
Omics Data and Their Integrative Analysis to Support Stratified Medicine in Neurodegenerative Diseases


Abstract
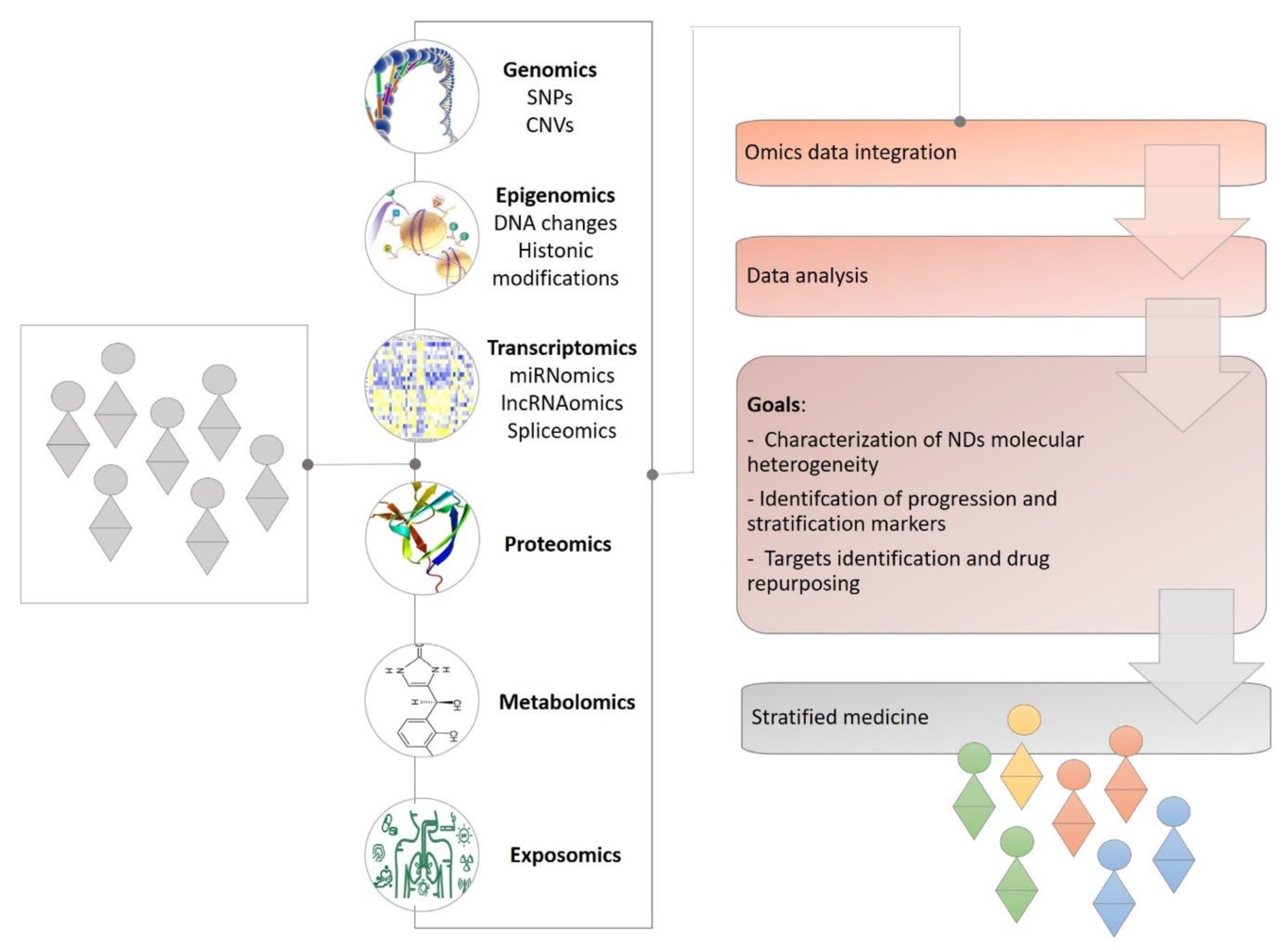
:1. Introduction
2. Alzheimer’s Disease
3. Parkinson’s Disease
4. Amyotrophic Lateral Sclerosis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kovacs, G.G. Molecular pathology of neurodegenerative diseases: Principles and practice. J. Clin. Pathol. 2019, 72, 725–735. [Google Scholar] [CrossRef]
- Das, S.; Zhang, Z.; Ang, L.C. Clinicopathological overlap of neurodegenerative diseases: A comprehensive review. J. Clin. Neurosci. Off. J. Neurosurg. Soc. Australas. 2020, 78, 30–33. [Google Scholar]
- Chen, W.-J.; Cheng, X.; Fu, Y.; Zhao, M.; McGinley, J.; Westenberger, A.; Xiong, Z.-Q. Rethinking monogenic neurological diseases. BMJ 2020, 371, m3752. [Google Scholar] [CrossRef]
- Erkkinen, M.G.; Kim, M.O.; Geschwind, M.D. Clinical Neurology and Epidemiology of the Major Neurodegenerative Diseases. Cold Spring Harbor Perspect. Biol. 2018, 10, a033118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Santiago, J.A.; Bottero, V.; Potashkin, J.A. Dissecting the Molecular Mechanisms of Neurodegenerative Diseases through Network Biology. Front. Aging Neurosci. 2017, 9, 166. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perrone, F.; Cacace, R.; van der Zee, J.; Van Broeckhoven, C. Emerging genetic complexity and rare genetic variants in neurodegenerative brain diseases. Genome Med. 2021, 13, 59. [Google Scholar] [CrossRef] [PubMed]
- Kiaei, M. New hopes and challenges for treatment of neurodegenerative disorders: Great opportunities for young neuroscientists. Basic Clin. Neurosci. 2013, 4, 3–4. [Google Scholar]
- Yiannopoulou, K.G.; Anastasiou, A.I.; Zachariou, V.; Pelidou, S.H. Reasons for Failed Trials of Disease-Modifying Treatments for Alzheimer Disease and Their Contribution in Recent Research. Biomedicines 2019, 7, 97. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ong, S.; Ling, J.; Ballantyne, A.; Lysaght, T.; Xafis, V. Perceptions of ‘Precision’ and ‘Personalised’ Medicine in Singapore and Associated Ethical Issues. Asian Bioeth. Rev. 2021, 13, 179–194. [Google Scholar] [CrossRef]
- Erikainen, S.; Chan, S. Contested futures: Envisioning “Personalized,” “Stratified,” and “Precision” medicine. New Genet. Soc. 2019, 38, 308–330. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Badhwar, A.; Haqqani, A.S. Biomarker potential of brain-secreted extracellular vesicles in blood in Alzheimer’s disease. Alzheimer’s Dement. 2020, 12, e12001. [Google Scholar] [CrossRef]
- Sancesario, G.M.; Toniolo, S.; Chiasserini, D.; Di Santo, S.G.; Zegeer, J.; Bernardi, G.; Musicco, M.; Caltagirone, C.; Parnetti, L.; Parnetti, S. The Clinical Use of Cerebrospinal Fluid Biomarkers for Alzheimer’s Disease Diagnosis: The Italian Selfie. J. Alzheimer’s Dis. 2017, 55, 1659–1666. [Google Scholar] [CrossRef]
- Manzoni, C.; Kia, D.A.; Vandrovcova, J.; Hardy, J.; Wood, N.W.; Lewis, P.A.; Ferrari, R. Genome, transcriptome and proteome: The rise of omics data and their integration in biomedical sciences. Brief. Bioinforma. 2018, 19, 286–302. [Google Scholar] [CrossRef]
- Hasin, Y.; Seldin, M.; Lusis, A. Multi-omics approaches to disease. Genome Biol. 2017, 18, 1–15. [Google Scholar] [CrossRef]
- Zielinski, J.M.; Luke, J.J.; Guglietta, S.; Krieg, C. High Throughput Multi-Omics Approaches for Clinical Trial Evaluation and Drug Discovery. Front. Immunol. 2021, 12, 590742. [Google Scholar] [CrossRef] [PubMed]
- Olivier, M.; Asmis, R.; Hawkins, G.A.; Howard, T.D.; Cox, L.A. The Need for Multi-Omics Biomarker Signatures in Precision Medicine. Int. J. Mol. Sci. 2019, 20, 4781. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, A.; Sidhu, J.; Goyal, A.; Tsao, J.W. Alzheimer Disease. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2020. [Google Scholar]
- Crews, L.; Masliah, E. Molecular mechanisms of neurodegeneration in Alzheimer’s disease. Hum. Mol. Genet. 2010, 19, R12–R20. [Google Scholar] [CrossRef] [PubMed]
- Bekris, L.M.; Yu, C.E.; Bird, T.D.; Tsuang, D.W. Genetics of Alzheimer disease. J. Geriatr. Psychiatr. Neurol. 2010, 23, 213–227. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nicolas, G.; Charbonnier, C.; Campion, D. From Common to Rare Variants: The Genetic Component of Alzheimer Disease. Hum. Hered. 2016, 81, 129–141. [Google Scholar] [CrossRef] [PubMed]
