
Distinct Fatty Acid Compositions of HDL Phospholipids Are Characteristic of Metabolic Syndrome and Premature Coronary Heart Disease—Family Study

 and
and
Abstract
:1. Introduction
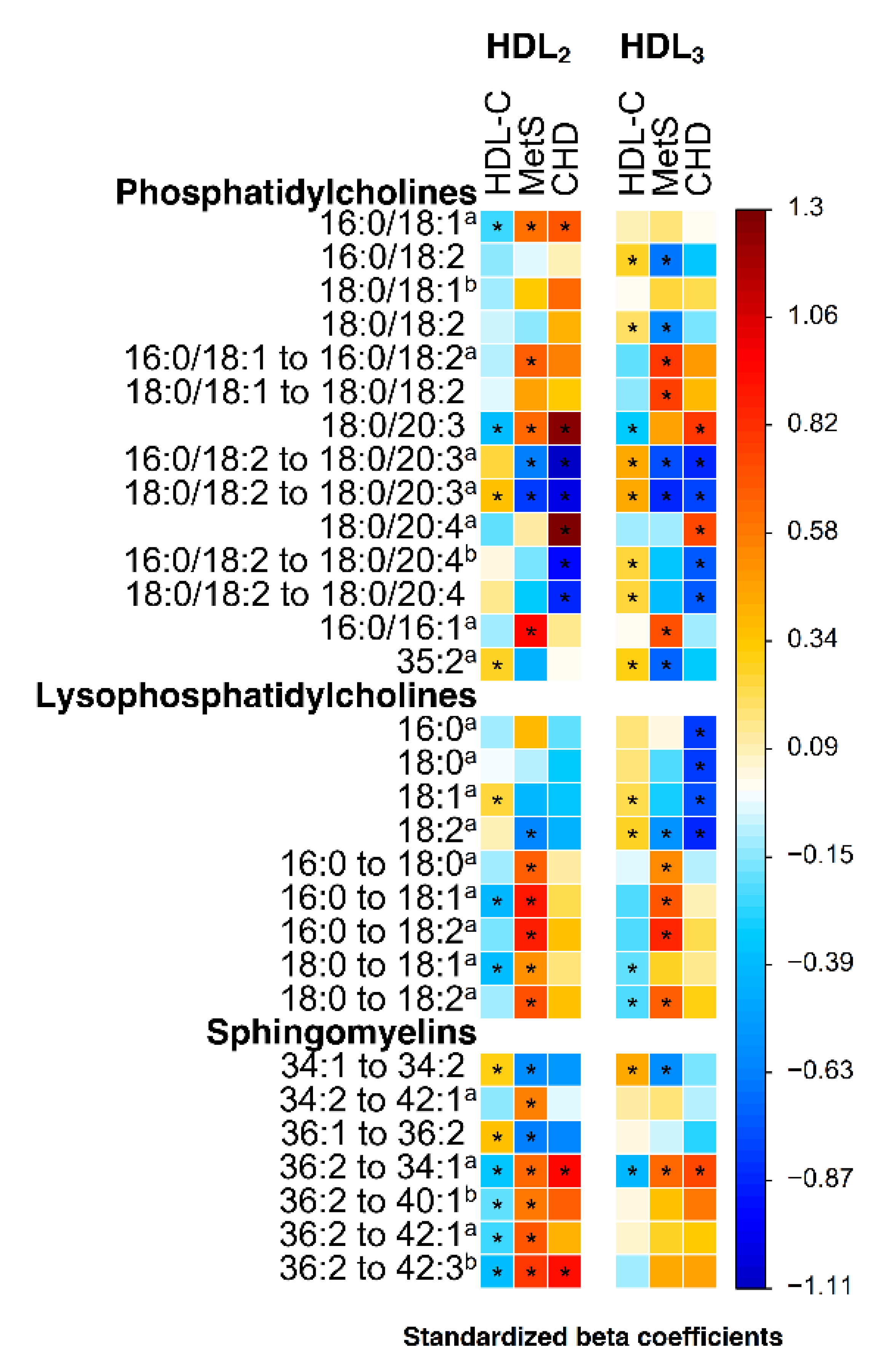
2. Results
2.1. Study Population
2.2. Lipidomic Associations of HDL Fractions with Metabolic Syndrome, HDL-C and Premature CHD
2.3. Fatty Acid Composition of Lysophosphatidylcholine and Phosphatidylcholine Species in Metabolic Syndrome
2.4. Phosphatidylcholines PC (18:0/20:3) and PC (18:0/20:4) in Metabolic Syndrome and Premature CHD
2.5. Sphingomyelins in Metabolic Syndrome and Premature CHD
2.6. Lipidomic Associations of Premature CHD Adjusted for Metabolic Syndrome Status, HDL-C, Other Parameters of Metabolic Syndrome or Cardiovascular Risk Factors
2.7. Phospholipids and Serum Free Fatty Acids
3. Discussion
4. Study Subjects, Materials and Methods
4.1. Study Subjects
4.2. Materials and Methods
4.2.1. Clinical Measurements
4.2.2. Blood Samples
4.2.3. Clinical Chemistry Measurements
4.2.4. Adiponectin Measurements
4.2.5. Isolation and Analysis of Chemical Composition of Lipoprotein Fractions
4.2.6. Lipidomic Analysis
4.2.7. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Gordon, T.; Castelli, W.P.; Hjortland, M.C.; Kannel, W.B.; Dawber, T.R. High Density Lipoprotein as a Protective Factor Against Coronary Heart Disease. the Framingham Study. Am. J. Med. 1977, 62, 707–714. [Google Scholar] [CrossRef]
- Haase, C.L.; Tybjaerg-Hansen, A.; Qayyum, A.A.; Schou, J.; Nordestgaard, B.G.; Frikke-Schmidt, R. LCAT, HDL Cholesterol and Ischemic Cardiovascular Disease: A Mendelian Randomization Study of HDL Cholesterol in 54,500 Individuals. J. Clin. Endocrinol. Metab. 2012, 97, 248. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Voight, B.F.; Peloso, G.M.; Orho-Melander, M.; Frikke-Schmidt, R.; Barbalic, M.; Jensen, M.K.; Hindy, G.; Holm, H.; Ding, E.L.; Johnson, T.; et al. Plasma HDL Cholesterol and Risk of Myocardial Infarction: A Mendelian Randomisation Study. Lancet 2012, 380, 572–580. [Google Scholar] [CrossRef] [Green Version]
- Holmes, M.V.; Asselbergs, F.W.; Palmer, T.M.; Drenos, F.; Lanktree, M.B.; Nelson, C.P.; Dale, C.E.; Padmanabhan, S.; Finan, C.; Swerdlow, D.I.; et al. Mendelian Randomization of Blood Lipids for Coronary Heart Disease. Eur. Heart J. 2015, 36, 539–550. [Google Scholar] [CrossRef] [Green Version]
- Kontush, A.; Lindahl, M.; Lhomme, M.; Calabresi, L.; Chapman, M.J.; Davidson, W.S. Structure of HDL: Particle Subclasses and Molecular Components. In Handbook of Experimental Pharmacology; von Eckardstein, A., Kardassis, D., Eds.; Springer: Cham, Switzerland, 2015; Volume 224, pp. 3–51. ISBN 978-3-319-09665-0. [Google Scholar]
- Rader, D.J.; Hovingh, G.K. HDL and Cardiovascular Disease. Lancet 2014, 384, 618–625. [Google Scholar] [CrossRef]
- Kontush, A.; Lhomme, M.; Chapman, M.J. Unraveling the Complexities of the HDL Lipidome. J. Lipid Res. 2013, 54, 2950–2963. [Google Scholar] [CrossRef] [Green Version]
