
Congenital Deletion of Nedd4-2 in Lung Epithelial Cells Causes Progressive Alveolitis and Pulmonary Fibrosis in Neonatal Mice

 , , , ,
, , , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
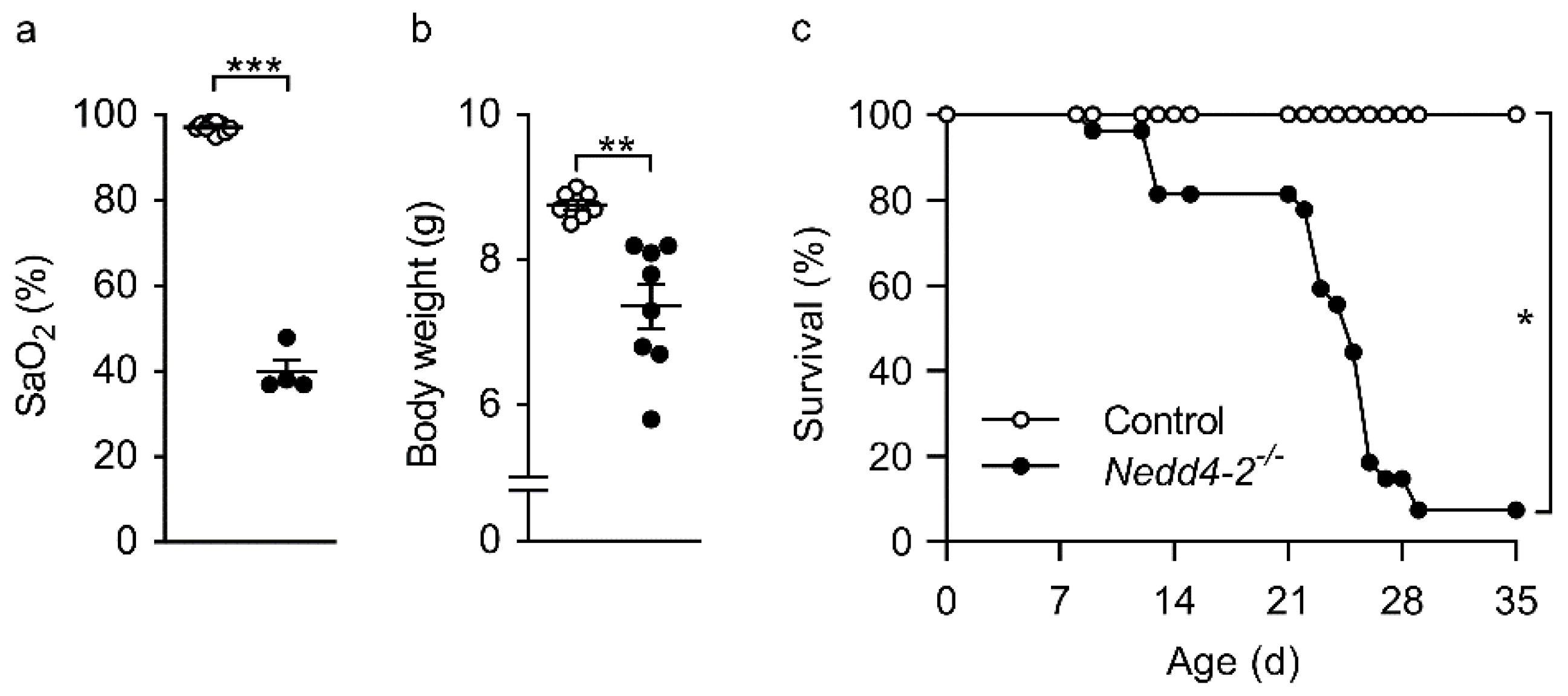
2.1. Congenital Deletion of Nedd4-2 in Lung Epithelial Cells Causes Severe Hypoxemia, Failure to Thrive and Early Mortality in Neonatal Mice
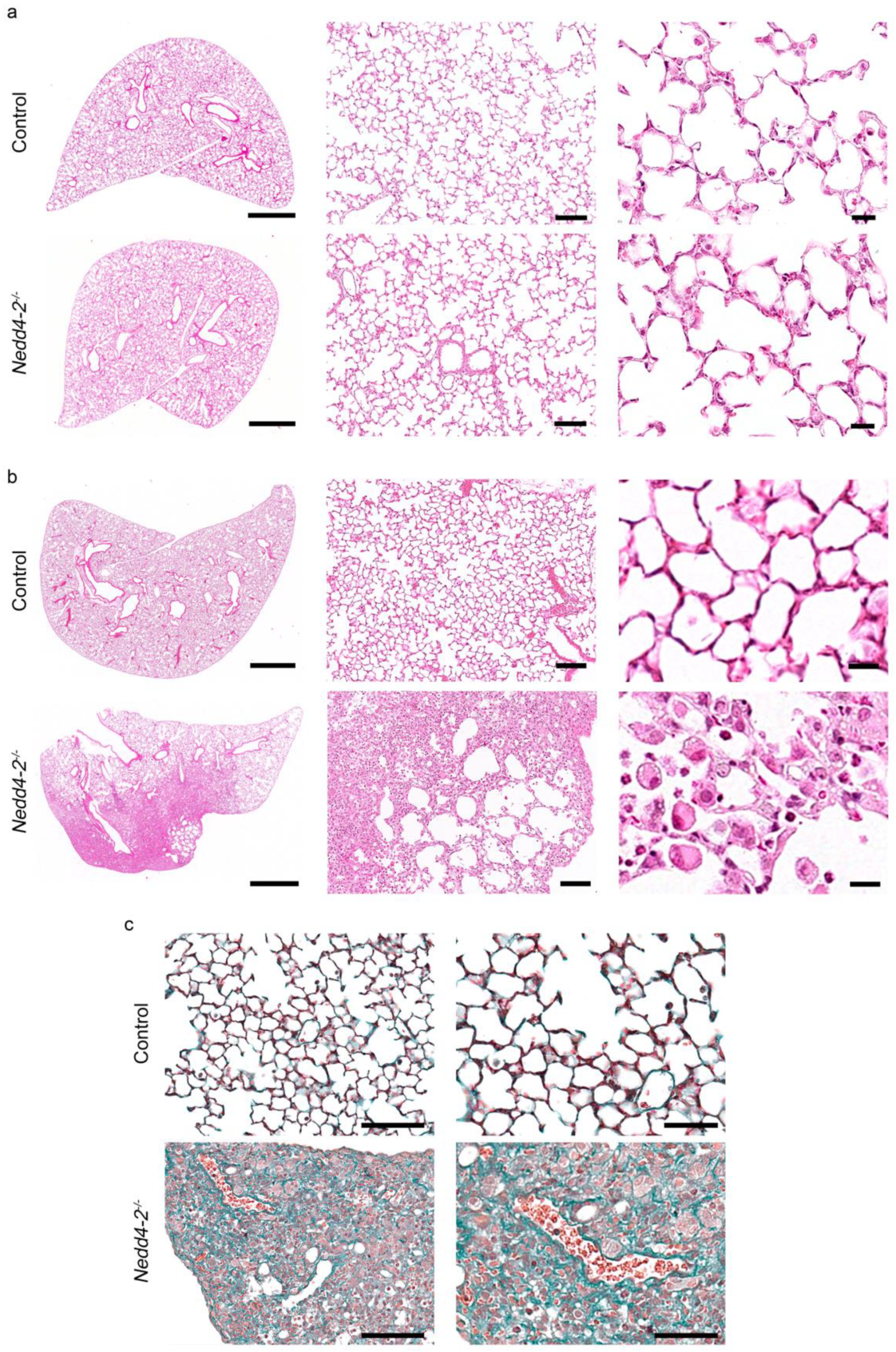
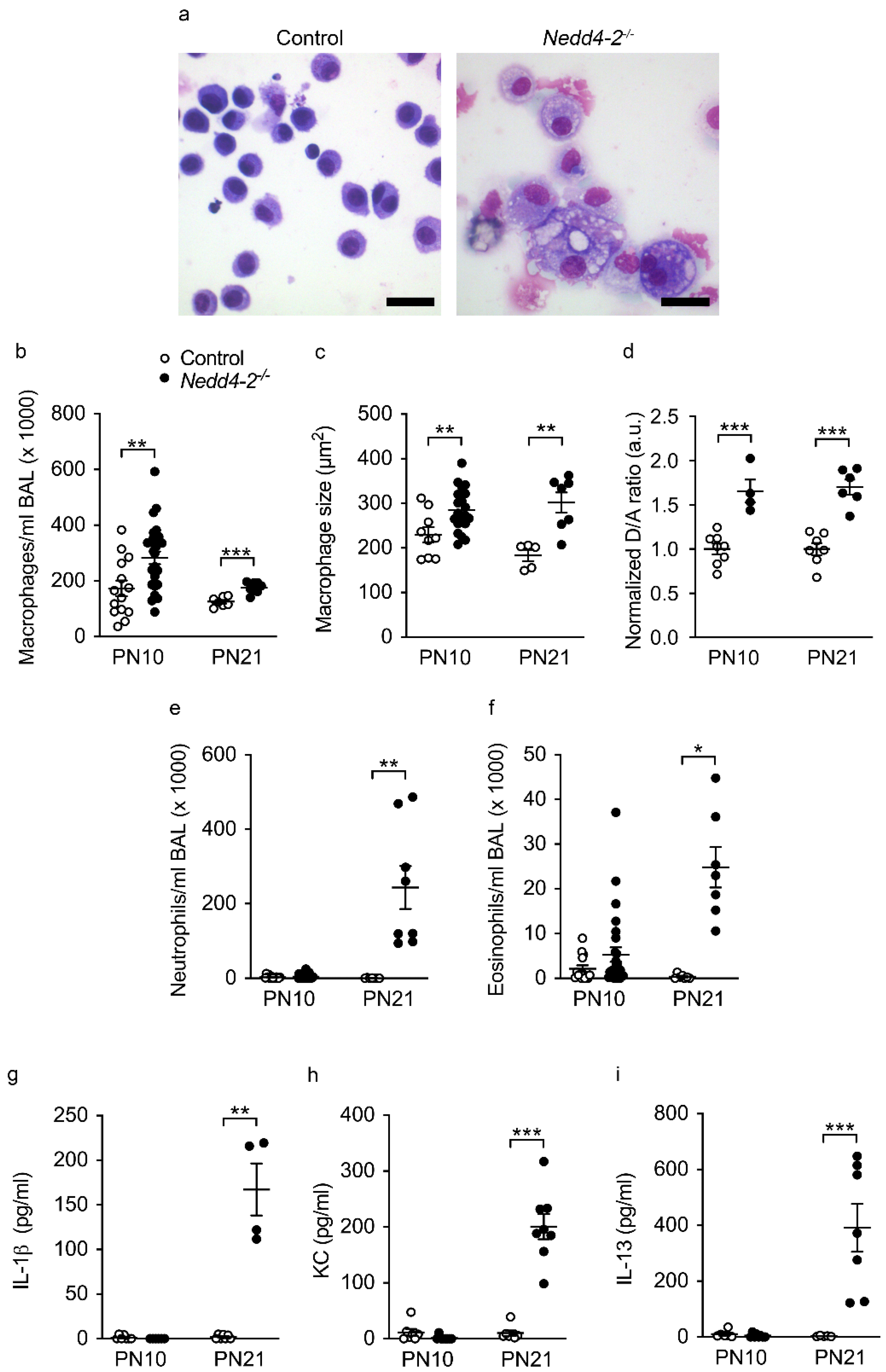
2.2. Congenital Deletion of Nedd4-2 in Lung Epithelial Cells Causes Alveolar Inflammation and Fibrosis in Neonatal Mice
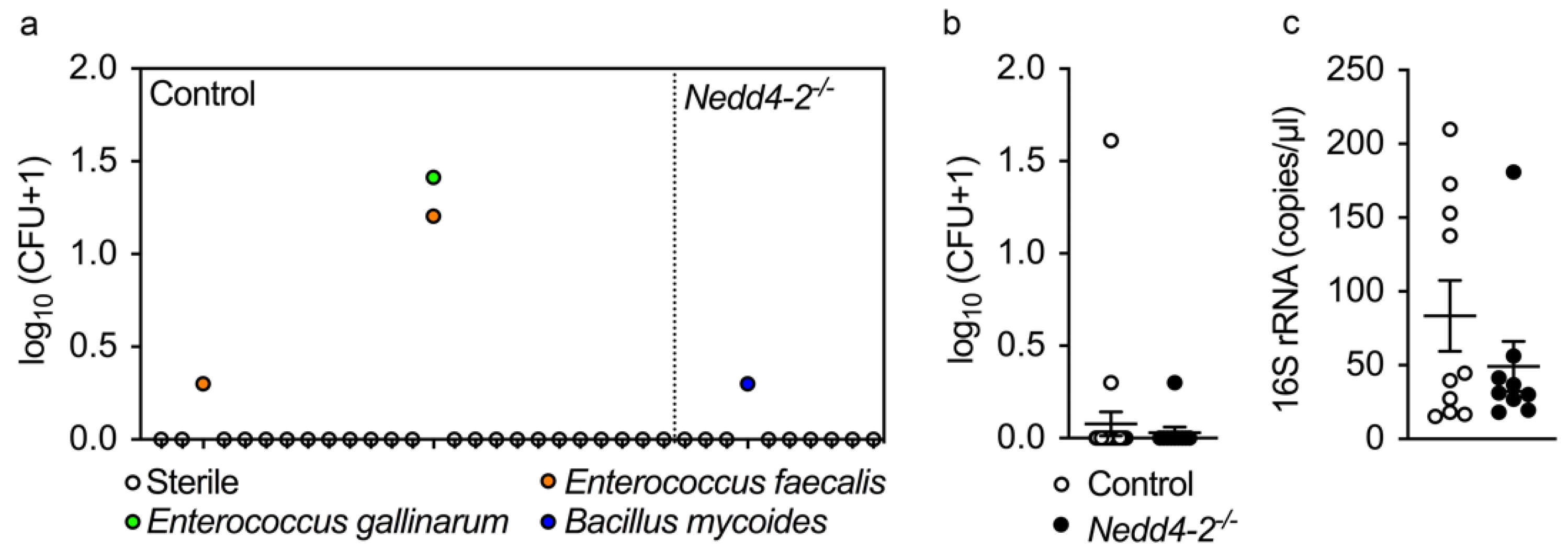
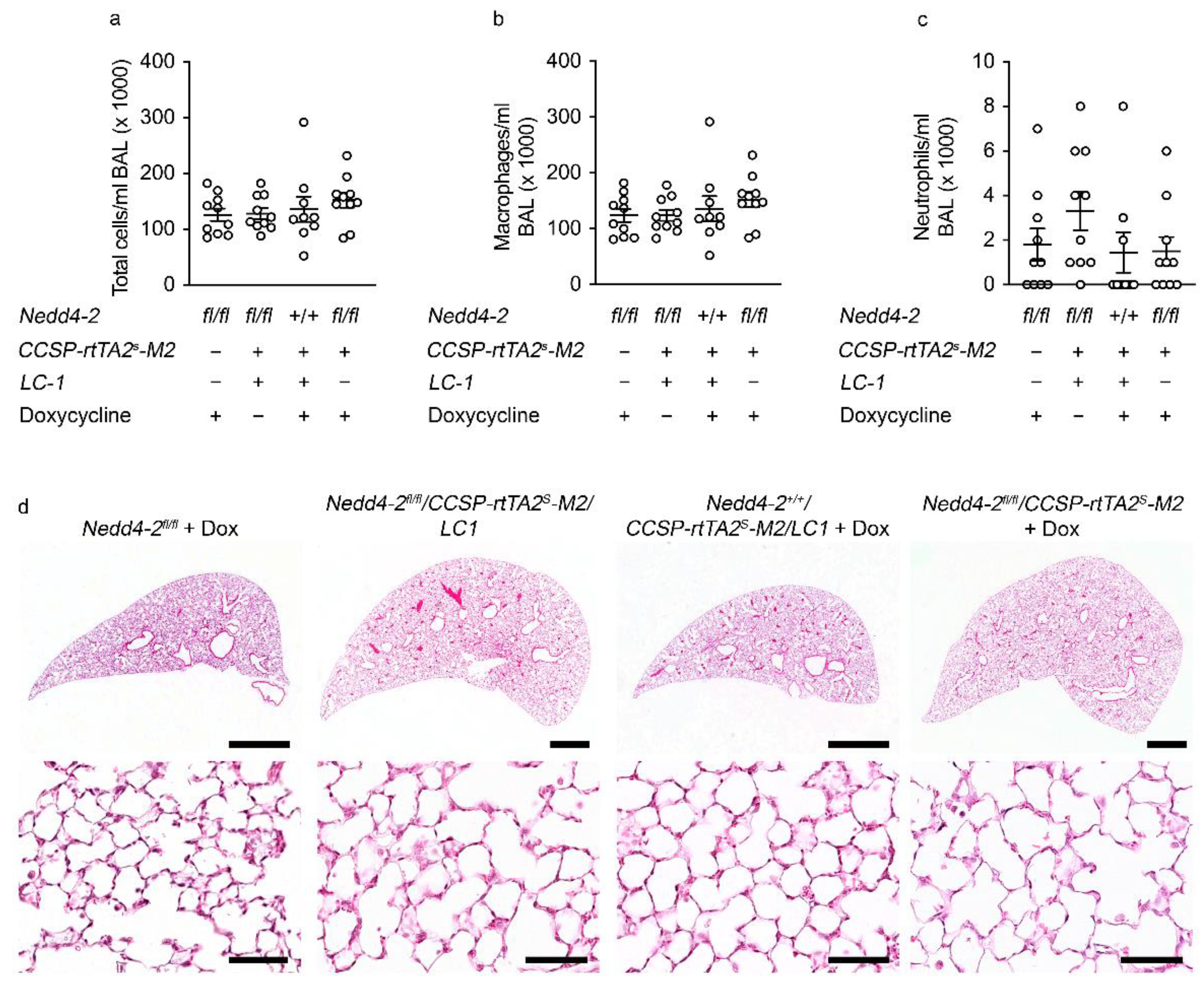
2.3. Development of Pneumonitis in Congenital Nedd4-2−/− Mice
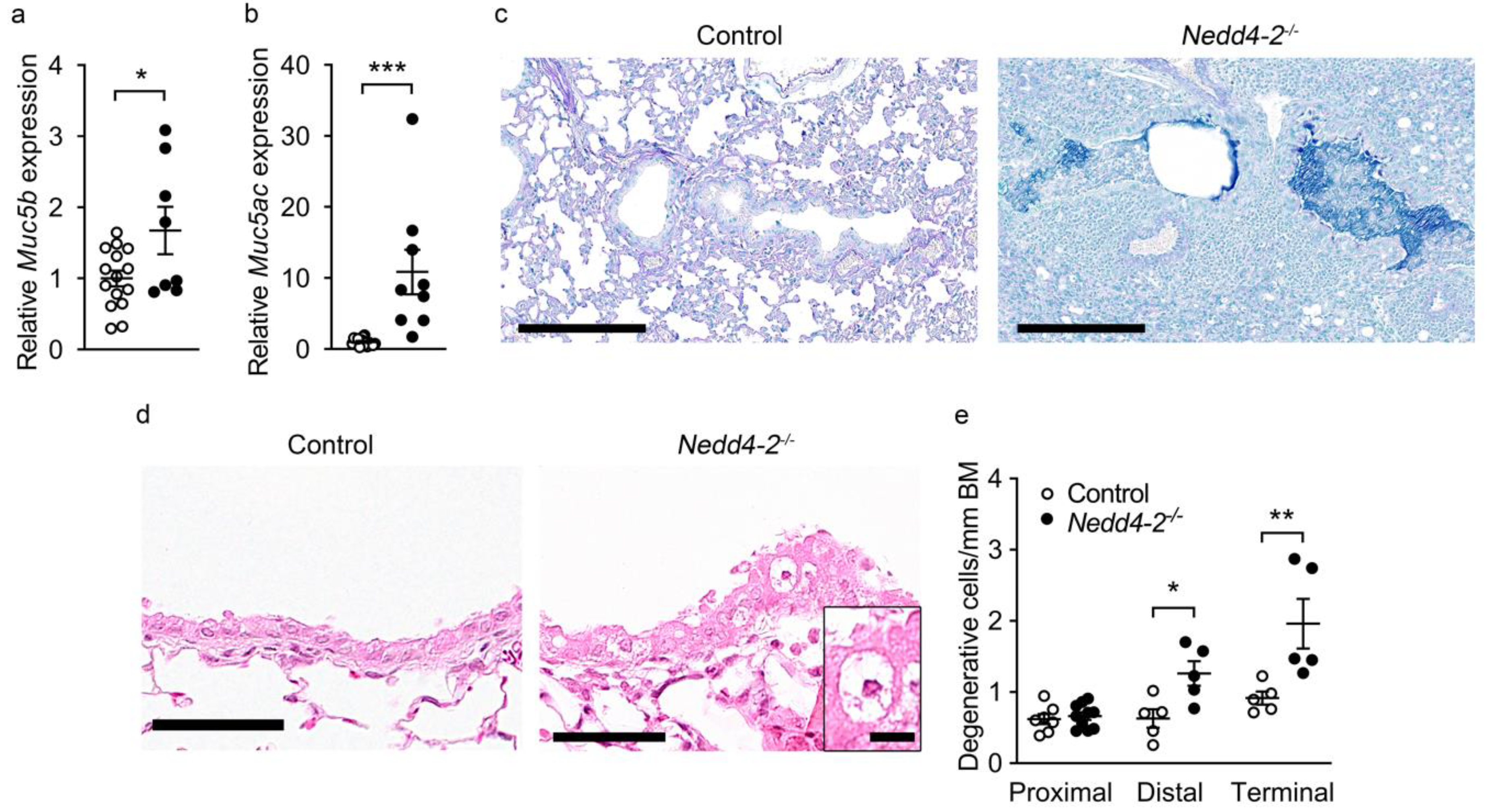
2.4. Congenital Deletion of Nedd4-2 in Lung Epithelial Cells Causes Mucus Plugging and Epithelial Necrosis in Distal and Terminal Airways in Neonatal Mice
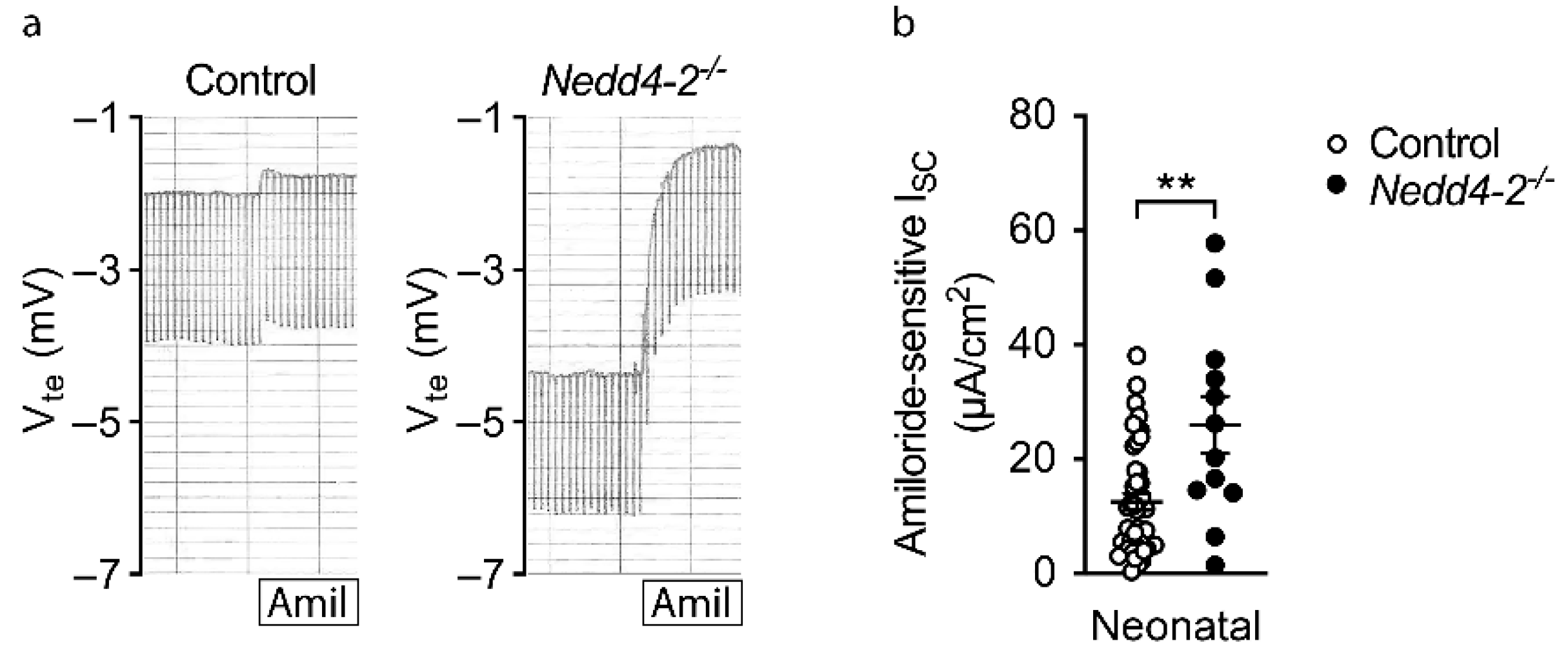
2.5. Increased ENaC Activity in Freshly Excised Airway Tissues of Congenital Nedd4-2−/− Mice
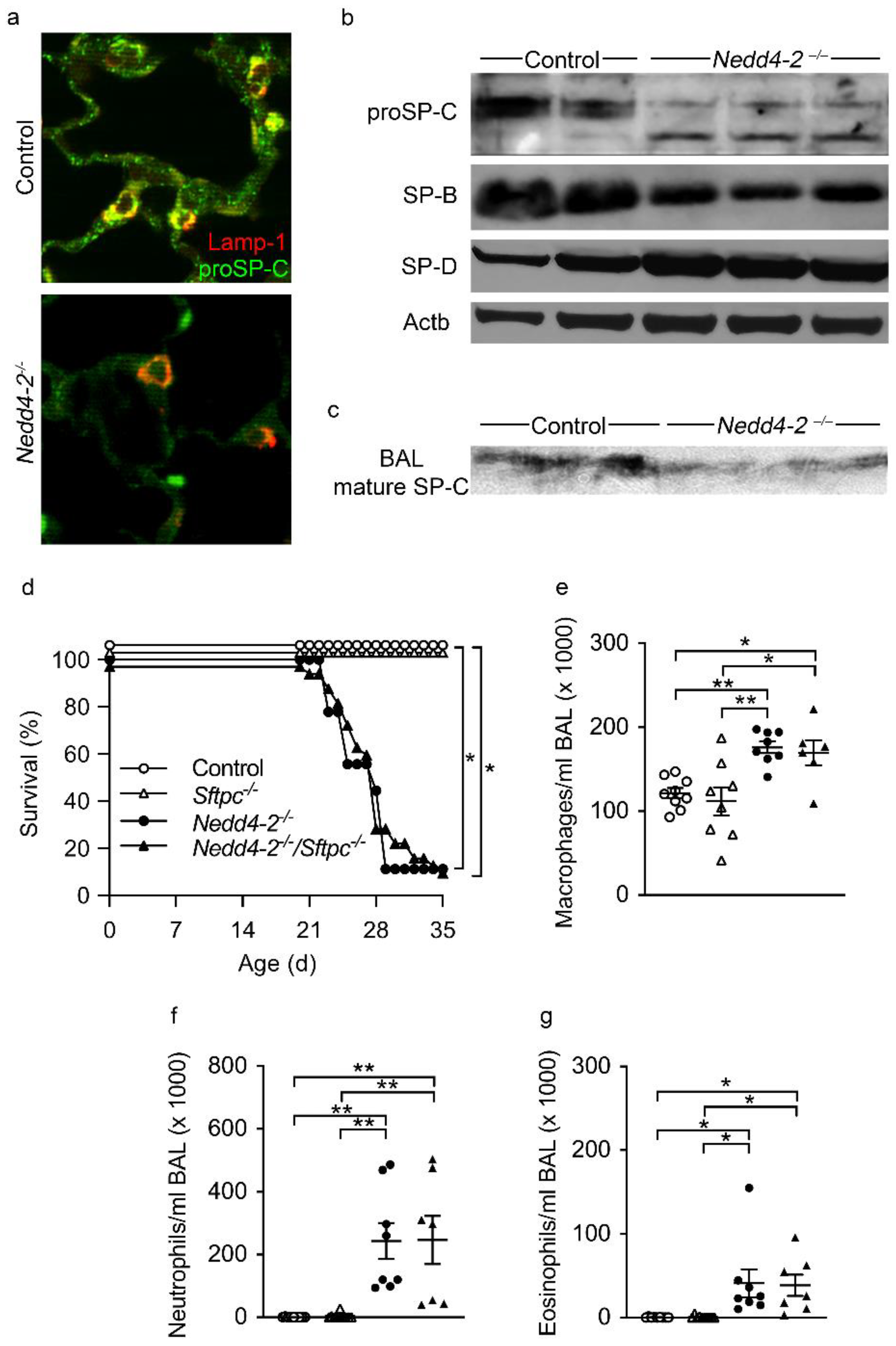
2.6. proSP-C Is Mistrafficked in Lung Epithelial Cells of Congenital Nedd4-2−/− Mice
3. Discussion
4. Materials and Methods
4.1. Experimental Animals
4.2. Measurement of Inflammatory Markers in BAL
4.3. Histology and Morphometry
4.4. Pulse Oximetry
4.5. Immunofluorescence Microscopy
4.6. SDS-PAGE and Immunoblotting
4.7. Electrogenic Ion Transport Measurements
4.8. mRNA Expression Analysis
4.9. Microbiology Studies
4.10. Statistical Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A


References
- Kotorashvili, A.; Russo, S.J.; Mulugeta, S.; Guttentag, S.; Beers, M.F. Anterograde Transport of Surfactant Protein C Proprotein to Distal Processing Compartments Requires PPDY-mediated Association with Nedd4 Ubiquitin Ligases. J. Biol. Chem. 2009, 284, 16667–16678. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Conkright, J.J.; Apsley, K.S.; Martin, E.P.; Ridsdale, R.; Rice, W.R.; Na, C.-L.; Yang, B.; Weaver, T.E. Nedd4-2–Mediated Ubiquitination Facilitates Processing of Surfactant Protein–C. Am. J. Respir. Cell Mol. Biol. 2010, 42, 181–189. [Google Scholar] [CrossRef] [PubMed]
- Gao, S.; Alarcon, C.; Sapkota, G.; Rahman, S.; Chen, P.Y.; Goerner, N.; Macias, M.J.; Erdjument-Bromage, H.; Tempst, P.; Massague, J. Ubiquitin ligase Nedd4L targets activated Smad2/3 to limit TGF-beta signaling. Mol. Cell 2009, 36, 457–468. [Google Scholar] [CrossRef] [Green Version]
