymond, C.K.; Hernandez, J.; Karr, R.; Hill, K.; Li, M. Collection of cell-free DNA for genomic analysis of solid tumors in a clinical laboratory setting. PLoS ONE 2017, 12, e0176241. [Google Scholar] [CrossRef]
- Forthun, R.B.; Hovland, R.; Schuster, C.; Puntervoll, H.; Brodal, H.P.; Namløs, H.M.; Aasheim, L.B.; Meza-Zepeda, L.A.; Gjertsen, B.T.; Knappskog, S.; et al. ctDNA detected by ddPCR reveals changes in tumour load in metastatic malignant melanoma treated with bevacizumab. Sci. Rep. 2019, 9, 1–15. [Google Scholar] [CrossRef] [PubMed]
- García-Romero, N.; Carrión-Navarro, J.; Areal-Hidalgo, P.; De Mendivil, A.O.; Asensi-Puig, A.; Madurga, R.; Núñez-Torres, R.; González-Neira, A.; Belda-Iniesta, C.; González-Rumayor, V.; et al. BRAF V600E Detection in Liquid Biopsies from Pediatric Central Nervous System Tumors. Cancers 2019, 12, 66. [Google Scholar] [CrossRef] [Green Version]
- Muralidharan, K.; Yekula, A.; Small, J.L.; Rosh, Z.S.; Kang, K.M.; Wang, L.; Lau, S.; Zhang, H.; Lee, H.; Bettegowda, C.; et al. TERT Promoter Mutation Analysis for Blood-Based Diagnosis and Monitoring of Gliomas. Clin. Cancer Res. 2021, 27, 169–178. [Google Scholar] [CrossRef] [PubMed]
- Parry, H.M.; Zuo, J.; Frumento, G.; Mirajkar, N.; Inman, C.; Edwards, E.; Griffiths, M.; Pratt, G.; Moss, P.A.H. Cytomegalovirus viral load within blood increases markedly in healthy people over the age of 70 years. Immun. Ageing 2016, 13, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shah, M.; Zorofchian, S.; Lan, C.; Duose, D.; Hu, P.; Esquenazi, Y. 5. Detecting mutations in cerebrospinal fluid: Liquid biopsy for diagnosis of central nervous system metastases. Cancer Genet. 2019, 233, S2–S3. [Google Scholar] [CrossRef]
- Escudero, L.; Llort, A.; Arias, A.; Diaz-Navarro, A.; Martínez-Ricarte, F.; Rubio-Perez, C.; Mayor, R.; Caratù, G.; Martínez-Sáez, E.; Vázquez-Méndez, É.; et al. Circulating tumour DNA from the cerebrospinal fluid allows the characterisation and monitoring of medulloblastoma. Nat. Commun. 2020, 11, 1–11. [Google Scholar] [CrossRef]
- Takayasu, T.; Shah, M.; Dono, A.; Yan, Y.; Borkar, R.; Putluri, N.; Zhu, J.-J.; Hama, S.; Yamasaki, F.; Tahara, H.; et al. Cerebrospinal fluid ctDNA and metabolites are informative biomarkers for the evaluation of CNS germ cell tumors. Sci. Rep. 2020, 10, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Capper, D.; Jones, D.T.W.; Sill, M.; Hovestadt, V.; Schrimpf, D.; Sturm, D.; Koelsche, C.; Sahm, F.; Chavez, L.; Reuss, D.E.; et al. DNA methylation-based classification of central nervous system tumours. Nature 2018, 555, 469–474. [Google Scholar] [CrossRef]
- Karimi, S.; Zuccato, J.A.; Mamatjan, Y.; Mansouri, S.; Suppiah, S.; Nassiri, F.; Diamandis, P.; Munoz, D.G.; Aldape, K.D.; Zadeh, G. The central nervous system tumor methylation classifier changes neuro-oncology practice for challenging brain tumor diagnoses and directly impacts patient care. Clin. EpiGenet. 2019, 11, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Shen, S.Y.; Burgener, J.M.; Bratman, S.V.; De Carvalho, D.D. Preparation of cfMeDIP-seq libraries for methylome profiling of plasma cell-free DNA. Nat. Protoc. 2019, 14, 2749–2780. [Google Scholar] [CrossRef]