- Yetukuri, L.; Söderlund, S.; Koivuniemi, A.; Seppänen-Laakso, T.; Niemelä, P.S.; Hyvönen, M.; Taskinen, M.R.; Vattulainen, I.; Jauhiainen, M.; Oresic, M. Composition and Lipid Spatial Distribution of HDL Particles in Subjects with Low and High HDL-Cholesterol. J. Lipid Res. 2010, 51, 2341–2351. [Google Scholar] [CrossRef] [Green Version]
- Laurila, P.P.; Surakka, I.; Sarin, A.P.; Yetukuri, L.; Hyötyläinen, T.; Söderlund, S.; Naukkarinen, J.; Tang, J.; Kettunen, J.; Mirel, D.B.; et al. Genomic, Transcriptomic, and Lipidomic Profiling Highlights the Role of Inflammation in Individuals with Low High-Density Lipoprotein Cholesterol. Arterioscler. Thromb. Vasc. Biol. 2013, 33, 847–857. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sutter, I.; Velagapudi, S.; Othman, A.; Riwanto, M.; Manz, J.; Rohrer, L.; Rentsch, K.; Hornemann, T.; Landmesser, U.; von Eckardstein, A. Plasmalogens of High-Density Lipoproteins (HDL) are Associated with Coronary Artery Disease and Anti-Apoptotic Activity of HDL. Atherosclerosis 2015, 241, 539–546. [Google Scholar] [CrossRef]
- Ståhlman, M.; Fagerberg, B.; Adiels, M.; Ekroos, K.; Chapman, J.M.; Kontush, A.; Boren, J. Dyslipidemia, but Not Hyperglycemia and Insulin Resistance, is Associated with Marked Alterations in the HDL Lipidome in Type 2 Diabetic Subjects in the DIWA Cohort: Impact on Small HDL Particles. Biochim. Biophys. Acta 2013, 1831, 1609–1617. [Google Scholar] [CrossRef]
- Denimal, D.; Nguyen, A.; Pais de Barros, J.P.; Bouillet, B.; Petit, J.M.; Verges, B.; Duvillard, L. Major Changes in the Sphingophospholipidome of HDL in Non-Diabetic Patients with Metabolic Syndrome. Atherosclerosis 2016, 246, 106–114. [Google Scholar] [CrossRef] [PubMed]
- Cardner, M.; Yalcinkaya, M.; Goetze, S.; Luca, E.; Balaz, M.; Hunjadi, M.; Hartung, J.; Shemet, A.; Krankel, N.; Radosavljevic, S.; et al. Structure-Function Relationships of HDL in Diabetes and Coronary Heart Disease. JCI Insight 2020, 5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jones, P.H.; Davidson, M.H.; Stein, E.A.; Bays, H.E.; McKenney, J.M.; Miller, E.; Cain, V.A.; Blasetto, J.W.; STELLAR Study Group. Comparison of the Efficacy and Safety of Rosuvastatin Versus Atorvastatin, Simvastatin, and Pravastatin Across Doses (STELLAR* Trial). Am. J. Cardiol. 2003, 92, 152–160. [Google Scholar] [CrossRef]
- Takeda, H.; Izumi, Y.; Tamura, S.; Koike, T.; Koike, Y.; Shiomi, M.; Bamba, T. Lipid Profiling of Serum and Lipoprotein Fractions in Response to Pitavastatin using an Animal Model of Familial Hypercholesterolemia. J. Proteome Res. 2020, 19, 1100–1108. [Google Scholar] [CrossRef] [PubMed]
- Albert, M.A.; Glynn, R.J.; Wolfert, R.L.; Ridker, P.M. The Effect of Statin Therapy on Lipoprotein Associated Phospholipase A2 Levels. Atherosclerosis 2005, 182, 193–198. [Google Scholar] [CrossRef]
- Orsoni, A.; Therond, P.; Tan, R.; Giral, P.; Robillard, P.; Kontush, A.; Meikle, P.J.; Chapman, M.J. Statin Action Enriches HDL3 in Polyunsaturated Phospholipids and Plasmalogens and Reduces LDL-Derived Phospholipid Hydroperoxides in Atherogenic Mixed Dyslipidemia. J. Lipid Res. 2016, 57, 2073–2087. [Google Scholar] [CrossRef] [Green Version]
