
Cardioprotective Effects of PPARβ/δ Activation against Ischemia/Reperfusion Injury in Rat Heart Are Associated with ALDH2 Upregulation, Amelioration of Oxidative Stress and Preservation of Mitochondrial Energy Production

 and
and
Abstract
:1. Introduction
2. Results
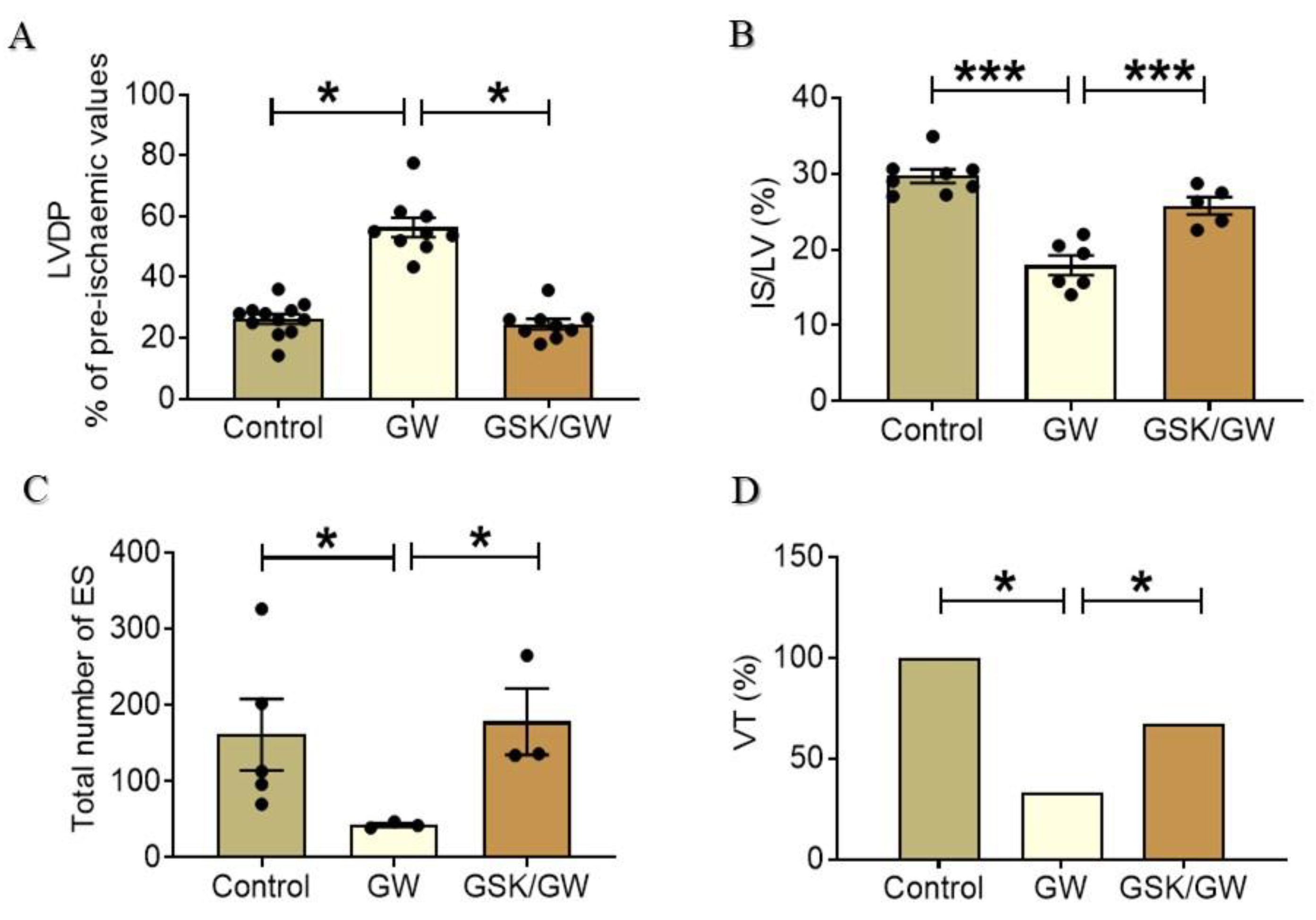
2.1. Administration of GW0742 Improves Myocardial Recovery and Reduces Infarct Size after I/R
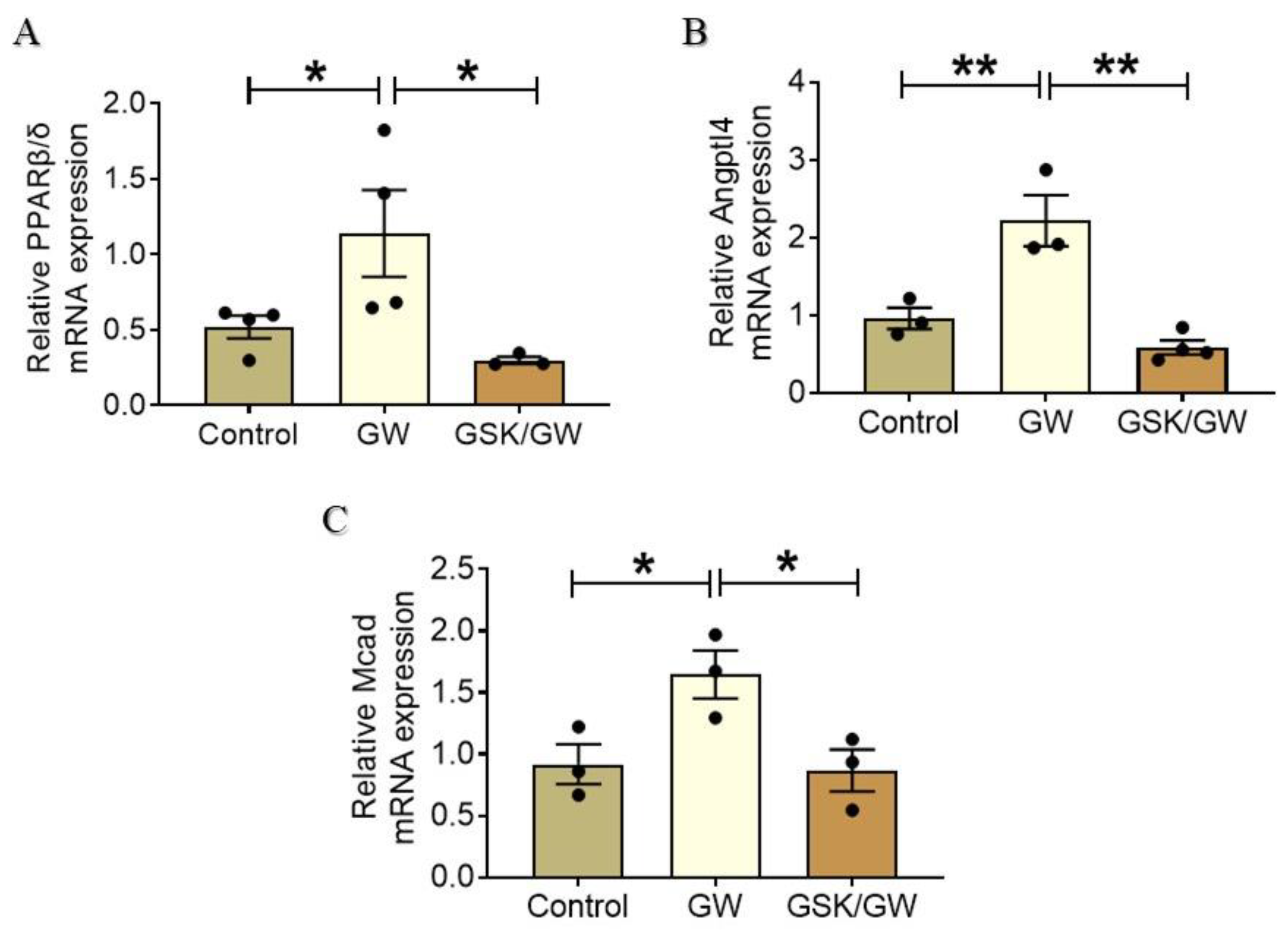
2.2. Administration of GW0742 Results in Activation of PPARβ/δ and Up-Regulation of Its Target Genes Angptl4 and Mcad
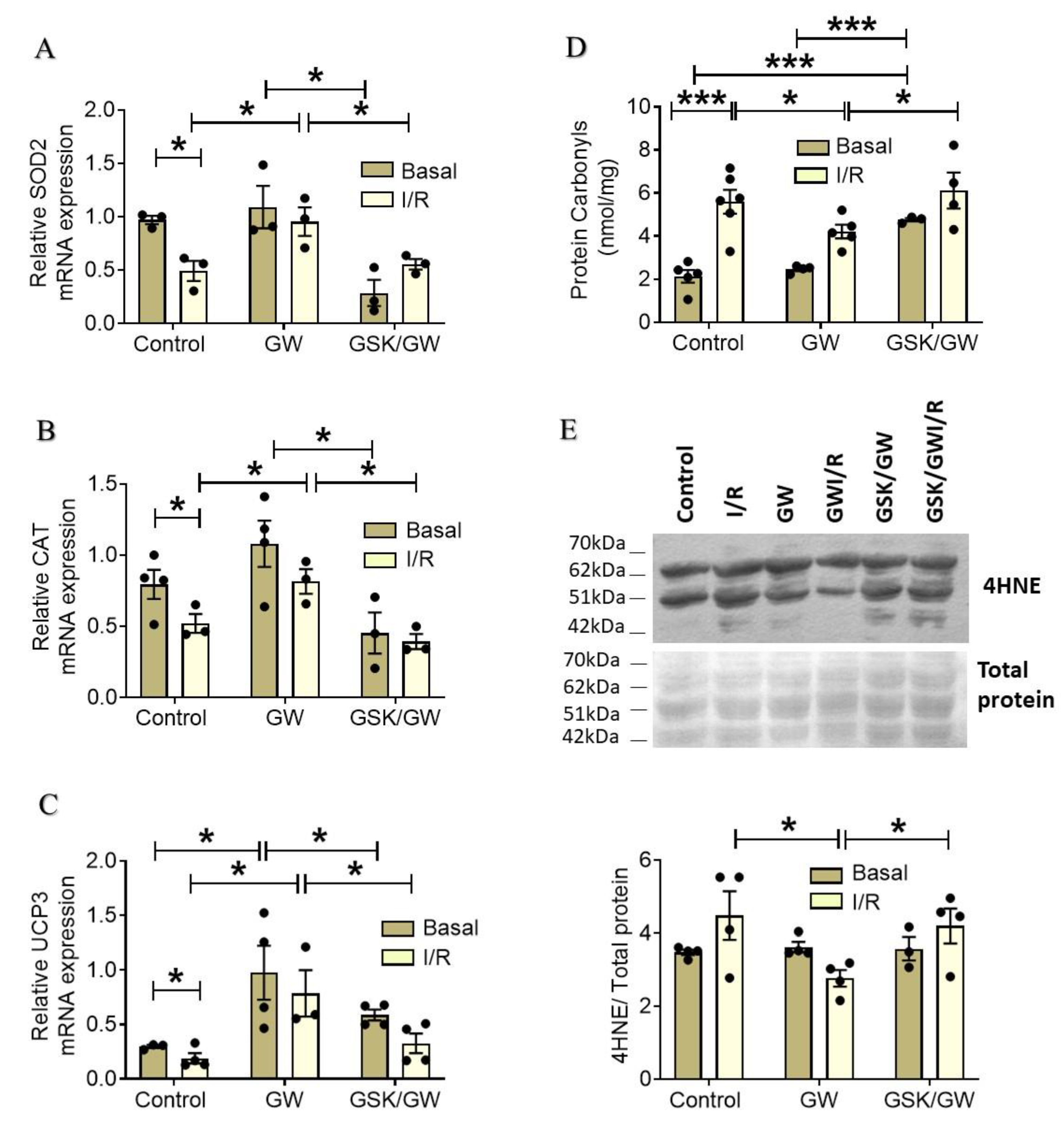
2.3. PPARβ/δ Upregulates Myocardial Antioxidant Enzymes and Ameliorates I/R-Induced Oxidative Stress
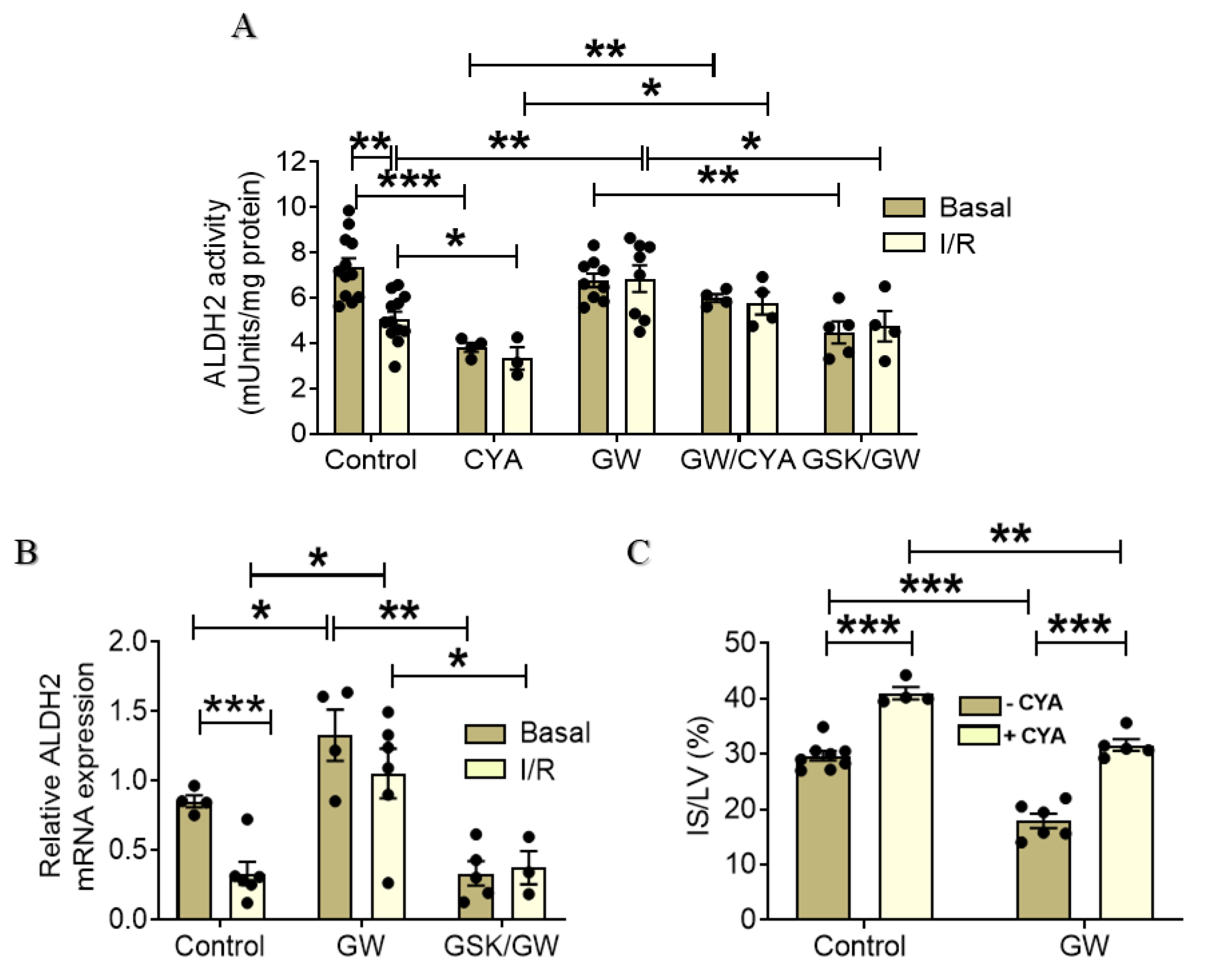
2.4. PPARβ/δ Upregulates and Activates ALDH2 in I/R Hearts Resulting in the Reduction of Infarct Size
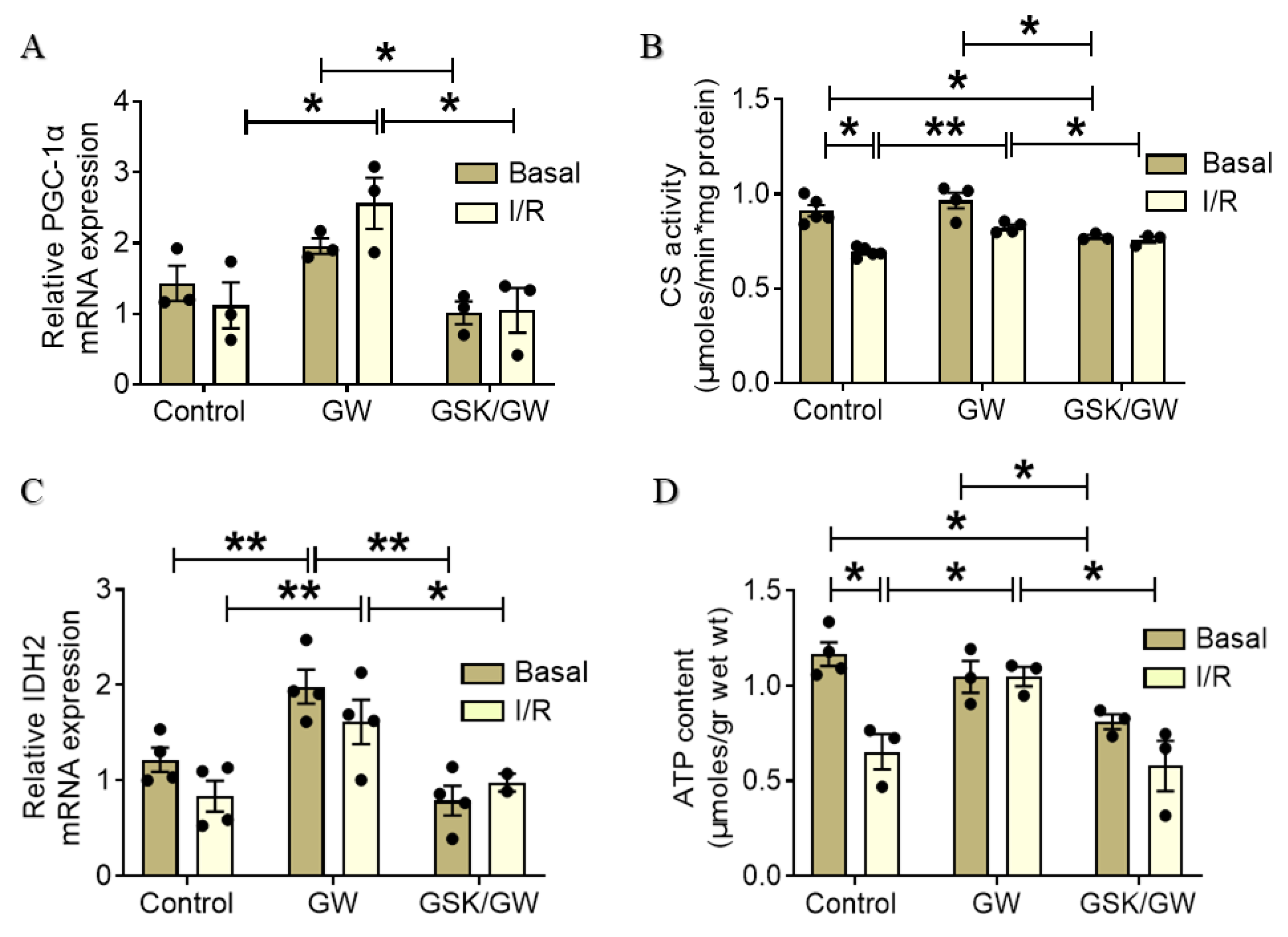
2.5. PPARβ/δ-Mediated Improvement of Mitochondrial Energy Production
3. Discussion
4. Materials and Methods
4.1. Animals
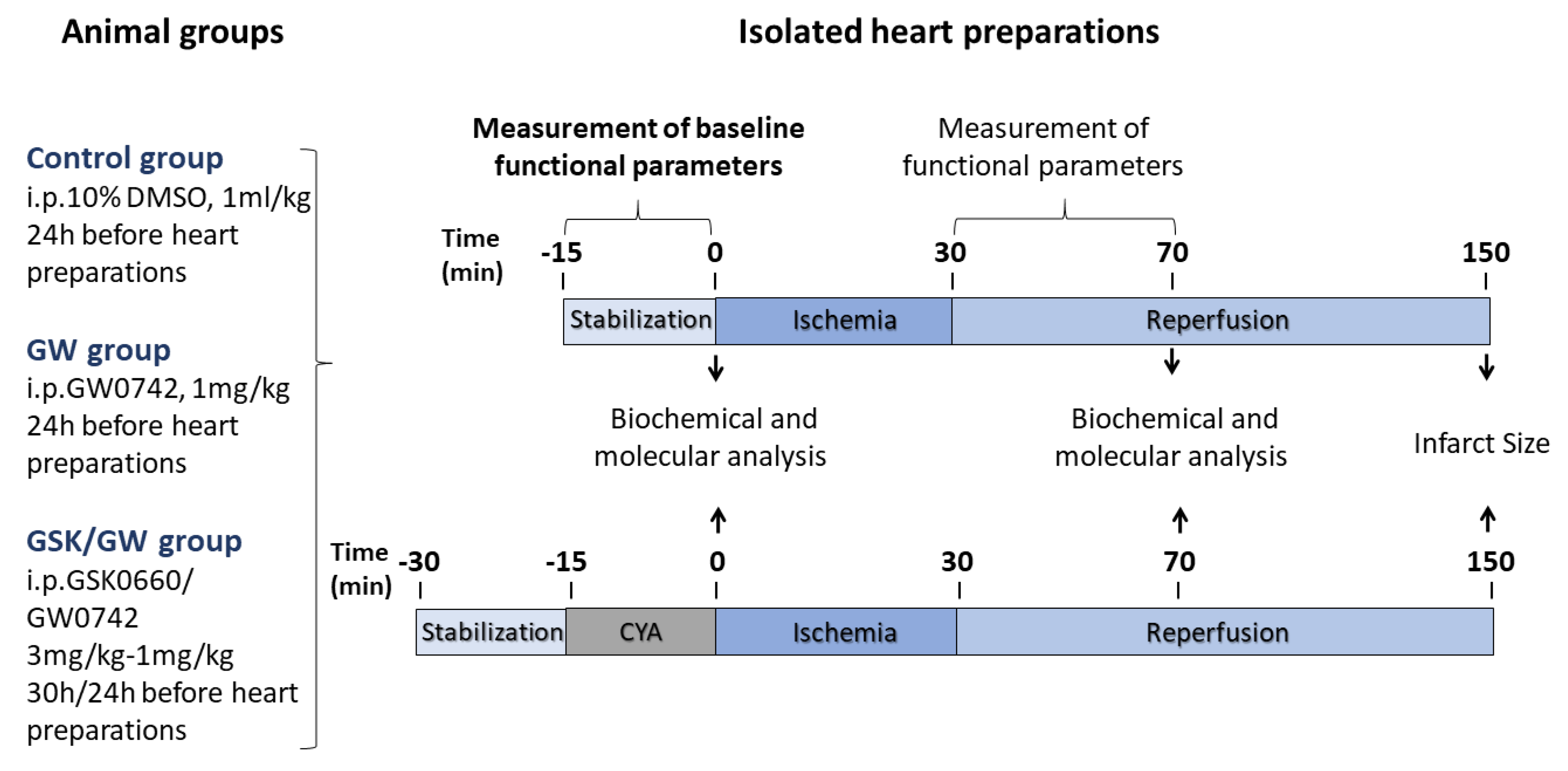
4.2. Drug Administration and Experimental Procedures for I/R in Isolated Hearts
4.3. Determination of IS
4.4. RNA Preparation and Quantitative Real-Time RT-PCR (qPCR)
4.5. Preparation of Whole Cell Protein Extracts
4.6. Western Blot
4.7. Determination of Protein Carbonyls
4.8. Measurement of ALDH2 and Citrate Synthase Enzymatic Activity
4.9. Determination of ATP Content
4.10. Statistical Analyses
Author Contributions
Funding