- Keren-Shaul, H.; Spinrad, A.; Weiner, A.; Matcovitch-Natan, O.; Dvir-Szternfeld, R.; Ulland, T.K.; David, E.; Baruch, K.; Lara-Astaiso, D.; Toth, B.; et al. A Unique Microglia Type Associated with Restricting Development of Alzheimer’s Disease. Cell 2017, 169, 1276–1290. [Google Scholar] [CrossRef]
- Fehlbaum-Beurdeley, P.; Sol, O.; Désiré, L.; Touchon, J.; Dantoine, T.; Vercelletto, M.; Gabelle, A.; Jarrige, A.-C.; Haddad, R.; Lemarié, J.C.; et al. Validation of AclarusDx™, a Blood-Based Transcriptomic Signature for the Diagnosis of Alzheimer’s Disease. J. Alzheimer’s Dis. 2012, 32, 169–181. [Google Scholar] [CrossRef] [PubMed]
- Thijssen, E.H.; La Joie, R.; Wolf, A.; Strom, A.; Wang, P.; Iaccarino, L.; Bourakova, V.; Cobigo, Y.; Heuer, H.; Spina, S.; et al. Diagnostic value of plasma phosphorylated tau181 in Alzheimer’s disease and frontotemporal lobar degeneration. Nat. Med. 2020, 26, 387–397. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Fang, J.; Bekris, L.M.; Kim, Y.H.; Pieper, A.A.; Leverenz, J.B.; Cummings, J.; Cheng, F. AlzGPS: A genome-wide positioning systems platform to catalyze multi-omics for Alzheimer’s drug discovery. Alzheimer’s Res. Ther. 2021, 13, 1–13. [Google Scholar]
- De Jager, P.L.; Ma, Y.; McCabe, C.; Xu, J.; Vardarajan, B.N.; Felsky, D.; Klein, H.-U.; White, C.C.; Peters, M.A.; Lodgson, B.; et al. A multi-omic atlas of the human frontal cortex for aging and Alzheimer’s disease research. Sci. Data 2018, 5, 1–13. [Google Scholar] [CrossRef]
- Clark, C.; Dayon, L.; Masoodi, M.; Bowman, G.L.; Popp, J. An integrative multi-omics approach reveals new central nervous system pathway alterations in Alzheimer’s disease. Alzheimer’s Res. Ther. 2021, 13, 1–9. [Google Scholar] [CrossRef]
- Xicota, L.; Ichou, F.; Lejeune, F.-X.; Colsch, B.; Tenenhaus, A.; Leroy, I.; Fontaine, G.; Lhomme, M.; Bertin, H.; Habert, M.-O.; et al. Multi-omics signature of brain amyloid deposition in asymptomatic individuals at-risk for Alzheimer’s disease: The INSIGHT-preAD study. EBioMedicine 2019, 47, 518–528. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nativio, R.; Lan, Y.; Donahue, G.; Sidoli, S.; Berson, A.; Srinivasan, A.R.; Shcherbakova, O.; Amlie-Wolf, A.; Nie, J.; Cui, X.; et al. An integrated multi-omics approach identifies epigenetic alterations associated with Alzheimer’s disease. Nat. Genet. 2020, 52, 1024–1035. [Google Scholar] [CrossRef]
- Bai, B.; Wang, X.; Li, Y.; Chen, P.C.; Yu, K.; Dey, K.K.; Yarbro, J.M.; Han, X.; Lutz, B.M.; Rao, S.; et al. Deep Multilayer Brain Proteomics Identifies Molecular Networks in Alzheimer’s Disease Progression. Neuron 2020, 105, 975–991. [Google Scholar] [CrossRef]
- Mueller, S.G.; Weiner, M.W.; Thal, L.J.; Petersen, R.C.; Jack, C.; Jagust, W.; Trojanowski, J.Q.; Toga, A.W.; Beckett, L. The Alzheimer’s disease neuroimaging initiative. Neuroimaging Clin. N. Am. 2005, 15, 869. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bennett, D.A.; Schneider, J.A.; Arvanitakis, Z.; Wilson, R.S. Overview and findings from the religious orders study. Curr. Alzheimer Res. 2012, 9, 628–645. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, Y.; Jun, G.R.; Zhang, X.; Chung, J.; Naj, A.C.; Chen, Y.; Bellenguez, C.; Hamilton-Nelson, K.; Martin, E.R.; Kunkle, B.W.; et al. Analysis of Whole-Exome Sequencing Data for Alzheimer Disease Stratified by APOE Genotype. JAMA Neurol. 2019, 76, 1099–1108. [Google Scholar] [CrossRef] [PubMed]
- Dagan, H.; Flashner-Abramson, E.; Vasudevan, S.; Jubran, M.R.; Cohen, E.; Kravchenko-Balasha, N. Exploring Alzheimer’s Disease Molecular Variability via Calculation of Personalized Transcriptional Signatures. Biomolecules 2020, 10, 503. [Google Scholar] [CrossRef] [PubMed]
- Milind, N.; Preuss, C.; Haber, A.; Ananda, G.; Mukherjee, S.; John, C.; Shapley, S.; Logsdon, B.A.; Crane, P.K.; Carter, G.W. Transcriptomic stratification of late-onset Alzheimer’s cases reveals novel genetic modifiers of disease pathology. PLoS Genet. 2020, 16, e1008775. [Google Scholar] [CrossRef] [PubMed]
- Neff, R.A.; Wang, M.; Vatansever, S.; Guo, L.; Ming, C.; Wang, Q.; Wang, E.; Horgusluoglu-Moloch, E.; Song, W.-m.; Li, A.; et al. Molecular subtyping of Alzheimer’s disease using RNA sequencing data reveals novel mechanisms and targets. Sci. Adv. 2021, 7, eabb5398. [Google Scholar] [CrossRef] [PubMed]
- Iqbal, K.; Flory, M.; Khatoon, S.; Soininen, H.; Pirttila, T.; Lehtovirta, M.; Alafuzoff, I.; Blennow, K.; Andreasen, N.; Vanmechelen, E.; et al. Subgroups of Alzheimer’s disease based on cerebrospinal fluid molecular markers. Ann. Neurol. 2005, 58, 748–757. [Google Scholar] [CrossRef] [PubMed]
