
Neuroprotective Effects of Thiamine and Precursors with Higher Bioavailability: Focus on Benfotiamine and Dibenzoylthiamine


Abstract
:1. Introduction
2. Properties and Mechanism of Action of Benfotiamine
2.1. Structure and Physico-Chemical Properties of BFT
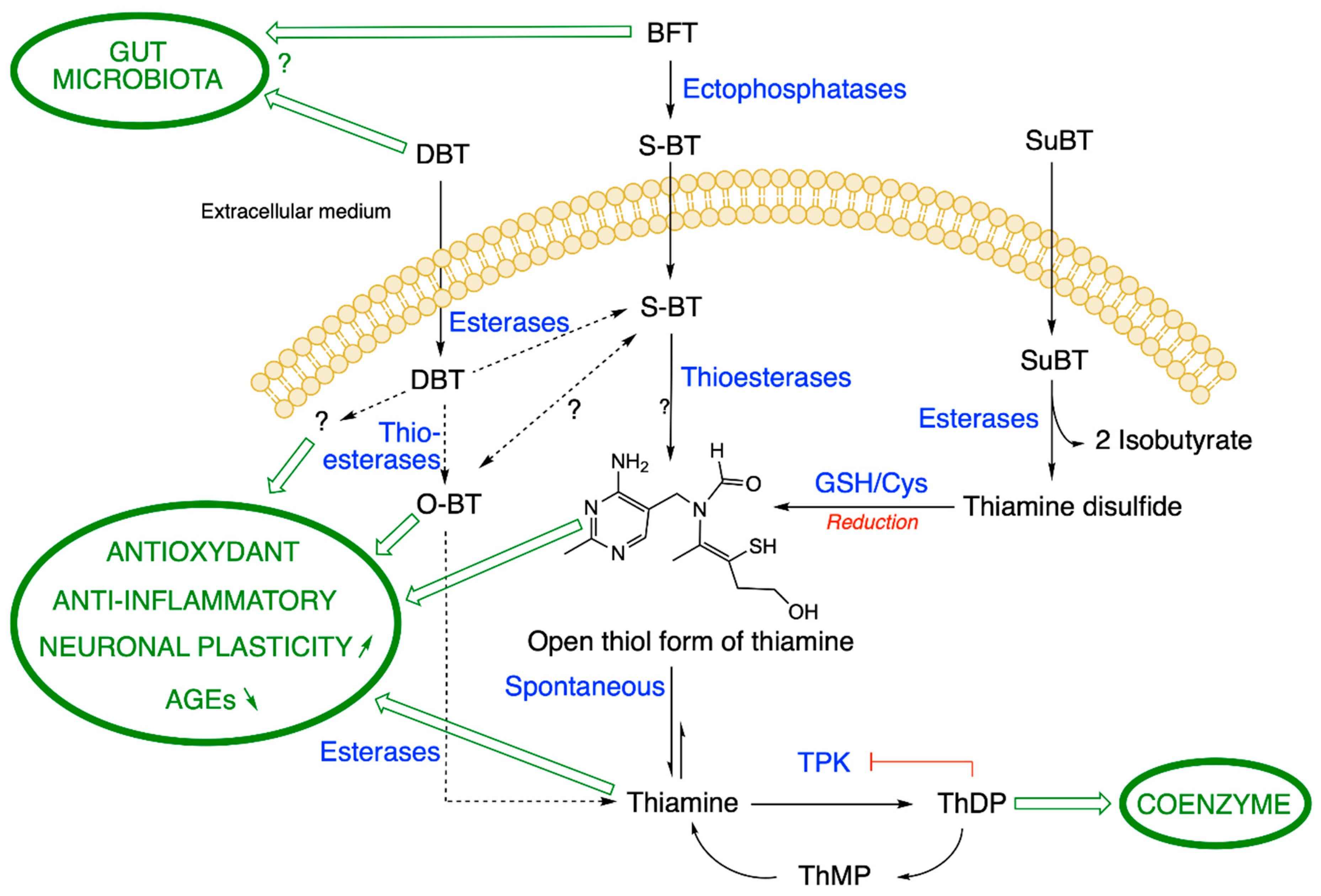
2.2. Metabolism of BFT
2.3. Overview of the Beneficial Effects of BFT Treatment in Animal Models and Humans
2.3.1. Beneficial Effects of BFT in Mouse Models of Brain Disorders
2.3.2. BFT Treatment in Clinical Studies of Patients with Mild AD
2.4. Mechanism of Action of Benfotiamine
2.4.1. Effects on Glucose Metabolism and Mitochondrial Function
2.4.2. Effects of BFT on Glycogen Synthase Kinase 3 (GSK3)
2.4.3. Possible Involvement of the PI3K/AKT Pathway in Neuroprotection by BFT
2.4.4. Effects of BFT on the Accumulation of AGEs
2.4.5. Antioxidant Effects of BFT
2.4.6. Anti-Inflammatory Effects of BFT
2.4.7. Effects of BFT on Glutamate Receptors, Synaptic Plasticity and Neurogenesis
3. Neuroprotective Properties of Dibenzoylthiamine
4. Conclusions and Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| AD | Alzheimer’s disease |
| AGE(s) | advanced glycation end product(s) |
| APP | amyloid precursor protein |
| DBT | dibenzoylthiamine |
| GSH | reduced glutathione |
| GSK | glycogen synthase kinase |
| LPS | lipopolysaccharide |
| O-BT | O-benzoylthiamine |
| ROS | reactive oxygen species |
| OGDHC | 2-oxoglutarate dehydrogenase complex |
| PDHC | pyruvate dehydrogenase complex |
| S-BT | S-benzoylthiamine |
| SuBT | sulbutiamine |
| TD | thiamine deficiency |
| ThDP | thiamine diphosphate |
| ThMP | thiamine monophosphate |
| TKT | transketolase |
| TPK | thiamine pyrophosphokinase |
| TTFD | thiamine tetrahydrofurfuryl disulfide |
References
- Jansen, B.C.P.; Donath, W.F. On the Isolation of Anti-Beriberi Vitamin. Proc. Kon. Ned. Akad. Wet. 1926, 29, 1390–1400. [Google Scholar]
- Grijns, G. Over Polyneuritis Gallinarum. I. Geneesk Tijdscht Ned. Ind. 1901, 41, 3–110. [Google Scholar]
- Peters, R.A. The Biochemical Lesion in Vitamin B1 Deficiency. Application of Modern Biochemical Analysis in Its Diagnosis. Lancet 1936, 1, 1161–1164. [Google Scholar] [CrossRef]
- Lohmann, K.; Schuster, P. Untersuchungen Über Die Cocarboxylase. Biochem. Z. 1937, 294, 188. [Google Scholar]
- McCandless, D.W. Thiamine Deficiency and Associated Clinical Disorders. In Contemporary Clinical Neuroscience, 1st ed.; Humana Press: Totowa, NJ, USA, 2010. [Google Scholar]
- Page, M.G.; Ankoma-Sey, V.; Coulson, W.F.; Bender, D.A. Brain Glutamate and Gamma-Aminobutyrate (GABA) Metabolism in Thiamin- Deficient Rats. Br. J. Nutr. 1989, 62, 245–253. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Plaitakis, A.; Hwang, E.C.; Woert, M.H.; Szilagyi, P.E.; Berl, S. Effect of Thiamin Deficiency on Brain Neurotransmitter Systems. Ann. N. Y. Acad. Sci. 1982, 378, 367–381. [Google Scholar] [CrossRef] [PubMed]
- Bettendorff, L. Basic Nutrition and Metabolism. In Present Knowledge in Nutrition, 11th ed.; Marriott, B., Birt, D.F., Stalling, V., Yates, A., Eds.; Academic Press: Cambridge, MA, USA, 2020; Volume 1, p. 676. ISBN 978-0-323-66162-1. [Google Scholar]