- I. S. Sobczak, A.; A. Blindauer, C.; J. Stewart, A. Changes in Plasma Free Fatty Acids Associated with Type-2 Diabetes. Nutrients 2019, 11, 2022. [Google Scholar] [CrossRef] [Green Version]
- Papathanasiou, A.; Kostara, C.; Cung, M.T.; Seferiadis, K.; Elisaf, M.; Bairaktari, E.; Goudevenos, I.A. Analysis of the Composition of Plasma Lipoproteins in Patients with Extensive Coronary Heart Disease using 1H NMR Spectroscopy. Hellenic J. Cardiol. 2008, 49, 72–78. [Google Scholar]
- Hopkins, P.N. Molecular Biology of Atherosclerosis. Physiol. Rev. 2013, 93, 1317–1542. [Google Scholar] [CrossRef]
- Csordas, A.; Wick, G.; Laufer, G.; Bernhard, D. An Evaluation of the Clinical Evidence on the Role of Inflammation and Oxidative Stress in Smoking-Mediated Cardiovascular Disease. Biomark Insights 2008, 3, 127–139. [Google Scholar] [CrossRef]
- Zhu, C.; Sawrey-Kubicek, L.; Beals, E.; Hughes, R.L.; Rhodes, C.H.; Sacchi, R.; Zivkovic, A.M. The HDL Lipidome is Widely Remodeled by Fast Food Versus Mediterranean Diet in 4 Days. Metabolomics 2019, 15, 114. [Google Scholar] [CrossRef] [Green Version]
- Le Barz, M.; Boulet, M.M.; Calzada, C.; Cheillan, D.; Michalski, M.C. Alterations of Endogenous Sphingolipid Metabolism in Cardiometabolic Diseases: Towards Novel Therapeutic Approaches. Biochimie 2020, 169, 133–143. [Google Scholar] [CrossRef] [PubMed]
- Pruzanski, W.; Stefanski, E.; de Beer, F.C.; de Beer, M.C.; Ravandi, A.; Kuksis, A. Comparative Analysis of Lipid Composition of Normal and Acute-Phase High Density Lipoproteins. J. Lipid Res. 2000, 41, 1035–1047. [Google Scholar] [CrossRef]
- Saltiel, A.R.; Olefsky, J.M. Inflammatory Mechanisms Linking Obesity and Metabolic Disease. J. Clin. Invest. 2017, 127, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Kulkarni, H.; Meikle, P.J.; Mamtani, M.; Weir, J.M.; Barlow, C.K.; Jowett, J.B.; Bellis, C.; Dyer, T.D.; Johnson, M.P.; Rainwater, D.L.; et al. Variability in Associations of Phosphatidylcholine Molecular Species with Metabolic Syndrome in Mexican-American Families. Lipids 2013, 48, 497–503. [Google Scholar] [CrossRef] [Green Version]
- Meikle, P.J.; Wong, G.; Barlow, C.K.; Weir, J.M.; Greeve, M.A.; MacIntosh, G.L.; Almasy, L.; Comuzzie, A.G.; Mahaney, M.C.; Kowalczyk, A.; et al. Plasma Lipid Profiling shows Similar Associations with Prediabetes and Type 2 Diabetes. PLoS ONE 2013, 8, e74341. [Google Scholar] [CrossRef] [Green Version]
- Schwab, U.; Seppänen-Laakso, T.; Yetukuri, L.; Agren, J.; Kolehmainen, M.; Laaksonen, D.E.; Ruskeepää, A.L.; Gylling, H.; Uusitupa, M.; Oresic, M.; et al. Triacylglycerol Fatty Acid Composition in Diet-Induced Weight Loss in Subjects with Abnormal Glucose Metabolism--the GENOBIN Study. PLoS ONE 2008, 3, e2630. [Google Scholar] [CrossRef] [PubMed]
