
Epigenetic Targeting of Histone Deacetylases in Diagnostics and Treatment of Depression



1
Department of Biochemistry, Inje University College of Medicine, Busan 47392, Korea
2
Division of Biological Sciences, Sookmyung Women’s University, Seoul 04310, Korea
3
Research Institute for Women’s Health, Sookmyung Women’s University, Seoul 04310, Korea
4
Department of Molecular and Life Science, College of Science and Convergence Technology, Hanyang University ERICA Campus, Ansan 15588, Korea
5
BK21 FOUR KNU Creative BioResearch Group, School of Life Sciences, College of National Sciences, Kyungpook National University, Daegu 41566, Korea
*
Authors to whom correspondence should be addressed.
Int. J. Mol. Sci. 2021, 22(10), 5398; https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22105398
Submission received: 26 April 2021
/
Revised: 17 May 2021
/
Accepted: 18 May 2021
/
Published: 20 May 2021
(This article belongs to the Special Issue Novel Strategies in the Development of New Therapies, Drug Substances and Drug Carriers)
Abstract
:Depression is a highly prevalent, disabling, and often chronic illness that places substantial burdens on patients, families, healthcare systems, and the economy. A substantial minority of patients are unresponsive to current therapies, so there is an urgent need to develop more broadly effective, accessible, and tolerable therapies. Pharmacological regulation of histone acetylation level has been investigated as one potential clinical strategy. Histone acetylation status is considered a potential diagnostic biomarker for depression, while inhibitors of histone deacetylases (HDACs) have garnered interest as novel therapeutics. This review describes recent advances in our knowledge of histone acetylation status in depression and the therapeutic potential of HDAC inhibitors.
1. Introduction
Depression is characterized by recurrent episodes of sadness and despondency (depressed mood) frequently accompanied by anhedonia, loss of appetite, reduced concentration and energy, excessive guilt, and recurrent suicidal ideation [1]. Despite treatment, more than 50% of patients experience recurrent episodes and approximately 80% of those with a history of two episodes experience another relapse [2]. Both the incidence and prevalence of depression are increasing, and depression is now a major global healthcare burden and cause of lost economic productivity [3]. Current treatment guidelines recommend modulators of monoaminergic transmission such as monoamine oxidase (MAO) inhibitors and specific serotonin reuptake inhibitors (SSRIs) as first-line therapy based on the theory that depression arises from abnormal monoaminergic transmission. However, despite the availability of many monoamine modulators, approximately 50% of patients are unresponsive to these treatments [4].
Indeed, the clinical diagnosis and treatment of depression based on the Diagnostic and Statistical Manual of Mental Disorders (DSM) or the wide-ranging International Statistical Classification of Diseases and Related Health Problems (ICD) have focused on observable behaviors (signs) and self-reported feelings and thoughts (symptoms). Classifying mental disorders according to clinical signs and symptoms has led to a limitation in reflecting the underlying pathophysiology, and to heterogeneity within groups diagnosed with the same psychiatric disease [5]. Thus, attempts have emerged to suggest the novel classification of mental disorders that reflects biological mechanisms, such as Research Domain Criteria (RDoC) and biological classification of mental disorders (BeCOME) study [6,7]. Furthermore, many studies have aimed to identify the pathomechanism of depression to overcome the limitations of other existing tools for its diagnosis and treatment.
In addition to the well-known monoaminergic neurotransmitter dysfunction, altered hypothalamic-pituitary-adrenal (HPA) axis activity, dysfunctional brain network activity, impaired neurotrophic factor signaling, and neuroinflammation have been implicated in depression and studied for potential diagnostic biomarkers and therapeutic targets [8,9,10]. Additionally, changes in brain structure [11,12], gastrointestinal factors [13,14], oxidative stress [15], and endocannabinoid system components [16] have also been implicated in depression [17]. In addition, correlation studies for the aforementioned biomarkers such as inflammatory factors and brain structural changes also have been conducted in depression [18,19]. Family, twin, and adoption studies suggest that genetic factors account for 30–40% of the variance in depression risk [20], but early genome-wide association studies (GWASs) failed to identify genetic variants strongly associated with depression, suggesting that genetic susceptibility is mediated by heterogeneous combinations of risk alleles [21,22,23]. However, recent GWASs have identified several genetic loci reproducibly associated with depression [24,25,26,27,28].
The remaining 60–70% of the variation in depression risk appears to be determined by environmental factors [29]. Environmental stressors such as physical, emotional, and sexual abuse, social rejection, and other early adverse experiences and stressful life events such as the death of a loved one, illness, injury, disability, and functional decline are demonstrated risk factors for depression [30,31,32]. Individual variations in susceptibility to such stimuli may be explained in part by genetic factors. Indeed, a gene-environment interaction model positing that penetrant and complex genetic predispositions interact with environmental factors to determine depression susceptibility is now widely accepted [33].
In this gene-environmental interaction model, epigenetic mechanisms act as a bridge between genes and environmental factors [34]. Epigenetics refers to “heritable, but reversible, regulation of various genomic functions mediated principally through changes in DNA methylation and chromatin structure” [35]. Thus, epigenetic mechanisms are the processes by which various types of cells within the same organism acquire unique transcriptional properties and functions during development [36]. This dynamic and reversible process also contributes to the transcriptional plasticity manifested by the neurons and glia in the brain. Therefore, it is associated with learning and memory, age-related neurodegeneration, cognitive and behavioral effects of early experiences, repeated drug exposure, chronic stress, prolonged changes in nutritional status, and exposure to environmental toxins [37]. The functional analyses of DNA methylation quantitative trait locus (meQTL) and non-coding RNA (ncRNA) in depression-associated single nucleotide polymorphisms (SNPs) revealed that alterations in DNA methylation and ncRNAs interact with genetic factors in depression, which underscores the importance of epigenetic regulation for depression [38]. Thus, the present review provides an overview of the impact of histone deacetylation on the pathophysiology of depression and the therapeutic potential of its modulation.
2. Histone Acetylation
Dynamic acetylation and deacetylation of histone lysine (Lys) residues control the packaging of genomic DNA, thereby influencing DNA replication, transcription, DNA repair, and cell cycle progression [39]. Histone acetyltransferase enzymes (HATs) catalyze the transfer of acetyl groups from acetyl CoA to the ε-amino groups of Lys residues within histones [40], while histone deacetylases (HDACs) remove these acetyl groups [41]. Thus, the balance between HAT and HDAC activities determines the net histone acetylation status of the genome. By dynamically modulating the interaction between histones and DNA at the local level, histone acetylation regulates the accessibility of gene promoters to various binding factors such as transcription factors. In addition, acetylation/deacetylation of non-histone proteins modulated by HATs and HDACs also regulates diverse cellular functions [42].
3. Histone Deacetylase (HDAC) Families and Classes
Human HDACs are traditionally divided into two families, the Zn2+-dependent amidohydrolases including class I, II, and IV HDACs and the NAD+-dependent class III SIRT enzymes (Table 1). To date, 18 HDACs have been identified in humans and are grouped by sequence homology and domain organization [43]. Class I HDACs share structural homology with the yeast transcriptional regulator Rpd3 and typically act as the catalytic subunit within a complex of cognate corepressors to inhibit transcription in the cell nucleus [44]. HDAC1 and 2 are present in NuRD, Sin3, NODE, CoREST, and MiDAC complexes, while HDAC3 is a component of SMRT and NCoR corepressor complexes [45,46]. In contrast, HDAC8 can function independently without forming a multiprotein complex [47].
Class II HDACs are highly homologous to yeast Hda1 and are subdivided into two groups [48]. Class IIa HDACs 4, 5, 7, and 9 each have a single catalytic domain and a unique adaptor domain including a transcription factor MEF2-binding motif [49], while class IIb HDACs 6 and 10 contain two catalytic domains, a ubiquitin-binding zinc finger domain and a leucine-rich repeat domain [50,51,52,53,54]. In contrast to class I HDACs, which are exclusively localized in the nucleus, class II enzymes can shuttle between the cytoplasm and nucleus in response to various regulatory cues [49].
HDAC11, a homolog of yeast Hos3, is the only member of Class IV [55]. It is primarily expressed in the brain, skeletal muscle, heart, testis, and kidney, suggesting specific functions in development, inflammation, metabolism [55].
Class III HDACs are homologous to yeast Sir2. Like other HDACs, Class III members are involved in transcriptional silencing but have a deoxyhypusine synthase-like NAD/FAD-binding domain clearly distinct from the catalytic domains of other HDAC classes [56]. Seven Sir2-like proteins (SIRT1-7), referred to as sirtuins, have been identified in humans [57]. These sirtuins possess additional domain(s) such as a mono-ADP-ribosyltransferase domain. SIRT1 has the strongest histone deacetylase activity among sirtuins, while SIRT5 shows weak deacetylase activity but robust lysine desuccinylase and demalonylase activities [58]. These enzymes are differentially localized to the nucleus (SIRT1, 2, 3, 6, and 7), cytoplasm (SIRT1 and 2), and mitochondria (SIRT3, 4, and 5) [43].
4. HDAC and Depression
Among the epigenetic mechanisms, the most well-studied for contributions to depression are DNA methylation mediated by DNA methyltransferases (DNMTs) and histone post-transcriptional modifications (PTMs), including acetylation/deacetylation. Associations between depression and DNA methylation have been suggested in many studies. For example, increased DNMT3A levels were found in the nucleus accumbens (NAc), the limbic region regulating reward behavior, in the postmortem brains of depressed patients, and in animal models of depression [59,60]. Data on DNA methylation age (DNAm age) derived from blood and brain tissues indicate that patients with depression displayed higher levels of epigenetic aging than those with normal subjects [61].
Along with DNA methylation, histone acetylation via HAT and deacetylation via HDAC are reported to be crucial for long-term stress adaptation and responses to antidepressant therapy [34]. Further, several studies have suggested a relationship between depression and histone deacetylation. Chronic social defeat stress transiently suppressed histone acetylation in the NAc of mice [62], while HDAC inhibition exerted antidepressant-like effects in animal models of stress-induced depression [62,63,64,65,66]. Moreover, the expression levels of HDAC2 and HDAC5 mRNAs in peripheral white blood cells were elevated in depressed patients compared to healthy controls [67]. Singh et al. [68,69] also reported the association between depression and HDAC6, which contributes to the stabilization of microtubules in the brain by regulating acetylation of α-tubulin. Interestingly, the effects of early-life stress (e.g., maternal separation) and subsequent environmental enrichment on depressive behavior and HDAC/DNMT activities in the hippocampus and prefrontal cortex (PFC) are sex-dependent, which supports sex differences in the prevalence of depression [70].
Diverse reports have suggested that sirtuins, categorized as class III HDACs, play several roles in the mammalian brain, such as modulating brain structure through axon elongation, outgrowth of neurites, and dendritic branching [71]. Among such sirtuin proteins, SIRT1 is associated with high-order brain function including synaptic plasticity and memory formation [72]. As a result of studies based on these reported functions of SIRT1, many researchers have demonstrated the relationship between SIRT1 and depression. For example, the expression of SIRT1 in peripheral blood was downregulated in depressed patients compared to healthy controls [73]. Furthermore, these results were reproduced in animal studies; altered activity of SIRT1 in the hippocampus and the NAc provoked depressive-like behaviors in animal models of depression [74,75].