- Whitfield, K.C.; Bourassa, M.W.; Adamolekun, B.; Bergeron, G.; Bettendorff, L.; Brown, K.H.; Cox, L.; Fattal-Valevski, A.; Fischer, P.R.; Frank, E.L.; et al. Thiamine Deficiency Disorders: Diagnosis, Prevalence, and a Roadmap for Global Control Programs. Ann N. Y. Acad. Sci. 2018, 1430, 3–43. [Google Scholar] [CrossRef] [PubMed]
- Smith, T.J.; Johnson, C.R.; Koshy, R.; Hess, S.Y.; Qureshi, U.A.; Mynak, M.L.; Fischer, P.R. Thiamine Deficiency Disorders: A Clinical Perspective. Ann. N. Y. Acad. Sci. 2020. [Google Scholar] [CrossRef]
- Calingasan, N.Y.; Chun, W.J.; Park, L.C.; Uchida, K.; Gibson, G.E. Oxidative Stress Is Associated with Region-Specific Neuronal Death during Thiamine Deficiency. J. Neuropathol. Exp. Neurol. 1999, 58, 946–958. [Google Scholar] [CrossRef]
- Karuppagounder, S.S.; Shi, Q.; Xu, H.; Gibson, G.E. Changes in Inflammatory Processes Associated with Selective Vulnerability Following Mild Impairment of Oxidative Metabolism. Neurobiol. Dis. 2007, 26, 353–362. [Google Scholar] [CrossRef] [Green Version]
- Hazell, A.S.; Faim, S.; Wertheimer, G.; Silva, V.R.; Marques, C.S. The Impact of Oxidative Stress in Thiamine Deficiency: A Multifactorial Targeting Issue. Neurochem. Int. 2013, 62, 796–802. [Google Scholar] [CrossRef] [PubMed]
- Gibson, G.E.; Hirsch, J.A.; Fonzetti, P.; Jordan, B.D.; Cirio, R.T.; Elder, J. Vitamin B1 (Thiamine) and Dementia. Ann. N. Y. Acad. Sci. 2016, 1367, 21–30. [Google Scholar] [CrossRef]
- Chandrakumar, A.; Bhardwaj, A.; W’t Jong, G. Review of Thiamine Deficiency Disorders: Wernicke Encephalopathy and Korsakoff Psychosis. J. Basic Clin. Physiol. Pharmacol. 2018, 30, 153–162. [Google Scholar] [CrossRef]
- Bettendorff, L. Thiamine in Excitable Tissues: Reflections on a Non-Cofactor Role. Metab. Brain Dis. 1994, 9, 183–209. [Google Scholar] [CrossRef]
- Bettendorff, L.; Wins, P. Thiamin Diphosphate in Biological Chemistry: New Aspects of Thiamin Metabolism, Especially Triphosphate Derivatives Acting Other than as Cofactors. FEBS J. 2009, 276, 2917–2925. [Google Scholar] [CrossRef] [PubMed]
- Aleshin, V.A.; Mkrtchyan, G.V.; Bunik, V.I. Mechanisms of Non-Coenzyme Action of Thiamine: Protein Targets and Medical Significance. Biochem. Biokhimiia 2019, 84, 829–850. [Google Scholar] [CrossRef] [PubMed]
- Said, H.M. Intestinal Absorption of Water-Soluble Vitamins in Health and Disease. Biochem. J. 2011, 437, 357–372. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Greenwood, J.; Love, E.R.; Pratt, O.E. Kinetics of Thiamine Transport across the Blood-Brain Barrier in the Rat. J. Physiol. 1982, 327, 95–103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lonsdale, D. A Review of the Biochemistry, Metabolism and Clinical Benefits of Thiamin(e) and Its Derivatives. Evid. Based Complementary Altern. Med. 2006, 3, 49–59. [Google Scholar] [CrossRef]
- Starling-Soares, B.; Carrera-Bastos, P.; Bettendorff, L. Role of the Synthetic B1 Vitamin Sulbutiamine on Health. J. Nutr. Metab. 2020, 2020, 9349063. [Google Scholar] [CrossRef]
- Lonsdale, D.; Shamberger, R.J.; Audhya, T. Treatment of Autism Spectrum Children with Thiamine Tetrahydrofurfuryl Disulfide: A Pilot Study. Neuroendocr. Lett. 2002, 23, 303–308. [Google Scholar]
- Hammes, H.P.; Du, X.; Edelstein, D.; Taguchi, T.; Matsumura, T.; Ju, Q.; Lin, J.; Bierhaus, A.; Nawroth, P.; Hannak, D.; et al. Benfotiamine Blocks Three Major Pathways of Hyperglycemic Damage and Prevents Experimental Diabetic Retinopathy. Nat. Med. 2003, 9, 294–299. [Google Scholar] [CrossRef]