- Meikle, P.J.; Wong, G.; Tsorotes, D.; Barlow, C.K.; Weir, J.M.; Christopher, M.J.; MacIntosh, G.L.; Goudey, B.; Stern, L.; Kowalczyk, A.; et al. Plasma Lipidomic Analysis of Stable and Unstable Coronary Artery Disease. Arterioscler. Thromb. Vasc. Biol. 2011, 31, 2723–2732. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sutter, I.; Klingenberg, R.; Othman, A.; Rohrer, L.; Landmesser, U.; Heg, D.; Rodondi, N.; Mach, F.; Windecker, S.; Matter, C.M.; et al. Decreased Phosphatidylcholine Plasmalogens--A Putative Novel Lipid Signature in Patients with Stable Coronary Artery Disease and Acute Myocardial Infarction. Atherosclerosis 2016, 246, 130–140. [Google Scholar] [CrossRef]
- Pietiläinen, K.H.; Rog, T.; Seppänen-Laakso, T.; Virtue, S.; Gopalacharyulu, P.; Tang, J.; Rodriguez-Cuenca, S.; Maciejewski, A.; Naukkarinen, J.; Ruskeepää, A.L.; et al. Association of Lipidome Remodeling in the Adipocyte Membrane with Acquired Obesity in Humans. PLoS Biol. 2011, 9, e1000623. [Google Scholar] [CrossRef] [Green Version]
- Fernandez, C.; Sandin, M.; Sampaio, J.L.; Almgren, P.; Narkiewicz, K.; Hoffmann, M.; Hedner, T.; Wahlstrand, B.; Simons, K.; Shevchenko, A.; et al. Plasma Lipid Composition and Risk of Developing Cardiovascular Disease. PLoS ONE 2013, 8, e71846. [Google Scholar] [CrossRef] [Green Version]
- Diaz, S.O.; Sanchez-Quesada, J.L.; de Freitas, V.; Leite-Moreira, A.; Barros, A.S.; Reis, A. Exploratory Analysis of Large-Scale Lipidome in Large Cohorts: Are we any Closer of Finding Lipid-Based Markers Suitable for CVD Risk Stratification and Management? Anal. Chim. Acta 2021, 1142, 189–200. [Google Scholar] [CrossRef] [PubMed]
- Zannis, V.I.; Fotakis, P.; Koukos, G.; Kardassis, D.; Ehnholm, C.; Jauhiainen, M.; Chroni, A. HDL biogenesis, remodeling, and catabolism. In Handbook of Experimental Pharmacology; von Eckardstein, A., Kardassis, D., Eds.; Springer: Cham, Switzerland, 2015; Volume 224, pp. 53–111. ISBN 978-3-319-09665-0. [Google Scholar]
- Warensjö, E.; Riserus, U.; Vessby, B. Fatty Acid Composition of Serum Lipids Predicts the Development of the Metabolic Syndrome in Men. Diabetologia 2005, 48, 1999–2005. [Google Scholar] [CrossRef] [Green Version]
- Perreault, M.; Roke, K.; Badawi, A.; Nielsen, D.E.; Abdelmagid, S.A.; El-Sohemy, A.; Ma, D.W.; Mutch, D.M. Plasma Levels of 14:0, 16:0, 16:1n-7, and 20:3n-6 are Positively Associated, but 18:0 and 18:2n-6 are Inversely Associated with Markers of Inflammation in Young Healthy Adults. Lipids 2014, 49, 255–263. [Google Scholar] [CrossRef]
- Bellis, C.; Kulkarni, H.; Mamtani, M.; Kent, J.W., Jr.; Wong, G.; Weir, J.M.; Barlow, C.K.; Diego, V.; Almeida, M.; Dyer, T.D.; et al. Human Plasma Lipidome is Pleiotropically Associated with Cardiovascular Risk Factors and Death. Circ. Cardiovasc. Genet. 2014, 7, 854–863. [Google Scholar] [CrossRef]
- Davidson, W.S.; Gillotte, K.L.; Lund-Katz, S.; Johnson, W.J.; Rothblat, G.H.; Phillips, M.C. The Effect of High Density Lipoprotein Phospholipid Acyl Chain Composition on the Efflux of Cellular Free Cholesterol. J. Biol. Chem. 1995, 270, 5882–5890. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rye, K.A.; Hime, N.J.; Barter, P.J. The Influence of Sphingomyelin on the Structure and Function of Reconstituted High Density Lipoproteins. J. Biol. Chem. 1996, 271, 4243–4250. [Google Scholar] [CrossRef] [Green Version]