5. HDAC and the Hypothalamic-Pituitary-Adrenal (HPA) Axis
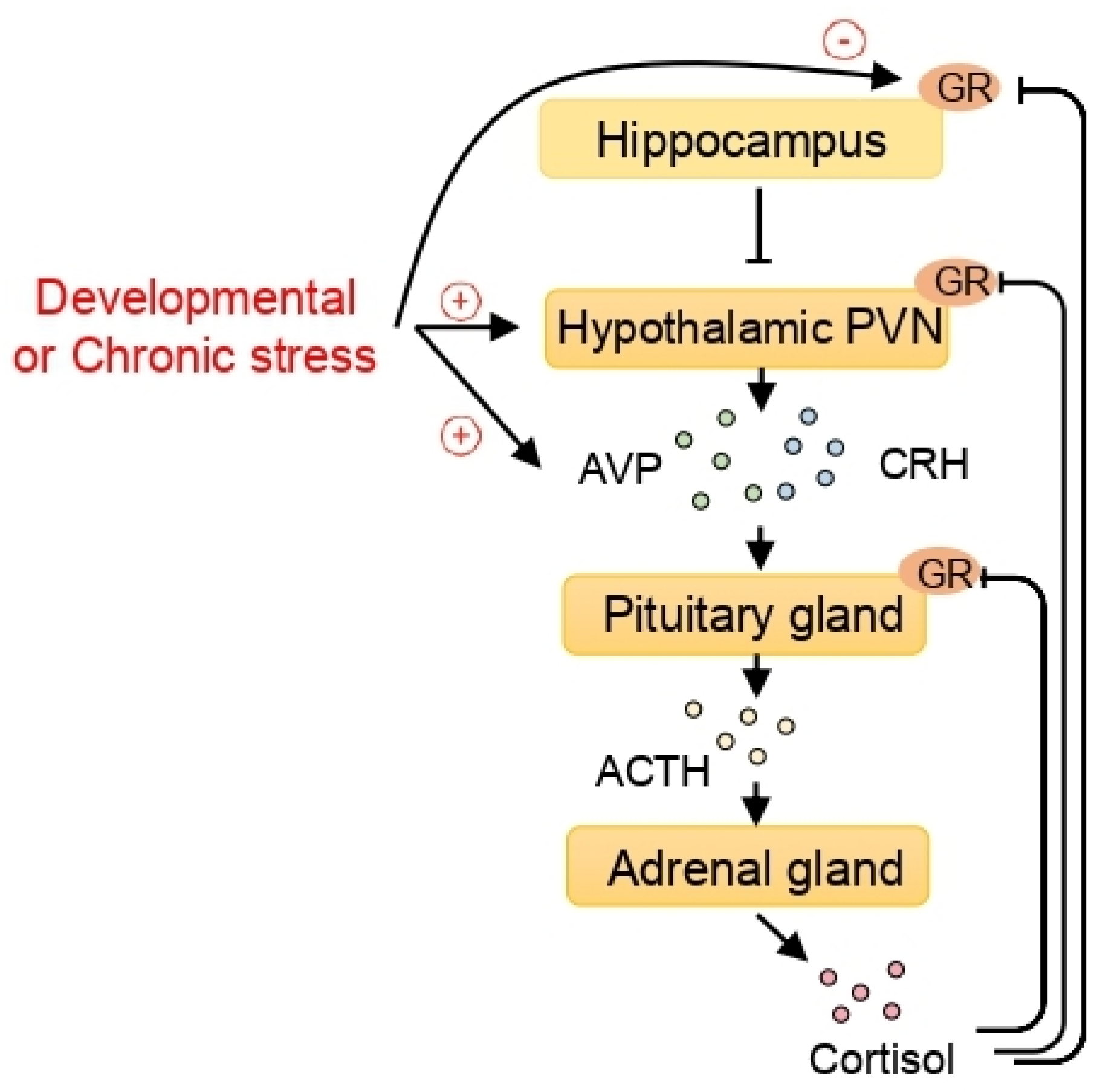
From the epigenetic perspective, stress is considered to be an important factor in the etiology of stress-related disorders such as depression and anxiety [76]. When exposed to social and physical stressors, the paraventricular nucleus (PVN) of the hypothalamus is stimulated to secrete both corticotrophin-releasing hormone (CRH) and arginine vasopressin (AVP) which stimulate the release of adrenocorticotropic hormone (ACTH) in the pituitary gland. Consequently, mainly cortisol in humans and corticosterone in rodents are produced in the adrenal cortex and released into the bloodstream, exerting their effects through glucocorticoid receptors (GRs) in each tissue. The activation of GRs in the PVN of the hypothalamus and pituitary corticotroph cells inhibits the hypothalamic release of CRH and AVP and contributes to the negative feedback regulation of the HPA axis [77,78,79,80]. Additionally, the hippocampus can contribute to feedback regulation of the HPA axis through GR signaling [81]. This regulation is important in handling challenging situations and maintaining homeostasis (Figure 1).
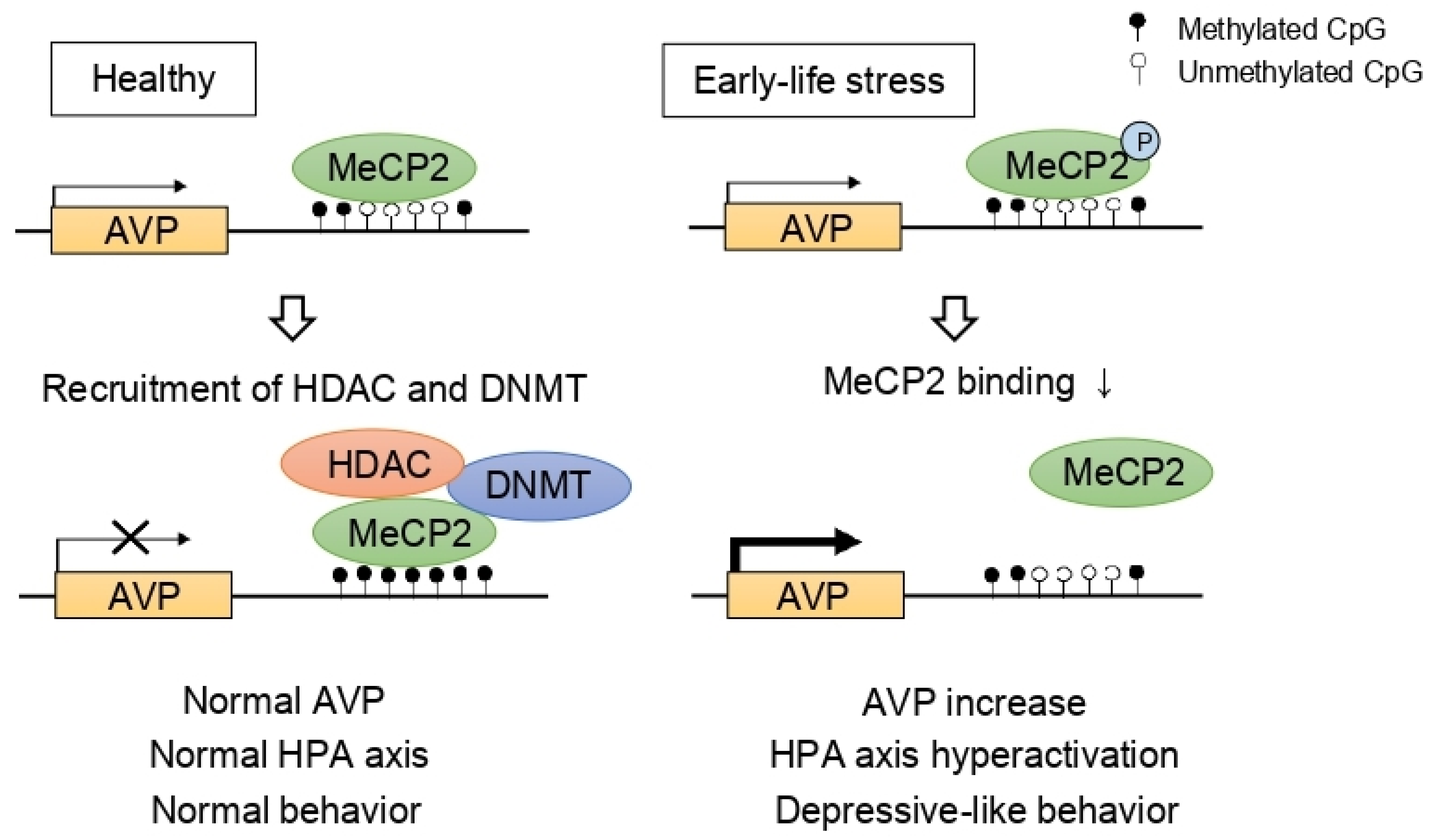
Stress, especially in chronic or developmentally critical periods (i.e., prenatal and postnatal periods), influences various epigenetic mechanisms including DNA methylations and histone modifications, leading to structural and regulatory changes and fine-tunes the neural circuitry [82,83,84]. For example, researchers reported that early-life stress influences HDAC expression in the mouse brain [85,86]. Given that the HPA axis is one of the main stress responses, many researchers investigated the epigenetic regulation of the HPA axis in depression and identified the indirect effects of HDACs on the HPA axis. Murgatroyd et al. [87] focused on AVP which was reported to be important in the regulation of mood behaviors [88]. The authors demonstrated that early-life stress, represented by maternal deprivation, modulated AVP expression dynamically in the PVN of the hypothalamus initially through methyl CpG binding protein 2 (MeCP2) phosphorylation and later by AVP enhancer hypomethylation [87]. Considering that MeCP2 forms a complex consisting of HDAC and DNMT, consequently inducing gene silencing, HDAC is considered as a modulator of the HPA axis (Figure 2).
Unlike AVP, CRH expression in the hypothalamus, another component of the HPA axis, was not changed by maternal deprivation [87]. However, GR expression in the hippocampus was influenced under early-life stress through epigenetic mechanisms. Maternal deprivation affected DNA methylation status in the promoter of GR exons in the hippocampus, which mediates the recruitment of HDAC-containing repressor complexes (e.g., HDAC5) to hypermethylated loci [89,90]. These effects of early-life stress on hippocampal GR were reversed by HDAC inhibitors such as trichostatin A.
6. HDAC and Brain-Derived Neurotrophic Factor
Brain-derived neurotrophic factor (BDNF) is a critical ligand guiding neurodevelopment and the ongoing neuroplastic processes required for behavioral adaptation, such as neurogenesis, synaptic plasticity, dendritic arborization, and pruning, and dendritic spine maturation [91,92]. Antidepressants and exercise increase endogenous BDNF in rodents, resulting in enhanced neurogenesis, reduced neuronal apoptosis, and inhibition of stress-induced depressive-like behaviors [92], while reduced BDNF is associated with depression as well as other neuropsychiatric and neurologic diseases such as Parkinson’s disease and Alzheimer’s disease [91]. Further, lower BDNF levels are observed in the PFC and the hippocampus of suicide victims compared to non-victims of suicide with or without depression [93].