- Nassiri, F.; Chakravarthy, A.; Feng, S.; Shen, S.Y.; Nejad, R.; Zuccato, J.A.; Voisin, M.R.; Patil, V.; Horbinski, C.; Aldape, K.; et al. Detection and discrimination of intracranial tumors using plasma cell-free DNA methylomes. Nat. Med. 2020, 26, 1044–1047. [Google Scholar] [CrossRef]
- Shen, S.Y.; Singhania, R.; Fehringer, G.; Chakravarthy, A.; Roehrl, M.H.A.; Chadwick, D.; Zuzarte, P.C.; Borgida, A.; Wang, T.T.; Li, T.; et al. Sensitive tumour detection and classification using plasma cell-free DNA methylomes. Nat. Cell Biol. 2018, 563, 579–583. [Google Scholar] [CrossRef] [PubMed]
- Nuzzo, P.V.; Berchuck, J.E.; Korthauer, K.; Spisak, S.; Nassar, A.H.; Abou Alaiwi, S. Detection of renal cell carcinoma using plasma and urine cell-free DNA methylomes. Nat. Med. 2020, 26, 1041–1043. [Google Scholar] [CrossRef] [PubMed]
- Sabedot, T.; Malta, T.; Snyder, J.; Nelson, K.; Wells, M.; Decarvalho, A.; Mukherjee, A.; Chitale, D.; Mosella, M.; Sokolov, A.; et al. A serum-based DNA methylation assay provides accurate detection of glioma. Neuro-Oncol. 2021. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Zhao, S.; Lee, M.; Yin, Y.; Li, J.; Zhou, Y.; Ballester, L.Y.; Esquenazi, Y.; Dashwood, R.H.; Davies, P.J.A.; et al. Reliable tumor detection by whole-genome methylation sequencing of cell-free DNA in cerebrospinal fluid of pediatric medulloblastoma. Sci. Adv. 2020, 6, eabb5427. [Google Scholar] [CrossRef] [PubMed]
- Jonas, S.; Izaurralde, E. Towards a molecular understanding of microRNA-mediated gene silencing. Nat. Rev. Genet. 2015, 16, 421–433. [Google Scholar] [CrossRef] [PubMed]
- Reddy, K.B. MicroRNA (miRNA) in cancer. Cancer Cell Int. 2015, 15, 1–6. [Google Scholar] [CrossRef] [Green Version]
- Turchinovich, A.; Weiz, L.; Langheinz, A.; Burwinkel, B. Characterization of extracellular circulating microRNA. Nucleic Acids Res. 2011, 39, 7223–7233. [Google Scholar] [CrossRef]
- Mitchell, P.S.; Parkin, R.K.; Kroh, E.M.; Fritz, B.R.; Wyman, S.K.; Pogosova-Agadjanyan, E.L.; Peterson, A.; Noteboom, J.; O’Briant, K.C.; Allen, A.; et al. Circulating microRNAs as stable blood-based markers for cancer detection. Proc. Natl. Acad. Sci. USA 2008, 105, 10513–10518. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chevillet, J.R.; Lee, I.; Briggs, H.A.; He, Y.; Wang, K. Issues and prospects of microRNA-based biomarkers in blood and other body fluids. Molecules 2014, 19, 6080–6105. [Google Scholar] [CrossRef]
- Tumilson, C.A.; Lea, R.W.; Alder, J.E.; Shaw, L. Circulating MicroRNA Biomarkers for Glioma and Predicting Response to Therapy. Mol. Neurobiol. 2014, 50, 545–558. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Li, P.; Li, A.; Jiang, W.; Wang, H.; Wang, J.; Xie, K. Plasma specific miRNAs as predictive biomarkers for diagnosis and prognosis of glioma. J. Exp. Clin. Cancer Res. 2012, 31, 97. [Google Scholar] [CrossRef] [Green Version]