- Rotin, D.; Staub, O. Nedd4-2 and the Regulation of Epithelial Sodium Transport. Front. Physiol. 2012, 3. [Google Scholar] [CrossRef] [Green Version]
- Mall, M.A. ENaC inhibition in cystic fibrosis: Potential role in the new era of CFTR modulator therapies. Eur. Respir. J. 2020, 56, 2000946. [Google Scholar] [CrossRef]
- Mall, M.A.; Button, B.; Johannesson, B.; Zhou, Z.; Livraghi, A.; Caldwell, R.A.; Schubert, S.C.; Schultz, C.; O’Neal, W.K.; Pradervand, S.; et al. Airway Surface Liquid Volume Regulation Determines Different Airway Phenotypes in Liddle Compared with ENaC-overexpressing Mice. J. Biol. Chem. 2010, 285, 26945–26955. [Google Scholar] [CrossRef] [Green Version]
- Aschner, Y.; Downey, G.P. Transforming Growth Factor-β: Master Regulator of the Respiratory System in Health and Disease. Am. J. Respir. Cell Mol. Biol. 2016, 54, 647–655. [Google Scholar] [CrossRef] [Green Version]
- Zhou, R.; Patel, S.V.; Snyder, P.M. Nedd4-2 catalyzes ubiquitination and degradation of cell surface ENaC. J. Biol. Chem. 2007, 282, 20207–20212. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duerr, J.; Leitz, D.H.W.; Szczygiel, M.; Dvornikov, D.; Fraumann, S.G.; Kreutz, C.; Zadora, P.K.; Seyhan Agircan, A.; Konietzke, P.; Engelmann, T.A.; et al. Conditional deletion of Nedd4-2 in lung epithelial cells causes progressive pulmonary fibrosis in adult mice. Nat. Commun. 2020, 11, 2012. [Google Scholar] [CrossRef]
- Boase, N.A.; Rychkov, G.Y.; Townley, S.L.; Dinudom, A.; Candi, E.; Voss, A.K.; Tsoutsman, T.; Semsarian, C.; Melino, G.; Koentgen, F.; et al. Respiratory distress and perinatal lethality in Nedd4-2-deficient mice. Nat. Commun. 2011, 2, 287. [Google Scholar] [CrossRef] [Green Version]
- Kimura, T.; Kawabe, H.; Jiang, C.; Zhang, W.; Xiang, Y.-Y.; Lu, C.; Salter, M.W.; Brose, N.; Lu, W.-Y.; Rotin, D. Deletion of the ubiquitin ligase Nedd4L in lung epithelia causes cystic fibrosis-like disease. Proc. Natl. Acad. Sci. USA 2011, 108, 3216–3221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duerr, J.; Gruner, M.; Schubert, S.C.; Haberkorn, U.; Bujard, H.; Mall, M.A. Use of a New-Generation Reverse Tetracycline Transactivator System for Quantitative Control of Conditional Gene Expression in the Murine Lung. Am. J. Respir. Cell Mol. Biol. 2011, 44, 244–254. [Google Scholar] [CrossRef]