- Toschi, N.; Lista, S.; Baldacci, F.; Cavedo, E.; Zetterberg, H.; Blennow, K.; Kilimann, I.; Teipel, S.J.; Melo Dos Santos, A.; Epelbaum, S.; et al. Biomarker-guided clustering of Alzheimer’s disease clinical syndromes. Neurobiol. Aging 2019, 83, 42–53. [Google Scholar] [CrossRef]
- Lerche, S.; Schulte, C.; Wurster, I.; Machetanz, G.; Roeben, B.; Zimmermann, M.; Deuschle, C.; Hauser, A.K.; Bohringer, J.; Krageloh-Mann, I.; et al. The Mutation Matters: CSF Profiles of GCase, Sphingolipids, alpha-Synuclein in PDGBA. Move. Disord. Off. J. Move. Disord. Soc. 2021. [Google Scholar] [CrossRef] [PubMed]
- Prasuhn, J.; Brüggemann, N.; Hessler, N.; Berg, D.; Gasser, T.; Brockmann, K.; Olbrich, D.; Ziegler, A.; König, I.R.; Klein, C.; et al. An omics-based strategy using coenzyme Q10 in patients with Parkinson’s disease: Concept evaluation in a double-blind randomized placebo-controlled parallel group trial. Neurol. Res. Pract. 2019, 1, 1–7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carling, P.J.; Mortiboys, H.; Green, C.; Mihaylov, S.; Sandor, C.; Schwartzentruber, A.; Taylor, R.; Wei, W.; Hastings, C.; Wong, S.; et al. Deep phenotyping of peripheral tissue facilitates mechanistic disease stratification in sporadic Parkinson’s disease. Prog. Neurobiol. 2020, 187, 101772. [Google Scholar] [CrossRef] [PubMed]
- Hipp, G.; Vaillant, M.; Diederich, N.J.; Roomp, K.; Satagopam, V.P.; Banda, P.; Sandt, E.; Mommaerts, K.; Schmitz, S.K.; Longhino, L.; et al. The Luxembourg Parkinson’s Study: A Comprehensive Approach for Stratification and Early Diagnosis. Front. Aging Neurosci. 2018, 10, 326. [Google Scholar] [CrossRef] [Green Version]
- Kia, D.A.; Zhang, D.; Guelfi, S.; Manzoni, C.; Hubbard, L.; Reynolds, R.H.; Botia, J.; Ryten, M.; Ferrari, R.; Lewis, P.A.; et al. Identification of Candidate Parkinson Disease Genes by Integrating Genome-Wide Association Study, Expression, and Epigenetic Data Sets. JAMA Neurol. 2021, 78, 464–472. [Google Scholar] [CrossRef] [PubMed]
- Aronica, E.; Baas, F.; Iyer, A.; ten Asbroek, A.L.; Morello, G.; Cavallaro, S. Molecular classification of amyotrophic lateral sclerosis by unsupervised clustering of gene expression in motor cortex. Neurobiol. Dis. 2015, 74, 359–376. [Google Scholar] [CrossRef] [Green Version]
- Morello, G.; Spampinato, A.G.; Cavallaro, S. Molecular Taxonomy of Sporadic Amyotrophic Lateral Sclerosis Using Disease-Associated Genes. Front. Neurol. 2017, 8, 152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tam, O.H.; Rozhkov, N.V.; Shaw, R.; Kim, D.; Hubbard, I.; Fennessey, S.; Propp, N.; Consortium, N.A.; Fagegaltier, D.; Harris, B.T.; et al. Postmortem Cortex Samples Identify Distinct Molecular Subtypes of ALS: Retrotransposon Activation, Oxidative Stress, and Activated Glia. Cell Rep. 2019, 29, 1164–1177. [Google Scholar] [CrossRef] [Green Version]
- Wuolikainen, A.; Andersen, P.M.; Moritz, T.; Marklund, S.L.; Antti, H. ALS patients with mutations in the SOD1 gene have an unique metabolomic profile in the cerebrospinal fluid compared with ALS patients without mutations. Mol. Genet. Metab. 2012, 105, 472–478. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.; Sandhu, D.; Konrad, C.; Roychoudhury, D.; Schwartz, B.I.; Cheng, R.R.; Bredvik, K.; Kawamata, H.; Calder, E.L.; Studer, L.; et al. Identification of a Distinct Metabolomic Subtype of Sporadic ALS Patients. bioRxiv 2018, 416396. [Google Scholar] [CrossRef]
- Devi, G.; Scheltens, P. Heterogeneity of Alzheimer’s disease: Consequence for drug trials? Alzheimer’s Res. Ther. 2018, 10, 122. [Google Scholar] [CrossRef] [Green Version]
- Ferreira, D.; Wahlund, L.O.; Westman, E. The heterogeneity within Alzheimer’s disease. Aging 2018, 10, 3058–3060. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Caselli, R.J. Age stratification corrects bias in estimated hazard of APOE genotype for Alzheimer’s disease. Alzheimers Dement. 2018, 4, 602–608. [Google Scholar] [CrossRef]
- Williams, T.; Borchelt, D.R.; Chakrabarty, P. Therapeutic approaches targeting Apolipoprotein E function in Alzheimer’s disease. Mol. Neurodegener. 2020, 15, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marioni, R.E.; Campbell, A.; Hagenaars, S.P.; Nagy, R.; Amador, C.; Hayward, C.; Porteous, D.J.; Visscher, P.M.; Deary, I.J. Genetic Stratification to Identify Risk Groups for Alzheimer’s Disease. J. Alzheimer’s Dis. 2017, 57, 275–283. [Google Scholar] [CrossRef] [Green Version]