- Roth, P.; Wischhusen, J.; Happold, C.; Chandran, P.A.; Hofer, S.; Eisele, G.; Weller, M.; Keller, A. A specific miRNA signature in the peripheral blood of glioblastoma patients. J. Neurochem. 2011, 118, 449–457. [Google Scholar] [CrossRef] [Green Version]
- D’Urso, P.I.; D’Urso, O.F.; Gianfreda, C.D.; Mezzolla, V.; Storelli, C.; Marsigliante, S. miR-15b and miR-21 as Circulating Biomarkers for Diagnosis of Glioma. Curr. Genomics. 2015, 16, 304. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ohno, M.; Matsuzaki, J.; Kawauchi, J.; Aoki, Y.; Miura, J.; Takizawa, S.; Kato, K.; Sakamoto, H.; Matsushita, Y.; Takahashi, M.; et al. Assessment of the Diagnostic Utility of Serum MicroRNA Classification in Patients With Diffuse Glioma. JAMA Netw. Open 2019, 2, e1916953. [Google Scholar] [CrossRef] [PubMed]
- Zhi, F.; Shao, N.; Wang, R.; Deng, D.; Xue, L.; Wang, Q.; Zhang, Y.; Shi, Y.; Xia, X.; Wang, S.; et al. Identification of 9 serum microRNAs as potential noninvasive biomarkers of human astrocytoma. Neuro-Oncol. 2014, 17, 383–391. [Google Scholar] [CrossRef]
- Regazzo, G.; Terrenato, I.; Spagnuolo, M.; Carosi, M.; Cognetti, G.; Cicchillitti, L.; Sperati, F.; Villani, V.; Carapella, C.; Piaggio, G.; et al. A restricted signature of serum miRNAs distinguishes glioblastoma from lower grade gliomas. J. Exp. Clin. Cancer Res. 2016, 35, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, J.; Li, L.; Jiang, C. Identification and Evaluation of Serum MicroRNA-29 Family for Glioma Screening. Mol. Neurobiol. 2014, 52, 1540–1546. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.; Pang, B.; Xin, T.; Guo, H.; Xing, Y.; Xu, S.; Pang, Q. Plasma miR-221/222 Family as Novel Descriptive and Prognostic Biomarkers for Glioma. Mol. Neurobiol. 2016, 53, 1452–1460. [Google Scholar] [CrossRef]
- Skog, J.; Würdinger, T.; Van Rijn, S.; Meijer, D.H.; Gainche, L.; Curry, W.T., Jr.; Carter, B.S.; Krichevsky, A.M.; Breakefield, X.O. Glioblastoma microvesicles transport RNA and proteins that promote tumour growth and provide diagnostic biomarkers. Nat. Cell Biol. 2008, 10, 1470–1476. [Google Scholar] [CrossRef] [PubMed]
- Qu, S.; Guan, J.; Liu, Y. Identification of microRNAs as novel biomarkers for glioma detection: A meta-analysis based on 11 articles. J. Neurol. Sci. 2015, 348, 181–187. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Q.; Liu, J.; Quan, J.; Liu, W.; Tan, H.; Li, W. MicroRNAs as potential biomarkers for the diagnosis of glioma: A systematic review and meta-analysis. Cancer Sci. 2018, 109, 2651–2659. [Google Scholar] [CrossRef] [Green Version]
- Zhi, F.; Shao, N.; Li, B.; Xue, L.; Deng, D.; Xu, Y.; Lan, Q.; Peng, Y.; Yang, Y. A serum 6-miRNA panel as a novel non-invasive biomarker for meningioma. Sci. Rep. 2016, 6, 32067. [Google Scholar] [CrossRef] [Green Version]
- Sato, J.; Shimomura, A.; Kawauchi, J.; Matsuzaki, J.; Yamamoto, Y.; Takizawa, S.; Sakamoto, H.; Ohno, M.; Narita, Y.; Ochiya, T.; et al. Brain metastasis-related microRNAs in patients with advanced breast cancer. PLoS ONE 2019, 14, e0221538. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Yuan, Y.; Cho, J.-H.; McClarty, S.; Baxter, D.; Galas, D.J. Comparing the MicroRNA Spectrum between Serum and Plasma. PLoS ONE 2012, 7, e41561. [Google Scholar] [CrossRef]