Institutional Review Board Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| 4-HNE | 4-hydroxy-2-nonenal |
| ALDH2 | aldehyde dehydrogenase 2 |
| Angptl4 | angiopoietin like peptide 4 |
| CS | citrate synthase |
| CYA | cyanamide |
| IDH2 | isocitrate dehydrogenase 2 |
| I/R | ischemia reperfusion |
| IS | infarct size |
| iNOS | inducible NO synthase |
| IRI | ischemia/reperfusion injury |
| LVDP | left ventricular developed pressure |
| Mcad | medium-chain acyl-CoA dehydrogenase |
| MDA | malondialdehyde |
| MI | myocardial infarction |
| MPTP | mitochondrial permeability transition pore |
| PGC1-α | peroxisome proliferator activated receptor gamma co-activator 1 alpha |
| PPARβ/δ | Peroxisome Proliferator Activated Receptor β/δ |
| ROS | reactive oxygen species |
| SOD2 | superoxide dismutase 2 |
| UCP3 | uncoupling protein 3 |
References
- Hausenloy, D.J.; Yellon, D.M. Myocardial ischemia-reperfusion injury: A neglected therapeutic target. J. Clin. Investig. 2013, 123, 92–100. [Google Scholar] [CrossRef]
- Davidson, S.M.; Ferdinandy, P.; Andreadou, I.; Bøtker, H.E.; Heusch, G.; Ibáñez, B.; Ovize, M.; Schulz, R.; Yellon, D.; Hausenloy, D.J.; et al. Multitarget Strategies to Reduce Myocardial Ischemia/Reperfusion Injury. J. Am. Coll. Cardiol. 2019, 73, 89–99. [Google Scholar] [CrossRef]
- Heusch, G. Myocardial ischaemia–reperfusion injury and cardioprotection in perspective. Nat. Rev. Cardiol. 2020, 17, 773–789. [Google Scholar] [CrossRef]