- Zerrad-Saadi, A.; Therond, P.; Chantepie, S.; Couturier, M.; Rye, K.A.; Chapman, M.J.; Kontush, A. HDL3-Mediated Inactivation of LDL-Associated Phospholipid Hydroperoxides is Determined by the Redox Status of Apolipoprotein A-I and HDL Particle Surface Lipid Rigidity: Relevance to Inflammation and Atherogenesis. Arterioscler. Thromb. Vasc. Biol. 2009, 29, 2169–2175. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yancey, P.G.; de la Llera-Moya, M.; Swarnakar, S.; Monzo, P.; Klein, S.M.; Connelly, M.A.; Johnson, W.J.; Williams, D.L.; Rothblat, G.H. High Density Lipoprotein Phospholipid Composition is a Major Determinant of the Bi-Directional Flux and Net Movement of Cellular Free Cholesterol Mediated by Scavenger Receptor BI. J. Biol. Chem. 2000, 275, 36596–36604. [Google Scholar] [CrossRef] [Green Version]
- Marmillot, P.; Patel, S.; Lakshman, M.R. Reverse Cholesterol Transport is Regulated by Varying Fatty Acyl Chain Saturation and Sphingomyelin Content in Reconstituted High-Density Lipoproteins. Metabolism 2007, 56, 251–259. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baker, P.W.; Rye, K.A.; Gamble, J.R.; Vadas, M.A.; Barter, P.J. Phospholipid Composition of Reconstituted High Density Lipoproteins Influences their Ability to Inhibit Endothelial Cell Adhesion Molecule Expression. J. Lipid Res. 2000, 41, 1261–1267. [Google Scholar] [CrossRef]
- Stadler, J.T.; Lackner, S.; Morkl, S.; Trakaki, A.; Scharnagl, H.; Borenich, A.; Wonisch, W.; Mangge, H.; Zelzer, S.; Meier-Allard, N.; et al. Obesity Affects HDL Metabolism, Composition and Subclass Distribution. Biomedicines 2021, 9, 242. [Google Scholar] [CrossRef]
- Wonisch, W.; Falk, A.; Sundl, I.; Winklhofer-Roob, B.M.; Lindschinger, M. Oxidative Stress Increases Continuously with BMI and Age with Unfavourable Profiles in Males. Aging Male 2012, 15, 159–165. [Google Scholar] [CrossRef]
- Herrington, W.; Lacey, B.; Sherliker, P.; Armitage, J.; Lewington, S. Epidemiology of Atherosclerosis and the Potential to Reduce the Global Burden of Atherothrombotic Disease. Circ. Res. 2016, 118, 535–546. [Google Scholar] [CrossRef]
- Paavola, T.; Kuusisto, S.; Jauhiainen, M.; Kakko, S.; Kangas-Kontio, T.; Metso, J.; Soininen, P.; Ala-Korpela, M.; Bloigu, R.; Hannuksela, M.L.; et al. Impaired HDL2-Mediated Cholesterol Efflux is Associated with Metabolic Syndrome in Families with Early Onset Coronary Heart Disease and Low HDL-Cholesterol Level. PLoS ONE 2017, 12, e0171993. [Google Scholar] [CrossRef]
- Alberti, K.G.; Zimmet, P.; Shaw, J.; IDF Epidemiology Task Force Consensus Group. The Metabolic Syndrome-A New Worldwide Definition. Lancet 2005, 366, 1059–1062. [Google Scholar] [CrossRef]