- Baraniskin, A.; Kuhnhenn, J.; Schlegel, U.; Maghnouj, A.; Zöllner, H.; Schmiegel, W.; Hahn, S.; Schroers, R. Identification of microRNAs in the cerebrospinal fluid as biomarker for the diagnosis of glioma. Neuro-Oncol. 2011, 14, 29–33. [Google Scholar] [CrossRef]
- Teplyuk, N.M.; Mollenhauer, B.; Gabriely, G.; Giese, A.; Kim, E.; Smolsky, M.; Kim, R.Y.; Saria, M.G.; Pastorino, S.; Kesari, S.; et al. MicroRNAs in cerebrospinal fluid identify glioblastoma and metastatic brain cancers and reflect disease activity. Neuro-Oncol. 2012, 14, 689–700. [Google Scholar] [CrossRef] [Green Version]
- Wei, D.; Wan, Q.; Li, L.; Jin, H.; Liu, Y.; Wang, Y.; Zhang, G. MicroRNAs as Potential Biomarkers for Diagnosing Cancers of Central Nervous System: A Meta-analysis. Mol. Neurobiol. 2014, 51, 1452–1461. [Google Scholar] [CrossRef]
- Maclellan, S.A.; Macaulay, C.; Lam, S.; Garnis, C. Pre-profiling factors influencing serum microRNA levels. BMC Clin. Pathol. 2014, 14, 27. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.H.; Lee, S.K.; Yoo, Y.C.; Park, N.H.; Park, D.B.; Yoo, J.S.; An, H.J.; Park, Y.M.; Cho, K.G. Proteome analysis of human cerebrospinal fluid as a diagnostic biomarker in patients with meningioma. Med Sci. Monit. 2012, 18, BR450–BR460. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Okamoto, H.; Li, J.; Vortmeyer, A.O.; Jaffe, H.; Lee, Y.-S.; Gläsker, S.; Sohn, T.-S.; Zeng, W.; Ikejiri, B.; Proescholdt, M.A.; et al. Comparative Proteomic Profiles of Meningioma Subtypes. Cancer Res. 2006, 66, 10199–10204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wulfkuhle, J.D.; Paweletz, C.P.; Steeg, P.S.; Petricoin, E.F.; Liotta, L. Proteomic approaches to the diagnosis, treatment, and monitoring of cancer. Adv. Exp. Med. Biol. 2003, 532, 59–68. [Google Scholar] [CrossRef] [PubMed]
- Kalinina, J.; Peng, J.; Ritchie, J.C.; Van Meir, E.G. Proteomics of gliomas: Initial biomarker discovery and evolution of technology. Neuro-Oncol. 2011, 13, 926–942. [Google Scholar] [CrossRef]
- Batabyal, S.K.; Ghosh, B.; Sengupta, S.; Ghosh, S.N.; Chatterjee, R. Cerebrospinal fluid and serum carcinoembryonic antigen in brain tumors. Neoplasma 2003, 50, 377–379. [Google Scholar]
- Moldrich, G.; Lange, P.; Strik, H. Carcinoembryonic antigen in the CSF of cancer patients—The value of intrathecal synthesis and correlation with IgA-diffusion dynamics. Acta Neurol. Belg. 2010, 110, 314–320. [Google Scholar] [PubMed]
- Nakagawa, H.; Kubo, S.; Murasawa, A.; Nakajima, S.; Nakajima, Y.; Izumoto, S.; Hayakawa, T. Measurements of CSF biochemical tumor markers in patients with meningeal carcinomatosis and brain tumors. J. Neuro-Oncol. 1992, 12, 111–120. [Google Scholar] [CrossRef]
- Wang, P.; Piao, Y.; Zhang, X.; Li, W.; Hao, X. The concentration of CYFRA 21-1, NSE and CEA in cerebro-spinal fluid can be useful indicators for diagnosis of meningeal carcinomatosis of lung cancer. Cancer Biomark. 2013, 13, 123–130. [Google Scholar] [CrossRef]