Expression of BDNF is influenced by environmental stimuli via histone modification at different promoter sites in distinct brain regions especially during development [94]. Prenatal stress exposure was reported to increase HDAC expression and decrease BDNF expression in the hippocampus, resulting in anxiety- and depression-like behaviors [95]. In addition to prenatal stress, early postnatal stress also induced changes in histone modification and an increase of HDAC in the hippocampus, leading to changes in BDNF expression and behavior in rodents [92]. Not only during development but stress during adulthood also up-regulated MeCP2 levels at the Bdnf promoter and Hdac5 expression in the hippocampus [96]. On the other hand, antidepressants and HDAC inhibitors (e.g., sodium butyrate, trichostatin A, and valproic acid) increased BDNF expression and it was associated with reduced DNA methylation and histone deacetylation around the Bdnf promoter region [97,98,99]. Similar to other HDACs, SIRT1 also can regulate BDNF expression through interaction with MeCP2 [100].
7. HDAC and Neuronal Plasticity
Experience-dependent neuronal plasticity, characterized by sustained changes in synaptic structure and strength, is the neurocellular basis for sensing, adapting, and responding to environmental changes, including stress [101,102]. Thus, it is not surprising that aberrant synaptic plasticity is associated with the pathophysiology of depression. Indeed, both preclinical models of depression and depressed patients exhibit abnormalities in factors that regulate synaptic plasticity [33,103,104]. One of the strongest factors disrupting normal neuronal plasticity is chronic stress, and severe or chronic stress can reduce the capacity of the brain to respond and adapt to stress, resulting in depression [102,105]. Stressors activate the HPA axis and consequently increase circulating glucocorticoid levels. Chronically elevated glucocorticoid decreases synaptic number, impairs plasticity, and leads to neuronal atrophy, resulting in disrupted neural circuitry within and among regions regulating mood, executive function, and cognition [101]. Moreover, glucocorticoid can alter gene transcription via epigenetic regulation of the GR [89].
Abnormal histone acetylation due to the imbalance between HAT and HDAC activities can also impair synaptic plasticity, thereby reducing cognitive capacity and inducing abnormal behaviors. For instance, histone lysine acetylation can enhance neuronal plasticity while activation of HDAC and concomitant deacetylation can impair neuronal plasticity [106]. The administration of the non-selective HDAC inhibitor sodium butyrate enhanced histone acetylation and long-term potentiation (LTP), a form of synaptic plasticity strongly implicated in learning and memory, and improved memory performance [107]. Conversely, HDAC2 overexpression reduced synaptic number and synaptic plasticity, resulting in long-lasting neural circuit abnormalities and memory impairment. These changes may occur via the inactivation of activity-dependent genes involved in synaptic plasticity. Further, these effects were reversed by the HDAC inhibitor suberoylanilide hydroxamic acid (SAHA) [108].
In addition to HDAC2, HDAC4 is also implicated in the regulation of neuronal plasticity. HDAC4 is a transcriptional repressor that can translocate from the neuronal cytoplasm to the nucleus, bind chromatin, and suppress the expression of transcription factors critical for synaptic plasticity and information processing such as myocyte enhancer factor 2A (MEF2A) and cAMP response element-binding protein (CREB) [109,110,111]. Brain-specific HDAC4 knockout in mice impaired hippocampus-dependent memory and long-term synaptic plasticity [112]. Chronic cocaine-induced promoter-specific change in HDAC3, which is known as a negative regulator of memory formation, in the NAc and interfering HDAC3 activity restored cocaine-induced synaptic plasticity [113]. In addition, SIRT1 knockout mice also exhibited impaired memory and hippocampal plasticity [72]. Taken together, these findings indicate that appropriate HDAC function is essential for synaptic and neuronal plasticity and that an abnormal shift in histone acetylation status can result in impaired neural plasticity and behavioral dysfunction.
8. Molecular Diagnosis of Depression: An Epigenetic Perspective
Studies on the pathophysiology of depression have identified several promising prognostic and diagnostic biomarkers, including factors associated with the HPA axis (e.g., CRH, ACTH, and cortisol), inflammatory factors (e.g., tumor necrosis factor (TNF)-α, interleukin (IL)-1β, IL-6, and C-reactive protein (CRP)), neurotrophic factors (e.g., BDNF and glial cell line-derived neurotrophic factor (GDNF)), insulin-like growth factor 1 (IGF-1), and changes in the area or volume of the hippocampus, amygdala, and PFC [17,114,115]. According to Kennis et al. [17], only cortisol in saliva was a significant biomarker for the onset/relapse/recurrence of depression, but careful interpretation is needed given the methodological heterogeneity among included studies.
In addition, several studies have identified the genes encoding the serotonin transporter (SLC6A4) [116,117], IL-1β [118,119,120], and FK506 binding protein 5 (FKBP5 or FKBP-51) [116,121] as potential genetic biomarkers for depression. The genetic loci related to depression (e.g., SNPs in LHPP, SIRT1 region) have also been revealed although there are differences between studies [24,26]. Furthermore, there are attempts to identify blood gene expression biomarkers and provide predictive information as well as precise and personalized diagnosis and treatment for depression [117,122]. Recently, researchers have attempted to integrate functional neuroimaging and genetic data (neuroimaging genetics) for depression. Buch et al. [123] found that polymorphisms of the serotonin transporter (5-HTTLPR) and BDNF genes were associated with structural and functional changes in the anterior cingulate cortex, amygdala, and hippocampus, regions of the mesocorticolimbic reward circuit strongly associated with behaviors impaired in depression [124]. These results provide a novel diagnostic strategy for depression and imply that genetic factors contribute to depression by modulating brain structure and function.
The diagnostic biomarkers associated with epigenetic regulation also have been attracted attention in various diseases including neuropsychiatric diseases [121,125]. For instance, an epigenome-wide association study by Jovanova et al. [126] identified the methylation of 3 CpG islands in blood associated with depression. Moreover, hypermethylation of BDNF and SLC6A4 genes have been found in depressed patients [127]. The local regions of histone acetylation may also serve as possible biomarkers for depression, as both animal and human postmortem studies have reported associations between histone modifications in brain tissue and depression. In addition, histone H3 lysine 27 trimethylation (H3K27me3) at the BDNF gene promoter IV of peripheral blood was downregulated in an antidepressant-responder group compared to a non-responder group [128]. Also, HDAC5 activity was significantly higher in peripheral leukocytes from drug-free depressive patients and normalized by antidepressant treatment [129]. The plasma levels of acetyl-L-carnitine (LAC), an acetylating agent that can pass through the blood–brain barrier, were decreased in depressed patients compared to control, where the degree of reduction in LAC was much greater in patients with treatment-resistant depression [130].
In recent years, diverse attempts have been conducted to visualize epigenetic factors and utilize them for diagnosis. For example, a positron emission tomography (PET) imaging study in human using [11C] Martinostat, the only selective tracer for class I/IIb HDAC in the central nervous system [131,132], demonstrated that [11C] Martinostat uptake in the dorsolateral PFC of patients with schizophrenia/schizoaffective disorder was lower compared to those of healthy controls, which is inconsistent with the results of postmortem studies [133]. Additionally, low [11C] Martinostat uptake was observed in the frontolimbic areas of patients with bipolar disorder compared with healthy controls [134]. Since no visualization studies have been published related to depression yet and it is still in its infancy, many additional studies are expected to be needed to apply them to a depression diagnosis.
9. Molecular Therapeutics of Depression: An Epigenetic Perspective
The current first-line therapies for depression are tricyclic antidepressants (TCAs), MAO inhibitors, and SSRIs, all of which target the dysfunction of monoaminergic transmission [115]. However, classical antidepressants such as TCAs (e.g., imipramine) and SSRIs (e.g., paroxetine, fluoxetine, and escitalopram) not only bind to monoamine transporters but also have indirect effects on both DNA methylation and histone PTM [135]. For example, the reduced DNA methylation at the Crh promoter and increased Crh mRNA expression in chronic social defeat stress-induced depression were reversed by chronic imipramine administration [136]. Additionally, the SSRI paroxetine was reported to inhibit DNMTs [98]. Chronic antidepressant administration was also found to increase acetylated histone H3 (AcH3) levels by reducing HDAC expression in several brain regions, including the NAc [137].
DNMT inhibitors are not approved as antidepressant drugs despite their documented antidepressant effects because modulation of global brain methylation can cause cognitive deficits [135]. However, HDAC inhibitors have been examined as novel therapeutics for treatment-resistant depression [34,138,139], and numerous preclinical studies have reported that various HDAC inhibitors exert antidepressant-like effects in animal models of stress-induced depression [62,63,66,140,141,142] (Table 2). In addition to the antidepressant effect, HDAC inhibitors promoted neuronal rewiring and recovery of motor functions after traumatic brain injury [143]. Also, HDAC inhibitors such as sodium butyrate and SAHA enhanced cognitive function, which may provide therapeutic options for depression that accompanies cognitive impairment [144,145,146]. A recent drug repositioning study for precise/personalized medicine in depression using bioinformatic analyses revealed that HDAC inhibitors such as trichostatin A and valproic acid as a new potential antidepressant drug [117].
While these results support the potential of HDAC inhibitors as novel therapeutic drugs for depression, their use in clinical practice is limited by severe side effects including thrombocytopenia and neutropenia [147,148]. Although several HDAC inhibitors, including vorinostat (SAHA), belinostat, panobinostat (LBH-589), romidepsin (FK2280), have been approved by the Food and Drug Agency (United Stated), the clinical application of these drugs is limited to certain forms of cancers (e.g., T-cell lymphoma and multiple myeloma) [149] and to date, there is no clinical trial evaluating the antidepressant effect of HDAC inhibitors in depression.
Apart from HDAC inhibitors, the acetylating agent LAC also has been reported to be a potential antidepressant that is mediated by neurotransmitter regulations such as serotonin and epigenetic regulation of key genes important for synaptic plasticity (e.g., BDNF and metabotropic glutamate receptor of class-2 (mGlu2)) [130]. Lactate, a metabolite produced by exercise, induced resilience to social defeat stress and reversed social avoidance behavior and anxiety by modulating the activity of HDAC2 and HDAC3 [150]. In addition, dihydrocaffeic acid (DHCA) and malvidin-3′-O-glucoside (Mal-gluc) induced a resilient state against social stress and attenuated depressive behaviors via epigenetic regulation [151]. In particular, Mal-gluc mediates the increase in histone acetylation of the Rac1 gene regulatory sequence through HDAC2 inhibition, and as a result, the modulation of synaptic plasticity occurs.