- Deterding, R.R.; DeBoer, E.M.; Cidon, M.J.; Robinson, T.E.; Warburton, D.; Deutsch, G.H.; Young, L.R. Approaching Clinical Trials in Childhood Interstitial Lung Disease and Pediatric Pulmonary Fibrosis. Am. J. Respir. Crit. Care Med. 2019, 200, 1219–1227. [Google Scholar] [CrossRef]
- Trojanek, J.B.; Cobos-Correa, A.; Diemer, S.; Kormann, M.; Schubert, S.C.; Zhou-Suckow, Z.; Agrawal, R.; Duerr, J.; Wagner, C.J.; Schatterny, J.; et al. Airway Mucus Obstruction Triggers Macrophage Activation and Matrix Metalloproteinase 12–Dependent Emphysema. Am. J. Respir. Cell Mol. Biol. 2014, 51, 709–720. [Google Scholar] [CrossRef]
- Cobos-Correa, A.; Trojanek, J.B.; Diemer, S.; Mall, M.A.; Schultz, C. Membrane-bound FRET probe visualizes MMP12 activity in pulmonary inflammation. Nat. Chem. Biol. 2009, 5, 628–630. [Google Scholar] [CrossRef]
- Fritzsching, B.; Zhou-Suckow, Z.; Trojanek, J.B.; Schubert, S.C.; Schatterny, J.; Hirtz, S.; Agrawal, R.; Muley, T.; Kahn, N.; Sticht, C.; et al. Hypoxic epithelial necrosis triggers neutrophilic inflammation via IL-1 receptor signaling in cystic fibrosis lung disease. Am. J. Respir. Crit. Care Med. 2015, 191, 902–913. [Google Scholar] [CrossRef] [Green Version]
- Mall, M.A.; Harkema, J.R.; Trojanek, J.B.; Treis, D.; Livraghi, A.; Schubert, S.; Zhou, Z.; Kreda, S.M.; Tilley, S.L.; Hudson, E.J.; et al. Development of Chronic Bronchitis and Emphysema in b-Epithelial Na+ Channel–Overexpressing Mice. Am. J. Respir. Crit. Care Med. 2008, 177, 730–742. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Z.; Treis, D.; Schubert, S.C.; Harm, M.; Schatterny, J.; Hirtz, S.; Duerr, J.; Boucher, R.C.; Mall, M.A. Preventive but Not Late Amiloride Therapy Reduces Morbidity and Mortality of Lung Disease in βENaC-overexpressing Mice. Am. J. Respir. Crit. Care Med. 2008, 178, 1245–1256. [Google Scholar] [CrossRef]
- Johannesson, B.; Hirtz, S.; Schatterny, J.; Schultz, C.; Mall, M.A. CFTR Regulates Early Pathogenesis of Chronic Obstructive Lung Disease in βENaC-Overexpressing Mice. PLoS ONE 2012, 7, e44059. [Google Scholar] [CrossRef] [PubMed]
- Eaton, D.C.; Malik, B.; Bao, H.F.; Yu, L.; Jain, L. Regulation of Epithelial Sodium Channel Trafficking by Ubiquitination. Proc. Am. Thorac. Soc. 2010, 7, 54–64. [Google Scholar] [CrossRef]
- Litao, M.K.; Hayes, D., Jr.; Chiwane, S.; Nogee, L.M.; Kurland, G.; Guglani, L. A novel surfactant protein C gene mutation associated with progressive respiratory failure in infancy. Pediatr. Pulmonol. 2016. [Google Scholar] [CrossRef] [PubMed]