- Pan, X.; Gong, N.; Zhao, J.; Yu, Z.; Gu, F.; Chen, J.; Sun, X.; Zhao, L.; Yu, M.; Xu, Z.; et al. Powerful Beneficial Effects of Benfotiamine on Cognitive Impairment and Beta-Amyloid Deposition in Amyloid Precursor Protein/Presenilin-1 Transgenic Mice. Brain 2010, 133, 1342–1351. [Google Scholar] [CrossRef] [PubMed]
- Tapias, V.; Jainuddin, S.; Ahuja, M.; Stack, C.; Elipenahli, C.; Vignisse, J.; Gerges, M.; Starkova, N.; Xu, H.; Starkov, A.A.; et al. Benfotiamine Treatment Activates the Nrf2/ARE Pathway and Is Neuroprotective in a Transgenic Mouse Model of Tauopathy. Hum. Mol. Genet. 2018, 27, 2874–2892. [Google Scholar] [CrossRef] [PubMed]
- Gibson, G.E.; Luchsinger, J.A.; Cirio, R.; Chen, H.; Franchino-Elder, J.; Hirsch, J.A.; Bettendorff, L.; Chen, Z.; Flowers, S.; Gerber, L.; et al. Benfotiamine and Cognitive Decline in Alzheimer’s Disease: Results of a Randomized Placebo-Controlled Phase IIa Clinical Trial. J. Alzheimers Dis. JAD 2020, 78, 989–1010. [Google Scholar] [CrossRef] [PubMed]
- Pan, X.; Chen, Z.; Fei, G.; Pan, S.; Bao, W.; Ren, S.; Guan, Y.; Zhong, C. Long-Term Cognitive Improvement after Benfotiamine Administration in Patients with Alzheimer’s Disease. Neurosci. Bull. 2016, 32, 591–596. [Google Scholar] [CrossRef]
- Sambon, M.; Gorlova, A.; Demelenne, A.; Alhama-Riba, J.; Coumans, B.; Lakaye, B.; Wins, P.; Fillet, M.; Anthony, D.C.; Strekalova, T.; et al. Dibenzoylthiamine Has Powerful Antioxidant and Anti-Inflammatory Properties in Cultured Cells and in Mouse Models of Stress and Neurodegeneration. Biomedicines 2020, 8, 361. [Google Scholar] [CrossRef]
- Loew, D. Pharmacokinetics of Thiamine Derivatives Especially of Benfotiamine. Int. J. Clin. Pharmacol. Ther. 1996, 34, 47–50. [Google Scholar]
- Lonsdale, D. Benfotiamine and Allithiamine Should Be Differentiated. Townsend Lett. Dr. Patients 2004, 257, 102. [Google Scholar]
- Fujiwara, M.; Watanabe, H.; Katsui, K. Allithiamine, a Newly Found Derivative of Vitamin B1. J. Biochem. 1954, 41, 29–39. [Google Scholar] [CrossRef]
- Sambon, M.; Napp, A.; Demelenne, A.; Vignisse, J.; Wins, P.; Fillet, M.; Bettendorff, L. Thiamine and Benfotiamine Protect Neuroblastoma Cells against Paraquat and β-Amyloid Toxicity by a Coenzyme-Independent Mechanism. Heliyon 2019, 5, e01710. [Google Scholar] [CrossRef] [Green Version]
- Sheng, L.; Cao, W.; Lin, P.; Chen, W.; Xu, H.; Zhong, C.; Yuan, F.; Chen, H.; Li, H.; Liu, C.; et al. Safety, Tolerability and Pharmacokinetics of Single and Multiple Ascending Doses of Benfotiamine in Healthy Subjects. Drug Des. Devel. Ther. 2021, 15, 1101–1110. [Google Scholar] [CrossRef] [PubMed]
- Bitsch, R.; Wolf, M.; Moller, J.; Heuzeroth, L.; Gruneklee, D. Bioavailability Assessment of the Lipophilic Benfotiamine as Compared to a Water-Soluble Thiamin Derivative. Ann. Nutr. Metab. 1991, 35, 292–296. [Google Scholar] [CrossRef]
- Gangolf, M.; Czerniecki, J.; Radermecker, M.; Detry, O.; Nisolle, M.; Jouan, C.; Martin, D.; Chantraine, F.; Lakaye, B.; Wins, P.; et al. Thiamine Status in Humans and Content of Phosphorylated Thiamine Derivatives in Biopsies and Cultured Cells. PLoS ONE 2010, 5, e13616. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, J.; Jonus, H.C.; Zastre, J.A.; Bartlett, M.G. Development of an IPRP-LC-MS/MS Method to Determine the Fate of Intracellular Thiamine in Cancer Cells. J. Chromatogr. B Analyt. Technol. Biomed. Life. Sci. 2019, 1124, 247–255. [Google Scholar] [CrossRef]