10. Conclusions
Depression is a common and disabling psychiatric disease with high recurrence rates and heterogeneous clinical manifestations, adding to treatment complexity and suggesting that depression is not a unitary disease entity. Indeed, numerous pathomechanisms likely contribute to depression, including abnormal epigenetic changes. Environmental stressors are the primary risk factors for depression, supporting contributions of epigenetic mechanisms to disease pathogenesis and progression. In this review, we summarized the latest knowledge on potential epigenetic mechanisms, especially histone acetylation/deacetylation, underlying disease pathophysiology, the utility of epigenetic markers for diagnosis, and the potential of epigenetic modulators, especially HDAC inhibitors, as therapeutics. Recent studies have shown that HDAC inhibition can upregulate BDNF expression, resulting in enhanced neural/synaptic plasticity, and exert an antidepressant-like effect on behavior. Conventional antidepressants targeting monoaminergic neurotransmission also modulate epigenetic mechanisms, further supporting the contributions of epigenetic dysregulation to the pathophysiology of depression. Thus, HDACs can be regarded as novel diagnostic and therapeutic targets for depression. However, further studies are needed to develop safe and effective HDAC inhibitors for clinical use.
Author Contributions
Conceptualization, H.-S.P.; writing, H.-S.P. and H.-Y.R.; review and editing, J.K., S.H.A., and H.-Y.R.; funding acquisition, H.-Y.R. All authors have read and agreed to the published version of the manuscript.
Funding
This research was supported by Kyungpook National University Research Fund, 2020.
Conflicts of Interest
The authors declare no conflict of interest.
References
- American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders, 5th ed.; American Psychiatric Association: Arlington, VA, USA, 2013. [Google Scholar]
- Burcusa, S.L.; Iacono, W.G. Risk for recurrence in depression. Clin. Psychol. Rev. 2007, 27, 959–985. [Google Scholar] [CrossRef] [Green Version]
- Liu, Q.; He, H.; Yang, J.; Feng, X.; Zhao, F.; Lyu, J. Changes in the global burden of depression from 1990 to 2017: Findings from the Global Burden of Disease study. J. Psychiatry Res. 2020, 126, 134–140. [Google Scholar] [CrossRef] [PubMed]
- Cipriani, A.; Furukawa, T.A.; Salanti, G.; Chaimani, A.; Atkinson, L.Z.; Ogawa, Y.; Levcht, S.; Ruhe, H.G.; Turner, E.H.; Higgins, J.P.T.; et al. Comparative efficacy and acceptability of 21 antidepressant drugs for the acute treatment of adults with major depressive disorder: A systematic review and network meta-analysis. Lancet 2018, 391, 1357–1366. [Google Scholar] [CrossRef] [Green Version]
- Calabro, M.; Fabbri, C.; Kasper, S.; Zohar, J.; Souery, D.; Montgomery, S.; Albani, D.; Forloni, G.; Ferentinos, P.; Rujescu, D.; et al. Research Domain Criteria (RDoC): A Perspective to Probe the Biological Background behind Treatment Efficacy in Depression. Curr. Med. Chem. 2021, 28, 1–23. [Google Scholar] [CrossRef] [PubMed]
- Insel, T.; Cuthbert, B.; Garvey, M.; Heinssen, R.; Pine, D.S.; Quinn, K.; Sanislow, C.; Wang, P. Research Domain Criteria (RDoC): Toward a New Classification Framework for Research on Mental Disorders. Am. J. Psychiatry 2010, 167, 748–751. [Google Scholar] [CrossRef] [Green Version]
- Bruckl, T.M.; Spoormaker, V.I.; Samann, P.G.; Brem, A.-K.; Henco, L.; Czamara, D.; Elbau, I.; Grandi, N.C.; Jollans, L.; Kuhnel, A.; et al. The biological classification of mental disorders (BeCOME) study: A protocol for an observational deep-phenotyping study for the identification of biological subtypes. BMC Psychiatry 2020, 20, 213. [Google Scholar] [CrossRef]
- Krishnan, V.; Nestler, E.J. The molecular neurobiology of depression. Nature 2008, 455, 894–902. [Google Scholar] [CrossRef]
- Hasler, G. Pathophysiology of depression: Do we have any solid evidence of interest to clinicians? World Psychiatry 2010, 9, 155–161. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krishnan, V.; Nestler, E.J. Linking Molecules to Mood: New Insight Into the Biology of Depression. Am. J. Psychiatry 2010, 167, 1305–1320. [Google Scholar] [CrossRef] [Green Version]
- Schlaepfer, T.E.; Cohen, M.X.; Frick, C.; Kosel, M.M.; Brodesser, D.; Axmacher, N.; Joe, A.Y.; Kreft, M.; Lenartz, D.; Sturm, V. Deep Brain Stimulation to Reward Circuitry Alleviates Anhedonia in Refractory Major Depression. Neuropsychopharmacology 2007, 33, 368–377. [Google Scholar] [CrossRef]
- Price, J.L.; Drevets, W.C. Neural circuits underlying the pathophysiology of mood disorders. Trends Cogn. Sci. 2012, 16, 61–71. [Google Scholar] [CrossRef]
- Clapp, M.; Aurora, N.; Herrera, L.; Bhatia, M.; Wilen, E.; Wakefield, S. Gut Microbiota’s Effect on Mental Health: The Gut-Brain Axis. Clin. Pract. 2017, 7, 131–136. [Google Scholar] [CrossRef] [PubMed]
- Wallace, C.J.K.; Milev, R. The effects of probiotics on depressive symptoms in humans: A systematic review. Ann. Gen. Psychiatry 2017, 16, 14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Black, C.N.; Bot, M.; Scheffer, P.G.; Penninx, B.W.J.H. Oxidative stress in major depressive and anxiety disorders, and the association with antidepressant use; results from a large adult cohort. Psychol. Med. 2017, 47, 936–948. [Google Scholar] [CrossRef] [Green Version]
- Navarrete, F.; Garcia-Gutierrez, M.S.; Jurado-Barba, R.; Rubio, G.; Gasparyan, A.; Austrich-Olivares, A.; Manzanares, J. Endocannabinoid System Components as Potential Biomarkers in Psychiatry. Front. Psychiatry 2020, 11, 315. [Google Scholar] [CrossRef] [PubMed]
- Kennis, M.; Gerritsen, L.; Van Dalen, M.; Williams, A.; Cuijpers, P.; Bockting, C. Prospective biomarkers of major depressive disorder: A systematic review and meta-analysis. Mol. Psychiatry 2020, 25, 321–338. [Google Scholar] [CrossRef] [Green Version]
- Opel, N.; Cearns, M.; Clark, S.; Toben, C.; Grotegerd, D.; Heindel, W.; Kugel, H.; Teuber, A.; Minnerup, H.; Berger, K.; et al. Large-scale evidence for an association between low-grade peripheral inflammation and brain structural alterations in major depression in the BiDirect study. J. Psychiatry Neurosci. 2019, 44, 423–431. [Google Scholar] [CrossRef] [Green Version]
- Green, C.; Shen, X.; Stevenson, A.J.; Conole, E.L.; Harris, M.A.; Barbu, M.C.; Hawkins, E.L.; Adams, M.J.; Hillary, R.F.; Lawrie, S.M.; et al. Structural brain correlates of serum and epigenetic markers of inflammation in major depressive disorder. Brain Behav. Immun. 2021, 92, 39–48. [Google Scholar] [CrossRef]
- Sullivan, P.F.; Neale, M.C.; Kendler, K.S. Genetic Epidemiology of Major Depression: Review and Meta-Analysis. Am. J. Psychiatry 2000, 157, 1552–1562. [Google Scholar] [CrossRef]
- Bosker, F.J.; Hartman, C.A.; Nolte, I.M.; Prins, B.P.; Terpstra, P.; Posthuma, D.; van Veen, T.; Willemsen, G.; DeRijk, R.H.; de Geus, E.J.; et al. Poor replication of candidate genes for major depressive disorder using genome-wide association data. Mol. Psychiatry 2010, 16, 516–532. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wray, N.R.; Pergadia, M.L.; Blackwood, D.H.R.; Penninx, B.W.J.H.; Gordon, S.D.; Nyholt, D.R.; Ripke, S.; MacIntyre, D.J.; McGhee, K.A.; Maclean, A.W.; et al. Genome-wide association study of major depressive disorder: New results, meta-analysis, and lessons learned. Mol. Psychiatry 2010, 17, 36–48. [Google Scholar] [CrossRef] [Green Version]
- Uher, R.; Investigators, G.; Investigators, M.; Investigators, S.D. Common Genetic Variation and Antidepressant Efficacy in Major Depressive Disorder: A Meta-Analysis of Three Genome-Wide Pharmacogenetic Studies. Am. J. Psychiatry 2013, 170, 207–217. [Google Scholar]
- Cai, N.; Bigdeli, T.B.; Kretzschmar, W.; Li, Y.; Liang, J.; Song, L.; Hu, J.; Li, Q.; Jin, W.; Hu, Z.; et al. Sparse whole-genome sequencing identifies two loci for major depressive disorder. Nature 2015, 523, 588–591. [Google Scholar] [CrossRef] [PubMed]
- Okbay, A.; Baselmans, B.M.L.; De Neve, J.E.; Turley, P.; Nivard, M.G.; Fontana, M.A.; Meddens, S.F.W.; Linner, R.K.; Rietveld, C.A.; Derringer, J.; et al. Genetic variants associated with subjective well-being, depressive symptoms, and neuroticism identified through genome-wide analyses. Nat. Genet. 2016, 48, 624–633. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hyde, C.L.; Nagle, M.W.; Tian, C.; Chen, X.; Paciga, S.A.; Wendland, J.R.; Tung, J.Y.; Hinds, D.A.; Perlis, R.H.; Winslow, A.R. Identification of 15 genetic loci associated with risk of major depression in individuals of European descent. Nat. Genet. 2016, 48, 1031–1036. [Google Scholar] [CrossRef] [PubMed]
- Lewis, C. Mega-Analysis of Genome-Wide Association Studies in Major Depressive Disorder: Mdd Working Group of the Psychiatric Genomics Consortium. Eur. Neuropsychopharm. 2017, 27, S119. [Google Scholar]
- Howard, D.M.; Adams, M.J.; Shirali, M.; Clarke, T.K.; Marioni, R.E.; Davies, G.; Coleman, J.R.I.; Alloza, C.; Shen, X.Y.; Barbu, M.C.; et al. Genome-wide association study of depression phenotypes in UK Biobank identifies variants in excitatory synaptic pathways. Nat. Commun. 2018, 9, 1470. [Google Scholar] [CrossRef] [Green Version]