- Talukder, M.H.; Zweier, J.L.; Periasamy, M. Targeting calcium transport in ischaemic heart disease. Cardiovasc. Res. 2009, 84, 345–352. [Google Scholar] [CrossRef] [Green Version]
- Xiao, M.; Zhong, H.; Xia, L.; Tao, Y.; Yin, H. Pathophysiology of mitochondrial lipid oxidation: Role of 4-hydroxynonenal (4-HNE) and other bioactive lipids in mitochondria. Free. Radic. Biol. Med. 2017, 111, 316–327. [Google Scholar] [CrossRef]
- Bugger, H.; Pfeil, K. Mitochondrial ROS in myocardial ischemia reperfusion and remodeling. Biochim. Biophys. Acta (BBA) Mol. Basis Dis. 2020, 1866, 165768. [Google Scholar] [CrossRef]
- Davidson, S.M.; Adameová, A.; Barile, L.; Cabrera-Fuentes, H.A.; Lazou, A.; Pagliaro, P.; Stensløkken, K.; Garcia-Dorado, D. Mitochondrial and mitochondrial-independent pathways of myocardial cell death during ischaemia and reperfusion injury. J. Cell. Mol. Med. 2020, 24, 3795–3806. [Google Scholar] [CrossRef] [Green Version]
- Lesnefsky, E.J.; Chen, Q.; Tandler, B.; Hoppel, C.L. Mitochondrial Dysfunction and Myocardial Ischemia-Reperfusion: Implications for Novel Therapies. Annu. Rev. Pharmacol. Toxicol. 2017, 57, 535–565. [Google Scholar] [CrossRef]
- Egea, J.; Fabregat, I.; Frapart, Y.M.; Ghezzi, P.; Görlach, A.; Kietzmann, T.; Kubaichuk, K.; Knaus, U.G.; Lopez, M.G.; Olaso-Gonzalez, G.; et al. European contribution to the study of ROS: A summary of the findings and prospects for the future from the COST action BM1203 (EU-ROS). Redox Biol. 2017, 13, 94–162. [Google Scholar] [CrossRef] [Green Version]