- Jonus, H.C.; Byrnes, C.C.; Kim, J.; Valle, M.L.; Bartlett, M.G.; Said, H.M.; Zastre, J.A. Thiamine Mimetics Sulbutiamine and Benfotiamine as a Nutraceutical Approach to Anticancer Therapy. Biomed. Pharm. Biomed. Pharm. 2020, 121, 109648. [Google Scholar] [CrossRef] [PubMed]
- Duclos, J.M.; Haake, P. Ring Opening of Thiamine Analogs. The Role of Ring Opening in Physiological Function. Biochemistry 1974, 13, 5358–5362. [Google Scholar] [CrossRef]
- Hurt, J.K.; Coleman, J.L.; Fitzpatrick, B.J.; Taylor-Blake, B.; Bridges, A.S.; Vihko, P.; Zylka, M.J. Prostatic Acid Phosphatase Is Required for the Antinociceptive Effects of Thiamine and Benfotiamine. PLoS ONE 2012, 7, e48562. [Google Scholar] [CrossRef] [Green Version]
- Volvert, M.L.; Seyen, S.; Piette, M.; Evrard, B.; Gangolf, M.; Plumier, J.C.; Bettendorff, L. Benfotiamine, a Synthetic S-Acyl Thiamine Derivative, Has Different Mechanisms of Action and a Different Pharmacological Profile than Lipid-Soluble Thiamine Disulfide Derivatives. BMC Pharmacol. 2008, 8, 10. [Google Scholar] [CrossRef] [Green Version]
- de Moraes, R.C.M.; Singulani, M.P.; de Gonçalves, A.C.; Portari, G.V.; da Silva Torrão, A. Oral Benfotiamine Reverts Cognitive Deficit and Increase Thiamine Diphosphate Levels in the Brain of a Rat Model of Neurodegeneration. Exp. Gerontol. 2020, 141, 111097. [Google Scholar] [CrossRef]
- Voskoboyev, A.I.; Ostrovsky, Y.M. Thiamin Pyrophosphokinase: Structure, Properties, and Role in Thiamin Metabolism. Ann. N. Y. Acad. Sci. 1982, 378, 161–176. [Google Scholar] [CrossRef] [PubMed]
- Thornalley, P.J. The Potential Role of Thiamine (Vitamin B1) in Diabetic Complications. Curr. Diabetes Rev. 2005, 1, 287–298. [Google Scholar] [CrossRef]
- Marchetti, V.; Menghini, R.; Rizza, S.; Vivanti, A.; Feccia, T.; Lauro, D.; Fukamizu, A.; Lauro, R.; Federici, M. Benfotiamine Counteracts Glucose Toxicity Effects on Endothelial Progenitor Cell Differentiation via Akt/FoxO Signaling. Diabetes 2006, 55, 2231–2237. [Google Scholar] [CrossRef] [Green Version]
- Wu, S.; Ren, J. Benfotiamine Alleviates Diabetes-Induced Cerebral Oxidative Damage Independent of Advanced Glycation End-Product, Tissue Factor and TNF-Alpha. Neurosci. Lett. 2006, 394, 158–162. [Google Scholar] [CrossRef]
- Ceylan-Isik, A.F.; Wu, S.; Li, Q.; Li, S.Y.; Ren, J. High-Dose Benfotiamine Rescues Cardiomyocyte Contractile Dysfunction in Streptozotocin-Induced Diabetes Mellitus. J. Appl. Physiol. 2006, 100, 150–156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Markova, N.; Bazhenova, N.; Anthony, D.C.; Vignisse, J.; Svistunov, A.; Lesch, K.-P.; Bettendorff, L.; Strekalova, T. Thiamine and Benfotiamine Improve Cognition and Ameliorate GSK-3β-Associated Stress-Induced Behaviours in Mice. Prog. Neuropsychopharmacol. Biol. Psychiatry 2017, 75, 148–156. [Google Scholar] [CrossRef] [PubMed]
- Vignisse, J.; Sambon, M.; Gorlova, A.; Pavlov, D.; Caron, N.; Malgrange, B.; Shevtsova, E.; Svistunov, A.; Anthony, D.C.; Markova, N.; et al. Thiamine and Benfotiamine Prevent Stress-Induced Suppression of Hippocampal Neurogenesis in Mice Exposed to Predation without Affecting Brain Thiamine Diphosphate Levels. Mol. Cell. Neurosci. 2017, 82, 126–136. [Google Scholar] [CrossRef]