- Mukherjee, S.; Mez, J.; Trittschuh, E.H.; Saykin, A.J.; Gibbons, L.E.; Fardo, D.W.; Wessels, M.; Bauman, J.; Moore, M.; Choi, S.E.; et al. Genetic data and cognitively defined late-onset Alzheimer’s disease subgroups. Mol. Psychiatr. 2018, 25, 2942–2951. [Google Scholar] [CrossRef] [Green Version]
- Mitelpunkt, A.; Galili, T.; Kozlovski, T.; Bregman, N.; Shachar, N.; Markus-Kalish, M.; Benjamini, Y. Novel Alzheimer’s disease subtypes identified using a data and knowledge driven strategy. Sci. Rep. 2020, 10, 1327. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nazarian, A.; Yashin, A.I.; Kulminski, A.M. Summary-Based Methylome-Wide Association Analyses Suggest Potential Genetically Driven Epigenetic Heterogeneity of Alzheimer’s Disease. J. Clin. Med. 2020, 9, 1489. [Google Scholar] [CrossRef]
- Oresic, M.; Lotjonen, J.; Soininen, H. Systems medicine and the integration of bioinformatic tools for the diagnosis of Alzheimer’s disease. Genome Med. 2009, 1, 83. [Google Scholar] [PubMed]
- Bredesen, D.E. Metabolic profiling distinguishes three subtypes of Alzheimer’s disease. Aging 2015, 7, 595–600. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wood, P.L.; Medicherla, S.; Sheikh, N.; Terry, B.; Phillipps, A.; Kaye, J.A.; Quinn, J.F.; Woltjer, R.L. Targeted Lipidomics of Fontal Cortex and Plasma Diacylglycerols (DAG) in Mild Cognitive Impairment and Alzheimer’s Disease: Validation of DAG Accumulation Early in the Pathophysiology of Alzheimer’s Disease. J. Alzheimer’s Dis. 2015, 48, 537–546. [Google Scholar] [CrossRef] [Green Version]
- Czech, C.; Berndt, P.; Busch, K.; Schmitz, O.; Wiemer, J.; Most, V.; Hampel, H.; Kastler, J.; Senn, H. Metabolite profiling of Alzheimer’s disease cerebrospinal fluid. PLoS ONE 2012, 7, e31501. [Google Scholar] [CrossRef] [PubMed]
- Bellec, P.; Dixon, R.A.; Li, L.; Masellis, M.; Duchesne, S.; Chertkow, H.; Black, S.E.; Sapkota, S.; McFall, G.P.; Badhwar, A. A multiomics approach to heterogeneity in Alzheimer’s disease: Focused review and roadmap. Brain J. Neurol. 2020, 143, 1315–1331. [Google Scholar]
- Wang, G.; Zhou, Y.; Huang, F.-J.; Tang, H.-D.; Xu, X.-H.; Liu, J.-J.; Wang, Y.; Deng, Y.-L.; Ren, R.-J.; Xu, W.; et al. Plasma Metabolite Profiles of Alzheimer’s Disease and Mild Cognitive Impairment. J. Proteome Res. 2014, 13, 2649–2658. [Google Scholar] [CrossRef] [PubMed]
- Greenland, J.C.; Williams-Gray, C.H.; Barker, R.A. The clinical heterogeneity of Parkinson’s disease and its therapeutic implications. Eur. J. Neurosci. 2019, 49, 328–338. [Google Scholar] [CrossRef] [PubMed]
- Cherian, A.; Divya, K.P. Genetics of Parkinson’s disease. Acta Neurol. Belg. 2020, 120, 1297–1305. [Google Scholar] [CrossRef]
- La Cognata, V.; Morello, G.; D’Agata, V.; Cavallaro, S. Copy number variability in Parkinson’s disease: Assembling the puzzle through a systems biology approach. Hum. Genet. 2017, 136, 13–37. [Google Scholar] [CrossRef] [Green Version]
- Krüger, R.; Klucken, J.; Weiss, D.; Tönges, L.; Kolber, P.; Unterecker, S.; Lorrain, M.; Baas, H.; Müller, T.; Riederer, P. Classification of advanced stages of Parkinson’s disease: Translation into stratified treatments. J. Neural Transm. 2017, 124, 1015–1027. [Google Scholar] [CrossRef]
- Mu, J.; Chaudhuri, K.R.; Bielza, C.; de Pedro-Cuesta, J.; Larrañaga, P.; Martinez-Martin, P. Parkinson’s Disease Subtypes Identified from Cluster Analysis of Motor and Non-motor Symptoms. Front. Aging Neurosci. 2017, 9, 301. [Google Scholar] [CrossRef] [PubMed]
- Lawton, M.; Ben-Shlomo, Y.; May, M.T.; Baig, F.; Barber, T.R.; Klein, J.C.; Swallow, D.M.A.; Malek, N.; Grosset, K.A.; Bajaj, N.; et al. Developing and validating Parkinson’s disease subtypes and their motor and cognitive progression. J. Neurol. Neurosurg. Psychiatr. 2018, 89, 1279–1287. [Google Scholar] [CrossRef] [PubMed]
- Marras, C.; Lang, A. Parkinson’s disease subtypes: Lost in translation? J. Neurol. Neurosurg. Psychiatr. 2012, 84, 409–415. [Google Scholar] [CrossRef] [PubMed]
- Fereshtehnejad, S.-M.; Postuma, R.B. Subtypes of Parkinson’s Disease: What Do They Tell Us About Disease Progression? Curr. Neurol. Neurosci. Rep. 2017, 17, 34. [Google Scholar] [CrossRef]