- Saveanu, R.V.; Nemeroff, C.B. Etiology of Depression: Genetic and Environmental Factors. Psychiatry Clin. N. Am. 2012, 35, 51–71. [Google Scholar] [CrossRef]
- Bruce, M.L. Psychosocial risk factors for depressive disorders in late life. Biol. Psychiatry 2002, 52, 175–184. [Google Scholar] [CrossRef]
- Cheptou, P.O.; Donohue, K. Epigenetics as a new avenue for the role of inbreeding depression in evolutionary ecology. Heredity 2013, 110, 205–206. [Google Scholar] [CrossRef] [Green Version]
- Shapero, B.G.; Black, S.K.; Liu, R.T.; Klugman, J.; Bender, R.E.; Abramson, L.Y.; Alloy, L.B. Stressful Life Events and Depression Symptoms: The Effect of Childhood Emotional Abuse on Stress Reactivity. J. Clin. Psychol. 2014, 70, 209–223. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, H.; Kennedy, P.J.; Nestler, E.J. Epigenetics of the Depressed Brain: Role of Histone Acetylation and Methylation. Neuropsychopharmacology 2013, 38, 124–137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, E.; Tsai, S.-J. Epigenetics and Depression: An Update. Psychiatry Investig. 2019, 16, 654–661. [Google Scholar] [CrossRef] [PubMed]
- Mill, J.; Petronis, A. Molecular studies of major depressive disorder: The epigenetic perspective. Mol. Psychiatry 2007, 12, 799–814. [Google Scholar] [CrossRef] [Green Version]
- O’Donnell, K.J.; Meaney, M.J. Epigenetics, Development, and Psychopathology. Annu. Rev. Clin. Psychol. 2020, 16, 327–350. [Google Scholar] [CrossRef] [Green Version]
- Meaney, M.J.; Ferguson-Smith, A.C. Epigenetic regulation of the neural transcriptome: The meaning of the marks. Nat. Neurosci. 2010, 13, 1313–1318. [Google Scholar] [CrossRef]
- Ciuculete, D.M.; Voisin, S.; Kular, L.; Jonsson, J.; Rask-Andersen, M.; Mwinyi, J.; Schioth, H.B. meQTL and ncRNA functional analyses of 102 GWAS-SNPs associated with depression implicate HACE1 and SHANK2 genes. Clin. Epigenetics 2020, 12, 99. [Google Scholar] [CrossRef]
- Wade, P.A.; Pruss, D.; Wolffe, A.P. Histone acetylation: Chromatin in action. Trends Biochem. Sci. 1997, 22, 128–132. [Google Scholar] [CrossRef]
- Roth, S.Y.; Denu, J.M.; Allis, C.D. Histone Acetyltransferases. Annu. Rev. Biochem. 2001, 70, 81–120. [Google Scholar] [CrossRef]
- Marks, P.A.; Miller, T.; Richon, V.M. Histone deacetylases. Curr. Opin. Pharmacol. 2003, 3, 344–351. [Google Scholar] [CrossRef]
- Narita, T.; Weinert, B.T.; Choudhary, C. Functions and mechanisms of non-histone protein acetylation. Nat. Rev. Mol. Cell Bio. 2019, 20, 156–174. [Google Scholar] [CrossRef] [PubMed]
- Seto, E.; Yoshida, M. Erasers of Histone Acetylation: The Histone Deacetylase Enzymes. Cold Spring Harb. Perspect. Biol. 2014, 6, a018713. [Google Scholar] [CrossRef] [Green Version]
- Taunton, J.; Hassig, C.A.; Schreiber, S.L. A Mammalian Histone Deacetylase Related to the Yeast Transcriptional Regulator Rpd3p. Science 1996, 272, 408–411. [Google Scholar] [CrossRef] [PubMed]
- Ayer, D.E. Histone deacetylases: Transcriptional repression with SINers and NuRDs. Trends Cell Biol. 1999, 9, 193–198. [Google Scholar] [CrossRef]
- Wen, Y.D.; Perissi, V.; Staszewski, L.M.; Yang, W.M.; Krones, A.; Glass, C.K.; Rosenfeld, M.G.; Seto, E. The histone deacetylase-3 complex contains nuclear receptor corepressors. Proc. Natl. Acad. Sci. USA 2000, 97, 7202–7207. [Google Scholar] [CrossRef] [Green Version]
- Hu, E.; Chen, Z.X.; Fredrickson, T.; Zhu, Y.; Kirkpatrick, R.; Zhang, G.-F.; Johanson, K.; Sung, C.-M.; Liu, R.G.; Winkler, J. Cloning and Characterization of a Novel Human Class I Histone Deacetylase That Functions as a Transcription Repressor. J. Biol. Chem. 2000, 275, 15254–15264. [Google Scholar] [CrossRef] [Green Version]
- Grozinger, C.M.; Hassig, C.A.; Schreiber, S.L. Three proteins define a class of human histone deacetylases related to yeast Hda1p. Proc. Natl. Acad. Sci. USA 1999, 96, 4868–4873. [Google Scholar] [CrossRef] [Green Version]
- Muslin, A.J.; Xing, H.M. 14-3-3 proteins: Regulation of subcellular localization by molecular interference. Cell. Signal 2000, 12, 703–709. [Google Scholar] [CrossRef]
- Grozinger, C.M.; Schreiber, S.L. Regulation of histone deacetylase 4 and 5 and transcriptional activity by 14-3-3-dependent cellular localization. Proc. Natl. Acad. Sci. USA 2000, 97, 7835–7840. [Google Scholar] [CrossRef] [Green Version]
- Wang, A.H.; Kruhlak, M.J.; Wu, J.; Bertos, N.R.; Vezmar, M.; Posner, B.I.; Bazett-Jones, D.P.; Yang, X.-J. Regulation of Histone Deacetylase 4 by Binding of 14-3-3 Proteins. Mol. Cell. Biol. 2000, 20, 6904–6912. [Google Scholar] [CrossRef] [Green Version]
- McKinsey, T.A.; Zhang, C.L.; Olson, E.N. Activation of the myocyte enhancer factor-2 transcription factor by calcium/calmodulin-dependent protein kinase-stimulated binding of 14-3-3 to histone deacetylase 5. Proc. Natl. Acad. Sci. USA 2000, 97, 14400–14405. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kao, H.-Y.; Downes, M.; Ordentlich, P.; Evans, R.M. Isolation of a novel histone deacetylase reveals that class I and class II deacetylases promote SMRT-mediated repression. Gene Dev. 2000, 14, 55–66. [Google Scholar]
- Zhang, H.; Okada, S.; Hatano, M.; Okabe, S.; Tokuhisa, T. A new functional domain of Bcl6 family that recruits histone deacetylases. Biochim. Biophys. Acta (BBA) Mol. Cell Res. 2001, 1540, 188–200. [Google Scholar] [CrossRef] [Green Version]
- Gao, L.; Cueto, M.A.; Asselbergs, F.; Atadja, P. Cloning and Functional Characterization of HDAC11, a Novel Member of the Human Histone Deacetylase Family. J. Biol. Chem. 2002, 277, 25748–25755. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brachmann, C.B.; Sherman, J.M.; Devine, S.E.; Cameron, E.E.; Pillus, L.; Boeke, J.D. The SIR2 gene family, conserved from bacteria to humans, functions in silencing, cell-cycle progression, and chromosome stability. Genes Dev. 1995, 9, 2888–2902. [Google Scholar] [CrossRef] [Green Version]
- Frye, R.A. Characterization of Five Human cDNAs with Homology to the Yeast SIR2 Gene: Sir2-like Proteins (Sirtuins) Metabolize NAD and May Have Protein ADP-Ribosyltransferase Activity. Biochem. Biophys. Res. Commun. 1999, 260, 273–279. [Google Scholar] [CrossRef] [PubMed]
- Du, J.; Zhou, Y.; Su, X.; Yu, J.J.; Khan, S.; Jiang, H.; Kim, J.; Woo, J.; Choi, B.H.; He, B.; et al. Sirt5 Is a NAD-Dependent Protein Lysine Demalonylase and Desuccinylase. Science 2011, 334, 806–809. [Google Scholar] [CrossRef] [Green Version]
- LaPlant, Q.; Vialou, V.; Covington, H.E., 3rd; Dumitriu, D.; Feng, J.; Warren, B.L.; Maze, I.; Dietz, D.M.; Watts, E.L.; Iniguez, S.D.; et al. Dnmt3a regulates emotional behavior and spine plasticity in the nucleus accumbens. Nat. Neurosci. 2010, 13, 1137–1143. [Google Scholar] [CrossRef] [Green Version]
- Hodes, G.E.; Pfau, M.L.; Purushothaman, I.; Ahn, H.F.; Golden, S.A.; Christoffel, D.J.; Magida, J.; Brancato, A.; Takahashi, A.; Flanigan, M.E.; et al. Sex Differences in Nucleus Accumbens Transcriptome Profiles Associated with Susceptibility versus Resilience to Subchronic Variable Stress. J. Neurosci. 2015, 35, 16362–16376. [Google Scholar] [CrossRef]
- Han, L.K.M.; Aghajani, M.; Clark, S.L.; Chan, R.F.; Hattab, M.W.; Shabalin, A.A.; Zhao, M.; Kumar, G.; Xie, L.Y.; Jansen, R.; et al. Epigenetic Aging in Major Depressive Disorder. Am. J. Psychiatry 2018, 175, 774–782. [Google Scholar] [CrossRef] [Green Version]
- Covington, H.E.; Maze, I.; LaPlant, Q.C.; Vialou, V.F.; Ohnishi, Y.N.; Berton, O.; Fass, D.M.; Renthal, W.; Rush, A.J.; Wu, E.Y.; et al. Antidepressant Actions of Histone Deacetylase Inhibitors. J. Neurosci. 2009, 29, 11451–11460. [Google Scholar] [CrossRef] [PubMed]
- Covington, H.E.; Vialou, V.F.; LaPlant, Q.; Ohnishi, Y.N.; Nestler, E.J. Hippocampal-dependent antidepressant-like activity of histone deacetylase inhibition. Neurosci. Lett. 2011, 493, 122–126. [Google Scholar] [CrossRef] [Green Version]
- Covington, H.E.; Maze, I.; Vialou, V.; Nestler, E.J. Antidepressant action of HDAC inhibition in the prefrontal cortex. Neuroscience 2015, 298, 329–335. [Google Scholar] [CrossRef] [Green Version]
- Schroeder, F.A.; Lin, C.L.; Crusio, W.E.; Akbarian, S. Antidepressant-Like Effects of the Histone Deacetylase Inhibitor, Sodium Butyrate, in the Mouse. Biol. Psychiatry 2007, 62, 55–64. [Google Scholar] [CrossRef] [PubMed]
- Tsankova, N.M.; Berton, O.; Renthal, W.; Kumar, A.; Neve, R.L.; Nestler, E.J. Sustained hippocampal chromatin regulation in a mouse model of depression and antidepressant action. Nat. Neurosci. 2006, 9, 519–525. [Google Scholar] [CrossRef]
- Hobara, T.; Uchida, S.; Otsuki, K.; Matsubara, T.; Funato, H.; Matsuo, K.; Suetsugi, M.; Watanabe, Y. Altered gene expression of histone deacetylases in mood disorder patients. J. Psychiatry Res. 2010, 44, 263–270. [Google Scholar] [CrossRef]