- Mulugeta, S.; Nguyen, V.; Russo, S.J.; Muniswamy, M.; Beers, M.F. A Surfactant Protein C Precursor Protein BRICHOS Domain Mutation Causes Endoplasmic Reticulum Stress, Proteasome Dysfunction, and Caspase 3 Activation. Am. J. Respir. Cell Mol. Biol. 2005, 32, 521–530. [Google Scholar] [CrossRef] [PubMed]
- Kurland, G.; Deterding, R.R.; Hagood, J.S.; Young, L.R.; Brody, A.S.; Castile, R.G.; Dell, S.; Fan, L.L.; Hamvas, A.; Hilman, B.C.; et al. An official American Thoracic Society clinical practice guideline: Classification, evaluation, and management of childhood interstitial lung disease in infancy. Am. J. Respir. Crit. Care Med. 2013, 188, 376–394. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nureki, S.I.; Tomer, Y.; Venosa, A.; Katzen, J.; Russo, S.J.; Jamil, S.; Barrett, M.; Nguyen, V.; Kopp, M.; Mulugeta, S.; et al. Expression of mutant Sftpc in murine alveolar epithelia drives spontaneous lung fibrosis. J. Clin. Investig. 2018, 128, 4008–4024. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Venosa, A.; Katzen, J.; Tomer, Y.; Kopp, M.; Jamil, S.; Russo, S.J.; Mulugeta, S.; Beers, M.F. Epithelial Expression of an Interstitial Lung Disease-Associated Mutation in Surfactant Protein-C Modulates Recruitment and Activation of Key Myeloid Cell Populations in Mice. J. Immunol. 2019, 202, 2760–2771. [Google Scholar] [CrossRef]
- Hawkins, A.; Guttentag, S.H.; Deterding, R.; Funkhouser, W.K.; Goralski, J.L.; Chatterjee, S.; Mulugeta, S.; Beers, M.F. A non-BRICHOS SFTPC mutant (SP-CI73T) linked to interstitial lung disease promotes a late block in macroautophagy disrupting cellular proteostasis and mitophagy. Am. J. Physiol. Lung Cell Mol. Physiol. 2015, 308, L33–L47. [Google Scholar] [CrossRef] [Green Version]
- Deutsch, G.H.; Young, L.R.; Deterding, R.R.; Fan, L.L.; Dell, S.D.; Bean, J.A.; Brody, A.S.; Nogee, L.M.; Trapnell, B.C.; Langston, C.; et al. Diffuse lung disease in young children: Application of a novel classification scheme. Am. J. Respir. Crit. Care Med. 2007, 176, 1120–1128. [Google Scholar] [CrossRef] [Green Version]
- Rice, A.; Tran-Dang, M.A.; Bush, A.; Nicholson, A.G. Diffuse lung disease in infancy and childhood: Expanding the chILD classification. Histopathology 2013, 63, 743–755. [Google Scholar] [CrossRef]