- Andreadou, I.; Daiber, A.; Baxter, G.F.; Brizzi, M.F.; Di Lisa, F.; Kaludercic, N.; Lazou, A.; Varga, Z.V.; Zuurbier, C.J.; Schulz, R.; et al. Influence of cardiometabolic comorbidities on myocardial function, infarction, and cardioprotection: Role of cardiac redox signaling. Free. Radic. Biol. Med. 2021, 166, 33–52. [Google Scholar] [CrossRef]
- Barteková, M.; Adameová, A.; Görbe, A.; Ferenczyová, K.; Pecháňová, O.; Lazou, A.; Dhalla, N.S.; Ferdinandy, P.; Giricz, Z. Natural and synthetic antioxidants targeting cardiac oxidative stress and redox signaling in cardiometabolic diseases. Free. Radic. Biol. Med. 2021, 169, 446–477. [Google Scholar] [CrossRef]
- Ravingerová, T.; Adameová, A.; Kelly, T.; Antonopoulou, E.; Pancza, D.; Ondrejčáková, M.; Khandelwal, V.K.M.; Čarnická, S.; Lazou, A. Changes in PPAR gene expression and myocardial tolerance to ischaemia: Relevance to pleiotropic effects of statins. Can. J. Physiol. Pharmacol. 2009, 87, 1028–1036. [Google Scholar] [CrossRef]
- Ravingerová, T.; Adameova, A.; Čarnická, S.; Nemcekova, M.; Kelly, T.; Matejíková, J.; Galatou, E.; Barlaka, E.; Lazou, A. The role of PPAR in myocardial response to ischemia in normal and diseased heart. Gen. Physiol. Biophys. 2012, 30, 329–341. [Google Scholar] [CrossRef] [Green Version]
- Ravingerová, T.; Čarnická, S.; Nemčeková, M.; Ledvényiová, V.; Adameová, A.; Kelly, T.; Barlaka, E.; Galatou, E.; Khandelwal, V.K.M.; Lazou, A. PPAR-alpha activation as a preconditioning-like intervention in rats in vivo confers myocardial protection against acute ischaemia–reperfusion injury: Involvement of PI3K–Akt. Can. J. Physiol. Pharmacol. 2012, 90, 1135–1144. [Google Scholar] [CrossRef] [PubMed]