- Khwaja, F.W.; Nolen, J.D.L.; Mendrinos, S.E.; Lewis, M.M.; Olson, J.J.; Pohl, J.; Van Meir, E.G.; Ritchie, J.C.; Brat, D.J. Proteomic analysis of cerebrospinal fluid discriminates malignant and nonmalignant disease of the central nervous system and identifies specific protein markers. Proteomics 2006, 6, 6277–6287. [Google Scholar] [CrossRef] [PubMed]
- Samuel, N.; Remke, M.; Rutka, J.T.; Raught, B.; Malkin, D. Proteomic analyses of CSF aimed at biomarker development for pediatric brain tumors. J. Neuro-Oncol. 2014, 118, 225–238. [Google Scholar] [CrossRef]
- Qaddoumi, I.; Sane, M.; Li, S.; Kocak, M.; Pai-Panandiker, A.; Harreld, J.; Klimo, P.; Wright, K.; Broniscer, A.; Gajjar, A. Diagnostic utility and correlation of tumor markers in the serum and cerebrospinal fluid of children with intracranial germ cell tumors. Child Nerv. Syst. 2012, 28, 1017–1024. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nishizaki, T.; Kajiwara, K.; Adachi, N.; Tsuha, M.; Nakayama, H.; Ohshita, N.; Ikeda, N.; Iwamoto, N.; Ito, H.; Suzuki, M. Detection of craniospinal dissemination of intracranial germ cell tumours based on serum and cerebrospinal fluid levels of tumour markers. J. Clin. Neurosci. 2001, 8, 27–30. [Google Scholar] [CrossRef]
- Watanabe, S.; Aihara, Y.; Kikuno, A.; Sato, T.; Komoda, T.; Kubo, O.; Amano, K.; Okada, Y.; Koyamaishi, Y. A Highly Sensitive and Specific Chemiluminescent Enzyme Immunoassay for Placental Alkaline Phosphatase in the Cerebrospinal Fluid of Patients with Intracranial Germinomas. Pediatr. Neurosurg. 2012, 48, 141–145. [Google Scholar] [CrossRef]
- Miyanohara, O.; Takeshima, H.; Kaji, M.; Hirano, H.; Sawamura, Y.; Kochi, M.; Kuratsu, J.-I. Diagnostic significance of soluble c-kit in the cerebrospinal fluid of patients with germ cell tumors. J. Neurosurg. 2002, 97, 177–183. [Google Scholar] [CrossRef] [Green Version]
- Schuhmann, M.; Zucht, H.; Nassimi, R.; Heine, G.; Schneekloth, C.; Stuerenburg, H.; Selle, H. Peptide screening of cerebrospinal fluid in patients with glioblastoma multiforme. Eur. J. Surg. Oncol. (EJSO) 2010, 36, 201–207. [Google Scholar] [CrossRef] [Green Version]
- MSaratsis, A.M.; Yadavilli, S.; Magge, S.; Rood, B.R.; Perez, J.; Hill, D.A. Insights into pediatric diffuse intrinsic pontine glioma through proteomic analysis of cerebrospinal fluid. Neuro Oncol. 2012, 14, 547–560. [Google Scholar] [CrossRef] [PubMed]
- Galati, D.; Di Noto, R.; Del Vecchio, L. Diagnostic strategies to investigate cerebrospinal fluid involvement in haematological malignancies. Leuk. Res. 2013, 37, 231–237. [Google Scholar] [CrossRef] [PubMed]
- Roy, S.; Josephson, S.A.; Fridlyand, J.; Karch, J.; Kadoch, C.; Karrim, J.; Damon, L.; Treseler, P.; Kunwar, S.; Shuman, M.A.; et al. Protein Biomarker Identification in the CSF of Patients With CNS Lymphoma. J. Clin. Oncol. 2008, 26, 96–105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gomes, H.R. Cerebrospinal fluid approach on neuro-oncology. Arq. Neuro-Psiquiatr. 2013, 71, 677–680. [Google Scholar] [CrossRef] [Green Version]