- Singh, H.; Wray, N.; Schappi, J.M.; Rasenick, M.M. Disruption of lipid-raft localized Galphas/tubulin complexes by antidepressants: A unique feature of HDAC6 inhibitors, SSRI and tricyclic compounds. Neuropsychopharmacology 2018, 43, 1481–1491. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, H.; Chmura, J.; Bhaumik, R.; Pandey, G.N.; Rasenick, M.M. Membrane-Associated α-Tubulin Is Less Acetylated in Postmortem Prefrontal Cortex from Depressed Subjects Relative to Controls: Cytoskeletal Dynamics, HDAC6, and Depression. J. Neurosci. 2020, 40, 4033–4041. [Google Scholar] [CrossRef]
- Borba, L.A.; Broseghini, L.D.; Manosso, L.M.; de Moura, A.B.; Botelho, M.E.M.; Arent, C.O.; Behenck, J.P.; Hilsendeger, A.; Kammer, L.H.; Valvassori, S.S.; et al. Environmental enrichment improves lifelong persistent behavioral and epigenetic changes induced by early-life stress. J. Psychiatry Res. 2021, 138, 107–116. [Google Scholar] [CrossRef]
- Herskovits, A.Z.; Guarente, L. SIRT1 in Neurodevelopment and Brain Senescence. Neuron 2014, 81, 471–483. [Google Scholar] [CrossRef] [Green Version]
- Michan, S.; Li, Y.; Chou, M.M.; Parrella, E.; Ge, H.; Long, J.M.; Allard, J.S.; Lewis, K.; Miller, M.; Xu, W.; et al. SIRT1 Is Essential for Normal Cognitive Function and Synaptic Plasticity. J. Neurosci. 2010, 30, 9695–9707. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kishi, T.; Yoshimura, R.; Kitajima, T.; Okochi, T.; Okumura, T.; Tsunoka, T.; Yamanouchi, Y.; Kinoshita, Y.; Kawashima, K.; Fukuo, Y.; et al. SIRT1 gene is associated with major depressive disorder in the Japanese population. J. Affect. Disord. 2010, 126, 167–173. [Google Scholar] [CrossRef] [PubMed]
- Abe-Higuchi, N.; Uchida, S.; Yamagata, H.; Higuchi, F.; Hobara, T.; Hara, K.; Kobayashi, A.; Watanabe, Y. Hippocampal Sirtuin 1 Signaling Mediates Depression-like Behavior. Biol. Psychiatry 2016, 80, 815–826. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, H.-D.; Hesterman, J.; Call, T.; Magazu, S.; Keeley, E.; Armenta, K.; Kronman, H.; Neve, R.L.; Nestler, E.J.; Ferguson, D. SIRT1 Mediates Depression-Like Behaviors in the Nucleus Accumbens. J. Neurosci. 2016, 36, 8441–8452. [Google Scholar] [CrossRef]
- Yang, L.; Zhao, Y.; Wang, Y.; Liu, L.; Zhang, X.; Li, B.; Cui, R. The Effects of Psychological Stress on Depression. Curr. Neuropharmacol. 2015, 13, 494–504. [Google Scholar] [CrossRef] [Green Version]
- Murgatroyd, C.; Spengler, D. Epigenetics of Early Child Development. Front. Psychiatry 2011, 2, 16. [Google Scholar] [CrossRef] [Green Version]
- Sandi, C.; Haller, J. Stress and the social brain: Behavioural effects and neurobiological mechanisms. Nat. Rev. Neurosci. 2015, 16, 290–304. [Google Scholar] [CrossRef] [Green Version]
- McEwen, B.S.; Bowles, N.P.; Gray, J.D.; Hill, M.N.; Hunter, R.G.; Karatsoreos, I.N.; Nasca, C. Mechanisms of stress in the brain. Nat. Neurosci. 2015, 18, 1353–1363. [Google Scholar] [CrossRef]
- Godoy, L.D.; Rossignoli, M.T.; Delfino-Pereira, P.; Garcia-Cairasco, N.; de Lima Umeoka, E.H. A Comprehensive Overview on Stress Neurobiology: Basic Concepts and Clinical Implications. Front. Behav. Neurosci. 2018, 12, 127. [Google Scholar] [CrossRef] [Green Version]
- Paul, S.; Jeon, W.K.; Bizon, J.L.; Han, J.S. Interaction of basal forebrain cholinergic neurons with the glucocorticoid system in stress regulation and cognitive impairment. Front. Aging Neurosci. 2015, 7, 43. [Google Scholar] [CrossRef] [Green Version]
- Murgatroyd, C.; Spengler, D. Epigenetic programming of the HPA axis: Early life decides. Stress 2011, 14, 581–589. [Google Scholar] [CrossRef] [PubMed]
- Stankiewicz, A.M.; Swiergiel, A.H.; Lisowski, P. Epigenetics of stress adaptations in the brain. Brain Res. Bull. 2013, 98, 76–92. [Google Scholar] [CrossRef] [Green Version]
- Klengel, T.; Binder, E.B. Epigenetics of Stress-Related Psychiatric Disorders and Gene x Environment Interactions. Neuron 2015, 86, 1343–1357. [Google Scholar] [CrossRef] [Green Version]
- Levine, A.; Worrell, T.R.; Zimnisky, R.; Schmauss, C. Early life stress triggers sustained changes in histone deacetylase expression and histone H4 modifications that alter responsiveness to adolescent antidepressant treatment. Neurobiol. Dis. 2012, 45, 488–498. [Google Scholar] [CrossRef] [Green Version]
- Tesone-Coelho, C.; Morel, L.J.; Bhatt, J.; Estevez, L.; Naudon, L.; Giros, B.; Zwiller, J.; Dauge, V. Vulnerability to opiate intake in maternally deprived rats: Implication of MeCP2 and of histone acetylation. Addict. Biol. 2013, 20, 120–131. [Google Scholar] [CrossRef] [PubMed]
- Murgatroyd, C.; Patchev, A.V.; Wu, Y.; Micale, V.; Bockmuhl, Y.; Fischer, D.; Holsboer, F.; Wotjak, C.T.; Almeida, O.F.; Spengler, D. Dynamic DNA methylation programs persistent adverse effects of early-life stress. Nat. Neurosci. 2009, 12, 1559–1566. [Google Scholar] [CrossRef] [PubMed]
- Neumann, I.D.; Landgraf, R. Balance of brain oxytocin and vasopressin: Implications for anxiety, depression, and social behaviors. Trends Neurosci. 2012, 35, 649–659. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.Y.; Labonte, B.; Wen, X.L.; Turecki, G.; Meaney, M.J. Epigenetic mechanisms for the early environmental regulation of hippocampal glucocorticoid receptor gene expression in rodents and humans. Neuropsychopharmacology 2013, 38, 111–123. [Google Scholar] [CrossRef] [Green Version]
- Seo, M.K.; Kim, S.G.; Seog, D.H.; Bahk, W.M.; Kim, S.H.; Park, S.W.; Lee, J.G. Effects of Early Life Stress on Epigenetic Changes of the Glucocorticoid Receptor 17 Promoter during Adulthood. Int. J. Mol. Sci. 2020, 21, 6331. [Google Scholar] [CrossRef]
- Nagahara, A.H.; Tuszynski, M.H. Potential therapeutic uses of BDNF in neurological and psychiatric disorders. Nat. Rev. Drug Discov. 2011, 10, 209–219. [Google Scholar] [CrossRef]
- Hing, B.; Sathyaputri, L.; Potash, J.B. A comprehensive review of genetic and epigenetic mechanisms that regulate BDNF expression and function with relevance to major depressive disorder. Am. J. Med. Genet. B 2018, 177, 143–167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Misztak, P.; Panczyszyn-Trzewik, P.; Nowak, G.; Sowa-Kucma, M. Epigenetic marks and their relationship with BDNF in the brain of suicide victims. PLoS ONE 2020, 15, e0239335. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.-W.; Chen, L.Y. Epigenetic Regulation of BDNF Gene during Development and Diseases. Int. J. Mol. Sci. 2017, 18, 571. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.; Fan, W.D.; Zhang, X.Q.; Dong, E.B. Gestational stress induces depressive-like and anxiety-like phenotypes through epigenetic regulation of BDNF expression in offspring hippocampus. Epigenetics 2016, 11, 150–162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seo, M.K.; Ly, N.N.; Lee, C.H.; Cho, H.Y.; Choi, C.M.; Nhu, L.H.; Lee, J.G.; Lee, B.J.; Kim, G.M.; Yoon, B.J.; et al. Early life stress increases stress vulnerability through BDNF gene epigenetic changes in the rat hippocampus. Neuropharmacology 2016, 105, 388–397. [Google Scholar] [CrossRef]
- Su, C.L.; Su, C.W.; Hsiao, Y.H.; Gean, P.W. Epigenetic regulation of BDNF in the learned helplessness-induced animal model of depression. J. Psychiatry Res. 2016, 76, 101–110. [Google Scholar] [CrossRef]
- Gassen, N.C.; Fries, G.R.; Zannas, A.S.; Hartmann, J.; Zschocke, J.; Hafner, K.; Carrillo-Roa, T.; Steinbacher, J.; Preissinger, S.N.; Hoeijmakers, L.; et al. Chaperoning epigenetics: FKBP51 decreases the activity of DNMT1 and mediates epigenetic effects of the antidepressant paroxetine. Sci. Signal. 2015, 8, ra119. [Google Scholar] [CrossRef]
- Varela, R.B.; Resende, W.R.; Dal-Pont, G.C.; Gava, F.F.; Tye, S.J.; Quevedo, J.; Valvassori, S.S. HDAC inhibitors reverse mania-like behavior and modulate epigenetic regulatory enzymes in an animal model of mania induced by Ouabain. Pharmacol. Biochem. Behav. 2020, 193, 172917. [Google Scholar] [CrossRef]
- Zocchi, L.; Sassone-Corsi, P. SIRT1-mediated deacetylation of MeCP2 contributes to BDNF expression. Epigenetics 2012, 7, 695–700. [Google Scholar] [CrossRef] [Green Version]
- Duman, R.S.; Aghajanian, G.K.; Sanacora, G.; Krysta, J.H. Synaptic plasticity and depression: New insights from stress and rapid-acting antidepressants. Nat. Med. 2016, 22, 238–249. [Google Scholar] [CrossRef] [Green Version]
- Uchida, S.; Yamagata, H.; Seki, T.; Watanabe, Y. Epigenetic mechanisms of major depression: Targeting neuronal plasticity. Psychiatry Clin. Neurosci. 2018, 72, 212–227. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pena, C.J.; Bagot, R.C.; Labonte, B.; Nestler, E.J. Epigenetic Signaling in Psychiatric Disorders. J. Mol. Biol. 2014, 426, 3389–3412. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Labonte, B.; Engmann, O.; Purushothaman, I.; Menard, C.; Wang, J.S.; Tan, C.F.; Scarpa, J.R.; Moy, G.; Loh, Y.H.E.; Cahill, M.; et al. Sex-specific transcriptional signatures in human depression. Nat. Med. 2017, 23, 1102–1111. [Google Scholar] [CrossRef] [PubMed]
- de Kloet, E.R.; Oitzl, M.S.; Joels, M. Stress and cognition: Are corticosteroids good or bad guys? Trends Neurosci. 1999, 22, 422–426. [Google Scholar] [CrossRef]