- Bonora, E.; Targher, G.; Alberiche, M.; Bonadonna, R.C.; Saggiani, F.; Zenere, M.B.; Monauni, T.; Muggeo, M. Homeostasis Model Assessment Closely Mirrors the Glucose Clamp Technique in the Assessment of Insulin Sensitivity: Studies in Subjects with various Degrees of Glucose Tolerance and Insulin Sensitivity. Diabetes Care 2000, 23, 57–63. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Havel, R.J.; Eder, H.A.; Bragdon, J.H. The Distribution and Chemical Composition of Ultracentrifugally Separated Lipoproteins in Human Serum. J. Clin. Invest. 1955, 34, 1345–1353. [Google Scholar] [CrossRef] [Green Version]
- Liinamaa, M.J.; Hannuksela, M.L.; Kesaniemi, Y.A.; Savolainen, M.J. Altered Transfer of Cholesteryl Esters and Phospholipids in Plasma from Alcohol Abusers. Arterioscler. Thromb. Vasc. Biol. 1997, 17, 2940–2947. [Google Scholar] [CrossRef] [PubMed]
- Niemi, J.; Makinen, V.P.; Heikkonen, J.; Tenkanen, L.; Hiltunen, Y.; Hannuksela, M.L.; Jauhiainen, M.; Forsblom, C.; Taskinen, M.R.; Kesaniemi, Y.A.; et al. Estimation of VLDL, IDL, LDL, HDL2, apoA-I, and apoB from the Friedewald Inputs--apoB and IDL, but Not LDL, are Associated with Mortality in Type 1 Diabetes. Ann. Med. 2009, 41, 451–461. [Google Scholar] [CrossRef] [PubMed]
- Davis, C.S. Statistical Methods for the Analysis of Repeated Measurements, 1st ed.; Springer: New York, NY, USA, 2002; ISBN 978-0-387-21573-0. [Google Scholar]
- Liang, H.Q.; Rye, K.A.; Barter, P.J. Dissociation of Lipid-Free Apolipoprotein A-I from High Density Lipoproteins. J. Lipid Res. 1994, 35, 1187–1199. [Google Scholar] [CrossRef]


{kind=link}
| Characteristics | All Subjects | Men | Without Statin Medication or CHD | ||||
|---|---|---|---|---|---|---|---|
| All | MetS | No MetS | Premature CHD 1 | No CHD 1 | MetS | No MetS | |
| N | 112 | 58 | 54 | 30 | 20 | 26 | 40 |
| Age (y) | 51 (40–58) | 55 (47–60) 4 | 45 (32–53) | 52 (49–57) | 52 (44–59) | 50 (40–60) 4 | 42 (30–51) |
| Women (n) | 54 (48) | 25 (43) | 29 (54) | 0 (0) | 0 (0) | 16 (62) | 25 (63) |
| CHD (n) | 39 (35) | 29 (50) 4 | 10 (19) | 30 (100) | 0 (0) | 0 (0) | 0 (0) |
| Premature CHD (n) | 35 (31) | 26 (45) 4 | 9 (17) | 30 (100) | 0 (0) | 0 | 0 |
| Age at CHD onset (y) | 50 (43–55) | 51 (44–55) | 47 (41–56) | 47 (42–51) | NA | NA | NA |
| MetS (n) | 58 (52) | 58 (100) | 0 (0) | 22 (73) | 10 (50) | 26 | 40 |
| Diabetes (n) | 10 (9) | 7 (12) | 3 (6) | 2 (7) | 1 (5) | 1 (4) | 1 (3) |
| Hypertension (n) | 38 (34) | 31 (53) 4 | 7 (13) | 14 (47) 4 | 1 (5) | 10 (39) 4 | 2 (5) |
| BMI (kg/m2) | 27.9 (24.9–30.5) | 29.6 (27.8–31.6) 4 | 25.1 (22.2–27.8) | 29.5 (27.5–31.2) 4 | 27.3 (25.1–28.4) | 28.6 (26.2–30.6) 4 | 25.1 (21.3–27.9) |
| Waist (cm) | 94 (85–102) | 101 (95–106) 4 | 88 (80–92) | 102 (97–107) 4 | 95 (88–99) | 95 (88–100) 4 | 86 (77–92) |
| Syst. BP (mm Hg) | 128 (115–141) | 132 (121–142) 4 | 122 (113–135) | 130 (115–140) | 130 (120–141) | 131 (119–142) | 121 (113–134) |
| ACE/ATII (n) | 36 (32) | 27 (47) 4 | 9 (17) | 17 (57) 4 | 1 (5) | 6 (23) | 2 (5) |
| Statin (n) | 42 (38) | 29 (50) 4 | 13 (24) | 27 (90) 4 | 1 (5) | 0 (0) | 0 (0) |
| Smoker (n) | 29 (26) | 17 (29) | 12 (22) | 10 (33) | 5 (25) | 7 (27) | 10 (25) |