- Ravingerová, T.; Čarnická, S.; Ledvényiová, V.; Barlaka, E.; Galatou, E.; Chytilová, A.; Mandíková, P.; Nemčeková, M.; Adameova, A.; Kolář, F.; et al. Upregulation of Genes Involved in Cardiac Metabolism Enhances Myocardial Resistance to Ischemia/Reperfusion in the Rat Heart. Physiol. Res. 2013, 62, S151–S163. [Google Scholar] [CrossRef] [PubMed]
- Ravingerová, T.; Ledvényiová-Farkašová, V.; Ferko, M.; Barteková, M.; Bernatova, I.; Pecháňová, O.; Adameová, A.; Kolář, F.; Lazou, A. Pleiotropic preconditioning-like cardioprotective effects of hypolipidemic drugs in acute ischemia–reperfusion in normal and hypertensive rats. Can. J. Physiol. Pharmacol. 2015, 93, 495–503. [Google Scholar] [CrossRef]
- Barlaka, E.; Galatou, E.; Mellidis, K.; Ravingerova, T.; Lazou, A. Role of Pleiotropic Properties of Peroxisome Proliferator-Activated Receptors in the Heart: Focus on the Nonmetabolic Effects in Cardiac Protection. Cardiovasc. Ther. 2016, 34, 37–48. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Q.; Long, Q. PPARδ, a Potential Therapeutic Target for Heart Disease. Nucl. Recept. Res. 2018, 5, 101375. [Google Scholar] [CrossRef]
- Barlaka, E.; Görbe, A.; Gáspár, R.; Pálóczi, J.; Ferdinandy, P.; Lazou, A. Activation of PPARβ/δ protects cardiac myocytes from oxidative stress-induced apoptosis by suppressing generation of reactive oxygen/nitrogen species and expression of matrix metalloproteinases. Pharmacol. Res. 2015, 95, 102–110. [Google Scholar] [CrossRef] [Green Version]