- Zetterberg, H.; Andreasson, U.; Blennow, K. CSF Antithrombin III and Disruption of the Blood-Brain Barrier. J. Clin. Oncol. 2009, 27, 2302–2303. [Google Scholar] [CrossRef]
- Brunn, A.; Montesinos-Rongen, M.; Strack, A.; Reifenberger, G.; Mawrin, C.; Schaller, C.; Deckert, M. Expression pattern and cellular sources of chemokines in primary central nervous system lymphoma. Acta Neuropathol. 2007, 114, 271–276. [Google Scholar] [CrossRef]
- Rubenstein, J.L.; Wong, V.S.; Kadoch, C.; Gao, H.-X.; Barajas, R.; Chen, L.; Josephson, S.A.; Scott, B.; Douglas, V.; Maiti, M.; et al. CXCL13 plus interleukin 10 is highly specific for the diagnosis of CNS lymphoma. Blood 2013, 121, 4740–4748. [Google Scholar] [CrossRef] [Green Version]
- Sasagawa, Y.; Akai, T.; Tachibana, O.; Iizuka, H. Diagnostic value of interleukin-10 in cerebrospinal fluid for diffuse large B-cell lymphoma of the central nervous system. J. Neuro-Oncol. 2014, 121, 177–183. [Google Scholar] [CrossRef] [PubMed]
- Rajagopal, M.U.; Hathout, Y.; MacDonald, T.J.; Kieran, M.W.; Gururangan, S.; Blaney, S.M. Proteomic profiling of cerebrospinal fluid identifies prostaglandin D2 synthase as a putative biomarker for pediatric medulloblastoma: A pediatric brain tumor consortium study. Proteomics 2011, 11, 935–943. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saso, L.; Leone, M.G.; Sorrentino, C.; Giacomelli, S.; Silvestrini, B.; Grima, J.; Li, J.C.H.; Samy, E.; Mruk, D.; Cheng, C.Y. Quantification of prostaglandin D synthetase in cerebrospinal fluid: A potential marker for brain tumor. Biochem. Mol. Boil. Int. 1998, 46, 643–656. [Google Scholar] [CrossRef] [PubMed]
- Desiderio, C.; D’Angelo, L.; Rossetti, D.V.; Iavarone, F.; Giardina, B.; Castagnola, M.; Massimi, L.; Tamburrini, G.; Di Rocco, C. Cerebrospinal fluid top-down proteomics evidenced the potential biomarker role of LVV- and VV-hemorphin-7 in posterior cranial fossa pediatric brain tumors. Proteomics 2012, 12, 2158–2166. [Google Scholar] [CrossRef] [PubMed]
- Figarella-Branger, D.; Dubois, C.; Chauvin, P.; De Victor, B.; Gentet, J.C.; Rougon, G. Correlation between polysialic-neural cell adhesion molecule levels in CSF and medulloblastoma outcomes. J. Clin. Oncol. 1996, 14, 2066–2072. [Google Scholar] [CrossRef] [PubMed]
- Kao, C.L.; Chiou, S.H.; Ho, D.M.T.; Chen, Y.J.; Liu, R.S.; Lo, C.W. Elevation of plasma and cerebrospinal fluid osteopontin levels in patients with atypical teratoid/rhabdoid tumor. Am. J. Clin. Pathol. 2005, 123, 297–304. [Google Scholar] [CrossRef]
- Gollapalli, K.; Ray, S.; Srivastava, R.; Renu, D.; Singh, P.; Dhali, S.; Dikshit, J.B.; Srikanth, R.; Moiyadi, A.; Srivastava, S. Investigation of serum proteome alterations in human glioblastoma multiforme. Proteomics 2012, 12, 2378–2390. [Google Scholar] [CrossRef]
- Iwadate, Y.; Sakaida, T.; Hiwasa, T.; Nagai, Y.; Ishikura, H.; Takiguchi, M.; Yamaura, A.; Schwartzenberg-Bar-Yoseph, F.; Armoni, M.; Karnieli, E. Molecular Classification and Survival Prediction in Human Gliomas Based on Proteome Analysis. Cancer Res. 2004, 64, 2496–2501. [Google Scholar] [CrossRef] [Green Version]