| Ex-smoker (n) | 32 (29) | 18 (31) | 14 (26) | 16 (53) | 7 (35) | 6 (23) | 7 (18) |
| Pack-years 2 | 1 (0–15) | 10 (0–26) 4 | 0 (0–10) | 23 (9–30) 4 | 10 (0–15) | 0 (0–12) | 0 (0–6) |
| Alcohol (doses 3/week) | 2 (1–6) | 2 (0–4) | 2 (1–7) | 3 (0–6) 4 | 5 (2–13) | 3 (1–9) | 2 (1–7) |
| Total C (mmol/L) | 4.50 (3.93–5.20) | 4.50 (3.90–5.38) | 4.60 (4.00–5.05) | 3.95 (3.48–4.40) 4 | 5.20 (4.93–5.98) | 5.40 (4.43–6.00) | 4.90 (4.30–5.30) |
| HDL-C (mmol/L) | 1.28 (1.08–1.54) | 1.14 (0.98–1.37) 4 | 1.41 (1.23–1.77) | 1.07 (0.85–1.17) 4 | 1.28 (1.08–1.67) | 1.26 (1.05–1.55) 4 | 1.51 (1.26–1.81) |
| LDL-C (mmol/L) | 2.70 (2.30–3.30) | 2.70 (2.30–3.48) | 2.75 (2.28–3.23) | 2.40 (1.95–2.90) 4 | 3.30 (2.90–4.00) | 3.35 (2.68–4.13) | 2.90 (2.53–3.50) |
| TG (mmol/L) | 1.12 (0.84–1.73) | 1.48 (1.10–2.13) 4 | 0.90 (0.64–1.11) | 1.37 (0.97–2.24) | 1.38 (0.63–2.14) | 1.71 (1.06–2.13) 4 | 0.89 (0.63–1.08) |
| FFA (mmol/L) | 0.51 (0.37–0.65) | 0.49 (0.41–0.63) | 0.53 (0.36–0.69) | 0.41 (0.32–0.54) | 0.46 (0.35–0.57) | 0.55 (0.44–0.69) | 0.53 (0.37–0.68) |
| Glucose (mmol/L) | 5.8 (5.4–6.5) | 6.2 (5.7–6.6) 4 | 5.4 (5.1–5.9) | 6.3 (5.7–6.7) | 5.8 (5.5–6.4) | 6.0 (5.7–6.4) 4 | 5.4 (5.1–5.7) |
| HOMA-IR | 2.1 (1.3–3.6) | 2.7 (2.0–4.5) 4 | 1.4 (1.1–2.3) | 3.2 (2.0–4.5) 4 | 1.5 (1.1–2.0) | 2.0 (1.5–2.8) 4 | 1.3 (1.0–2.1) |
| Total adip. (mg/L) | 7.6 (5.3–10.6) | 6.2 (4.5–8.9) 4 | 8.7 (6.9–12.8) | 5.3 (3.9–7.3) 4 | 6.8 (4.9–10.2) | 7.4 (5.2–12.2) | 8.2 (6.8–12.6) |
| HMW adip. (mg/L) | 2.4 (1.3–4.6) | 1.7 (1.1–3.2) 4 | 3.1 (1.9–5.7) | 1.4 (1.1–2.6) | 1.9 (1.1–4.0) | 2.2 (1.2–4.4) | 3.0 (1.9–5.5) |
| ALT (U/L) | 25 (17–33) | 29 (21–38) 4 | 22 (16–29) | 31 (25–41) | 27 (20–34) | 28 (19–34) 4 | 19 (14–27) |
| Creatinine (µmol/L) | 66 (59–72) | 66 (58–72) | 66 (61–71) | 69 (61–74) | 72 (66–74) | 63 (56–71) | 64 (59–70) |
| Adjusted for | Phosphatidylcholines | Sphingomyelins | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| 16:0/18:2 to 18:0/20:3 | 18:0/18:2 to 18:0/20:3 | 16:0/18:2 to 18:0/20:4 | 36:2 to 34:1 | 36:2 to 42:3 | |||||||
| HDL2 4 | HDL3 4 | HDL2 4 | HDL3 4 | HDL2 5 | HDL3 5 | HDL2 4 | HDL3 4 | HDL2 5 | HDL3 4 | ||
| MetS | β1 | −1.06 3 | −0.79 2 | −0.91 3 | −0.67 1 | −1.01 3 | −0.80 2 | 0.91 2 | 0.66 1 | 0.74 3 | 0.37 |
| CI | −1.53–−0.59 | −1.30–−0.28 | −1.37–−0.46 | −1.19–−0.15 | −1.52–−0.51 | −1.32–−0.27 | 0.40–1.43 | 0.14–1.17 | 0.34–1.15 | −0.20 –0.94 | |
| β2 | −0.21 | −0.30 | −0.46 | −0.50 | 0.28 | 0.18 | 0.13 | 0.32 | 0.72 3 | 0.40 | |
| CI | −0.73–0.30 | −0.83 –0.23 | −0.96–0.03 | −1.06–0.06 | −0.31–0.88 | −0.36–0.73 | −0.33–0.58 | −0.21–0.84 | 0.33–1.11 | −0.15–0.95 | |
| HDL-C 4 | β1 | −0.83 2 | −0.49 | −0.63 2 | −0.35 | −1.17 3 | −0.70 1 | 0.84 2 | 0.36 | 0.68 1 | 0.38 |
| CI | −1.31–−0.35 | −1.03–−0.04 | −1.06–−0.20 | −0.90–0.20 | −1.77–−0.58 | −1.25–−0.15 | 0.24–1.43 | −0.20–0.91 | 0.11–1.25 | −0.24–1.01 | |
| TG 5 | β1 | −1.05 3 | −0.79 2 | −0.93 3 | −0.68 2 | −1.01 3 | −0.77 2 | 0.90 2 | 0.66 1 | 0.80 3 | 0.42 |