- Cheng, L.; Ding, G.; Qin, Q.; Huang, Y.; Lewis, W.; He, N.; Evans, R.M.; Schneider, M.D.; Brako, F.A.; Xiao, Y.; et al. Cardiomyocyte-restricted peroxisome proliferator-activated receptor-δ deletion perturbs myocardial fatty acid oxidation and leads to cardiomyopathy. Nat. Med. 2004, 10, 1245–1250. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Liu, J.; Li, Y.; Wu, S.; Luo, J.; Yang, H.; Subbiah, R.; Chatham, J.; Zhelyabovska, O.; Yang, Q. Peroxisome prolif-erator-activated receptor {delta} is an essential transcriptional regulator for mitochondrial protection and biogenesis in adult heart. Circ. Res. 2010, 106, 911–919. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Georgiadi, A.; Lichtenstein, L.; Degenhardt, T.; Boekschoten, M.V.; Van Bilsen, M.; Desvergne, B.; Müller, M.; Kersten, S. Induction of Cardiac Angptl4 by Dietary Fatty Acids Is Mediated by Peroxisome Proliferator-Activated Receptor β/δ and Protects Against Fatty Acid–Induced Oxidative Stress. Circ. Res. 2010, 106, 1712–1721. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dijk, W.; Kersten, S. Regulation of lipid metabolism by angiopoietin-like proteins. Curr. Opin. Lipidol. 2016, 27, 249–256. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perrino, C.; Schiattarella, G.; Sannino, A.; Pironti, G.; Petretta, M.P.; Cannavo, A.; Gargiulo, G.; Ilardi, F.; Magliulo, F.; Franzone, A.; et al. Genetic Deletion of Uncoupling Protein 3 Exaggerates Apoptotic Cell Death in the Ischemic Heart Leading to Heart Failure. J. Am. Hear. Assoc. 