- Akers, J.C.; Ramakrishnan, V.; Kim, R.; Skog, J.; Nakano, I.; Pingle, S. miR-21 in the Extracellular Vesicles (EVs) of Cerebrospinal Fluid (CSF): A Platform for Glioblastoma Biomarker Development. PLoS ONE 2013, 8, e78115. [Google Scholar] [CrossRef] [PubMed]
- Akers, J.C.; Gonda, D.; Kim, R.; Carter, B.S.; Chen, C.C. Biogenesis of extracellular vesicles (EV): Exosomes, microvesicles, retrovirus-like vesicles, and apoptotic bodies. J. Neuro-Oncol. 2013, 113, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, B.; Xu, K.; Zheng, X.; Chen, T.; Wang, J.; Song, Y.; Shao, Y.; Zheng, S. Application of exosomes as liquid biopsy in clinical diagnosis. Signal Transduct. Target. Ther. 2020, 5, 1–14. [Google Scholar] [CrossRef]
- Santiago-Dieppa, D.R.; Steinberg, J.; Gonda, D.; Cheung, V.J.; Carter, B.S.; Chen, C.C. Extracellular vesicles as a platform for “liquid biopsy” in glioblastoma patients. Expert Rev. Mol. Diagn. 2014, 14, 819–825. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giallombardo, M.; Borrás, J.C.; Castiglia, M.; Van Der Steen, N.; Mertens, I.; Pauwels, P.; Peeters, M.; Rolfo, C. Exosomal miRNA Analysis in Non-small Cell Lung Cancer (NSCLC) Patients’ Plasma Through qPCR: A Feasible Liquid Biopsy Tool. J. Vis. Exp. 2016. [Google Scholar] [CrossRef] [Green Version]
- Srivastava, A.; Moxley, K.; Ruskin, R.; Dhanasekaran, D.N.; Zhao, Y.D.; Ramesh, R. A Non-invasive Liquid Biopsy Screening of Urine-Derived Exosomes for miRNAs as Biomarkers in Endometrial Cancer Patients. AAPS J. 2018, 20, 82. [Google Scholar] [CrossRef]
- Wang, S.; Yang, Y.; Sun, L.; Qiao, G.; Song, Y.; Liu, B. Exosomal MicroRNAs as Liquid Biopsy Biomarkers in Hepatocellular Carcinoma. OncoTargets Ther. 2020, 13, 2021–2030. [Google Scholar] [CrossRef] [Green Version]
- Otake, K.; Kamiguchi, H.; Hirozane, Y. Identification of biomarkers for amyotrophic lateral sclerosis by comprehensive analysis of exosomal mRNAs in human cerebrospinal fluid. BMC Med Genom. 2019, 12, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hyun, K.-A.; Gwak, H.; Lee, J.; Kwak, B.; Jung, H.-I. Salivary Exosome and Cell-Free DNA for Cancer Detection. Micromachines 2018, 9, 340. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cai, X.; Janku, F.; Zhan, Q.; Fan, J.-B. Accessing Genetic Information with Liquid Biopsies. Trends Genet. 2015, 31, 564–575. [Google Scholar] [CrossRef] [PubMed]
- Hoshino, A.; Costa-Silva, B.; Shen, T.-L.; Rodrigues, G.; Hashimoto, A.; Mark, M.T.; Molina, H.; Kohsaka, S.; Di Giannatale, A.; Ceder, S.; et al. Tumour exosome integrins determine organotropic metastasis. Nature 2015, 527, 329–335. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Allenson, K.; Castillo, J.; Lucas, F.A.S.; Scelo, G.; Kim, D.U.; Bernard, V.; Davis, G.; Kumar, T.; Katz, M.; Overman, M.J.; et al. High prevalence of mutantKRAS in circulating exosome-derived DNA from early-stage pancreatic cancer patients. Ann. Oncol. 2017, 28, 741–747. [Google Scholar] [CrossRef] [PubMed]