- Engelmann, T.A.; Knudsen, L.; Leitz, D.H.W.; Duerr, J.; Beers, M.F.; Mall, M.A.; Ochs, M. Linking fibrotic remodeling and ultrastructural alterations of alveolar epithelial cells after deletion of Nedd4-2. Int. J. Mol. Sci. 2021. manuscript submitted. [Google Scholar]
- Mall, M.; Bleich, M.; Greger, R.; Schreiber, R.; Kunzelmann, K. The amiloride inhibitable Na+ conductance is reduced by CFTR in normal but not in cystic fibrosis airways. J. Clin. Investig. 1998, 102, 15–21. [Google Scholar] [CrossRef]
- Mall, M.; Grubb, B.R.; Harkema, J.R.; O’Neal, W.K.; Boucher, R.C. Increased airway epithelial Na+ absorption produces cystic fibrosis-like lung disease in mice. Nat. Med. 2004, 10, 487–493. [Google Scholar] [CrossRef]
- Mall, M.A. Role of cilia, mucus, and airway surface liquid in mucociliary dysfunction: Lessons from mouse models. J. Aerosol. Med. Pulm. Drug Deliv. 2008, 21, 13–24. [Google Scholar] [CrossRef]
- Boucher, R.C. Evidence for airway surface dehydration as the initiating event in CF airway disease. J. Intern. Med. 2007, 261, 5–16. [Google Scholar] [CrossRef]
- Evans, C.M.; Fingerlin, T.E.; Schwarz, M.I.; Lynch, D.; Kurche, J.; Warg, L.; Yang, I.V.; Schwartz, D.A. Idiopathic Pulmonary Fibrosis: A Genetic Disease That Involves Mucociliary Dysfunction of the Peripheral Airways. Physiol. Rev. 2016, 96, 1567–1591. [Google Scholar] [CrossRef] [PubMed]
- Button, B.; Cai, L.H.; Ehre, C.; Kesimer, M.; Hill, D.B.; Sheehan, J.K.; Boucher, R.C.; Rubinstein, M. A periciliary brush promotes the lung health by separating the mucus layer from airway epithelia. Science 2012, 337, 937–941. [Google Scholar] [CrossRef] [Green Version]
- Hancock, L.A.; Hennessy, C.E.; Solomon, G.M.; Dobrinskikh, E.; Estrella, A.; Hara, N.; Hill, D.B.; Kissner, W.J.; Markovetz, M.R.; Grove Villalon, D.E.; et al. Muc5b overexpression causes mucociliary dysfunction and enhances lung fibrosis in mice. Nat. Commun. 2018, 9, 5363. [Google Scholar] [CrossRef] [PubMed]
- Livraghi-Butrico, A.; Wilkinson, K.J.; Volmer, A.S.; Gilmore, R.C.; Rogers, T.D.; Caldwell, R.A.; Burns, K.A.; Esther, C.R., Jr.; Mall, M.A.; Boucher, R.C.; et al. Lung disease phenotypes caused by overexpression of combinations of α-, β-, and γ-subunits of the epithelial sodium channel in mouse airways. Am. J. Physiol. Lung Cell Mol. Physiol. 2018, 314, L318–L331. [Google Scholar] [CrossRef]
- Kaltofen, T.; Haase, M.; Thome, U.H.; Laube, M. Male Sex is Associated with a Reduced Alveolar Epithelial Sodium Transport. PLoS ONE 2015, 10, e0136178. [Google Scholar] [CrossRef] [PubMed]