- Gorlova, A.; Pavlov, D.; Anthony, D.C.; Ponomarev, E.; Sambon, M.; Proshin, A.; Shafarevich, I.; Babaevskaya, D.; Lesch, K.-P.; Bettendorff, L.; et al. Thiamine and Benfotiamine Counteract Ultrasound-Induced Aggression, Normalize AMPA Receptor Expression and Plasticity Markers, and Reduce Oxidative Stress in Mice. Neuropharmacology 2019, 156, 107543. [Google Scholar] [CrossRef] [PubMed]
- Nolan, K.A.; Black, R.S.; Sheu, K.F.; Langberg, J.; Blass, J.P. A Trial of Thiamine in Alzheimer’s Disease. Arch. Neurol. 1991, 48, 81–83. [Google Scholar] [CrossRef]
- Blass, J.P.; Gleason, P.; Brush, D.; DiPonte, P.; Thaler, H. Thiamine and Alzheimer’s Disease. A Pilot Study. Arch. Neurol. 1988, 45, 833–835. [Google Scholar] [CrossRef]
- Mimori, Y.; Katsuoka, H.; Nakamura, S. Thiamine Therapy in Alzheimer’s Disease. Metab. Brain Dis. 1996, 11, 89–94. [Google Scholar] [CrossRef] [PubMed]
- Brownlee, M. The Pathobiology of Diabetic Complications: A Unifying Mechanism. Diabetes 2005, 54, 1615–1625. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gibson, G.E.; Blass, J.P. Thiamine-Dependent Processes and Treatment Strategies in Neurodegeneration. Antioxid. Redox Signal. 2007, 9, 1605–1619. [Google Scholar] [CrossRef] [PubMed]
- Steen, E.; Terry, B.M.; Rivera, E.J.; Cannon, J.L.; Neely, T.R.; Tavares, R.; Xu, X.J.; Wands, J.R.; de la Monte, S.M. Impaired Insulin and Insulin-like Growth Factor Expression and Signaling Mechanisms in Alzheimer’s Disease--Is This Type 3 Diabetes? J. Alzheimers Dis. JAD 2005, 7, 63–80. [Google Scholar] [CrossRef] [Green Version]
- Butterworth, R.F.; Besnard, A.M. Thiamine-Dependent Enzyme Changes in Temporal Cortex of Patients with Alzheimer’s Disease. Metab. Brain Dis. 1990, 5, 179–184. [Google Scholar] [CrossRef]
- Mastrogiacomo, F.; Bergeron, C.; Kish, S.J. Brain Alpha-Ketoglutarate Dehydrogenase Complex Activity in Alzheimer’s Disease. J. Neurochem. 1993, 61, 2007–2014. [Google Scholar] [CrossRef]
- Mastrogiacomo, F.; Bettendorff, L.; Grisar, T.; Kish, S.J. Brain Thiamine, Its Phosphate Esters, and Its Metabolizing Enzymes in Alzheimer’s Disease. Ann. Neurol. 1996, 39, 585–591. [Google Scholar] [CrossRef]
- Karuppagounder, S.S.; Xu, H.; Shi, Q.; Chen, L.H.; Pedrini, S.; Pechman, D.; Baker, H.; Beal, M.F.; Gandy, S.E.; Gibson, G.E. Thiamine Deficiency Induces Oxidative Stress and Exacerbates the Plaque Pathology in Alzheimer’s Mouse Model. Neurobiol. Aging 2009, 30, 1587–1600. [Google Scholar] [CrossRef] [Green Version]
- Lin, M.T.; Beal, M.F. Mitochondrial Dysfunction and Oxidative Stress in Neurodegenerative Diseases. Nature 2006, 443, 787–795. [Google Scholar] [CrossRef]
- Johri, A.; Beal, M.F. Mitochondrial Dysfunction in Neurodegenerative Diseases. J. Pharmacol. Exp. Ther. 2012, 342, 619–630. [Google Scholar] [CrossRef] [Green Version]
- Liang, W.S.; Reiman, E.M.; Valla, J.; Dunckley, T.; Beach, T.G.; Grover, A.; Niedzielko, T.L.; Schneider, L.E.; Mastroeni, D.; Caselli, R.; et al. Alzheimer’s Disease Is Associated with Reduced Expression of Energy Metabolism Genes in Posterior Cingulate Neurons. Proc. Natl. Acad. Sci. USA 2008, 105, 4441–4446. [Google Scholar] [CrossRef] [Green Version]