- Guan, Z.H.; Giustetto, M.; Lomvardas, S.; Kim, J.H.; Miniaci, M.C.; Schwartz, J.H.; Thanos, D.; Kandel, E.R. Integration of Long-Term-Memory-Related Synaptic Plasticity Involves Bidirectional Regulation of Gene Expression and Chromatin Structure. Cell 2002, 111, 483–493. [Google Scholar] [CrossRef] [Green Version]
- Levenson, J.M.; O’Riordan, K.J.; Brown, K.D.; Trinh, M.A.; Molfese, D.L.; Sweatt, J.D. Regulation of Histone Acetylation during Memory Formation in the Hippocampus. J. Biol. Chem. 2004, 279, 40545–40559. [Google Scholar] [CrossRef] [Green Version]
- Guan, J.-S.; Haggarty, S.J.; Giacometti, E.; Dannenberg, J.-H.; Joseph, N.; Gao, J.; Nieland, T.J.F.; Zhou, Y.; Wang, X.; Mazitschek, R.; et al. HDAC2 negatively regulates memory formation and synaptic plasticity. Nature 2009, 459, 55–60. [Google Scholar] [CrossRef]
- Bolger, T.A.; Yao, T.P. Intracellular trafficking of histone deacetylase 4 regulates neuronal cell death. J. Neurosci. 2005, 25, 9544–9553. [Google Scholar] [CrossRef] [Green Version]
- Chen, B.; Cepko, C.L. HDAC4 Regulates Neuronal Survival in Normal and Diseased Retinas. Science 2009, 323, 256–259. [Google Scholar] [CrossRef] [Green Version]
- Sando, R.; Gounko, N.; Pieraut, S.; Liao, L.J.; Yates, J.; Maximov, A. HDAC4 Governs a Transcriptional Program Essential for Synaptic Plasticity and Memory. Cell 2012, 151, 821–834. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, M.S.; Akhtar, M.W.; Adachi, M.; Mahgoub, M.; Bassel-Duby, R.; Kavalali, E.T.; Olson, E.N.; Monteggia, L.M. An Essential Role for Histone Deacetylase 4 in Synaptic Plasticity and Memory Formation. J. Neurosci. 2012, 32, 10879–10886. [Google Scholar] [CrossRef] [Green Version]
- Campbell, R.R.; Kramar, E.A.; Pham, L.; Beardwood, J.H.; Augustynski, A.S.; Lopez, A.J.; Chitnis, O.S.; Delima, G.; Banihani, J.; Matheos, D.P.; et al. HDAC3 Activity within the Nucleus Accumbens Regulates Cocaine-Induced Plasticity and Behavior in a Cell-Type-Specific Manner. J. Neurosci. 2021, 41, 2814–2827. [Google Scholar] [CrossRef] [PubMed]
- Schmaal, L.; Pozzi, E.; Ho, T.C.; van Velzen, L.S.; Veer, I.M.; Opel, N.; Van Someren, E.J.W.; Han, L.K.M.; Aftanas, L.; Aleman, A.; et al. ENIGMA MDD: Seven years of global neuroimaging studies of major depression through worldwide data sharing. Transl. Psychiatry 2020, 10, 172. [Google Scholar] [CrossRef]
- Erjavec, G.N.; Sagud, M.; Perkovic, M.N.; Strac, D.S.; Konjevod, M.; Tudor, L.; Uzun, S.; Pivac, N. Depression: Biological markers and treatment. Prog. Neuro Psychopharmacol. Biol. Psychiatry 2021, 105, 110139. [Google Scholar] [CrossRef]
- Gadad, B.S.; Jha, M.K.; Czysz, A.; Furman, J.L.; Mayes, T.L.; Emslie, M.P.; Trivedi, M.H. Peripheral biomarkers of major depression and antidepressant treatment response: Current knowledge and future outlooks. J. Affect. Disord. 2018, 233, 3–14. [Google Scholar] [CrossRef] [PubMed]
- Le-Niculescu, H.; Roseberry, K.; Gill, S.S.; Levey, D.F.; Phalen, P.L.; Mullen, J.; Williams, A.; Bhairo, S.; Voegtline, T.; Davis, H.; et al. Precision medicine for mood disorders: Objective assessment, risk prediction, pharmacogenomics, and repurposed drugs. Mol. Psychiatry 2021, 1–29. [Google Scholar] [CrossRef]
- Himmerich, H.; Milenovic, S.; Fulda, S.; Plumakers, B.; Sheldrick, A.J.; Michel, T.M.; Kircher, T.; Rink, L. Regulatory T cells increased while IL-1 beta decreased during antidepressant therapy. J. Psychiatr. Res. 2010, 44, 1052–1057. [Google Scholar] [CrossRef] [PubMed]
- Cattaneo, A.; Gennarelli, M.; Uher, R.; Breen, G.; Farmer, A.; Aitchison, K.J.; Craig, I.W.; Anacker, C.; Zunsztain, P.A.; McGuffin, P.; et al. Candidate Genes Expression Profile Associated with Antidepressants Response in the GENDEP Study: Differentiating between Baseline ‘Predictors’ and Longitudinal ‘Targets’. Neuropsychopharmacology 2013, 38, 377–385. [Google Scholar] [CrossRef]
- Cattaneo, A.; Ferrari, C.; Uher, R.; Bocchio-Chiavetto, L.; Riva, M.A.; Pariante, C.M. Absolute Measurements of Macrophage Migration Inhibitory Factor and Interleukin-1-beta mRNA Levels Accurately Predict Treatment Response in Depressed Patients. Int. J. Neuropsychoph. 2016, 19, pyw045. [Google Scholar] [CrossRef] [Green Version]
- Belzeaux, R.; Lin, R.X.; Ju, C.; Chay, M.A.; Fiori, L.M.; Lutz, P.E.; Turecki, G. Transcriptomic and epigenomic biomarkers of antidepressant response. J. Affect. Disord. 2018, 233, 36–44. [Google Scholar] [CrossRef]
- Le-Niculescu, H.; Kurian, S.M.; Yehyawi, N.; Dike, C.; Patel, S.D.; Edenberg, H.J.; Tsuang, M.T.; Salomon, D.R.; Nurnberger, J.I., Jr.; Niculescu, A.B. Identifying blood biomarkers for mood disorders using convergent functional genomics. Mol. Psychiatry 2009, 14, 156–174. [Google Scholar] [CrossRef] [PubMed]
- Buch, A.M.; Liston, C. Dissecting diagnostic heterogeneity in depression by integrating neuroimaging and genetics. Neuropsychopharmacology 2021, 46, 156–175. [Google Scholar] [CrossRef] [PubMed]
- Miller, E.K.; Cohen, J.D. An Integrative Theory of Prefrontal Cortex Function. Annu. Rev. Neurosci. 2001, 24, 167–202. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mueller, T.M.; Meador-Woodruff, J.H. Post-translational protein modifications in schizophrenia. NPJ Schizophr. 2020, 6, 1–16. [Google Scholar] [CrossRef]
- Jovanova, O.S.; Nedeljkovic, I.; Spieler, D.; Walker, R.M.; Liu, C.Y.; Luciano, M.; Bressler, J.; Brody, J.; Drake, A.J.; Evans, K.L.; et al. DNA Methylation Signatures of Depressive Symptoms in Middle-aged and Elderly Persons Meta-analysis of Multiethnic Epigenome-wide Studies. JAMA Psychiatry 2018, 75, 949–959. [Google Scholar] [CrossRef] [Green Version]
- Li, M.; D’Arcy, C.; Li, X.; Zhang, T.; Joober, R.; Meng, X. What do DNA methylation studies tell us about depression? A systematic review. Transl. Psychiatry 2019, 9, 68. [Google Scholar] [CrossRef] [Green Version]
- Lopez, J.P.; Mamdani, F.; Labonte, B.; Beaulieu, M.M.; Yang, J.P.; Berlim, M.T.; Ernst, C.; Turecki, G. Epigenetic regulation of BDNF expression according to antidepressant response. Mol. Psychiatry 2013, 18, 398–399. [Google Scholar] [CrossRef] [Green Version]
- Iga, J.; Ueno, S.; Yamauchi, K.; Numata, S.; Kinouchi, S.; Tayoshi-Shibuya, S.; Song, H.W.; Ohmori, T. Altered HDAC5 and CREB mRNA expressions in the peripheral leukocytes of major depression. Prog. Neuro Psychopharmacol. Biol. Psychiatry 2007, 31, 628–632. [Google Scholar] [CrossRef]
- Nasca, C.; Bigio, B.; Lee, F.S.; Young, S.P.; Kautz, M.M.; Albright, A.; Beasley, J.; Millington, D.S.; Mathé, A.A.; Kocsis, J.H.; et al. Acetyl-l-carnitine deficiency in patients with major depressive disorder. Proc. Natl. Acad. Sci. USA 2018, 115, 8627–8632. [Google Scholar] [CrossRef] [Green Version]
- Wey, H.-Y.; Wang, C.; Schroeder, F.A.; Logan, J.; Price, J.C.; Hooker, J.M. Kinetic Analysis and Quantification of [11C]Martinostat for in Vivo HDAC Imaging of the Brain. ACS Chem. Neurosci. 2015, 6, 708–715. [Google Scholar] [CrossRef] [Green Version]
- Wey, H.Y.; Gilbert, T.M.; Zurcher, N.R.; She, A.; Bhanot, A.; Taillon, B.D.; Schroeder, F.A.; Wang, C.; Haggarty, S.J.; Hooker, J.M. Insights into neuroepigenetics through human histone deacetylase PET imaging. Sci. Transl. Med. 2016, 8, 351ra106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gilbert, T.M.; Zürcher, N.R.; Wu, C.J.; Bhanot, A.; Hightower, B.G.; Kim, M.; Albrecht, D.S.; Wey, H.-Y.; Schroeder, F.A.; Rodriguez-Thompson, A.; et al. PET neuroimaging reveals histone deacetylase dysregulation in schizophrenia. J. Clin. Investig. 2019, 129, 364–372. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tseng, C.-E.J.; Gilbert, T.M.; Catanese, M.C.; Hightower, B.G.; Peters, A.T.; Parmar, A.J.; Kim, M.; Wang, C.; Roffman, J.L.; Brown, H.E.; et al. In vivo human brain expression of histone deacetylases in bipolar disorder. Transl. Psychiatry 2020, 10, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Toth, M. Epigenetic Neuropharmacology: Drugs Affecting the Epigenome in the Brain. Annu. Rev. Pharmacol. Toxicol. 2021, 61, 181–201. [Google Scholar] [CrossRef] [PubMed]
- Elliott, E.; Ezra-Nevo, G.; Regev, L.; Neufeld-Cohen, A.; Chen, A. Resilience to social stress coincides with functional DNA methylation of the Crf gene in adult mice. Nat. Neurosci. 2010, 13, 1351–1353. [Google Scholar] [CrossRef]
- Ookubo, M.; Kanai, H.; Aoki, H.; Yamada, N. Antidepressants and mood stabilizers effects on histone deacetylase expression in C57BL/6 mice: Brain region specific changes. J. Psychiatry Res. 2013, 47, 1204–1214. [Google Scholar] [CrossRef]
- Deussing, J.M.; Jakovcevski, M. Histone Modifications in Major Depressive Disorder and Related Rodent Models. Adv. Exp. Med. Biol. 2017, 978, 169–183. [Google Scholar] [CrossRef]
- Fuchikami, M.; Yamamoto, S.; Morinobu, S.; Okada, S.; Yamawaki, Y.; Yamawaki, S. The potential use of histone deacetylase inhibitors in the treatment of depression. Prog. Neuro Psychopharmacol. Biol. Psychiatry 2016, 64, 320–324. [Google Scholar] [CrossRef] [Green Version]