- Lewis, S.J.G. Heterogeneity of Parkinson’s disease in the early clinical stages using a data driven approach. J. Neurol. Neurosurg. Psychiatr. 2005, 76, 343–348. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beeler, J.A.; Erro, R.; Vitale, C.; Amboni, M.; Picillo, M.; Moccia, M.; Longo, K.; Santangelo, G.; De Rosa, A.; Allocca, R.; et al. The Heterogeneity of Early Parkinson’s Disease: A Cluster Analysis on Newly Diagnosed Untreated Patients. PLoS ONE 2013, 8, e70244. [Google Scholar]
- De Pablo-Fernández, E.; Lees, A.J.; Holton, J.L.; Warner, T.T. Prognosis and Neuropathologic Correlation of Clinical Subtypes of Parkinson Disease. JAMA Neurol. 2019, 76, 470. [Google Scholar] [CrossRef]
- Von Linstow, C.U.; Gan-Or, Z.; Brundin, P. Precision medicine in Parkinson’s disease patients with LRRK2 and GBA risk variants—Let’s get even more personal. Transl. Neurodegener. 2020, 9, 1–10. [Google Scholar]
- Gasser, T. Usefulness of Genetic Testing in PD and PD Trials: A Balanced Review. J. Parkinson’s Dis. 2015, 5, 209–215. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Masellis, M.; Collinson, S.; Freeman, N.; Tampakeras, M.; Levy, J.; Tchelet, A.; Eyal, E.; Berkovich, E.; Eliaz, R.E.; Abler, V.; et al. Dopamine D2 receptor gene variants and response to rasagiline in early Parkinson’s disease: A pharmacogenetic study. Brain J. Neurol. 2016, 139, 2050–2062. [Google Scholar] [CrossRef] [Green Version]
- Weiss, D.; Herrmann, S.; Wang, L.; Schulte, C.; Brockmann, K.; Plewnia, C.; Gasser, T.; Sharma, M.; Gharabaghi, A.; Krüger, R. Alpha-synuclein gene variants may predict neurostimulation outcome. Move. Disord. 2016, 31, 601–603. [Google Scholar] [CrossRef] [PubMed]
- Petrucci, S.; Ginevrino, M.; Trezzi, I.; Monfrini, E.; Ricciardi, L.; Albanese, A.; Avenali, M.; Barone, P.; Bentivoglio, A.R.; Bonifati, V.; et al. GBA-Related Parkinson’s Disease: Dissection of Genotype-Phenotype Correlates in a Large Italian Cohort. Move. Disord. Off. J. Move. Disord. Soc. 2020, 35, 2106–2111. [Google Scholar] [CrossRef]
- Rosen, A.; Zeger, S.L. Precision medicine: Discovering clinically relevant and mechanistically anchored disease subgroups at scale. J. Clin. Investig. 2019, 129, 944–945. [Google Scholar] [CrossRef] [Green Version]
- Brown, R.H.; Longo, D.L.; Al-Chalabi, A. Amyotrophic Lateral Sclerosis. N. Engl. J. Med. 2017, 377, 162–172. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chiò, A.; Logroscino, G.; Traynor, B.J.; Collins, J.; Simeone, J.C.; Goldstein, L.A.; White, L.A. Global Epidemiology of Amyotrophic Lateral Sclerosis: A Systematic Review of the Published Literature. Neuroepidemiology 2013, 41, 118–130. [Google Scholar] [CrossRef] [Green Version]
- Ryan, M.; Heverin, M.; Doherty, M.A.; Davis, N.; Corr, E.M.; Vajda, A.; Pender, N.; McLaughlin, R.; Hardiman, O. Determining the incidence of familiality in ALS. Neurol. Genet. 2018, 4, e239. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhandari, R.; Kuhad, A.; Kuhad, A. Edaravone: A new hope for deadly amyotrophic lateral sclerosis. Drugs Today 2018, 54, 349. [Google Scholar] [CrossRef] [PubMed]
- Dash, R.P.; Babu, R.J.; Srinivas, N.R. Two Decades-Long Journey from Riluzole to Edaravone: Revisiting the Clinical Pharmacokinetics of the Only Two Amyotrophic Lateral Sclerosis Therapeutics. Clin. Pharmacokineti. 2018, 57, 1385–1398. [Google Scholar] [CrossRef] [PubMed]
- Jaiswal, M.K. Riluzole and edaravone: A tale of two amyotrophic lateral sclerosis drugs. Med. Res. Rev. 2018, 39, 733–748. [Google Scholar] [CrossRef] [PubMed]
- Petrov, D.; Mansfield, C.; Moussy, A.; Hermine, O. ALS Clinical Trials Review: 20 Years of Failure. Are We Any Closer to Registering a New Treatment? Front. Aging Neurosci. 2017, 9, 68. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sawada, H. Clinical efficacy of edaravone for the treatment of amyotrophic lateral sclerosis. Expert Opin. Pharmacother. 2017, 18, 735–738. [Google Scholar] [CrossRef]
- Taylor, J.P.; Brown, R.H.; Cleveland, D.W. Decoding ALS: From genes to mechanism. Nature 2016, 539, 197–206. [Google Scholar] [CrossRef] [Green Version]
- Krokidis, M.G. Transcriptomics in amyotrophic lateral sclerosis. Front. Biosci. 2018, 10, 103–121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vijayakumar, U.G.; Milla, V.; Cynthia Stafford, M.Y.; Bjourson, A.J.; Duddy, W.; Duguez, S.M.-R. A Systematic Review of Suggested Molecular Strata, Biomarkers and Their Tissue Sources in ALS. Front. Neurol. 2019, 10, 400. [Google Scholar] [CrossRef] [Green Version]
- Morello, G.; Guarnaccia, M.; Spampinato, A.G.; Salomone, S.; D’Agata, V.; Conforti, F.L.; Aronica, E.; Cavallaro, S. Integrative multi-omic analysis identifies new drivers and pathways in molecularly distinct subtypes of ALS. Sci. Rep. 2019, 9, 9968. [Google Scholar] [CrossRef]