- Lee, T.; Lee, H. Shared Blood Transcriptomic Signatures between Alzheimer’s Disease and Diabetes Mellitus. Biomedicines 2021, 9, 34. [Google Scholar] [CrossRef] [PubMed]
- Beurel, E.; Grieco, S.F.; Jope, R.S. Glycogen Synthase Kinase-3 (GSK3): Regulation, Actions, and Diseases. Pharmacol. Ther. 2015, 148, 114–131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaidanovich-Beilin, O.; Woodgett, J.R. GSK-3: Functional Insights from Cell Biology and Animal Models. Front. Mol. Neurosci. 2011, 4, 40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Woodgett, J.R. Molecular Cloning and Expression of Glycogen Synthase Kinase-3/Factor A. EMBO J. 1990, 9, 2431–2438. [Google Scholar] [CrossRef] [PubMed]
- Perez-Costas, E.; Gandy, J.C.; Melendez-Ferro, M.; Roberts, R.C.; Bijur, G.N. Light and Electron Microscopy Study of Glycogen Synthase Kinase-3beta in the Mouse Brain. PLoS ONE 2010, 5, e8911. [Google Scholar] [CrossRef]
- Peineau, S.; Taghibiglou, C.; Bradley, C.; Wong, T.P.; Liu, L.; Lu, J.; Lo, E.; Wu, D.; Saule, E.; Bouschet, T.; et al. LTP Inhibits LTD in the Hippocampus via Regulation of GSK3beta. Neuron 2007, 53, 703–717. [Google Scholar] [CrossRef] [Green Version]
- Pardo, M.; Abrial, E.; Jope, R.S.; Beurel, E. GSK3β Isoform-Selective Regulation of Depression, Memory and Hippocampal Cell Proliferation. Genes Brain Behav. 2016, 15, 348–355. [Google Scholar] [CrossRef]
- Cross, D.A.; Alessi, D.R.; Cohen, P.; Andjelkovich, M.; Hemmings, B.A. Inhibition of Glycogen Synthase Kinase-3 by Insulin Mediated by Protein Kinase B. Nature 1995, 378, 785–789. [Google Scholar] [CrossRef]
- Beaulieu, J.-M.; Gainetdinov, R.R.; Caron, M.G. Akt/GSK3 Signaling in the Action of Psychotropic Drugs. Annu. Rev. Pharmacol. Toxicol. 2009, 49, 327–347. [Google Scholar] [CrossRef] [Green Version]
- Hooper, C.; Killick, R.; Lovestone, S. The GSK3 Hypothesis of Alzheimer’s Disease. J. Neurochem. 2008, 104, 1433–1439. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takashima, A. GSK-3 Is Essential in the Pathogenesis of Alzheimer’s Disease. J. Alzheimers Dis. 2006, 9, 309–317. [Google Scholar] [CrossRef] [PubMed]
- Rockenstein, E.; Torrance, M.; Adame, A.; Mante, M.; Bar-on, P.; Rose, J.B.; Crews, L.; Masliah, E. Neuroprotective Effects of Regulators of the Glycogen Synthase Kinase-3beta Signaling Pathway in a Transgenic Model of Alzheimer’s Disease Are Associated with Reduced Amyloid Precursor Protein Phosphorylation. J. Neurosci. Off. J. Soc. Neurosci. 2007, 27, 1981–1991. [Google Scholar] [CrossRef]
- Ly, P.T.T.; Wu, Y.; Zou, H.; Wang, R.; Zhou, W.; Kinoshita, A.; Zhang, M.; Yang, Y.; Cai, F.; Woodgett, J.; et al. Inhibition of GSK3β-Mediated BACE1 Expression Reduces Alzheimer-Associated Phenotypes. J. Clin. Investig. 2013, 123, 224–235. [Google Scholar] [CrossRef] [Green Version]
- Kuhla, A.; Ludwig, S.C.; Kuhla, B.; Münch, G.; Vollmar, B. Advanced Glycation End Products Are Mitogenic Signals and Trigger Cell Cycle Reentry of Neurons in Alzheimer’s Disease Brain. Neurobiol. Aging 2015, 36, 753–761. [Google Scholar] [CrossRef]
- Langlais, P.J.; Anderson, G.; Guo, S.X.; Bondy, S.C. Increased Cerebral Free Radical Production during Thiamine Deficiency. Metab. Brain Dis. 1997, 12, 137–143. [Google Scholar] [CrossRef] [PubMed]