- Möhrmann, L.; Huang, H.J.; Hong, D.S.; Tsimberidou, A.M.; Funda, M.-B.; Piha-Paul, S.A.; Subbiah, V.; Karp, D.D.; Naing, A.; Krug, A.; et al. Liquid Biopsies Using Plasma Exosomal Nucleic Acids and Plasma Cell-Free DNA Compared with Clinical Outcomes of Patients with Advanced Cancers. Clin. Cancer Res. 2018, 24, 181–188. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shao, H.; Chung, J.; Balaj, L.; Charest, A.; Bigner, D.D.; Carter, B.S. Protein typing of circulating microvesicles allows real-time monitoring of glioblastoma therapy. Nat. Med. 2012, 18, 1835. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.W.; Balaj, L.; Liau, L.M.; Samuels, M.L.; Kotsopoulos, S.K.; Maguire, C.A.; LoGuidice, L.; Soto, H.; Garrett, M.; Zhu, L.D.; et al. BEAMing and Droplet Digital PCR Analysis of Mutant IDH1 mRNA in Glioma Patient Serum and Cerebrospinal Fluid Extracellular Vesicles. Mol. Ther. Nucleic Acids 2013, 2, e109. [Google Scholar] [CrossRef] [PubMed]
- Gan, H.K.; Kaye, A.H.; Luwor, R.B. The EGFRvIII variant in glioblastoma multiforme. J. Clin. Neurosci. 2009, 16, 748–754. [Google Scholar] [CrossRef]
- Figueroa, J.M.; Skog, J.; Akers, J.; Li, H.; Komotar, R.; Jensen, R.; Ringel, F.; Yang, I.; Kalkanis, S.; Thompson, R.; et al. Detection of wild-type EGFR amplification and EGFRvIII mutation in CSF-derived extracellular vesicles of glioblastoma patients. Neuro-Oncol. 2017, 19, 1494–1502. [Google Scholar] [CrossRef]
- Masoudi, M.S.; Mehrabian, E.; Mirzaei, H. MiR-21: A key player in glioblastoma pathogenesis. J. Cell. Biochem. 2017, 119, 1285–1290. [Google Scholar] [CrossRef] [PubMed]
- Momen-Heravi, F.; Balaj, L.; Alian, S.; Tigges, J.; Toxavidis, V.; Ericsson, M. Alternative methods for characterization of extracellular vesicles. Front. Physiol. 2012, 3, 354. [Google Scholar] [CrossRef] [Green Version]
- Balakrishnan, A.; Roy, S.; Fleming, T.; Leong, H.S.; Schuurmans, C. The Emerging Role of Extracellular Vesicles in the Glioma Microenvironment: Biogenesis and Clinical Relevance. Cancers 2020, 12, 1964. [Google Scholar] [CrossRef] [PubMed]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bunda, S.; Zuccato, J.A.; Voisin, M.R.; Wang, J.Z.; Nassiri, F.; Patil, V.; Mansouri, S.; Zadeh, G. Liquid Biomarkers for Improved Diagnosis and Classification of CNS Tumors. Int. J. Mol. Sci. 2021, 22, 4548. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22094548
Bunda S, Zuccato JA, Voisin MR, Wang JZ, Nassiri F, Patil V, Mansouri S, Zadeh G. Liquid Biomarkers for Improved Diagnosis and Classification of CNS Tumors. International Journal of Molecular Sciences. 2021; 22(9):4548. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22094548
Chicago/Turabian StyleBunda, Severa, Jeffrey A. Zuccato, Mathew R. Voisin, Justin Z. Wang, Farshad Nassiri, Vikas Patil, Sheila Mansouri, and Gelareh Zadeh. 2021. "Liquid Biomarkers for Improved Diagnosis and Classification of CNS Tumors" International Journal of Molecular Sciences 22, no. 9: 4548. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22094548