- Jia, J.; Conlon, T.M.; Ballester Lopez, C.; Seimetz, M.; Bednorz, M.; Zhou-Suckow, Z.; Weissmann, N.; Eickelberg, O.; Mall, M.A.; Yildirim, A. Cigarette smoke causes acute airway disease and exacerbates chronic obstructive lung disease in neonatal mice. Am. J. Physiol. Lung Cell Mol. Physiol. 2016, 311, L602–L610. [Google Scholar] [CrossRef]
- Balázs, A.; Mall, M.A. Mucus obstruction and inflammation in early cystic fibrosis lung disease: Emerging role of the IL-1 signaling pathway. Pediatric pulmonology 2019, 54 (Suppl. 3), S5–S12. [Google Scholar] [CrossRef] [Green Version]
- Montgomery, S.T.; Dittrich, A.S.; Garratt, L.W.; Turkovic, L.; Frey, D.L.; Stick, S.M.; Mall, M.A.; Kicic, A. Interleukin-1 is associated with inflammation and structural lung disease in young children with cystic fibrosis. J. Cyst. Fibros. 2018, 17, 715–722. [Google Scholar] [CrossRef]
- Montgomery, S.T.; Mall, M.A.; Kicic, A.; Stick, S.M. Hypoxia and sterile inflammation in cystic fibrosis airways: Mechanisms and potential therapies. Eur. Respir. J. 2017, 49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perl, A.-K.T.; Tichelaar, J.W.; Whitsett, J.A. Conditional gene expression in the respiratory epithelium of the mouse. Transgenic Res. 2002, 11, 21–29. [Google Scholar] [CrossRef] [PubMed]
- Manning, J.A.; Shah, S.S.; Nikolic, A.; Henshall, T.L.; Khew-Goodall, Y.; Kumar, S. The ubiquitin ligase NEDD4-2/NEDD4L regulates both sodium homeostasis and fibrotic signaling to prevent end-stage renal disease. Cell Death Dis. 2021, 12, 398. [Google Scholar] [CrossRef] [PubMed]
- Henshall, T.L.; Manning, J.A.; Alfassy, O.S.; Goel, P.; Boase, N.A.; Kawabe, H.; Kumar, S. Deletion of Nedd4-2 results in progressive kidney disease in mice. Cell Death Differ. 2017, 24, 2150–2160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, C.; Kawabe, H.; Rotin, D. The ubiquitin ligase Nedd4L regulates the Na/K/2Cl co-transporter NKCC1/SLC12A2 in the colon. J. Biol. Chem. 2017. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goel, P.; Manning, J.A.; Kumar, S. NEDD4-2 (NEDD4L): The ubiquitin ligase for multiple membrane proteins. Gene 2015, 557, 1–10. [Google Scholar] [CrossRef]