- Uchida, S.; Hara, K.; Kobayashi, A.; Otsuki, K.; Yamagata, H.; Hobara, T.; Suzuki, T.; Miyata, N.; Watanabe, Y. Epigenetic Status of Gdnf in the Ventral Striatum Determines Susceptibility and Adaptation to Daily Stressful Events. Neuron 2011, 69, 359–372. [Google Scholar] [CrossRef] [Green Version]
- Golden, S.A.; Christoffel, D.J.; Heshmati, M.; Hodes, G.E.; Magida, J.; Davis, K.; Cahill, M.E.; Dias, C.; Ribeiro, E.; Ables, J.L.; et al. Epigenetic regulation of RAC1 induces synaptic remodeling in stress disorders and depression. Nat. Med. 2013, 19, 337–344. [Google Scholar] [CrossRef] [Green Version]
- Han, A.; Sung, Y.-B.; Chung, S.-Y.; Kwon, M.-S. Possible additional antidepressant-like mechanism of sodium butyrate: Targeting the hippocampus. Neuropharmacology 2014, 81, 292–302. [Google Scholar] [CrossRef]
- Sada, N.; Fujita, Y.; Mizuta, N.; Ueno, M.; Furukawa, T.; Yamashita, T. Inhibition of HDAC increases BDNF expression and promotes neuronal rewiring and functional recovery after brain injury. Cell Death Dis. 2020, 11, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Athira, K.V.; Madhana, R.M.; Bais, A.K.; Singh, V.B.; Malik, A.; Sinha, S.; Lahkar, M.; Kumar, P.; Samudrala, P.K. Cognitive Improvement by Vorinostat through Modulation of Endoplasmic Reticulum Stress in a Corticosterone-Induced Chronic Stress Model in Mice. ACS Chem. Neurosci. 2020, 11, 2649–2657. [Google Scholar] [CrossRef]
- Ershadi, A.S.B.; Amini-Khoei, H.; Hosseini, M.-J.; Dehpour, A.R. SAHA Improves Depressive Symptoms, Cognitive Impairment and Oxidative Stress: Rise of a New Antidepressant Class. Neurochem. Res. 2021, 46, 1252–1263. [Google Scholar] [CrossRef] [PubMed]
- Vinarskaya, A.K.; Balaban, P.M.; Roshchin, M.V.; Zuzina, A.B. Sodium butyrate as a selective cognitive enhancer for weak or impaired memory. Neurobiol. Learn. Mem. 2021, 180, 107414. [Google Scholar] [CrossRef] [PubMed]
- Prince, H.M.; Bishton, M.J.; Harrison, S.J. Clinical Studies of Histone Deacetylase Inhibitors. Clin. Cancer Res. 2009, 15, 3958–3969. [Google Scholar] [CrossRef] [Green Version]
- Subramanian, S.; Bates, S.E.; Wright, J.J.; Espinoza-Delgado, I.; Piekarz, R.L. Clinical Toxicities of Histone Deacetylase Inhibitors. Pharmaceuticals 2010, 3, 2751–2767. [Google Scholar] [CrossRef] [Green Version]
- Majchrzak-Celinska, A.; Warych, A.; Szoszkiewicz, M. Novel Approaches to Epigenetic Therapies: From Drug Combinations to Epigenetic Editing. Genes 2021, 12, 208. [Google Scholar] [CrossRef]
- Karnib, N.; El-Ghandour, R.; El Hayek, L.; Nasrallah, P.; Khalifeh, M.; Barmo, N.; Jabre, V.; Ibrahim, P.; Bilen, M.; Stephan, J.; et al. Lactate is an antidepressant that mediates resilience to stress by modulating the hippocampal levels and activity of histone deacetylases. Neuropsychopharmacology 2019, 44, 1152–1162. [Google Scholar] [CrossRef]
- Wang, J.; Hodes, G.E.; Zhang, H.; Zhang, S.; Zhao, W.; Golden, S.A.; Bi, W.; Menard, C.; Kana, V.; Leboeuf, M.; et al. Epigenetic modulation of inflammation and synaptic plasticity promotes resilience against stress in mice. Nat. Commun. 2018, 9, 477. [Google Scholar] [CrossRef] [Green Version]
Figure 1.
The epigenetic effect of stress on the hypothalamic-pituitary-adrenal (HPA) axis and the epigenetic regulation of arginine vasopressin (AVP) expression. When exposed to stress, corticotrophin-releasing hormone (CRH) and AVP, released from the paraventricular nucleus (PVN) of the hypothalamus, stimulate the pituitary gland to secrete adrenocorticotropic hormone (ACTH). The adrenal glands, activated by ACTH, secrete cortisol. Cortisol exerts its function by binding to the glucocorticoid receptors (GRs). In turn, the GRs in the pituitary gland, the hypothalamic PVN, and the hippocampus play important roles in the feedback regulation of the HPA axis. Developmental or chronic stress, which can program the HPA axis, increases AVP expression and decreases hippocampal GR through epigenetic mechanisms including histone deacetylases (HDACs).
Figure 1.
The epigenetic effect of stress on the hypothalamic-pituitary-adrenal (HPA) axis and the epigenetic regulation of arginine vasopressin (AVP) expression. When exposed to stress, corticotrophin-releasing hormone (CRH) and AVP, released from the paraventricular nucleus (PVN) of the hypothalamus, stimulate the pituitary gland to secrete adrenocorticotropic hormone (ACTH). The adrenal glands, activated by ACTH, secrete cortisol. Cortisol exerts its function by binding to the glucocorticoid receptors (GRs). In turn, the GRs in the pituitary gland, the hypothalamic PVN, and the hippocampus play important roles in the feedback regulation of the HPA axis. Developmental or chronic stress, which can program the HPA axis, increases AVP expression and decreases hippocampal GR through epigenetic mechanisms including histone deacetylases (HDACs).

Figure 2.
Epigenetic programming of arginine vasopressin (AVP). In normal conditions, AVP expression is repressed by methyl CpG binding protein 2 (MeCP2), DNA methyltransferase (DNMT), and histone deacetylase (HDAC) complex. However, early-life stress induces MeCP2 phosphorylation, inhibiting the recruitment of DNMT and HDAC consequently leading to hypomethylation at the AVP enhancer. As a result, increased AVP levels contribute to hyperactivation of the HPA axis and depressive-like behaviors.
Figure 2.
Epigenetic programming of arginine vasopressin (AVP). In normal conditions, AVP expression is repressed by methyl CpG binding protein 2 (MeCP2), DNA methyltransferase (DNMT), and histone deacetylase (HDAC) complex. However, early-life stress induces MeCP2 phosphorylation, inhibiting the recruitment of DNMT and HDAC consequently leading to hypomethylation at the AVP enhancer. As a result, increased AVP levels contribute to hyperactivation of the HPA axis and depressive-like behaviors.


{kind=link}
{kind=link}
Table 1.
HDAC classification.
| Class | Protein (S. cerevisiae) | Protein (Human) | Subcellular Localization |
|---|---|---|---|
| Class I | Rpd3 | HDAC1 | Nucleus |
| HDAC2 | Nucleus | ||
| HDAC3 | Nucleus | ||
| HDAC8 | Nucleus | ||
| Class IIa | Hda1 | HDAC4 | Nucleus/cytoplasm |
| HDAC5 | Nucleus/cytoplasm | ||
| HDAC7 | Nucleus/cytoplasm | ||
| HDAC9 | Nucleus/cytoplasm | ||
| Class IIb | Hda1 | HDAC6 | Cytoplasm |
| HDAC10 | Cytoplasm | ||
| Class IV | Hos3 | HDAC11 | Nucleus/cytoplasm |
| Class III | Sir2 | SIRT1 | Nucleus/cytoplasm |
| SIRT2 | Nucleus/cytoplasm | ||
| SIRT3 | Nucleus/mitochondria | ||
| SIRT4 | Mitochondria | ||
| SIRT5 | Mitochondria | ||
| SIRT6 | Nucleus | ||
| SIRT7 | Nucleus |
Table 2.
Summary of the antidepressant actions of HDAC inhibitor in animal model.
| HDAC Inhibitor | Animal Model | Measurement of Antidepressant Effect | Molecular Mechanisms of Action | Ref. |
|---|---|---|---|---|
| MS-275 | Chronic social defeat stress | Social avoidance, sucrose preference, FST | acH3 ↑ in the NAc | [62] |
| Chronic social defeat stress | Sucrose preference test, social avoidance (combined with social enrichment) | acH3 ↑ in the hippocampus | [63] | |
| Chronic social defeat stress | Social avoidance, FST | acH3 ↑ in the mPFC | [64] | |
| Chronic social defeat stress | Social avoidance | Rac1 ↑ in the NAc synapse structural plasticity normalization | [141] | |
| SAHA | Chronic social defeat stress | Social avoidance, sucrose preference, FST | acH3 ↑ in the NAc | [62] |
| Chronic unpredictable mild stress | Social interaction, sucrose preference test, novelty-suppressed test, FST | HDAC2 inhibition, Gdnf ↑ in the NAc | [140] | |
| Sodium butyrate | Behavioral despair paradigm | TST | acH3 ↑ in the hippocampus, Bdnf↑ in the frontal cortex | [65] |
| Chronic social defeat stress | Social avoidance | HDAC5 inhibition, acH3 ↑ in Bdnf gene P3, P4 promotor | [66] | |
| Chronic restraint stress | Sucrose preference test, Light/dark test, TST, FST | HDAC2 ↑, pCREB ↑, AcH3 ↑, BDNF ↑ in the hippocampus | [142] |
BDNF, brain-derived neurotrophic factor; CREB, cAMP response element-binding protein; FST, forced swim test; GDNF, glial cell-derived neurotrophic factor; HDAC, histone deacetylase; mPFC, medial prefrontal cortex; NAc, nucleus accumbens; Rac1, Rac family small GTPase 1; SAHA, suberoylanilide hydroxamic acid; TST, tail suspension test; ↑ increase.
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Park, H.-S.; Kim, J.; Ahn, S.H.; Ryu, H.-Y. Epigenetic Targeting of Histone Deacetylases in Diagnostics and Treatment of Depression. Int. J. Mol. Sci. 2021, 22, 5398. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22105398
AMA Style
Park H-S, Kim J, Ahn SH, Ryu H-Y. Epigenetic Targeting of Histone Deacetylases in Diagnostics and Treatment of Depression. International Journal of Molecular Sciences. 2021; 22(10):5398. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22105398
Chicago/Turabian StylePark, Hyun-Sun, Jongmin Kim, Seong Hoon Ahn, and Hong-Yeoul Ryu. 2021. "Epigenetic Targeting of Histone Deacetylases in Diagnostics and Treatment of Depression" International Journal of Molecular Sciences 22, no. 10: 5398. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22105398
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.