2013, 2, 000086. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, C.-H.; Budas, G.R.; Churchill, E.N.; Disatnik, M.-H.; Hurley, T.; Mochly-Rosen, D. Activation of Aldehyde Dehydrogenase-2 Reduces Ischemic Damage to the Heart. Science 2008, 321, 1493–1495. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, C.-H.; Ferreira, J.C.B.; Gross, E.R.; Mochly-Rosen, D. Targeting Aldehyde Dehydrogenase 2: New Therapeutic Opportunities. Physiol. Rev. 2014, 94, 1–34. [Google Scholar] [CrossRef] [Green Version]
- Koppaka, V.; Thompson, D.C.; Chen, Y.; Ellermann, M.; Nicolaou, K.C.; Juvonen, R.O.; Petersen, D.; Deitrich, R.A.; Hurley, T.; Vasiliou, V. Aldehyde Dehydrogenase Inhibitors: A Comprehensive Review of the Pharmacology, Mechanism of Action, Substrate Specificity, and Clinical Application. Pharmacol. Rev. 2012, 64, 520–539. [Google Scholar] [CrossRef] [Green Version]
- Choksi, K.; Boylston, W.; Rabek, J.; Widger, W.; Papaconstantinou, J. Oxidatively damaged proteins of heart mitochondrial electron transport complexes. Biochim. Biophys. Acta (BBA) Mol. Basis Dis. 2004, 1688, 95–101. [Google Scholar] [CrossRef] [Green Version]
- Arany, Z.; He, H.; Lin, J.; Hoyer, K.; Handschin, C.; Toka, O.; Ahmad, F.; Matsui, T.; Chin, S.; Wu, P.-H.; et al. Transcriptional coactivator PGC-1α controls the energy state and contractile function of cardiac muscle. Cell Metab. 2005, 1, 259–271. [Google Scholar] [CrossRef] [Green Version]