| CI | −1.53–−0.58 | −1.29–−0.29 | −1.39–−0.46 | −1.20–−0.17 | −1.47–−0.54 | −1.27–−0.27 | 0.37–1.42 | 0.15–1.18 | 0.36–1.23 | −0.18–1.01 | |
| VLDL-TG 5 | β1 | −1.02 3 | −0.75 2 | −0.88 3 | −0.63 1 | −1.03 3 | −0.77 2 | 0.88 2 | 0.63 1 | 0.77 2 | 0.40 |
| CI | −1.48–−0.55 | −1.26–−0.24 | −1.34–−0.42 | −1.15–−0.11 | −1.50–−0.56 | −1.27–−0.27 | 0.34–1.41 | 0.11–1.15 | 0.30–1.24 | −0.21–1.01 | |
| VLDL protein 5 | β1 | −1.00 3 | −0.74 2 | −0.86 3 | −0.63 1 | −1.03 3 | −0.75 2 | 0.86 2 | 0.63 1 | 0.77 2 | 0.39 |
| CI | −1.46–−0.55 | −1.22–−0.25 | −1.30–−0.43 | −1.11–−0.14 | −1.51–−0.55 | −1.25–−0.25 | 0.34–1.38 | 0.12–1.13 | 0.32–1.22 | −0.20–0.97 | |
| Adipon. 4 | β1 | −0.96 3 | −0.71 2 | −0.86 2 | −0.60 1 | −1.00 3 | −0.74 2 | 0.90 2 | 0.61 1 | 0.82 2 | 0.44 |
| CI | −1.42–−0.50 | −1.23–−0.19 | −1.35–−0.37 | −1.13–−0.06 | −1.47–−0.53 | −1.24–−0.25 | 0.32–1.48 | 0.08–1.14 | 0.24–1.40 | −0.16–1.03 | |
| HMW adipon. 4 | β1 | −0.97 3 | −0.66 1 | −0.87 3 | −0.62 1 | −0.98 3 | −0.70 2 | 0.86 2 | 0.75 2 | 0.82 2 | 0.48 |
| CI | −1.44–−0.51 | −1.17–−0.15 | −1.35–−0.39 | −1.14–−0.10 | −1.47–−0.48 | −1.22–−0.18 | 0.31–1.42 | 0.24–1.25 | 0.28–1.35 | −0.11–1.06 | |
| Waist 5 | β1 | −0.97 3 | −0.73 2 | −0.83 3 | −0.58 1 | −0.94 3 | −0.77 2 | 0.88 3 | 0.66 1 | 0.59 2 | 0.37 |
| CI | −1.41–−0.52 | −1.22–−0.24 | −1.29–−0.37 | −1.09–−0.07 | −1.46–−0.42 | −1.34–−0.20 | 0.39–1.37 | 0.13–1.18 | 0.17–1.01 | −0.24–0.98 | |
| HOMA-IR 4 | β1 | −0.99 2 | −0.71 1 | −0.85 2 | −0.63 | −1.10 3 | −0.87 2 | 0.92 2 | 0.84 2 | 0.55 1 | 0.44 |
| CI | −1.55–−0.42 | −1.29–−0.12 | −1.40–−0.30 | −1.25–0.00 | −1.64–−0.57 | −1.41–−0.33 | 0.39–1.46 | 0.24–1.45 | 0.04–1.07 | −0.24–1.12 | |
| Smoker | β1 | −1.06 3 | −0.82 2 | −0.99 3 | −0.75 2 | −0.92 3 | −0.72 2 | 0.96 3 | 0.75 2 | 0.91 3 | 0.48 |
| CI | −1.53–−0.59 | −1.30–−0.33 | −1.49–−0.49 | −1.26–−0.23 | −1.37–−0.48 | −1.20–−0.24 | 0.45–1.47 | 0.22–1.29 | 0.42–1.39 | −0.09–1.05 | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Paavola, T.; Bergmann, U.; Kuusisto, S.; Kakko, S.; Savolainen, M.J.; Salonurmi, T. Distinct Fatty Acid Compositions of HDL Phospholipids Are Characteristic of Metabolic Syndrome and Premature Coronary Heart Disease—Family Study. Int. J. Mol. Sci. 2021, 22, 4908. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22094908
Paavola T, Bergmann U, Kuusisto S, Kakko S, Savolainen MJ, Salonurmi T. Distinct Fatty Acid Compositions of HDL Phospholipids Are Characteristic of Metabolic Syndrome and Premature Coronary Heart Disease—Family Study. International Journal of Molecular Sciences. 2021; 22(9):4908. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22094908
Chicago/Turabian StylePaavola, Timo, Ulrich Bergmann, Sanna Kuusisto, Sakari Kakko, Markku J. Savolainen, and Tuire Salonurmi. 2021. "Distinct Fatty Acid Compositions of HDL Phospholipids Are Characteristic of Metabolic Syndrome and Premature Coronary Heart Disease—Family Study" International Journal of Molecular Sciences 22, no. 9: 4908. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22094908