- La Cognata, V.; Gentile, G.; Aronica, E.; Cavallaro, S. Splicing Players Are Differently Expressed in Sporadic Amyotrophic Lateral Sclerosis Molecular Clusters and Brain Regions. Cells 2020, 9, 159. [Google Scholar] [CrossRef] [Green Version]
- Lanznaster, D.; de Assis, D.R.; Corcia, P.; Pradat, P.-F.; Blasco, H. Metabolomics Biomarkers: A Strategy Toward Therapeutics Improvement in ALS. Front. Neurol. 2018, 9, 1126. [Google Scholar] [CrossRef] [Green Version]
- Jääskeläinen, O.; Solje, E.; Hall, A.; Katisko, K.; Korhonen, V.; Tiainen, M.; Kangas, A.J.; Helisalmi, S.; Pikkarainen, M.; Koivisto, A.; et al. Low Serum High-Density Lipoprotein Cholesterol Levels Associate with the C9orf72 Repeat Expansion in Frontotemporal Lobar Degeneration Patients. J. Alzheimer’s Dis. 2019, 72, 127–137. [Google Scholar] [CrossRef] [Green Version]
- Guillemin, G.J.; Blasco, H.; Patin, F.; Descat, A.; Garçon, G.; Corcia, P.; Gelé, P.; Lenglet, T.; Bede, P.; Meininger, V.; et al. A pharmaco-metabolomics approach in a clinical trial of ALS: Identification of predictive markers of progression. PLoS ONE 2018, 13, e0198116. [Google Scholar]
- Bjornevik, K.; Zhang, Z.; O’Reilly, É.J.; Berry, J.D.; Clish, C.B.; Deik, A.; Jeanfavre, S.; Kato, I.; Kelly, R.S.; Kolonel, L.N.; et al. Prediagnostic plasma metabolomics and the risk of amyotrophic lateral sclerosis. Neurology 2019, 92, e2089–e2100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morello, G.; Cavallaro, S. Transcriptional analysis reveals distinct subtypes in amyotrophic lateral sclerosis: Implications for personalized therapy. Future Med. Chem. 2015, 7, 1335–1359. [Google Scholar] [CrossRef] [Green Version]
- Morello, G.; Conforti, F.L.; Parenti, R.; D’Agata, V.; Cavallaro, S. Selection of Potential Pharmacological Targets in ALS Based on Whole- Genome Expression Profiling. Curr. Med. Chem. 2015, 22, 2004–2021. [Google Scholar] [CrossRef] [PubMed]
- Apolloni, S.; Amadio, S.; Fabbrizio, P.; Morello, G.; Spampinato, A.G.; Latagliata, E.C.; Salvatori, I.; Proietti, D.; Ferri, A.; Madaro, L.; et al. Histaminergic transmission slows progression of amyotrophic lateral sclerosis. J. Cachexia Sarcopenia Muscle 2019, 10, 872–893. [Google Scholar] [CrossRef] [Green Version]
- Apolloni, S.; Fabbrizio, P.; Amadio, S.; Napoli, G.; Verdile, V.; Morello, G.; Iemmolo, R.; Aronica, E.; Cavallaro, S.; Volonte, C. Histamine Regulates the Inflammatory Profile of SOD1-G93A Microglia and the Histaminergic System Is Dysregulated in Amyotrophic Lateral Sclerosis. Front. Immunol. 2017, 8, 1689. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Volonte, C.; Morello, G.; Spampinato, A.G.; Amadio, S.; Apolloni, S.; D’Agata, V.; Cavallaro, S. Omics-based exploration and functional validation of neurotrophic factors and histamine as therapeutic targets in ALS. Ageing Res. Rev. 2020, 62, 101121. [Google Scholar] [CrossRef]
- Ruffini, N.; Klingenberg, S.; Schweiger, S.; Gerber, S. Common Factors in Neurodegeneration: A Meta-Study Revealing Shared Patterns on a Multi-Omics Scale. Cells 2020, 9, 2642. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Study (Year) | Sample | Omics Technique | Main Findings | Ref. | |
|---|---|---|---|---|---|
| AD | Nativio et al. (2020) | Postmortem human brain samples (lateral temporal lobe, Brodmann area 21 or 20) of AD patients (n = 12; mean age = 68), cognitively healthy older individuals (n = 10; mean age = 68) and healthy younger individuals (n = 8, mean age = 52) obtained from the Center for Neurodegenerative Disease Research brain bank at the University of Pennsylvania. | Transcriptomics, proteomics and epigenomics | Multi-omics analysis revealed that AD involves a reconfiguration of the epigenome, wherein H3K27ac and H3K9ac affect disease pathways by dysregulating transcription and chromatin–gene feedback loops. | [28] |
| Xicota et al. (2019) | Blood and plasma samples from 48 individuals amyloid positive and 48 amyloid negative (enrolled at the Pitié-Salpêtrière University Hospital, Paris, France). | Transcriptomics (RNA-sequencing), metabolomics and lipidomics using liquid chromatography-mass spectrometry | This study suggests a potential blood omics signature for the prediction of amyloid positivity in asymptomatic at-risk subjects. | [27] | |