- Tebay, L.E.; Robertson, H.; Durant, S.T.; Vitale, S.R.; Penning, T.M.; Dinkova-Kostova, A.T.; Hayes, J.D. Mechanisms of Activation of the Transcription Factor Nrf2 by Redox Stressors, Nutrient Cues, and Energy Status and the Pathways through Which It Attenuates Degenerative Disease. Free Radic. Biol. Med. 2015, 88, 108–146. [Google Scholar] [CrossRef] [Green Version]
- Bozic, I.; Savic, D.; Stevanovic, I.; Pekovic, S.; Nedeljkovic, N.; Lavrnja, I. Benfotiamine Upregulates Antioxidative System in Activated BV-2 Microglia Cells. Front. Cell. Neurosci. 2015, 9, 351. [Google Scholar] [CrossRef] [Green Version]
- Hansen, D.V.; Hanson, J.E.; Sheng, M. Microglia in Alzheimer’s Disease. J. Cell Biol. 2018, 217, 459–472. [Google Scholar] [CrossRef]
- Hazell, A.S.; Butterworth, R.F. Update of Cell Damage Mechanisms in Thiamine Deficiency: Focus on Oxidative Stress, Excitotoxicity and Inflammation. Alcohol. Alcohol. 2009, 44, 141–147. [Google Scholar] [CrossRef] [Green Version]
- Todd, K.G.; Butterworth, R.F. Early Microglial Response in Experimental Thiamine Deficiency: An Immunohistochemical Analysis. Glia 1999, 25, 190–198. [Google Scholar] [CrossRef]
- Sanchez-Ramirez, G.M.; Caram-Salas, N.L.; Rocha-Gonzalez, H.I.; Vidal-Cantu, G.C.; Medina-Santillan, R.; Reyes-Garcia, G.; Granados-Soto, V. Benfotiamine Relieves Inflammatory and Neuropathic Pain in Rats. Eur. J. Pharmacol. 2006, 530, 48–53. [Google Scholar] [CrossRef] [PubMed]
- Yadav, U.C.S.; Kalariya, N.M.; Srivastava, S.K.; Ramana, K.V. Protective Role of Benfotiamine, a Fat-Soluble Vitamin B1 Analogue, in Lipopolysaccharide-Induced Cytotoxic Signals in Murine Macrophages. Free Radic. Biol. Med. 2010, 48, 1423–1434. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bozic, I.; Savic, D.; Laketa, D.; Bjelobaba, I.; Milenkovic, I.; Pekovic, S.; Nedeljkovic, N.; Lavrnja, I. Benfotiamine Attenuates Inflammatory Response in LPS Stimulated BV-2 Microglia. PLoS ONE 2015, 10, e0118372. [Google Scholar] [CrossRef] [Green Version]
- Shoeb, M.; Ramana, K.V. Anti-Inflammatory Effects of Benfotiamine Are Mediated through the Regulation of the Arachidonic Acid Pathway in Macrophages. Free Radic. Biol. Med. 2012, 52, 182–190. [Google Scholar] [CrossRef] [Green Version]
- Vatsalya, V.; Li, F.; Frimodig, J.; Gala, K.S.; Srivastava, S.; Kong, M.; Ramchandani, V.A.; Feng, W.; Zhang, X.; McClain, C.J. Repurposing Treatment of Wernicke-Korsakoff Syndrome for Th-17 Cell Immune Storm Syndrome and Neurological Symptoms in COVID-19: Thiamine Efficacy and Safety, In-Vitro Evidence and Pharmacokinetic Profile. Front. Pharmacol. 2021, 11, 598128. [Google Scholar] [CrossRef]
- Clayton, D.A.; Grosshans, D.R.; Browning, M.D. Aging and Surface Expression of Hippocampal NMDA Receptors. J. Biol. Chem. 2002, 277, 14367–14369. [Google Scholar] [CrossRef] [Green Version]
- Heywood, R.; Wood, J.D.; Majeed, S.K. Tumorigenic and Toxic Effect of O,S-Dibenzoyl Thiamine Hydrochloride in Prolonged Dietary Administration to Rats. Toxicol. Lett. 1985, 26, 53–58. [Google Scholar] [CrossRef]
- Ketola, H.G.; Isaacs, G.R.; Robins, J.S.; Lloyd, R.C. Effectiveness and Retention of Thiamine and Its Analogs Administered to Steelhead and Landlocked Atlantic Salmon. J. Aquat. Anim. Health 2008, 20, 29–38. [Google Scholar] [CrossRef]