- Kuratomi, G.; Komuro, A.; Goto, K.; Shinozaki, M.; Miyazawa, K.; Miyazono, K.; Imamura, T. NEDD4-2 (neural precursor cell expressed, developmentally down-regulated 4-2) negatively regulates TGF-beta (transforming growth factor-beta) signalling by inducing ubiquitin-mediated degradation of Smad2 and TGF-beta type I receptor. Biochem. J. 2005, 386, 461–470. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Persaud, A.; Alberts, P.; Amsen, E.M.; Xiong, X.; Wasmuth, J.; Saadon, Z.; Fladd, C.; Parkinson, J.; Rotin, D. Comparison of substrate specificity of the ubiquitin ligases Nedd4 and Nedd4-2 using proteome arrays. Mol. Syst. Biol. 2009, 5, 333. [Google Scholar] [CrossRef] [PubMed]
- Baron, U.; Freundlieb, S.; Gossen, M.; Bujard, H. Co-regulation of two gene activities by tetracycline via a bidirectional promoter. Nucleic Acids Res. 1995, 23, 3605–3606. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schoenig, K.; Schwenk, F.; Rajewsky, K.; Hermann, B. Stringent doxycycline dependent control of CRE recombinase in vivo. Nucleic Acids Res. 2002, 30, e134. [Google Scholar] [CrossRef] [Green Version]
- Glasser, S.W.; Burhans, M.S.; Korfhagen, T.R.; Na, C.-L.; Sly, P.D.; Ross, G.F.; Ikegami, M.; Whitsett, J.A. Altered stability of pulmonary surfactant in SP-C-deficient mice. Proc. Natl. Acad. Sci. USA 2001, 98, 6366–6371. [Google Scholar] [CrossRef] [Green Version]
- Schindelin, J.; Arganda-Carreras, I.; Frise, E.; Kaynig, V.; Longair, M.; Pietzsch, T.; Preibisch, S.; Rueden, C.; Saalfeld, S.; Schmid, B.; et al. Fiji: An open-source platform for biological-image analysis. Nat. Methods 2012, 9, 676–682. [Google Scholar] [CrossRef] [Green Version]
- Schindelin, J.; Rueden, C.T.; Hiner, M.C.; Eliceiri, K.W. The ImageJ ecosystem: An open platform for biomedical image analysis. Mol. Reprod. Dev. 2015, 82, 518–529. [Google Scholar] [CrossRef] [Green Version]
- Evans, C.M.; Williams, O.W.; Tuvim, M.J.; Nigam, R.; Mixides, G.P.; Blackburn, M.R.; DeMayo, F.J.; Burns, A.R.; Smith, C.; Reynolds, S.D.; et al. Mucin is produced by clara cells in the proximal airways of antigen-challenged mice. Am. J. Respir. Cell Mol. Biol. 2004, 31, 382–394. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beers, M.F.; Kim, C.Y.; Dodia, C.; Fisher, A.B. Localization, synthesis, and processing of surfactant protein SP-C in rat lung analyzed by epitope-specific antipeptide antibodies. J. Biol. Chem. 1994, 269, 20318–20328. [Google Scholar] [CrossRef]
- Beers, M.F.; Lomax, C. Synthesis and processing of hydrophobic surfactant protein C by isolated rat type II cells. Am. J. Physiol. 1995, 269, L744–L753. [Google Scholar] [CrossRef]