| Clark et al. (2020) | Cerebrospinal fluid of 120 individuals, aged 55 or older, including subjects with normal cognition, mild cognitive impairment (MCI) or mild AD dementia were enrolled at the University Hospital of Lausanne, Switzerland. | Genetics, proteomics, metabolomics, lipidomics, one-carbon metabolism and neuroinflammation markers | Multi-omics integration identified five major dimensions of heterogenicity, explaining the variance within the cohort and differentially associated with AD. The analysis also identified combinations of a group of molecules that significantly improved the prediction of both AD and cognitive decline. | [26] | |
| Ma et al. (2019) | 10,441 unrelated non-Hispanic white individuals (5522 with AD, 4919 cognitively normal controls) in the Alzheimer’s Disease Sequencing Project case-control WES data set. | Genomics (whole-exome sequencing), genome-wide association analyses | This study highlighting the possibility to stratify AD patients based on their APOE genotype. In fact, the APOE ε4 allele shows a dose-dependent relationship with increased risk for late-onset and sporadic cases of AD, while the inheritance of the ∊2 allele is protective. | [32] | |
| Dagan et al. (2020) | 951 brain samples, obtained from up to 17 brain regions of 85 AD patients with varying severities of AD neuropathology and 22 non-demented subjects. All subjects ranged from 60 to 100 years of age. | Transcriptomics (Expression array) | The authors identified different altered transcriptional signatures characterized AD samples vs non-demented samples and specific transcriptional signatures associated with different subsets of AD patients, demonstrating the high molecular variability and complexity of gene expression in AD. | [33] | |
| Milind et al. (2020) | Post-mortem brain from 2114 human samples from three cohorts of patients with late-onset AD (including 312 North American Caucasian patients and 987 individuals from across the United States). | Genomics (whole-genome sequencing), transcriptomics (RNA-Sequencing) | The authors identified different molecular subtypes of late-onset AD patients associated with specific biological pathways and molecular processes. | [34] | |
| Neff et al. (2021) | 1543 transcriptomes across five brain regions in two AD cohorts (the Mount Sinai/JJ Peters VA Medical Center Brain Bank (MSBB-AD) and the Religious Orders Study–Memory and Aging Project). | Transcriptomics (RNA-Sequencing) | The authors identified three major molecular subtypes of AD corresponding to different combinations of multiple dysregulated pathways and subtype-specific drivers. | [35] | |
| Iqbal et al. (2005) | CSF samples of 468 clinically diagnosed Finnish and Swedish Alzheimer’s disease patients (N = 353) or non-Alzheimer’s subjects (N = 115) (mean age = 70) | Proteomics | The authors identified five AD subgroups based on CSF levels of Aβ1-42, tau, and ubiquitin; each subgroup presented a different clinical profile. | [36] | |
| Toschi et al. (2019) | CSF samples from 113 participants (20 healthy controls, 36 subjective memory complainers, 20 mild cognitive impairment, and 37 AD dementia). The multicenter cross-sectional study includes subjects from France, Germany and Sweden. All subjects ranged from 60 to 77 years of age. | Proteomics | The authors found a set of biologically defined clusters not significantly linked to the clinical diagnosis but exclusively based on core biological fluid markers which reflect distinct pathomechanistic alterations associated with the disease (i.e., brain Ab accumulation and neurofibrillary pathology, neuro-inflammation, axonal damage, and neurodegeneration). | [37] | |
| PD | Lerche et al. (2021) | CSF samples from 516 PD patients (102 PDGBA, 414 PDGBA_wildtype). The multicenter cross-sectional study includes subjects from United States, Europe, Israel, and Australia. | Genetics, proteomics, metabolomics | The authors demonstrated that variants in the glucocerebrosidase gene (GBA) may allow patient stratification for clinical trials merely based on mutation status and that might serve as a biochemical read-out for target engagement. | [38] |
| Prasuhn et al. (2019) | The study is ongoing. So far, >950 PD patients have been included. | Genomics, genome-wide association study | This study focuses on genetically stratified subgroups of Parkinson’s disease patients (PD) with enrichment of risk variants in mitochondrial genes, assuming that individuals with a “higher mitochondrial burden” will likely respond to coenzyme Q10. | [39] | |