- Safavi, M.; Hosseini-Sharifabad, A.; Seyed-Yousefi, Y.; Rabbani, M. Protective Effects of Citicoline and Benfotiamine Each Alone and in Combination on Streptozotocin-Induced Memory Impairment in Mice. Clin. Psychopharmacol. Neurosci. Off. Sci. J. Korean Coll. Neuropsychopharmacol. 2020, 18, 81–92. [Google Scholar] [CrossRef]
- Putnam, E.E.; Goodman, A.L. B Vitamin Acquisition by Gut Commensal Bacteria. PLoS Pathog. 2020, 16, e1008208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Costliow, Z.A.; Degnan, P.H. Thiamine Acquisition Strategies Impact Metabolism and Competition in the Gut Microbe Bacteroides Thetaiotaomicron. mSystems 2017, 2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fung, T.C.; Olson, C.A.; Hsiao, E.Y. Interactions between the Microbiota, Immune and Nervous Systems in Health and Disease. Nat. Neurosci. 2017, 20, 145–155. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Properties | BFT | DBT |
|---|---|---|
| Physicochemical properties | ||
| Solubility in organic solvents | No | Yes |
| Solubility in aqueous solutions | Yes (pH > 8) | Yes (pH < 6) |
| Metabolization | ||
| Enzymes [29,33] | Ectophosphatases/thioesterases | Estererases/thioesterases |
| Main metabolites [29,33] | Thiamine, S-BT | Thiamine, O-BT (?) |
| Pharmacological effects | ||
| Antioxidant effects (Nrf2-independent) | Yes (≤50 µM) [33] | Yes (≤50 µM) (↑ GSH and NADPH) [29] |
| Anti-inflammatory effects (probably via NF-κB) | ↓ iNOS and TNF-α [29,86] | ↓ iNOS and TNF-α [29] |
| Anti-AGEs effects | ↓ In blood of AD patients [27] | Not tested |
| Neuroprotective effects in neurodegenerative diseases and models | Slows down cognitive decline in AD patients [27,28] Decreases β-amyloid load and tauopathy in mouse model of AD [25] | Not tested in AD, but arrests motor dysfunction in a mouse model of amyotrophic lateral sclerosis, and relieves depressive-like behavior in mice submitted to chronic ultrasound stress |
| Effects on neuronal plasticity | ↑ NMDAR AMPAR expression 1 ↑ Neurogenesis 2 | Not tested |
| Possible molecular targets | ||
| TKT | Possibly increased activity [24] | No effect [29] |
| GSK3β | Inhibition by phosphorylation [25,42] Reduced expression [48] | Not tested |
| PI3K/AKT pathway | Activation [46] | Not tested |
| Nrf-2 | At concentrations > 100 µM [26] | Not tested |
| NF-κB | Inhibits LPS-induced nuclear translocation [29,86] | Inhibits LPS-induced nuclear translocation [29] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sambon, M.; Wins, P.; Bettendorff, L. Neuroprotective Effects of Thiamine and Precursors with Higher Bioavailability: Focus on Benfotiamine and Dibenzoylthiamine. Int. J. Mol. Sci. 2021, 22, 5418. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22115418
Sambon M, Wins P, Bettendorff L. Neuroprotective Effects of Thiamine and Precursors with Higher Bioavailability: Focus on Benfotiamine and Dibenzoylthiamine. International Journal of Molecular Sciences. 2021; 22(11):5418. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22115418
Chicago/Turabian StyleSambon, Margaux, Pierre Wins, and Lucien Bettendorff. 2021. "Neuroprotective Effects of Thiamine and Precursors with Higher Bioavailability: Focus on Benfotiamine and Dibenzoylthiamine" International Journal of Molecular Sciences 22, no. 11: 5418. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22115418