- Beers, M.F.; Bates, S.R.; Fisher, A.B. Differential extraction for the rapid purification of bovine surfactant protein B. Am. J. Physiol. 1992, 262, L773–L778. [Google Scholar] [CrossRef]
- Cao, Y.; Tao, J.Q.; Bates, S.R.; Beers, M.F.; Haczku, A. IL-4 induces production of the lung collectin surfactant protein-D. J. Allergy. Clin. Immunol. 2004, 113, 439–444. [Google Scholar] [CrossRef] [PubMed]
- Grubb, B.; Paradiso, A.; Boucher, R. Anomalies in ion transport in CF mouse tracheal epithelium. Am. J. Physiol. 1994, 267, C293–C300. [Google Scholar] [CrossRef]
- Anagnostopoulou, P.; Dai, L.; Schatterny, J.; Hirtz, S.; Duerr, J.; Mall, M.A. Allergic airway inflammation induces a pro-secretory epithelial ion transport phenotype in mice. Eur. Respir. J. 2010, 36, 1436–1447. [Google Scholar] [CrossRef] [PubMed]
- Pfaffl, M.W. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res. 2001, 29, e45. [Google Scholar] [CrossRef] [PubMed]
- Nonnenmacher, C.; Dalpke, A.; Rochon, J.; Flores-de-Jacoby, L.; Mutters, R.; Heeg, K. Real-time polymerase chain reaction for detection and quantification of bacteria in periodontal patients. J. Periodontol. 2005, 76, 1542–1549. [Google Scholar] [CrossRef] [PubMed]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Leitz, D.H.W.; Duerr, J.; Mulugeta, S.; Seyhan Agircan, A.; Zimmermann, S.; Kawabe, H.; Dalpke, A.H.; Beers, M.F.; Mall, M.A. Congenital Deletion of Nedd4-2 in Lung Epithelial Cells Causes Progressive Alveolitis and Pulmonary Fibrosis in Neonatal Mice. Int. J. Mol. Sci. 2021, 22, 6146. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22116146
Leitz DHW, Duerr J, Mulugeta S, Seyhan Agircan A, Zimmermann S, Kawabe H, Dalpke AH, Beers MF, Mall MA. Congenital Deletion of Nedd4-2 in Lung Epithelial Cells Causes Progressive Alveolitis and Pulmonary Fibrosis in Neonatal Mice. International Journal of Molecular Sciences. 2021; 22(11):6146. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22116146
Chicago/Turabian StyleLeitz, Dominik H. W., Julia Duerr, Surafel Mulugeta, Ayça Seyhan Agircan, Stefan Zimmermann, Hiroshi Kawabe, Alexander H. Dalpke, Michael F. Beers, and Marcus A. Mall. 2021. "Congenital Deletion of Nedd4-2 in Lung Epithelial Cells Causes Progressive Alveolitis and Pulmonary Fibrosis in Neonatal Mice" International Journal of Molecular Sciences 22, no. 11: 6146. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22116146