| Carling et al. (2020) | Skin fibroblasts of 100 sporadic PD patients (sPD) and 50 age-matched controls (age in years ± standard deviation (SD): sPD patients 61 ± 10.7 years; controls, 61 ± 13.1 years) from the Oxford Parkinson’s Disease Centre Discovery cohort and Sheffield Teaching Hospitals in UK. | Transcriptomics (RNA-sequencing), genomics, proteomics. | The authors identified distinct subgroups with mitochondrial (mito-sPD) or lysosomal (lyso-sPD) dysfunctions, sustaining the utility of using skin fibroblasts to undertake mechanistically rather than clinically defined sPD subgroups. | [40] | |
| Hipp et al. (2018) | The study is ongoing. So far, 498 patients and 520 healthy control have been included. The study includes all patients with parkinsonism in Luxembourg and the surrounding ‘Greater Region’ (including the German, French, and Belgian border regions). | Genomics, genotyping, transcriptomics, metabolomics/proteomics | The authors envision the Luxembourg Parkinson’s study as an important research platform for defining early diagnosis and progression markers that translate into stratified treatment approaches. The study is ongoing. | [41] | |
| Kia et al. (2021) | GWAS: 26,035 PD patients and 403,190 controls of European ancestry; eQTL Data: 134 control individuals (frontal cortex, temporal cortex, occipital cortex, hippocampus, thalamus, putamen, substantia nigra, medulla, cerebellum, and white matter); genome-wide methylation: substantia nigra and the frontal cortex of 134 individuals with PD from the Parkinson Disease UK Brain Bank. | Genome-wide association study, genomics, transcriptomics, epigenomics | The authors identified candidate genes whose change in expression, splicing or methylation are associated with the risk of PD. Interaction network analyses also highlighted the functional pathways and cell types in which these candidate genes have an important role. | [42] | |
| ALS | Aronica et al. (2015); Morello et al. (2019) | Post-mortem motor cortex from caucasian SALS patients (31, mean patient age = 57)) and control individuals (10, mean patient age = 55 years). | Transcriptomics (gene expression array), genomics | The authors demonstrated the utility of an integrative multi-omics molecular classification of ALS, by stratifying the genomes and transcriptomes of SALS postmortem cortex samples into two distinct molecular subtypes (sALS1 and sALS2) characterized by different combinations of genes and pathways. | [43,44] |
| Tam et al. (2019) | Frontal cortex samples from 77 ALS patients and 18 neurological and non-neurological controls from the NYGC ALS Consortium. | Transcriptomics (RNA-sequencing), genomics, proteomics | Unbiased machine learning algorithms identified three distinct ALS patient molecular subtypes representing both ALS disease-implicated signatures as well as additional correlated pathways. | [45] | |
| Wuolikainen et al. (2012) | Cerebrospinal fluid (CSF) from 16 ALS patients with 6 different mutations in the SOD1 gene compared with ALS-patients without mutations | Metabolomics (GC-TOFMS platform) | The authors found that patients with SOD1 mutations have a distinct metabolic profile in CSF and highlight the utility of metabolomics signature to distinguish ALS entity | [46] | |
| Chen et al. (2018) | 77 ALS -derived dermal fibroblast lines and 45 age/sex-matched controls. | Metabolomics (LC-QTOF platform) | The authors emphasize that sporadic ALS patients can be stratified into metabotypes, helping future development of personalized medicine. | [47] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
La Cognata, V.; Morello, G.; Cavallaro, S. Omics Data and Their Integrative Analysis to Support Stratified Medicine in Neurodegenerative Diseases. Int. J. Mol. Sci. 2021, 22, 4820. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22094820
La Cognata V, Morello G, Cavallaro S. Omics Data and Their Integrative Analysis to Support Stratified Medicine in Neurodegenerative Diseases. International Journal of Molecular Sciences. 2021; 22(9):4820. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22094820
Chicago/Turabian StyleLa Cognata, Valentina, Giovanna Morello, and Sebastiano Cavallaro. 2021. "Omics Data and Their Integrative Analysis to Support Stratified Medicine in Neurodegenerative Diseases" International Journal of Molecular Sciences 22, no. 9: 4820. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22094820