- Yue, T.-L.; Nerurkar, S.S.; Bao, W.; Jucker, B.M.; Sarov-Blat, L.; Steplewski, K.; Ohlstein, E.H.; Willette, R.N. In Vivo Activation of Peroxisome Proliferator-Activated Receptor-δ Protects the Heart from Ischemia/Reperfusion Injury in Zucker Fatty Rats. J. Pharmacol. Exp. Ther. 2008, 325, 466–474. [Google Scholar] [CrossRef] [Green Version]
- Kapoor, A.; Collino, M.; Castiglia, S.; Fantozzi, R.; Thiemermann, C. Activation of Peroxisome Proliferator-Activated Receptor-β/δ Attenuates Myocardial Ischemia/Reperfusion Injury in the Rat. Shock 2010, 34, 117–124. [Google Scholar] [CrossRef] [PubMed]
- Galatou, E.; Kelly, T.; Lazou, A. The PPARβ/δ agonist GW0742 modulates signaling pathways associated with cardiac myocyte growth via a non-genomic redox mechanism. Mol. Cell. Biochem. 2014, 395, 145–154. [Google Scholar] [CrossRef] [PubMed]
- Ali, F.Y.; Davidson, S.J.; Moraes, L.A.; Traves, S.L.; Paul-Clark, M.; Bishop-Bailey, D.; Warner, T.D.; Mitchell, J.A. Role of nuclear receptor signaling in platelets: Antithrombotic effects of PPARβ. FASEB J. 2005, 20, 326–328. [Google Scholar] [CrossRef]
- Han, J.-K.; Lee, H.-S.; Yang, H.-M.; Hur, J.; Jun, S.-I.; Kim, J.-Y.; Cho, C.-H.; Koh, G.-Y.; Peters, J.M.; Park, K.-W.; et al. Peroxisome Proliferator-Activated Receptor-Agonist Enhances Vasculogenesis by Regulating Endothelial Progenitor Cells Through Genomic and Nongenomic Activations of the Phosphatidylinositol 3-Kinase/Akt Pathway. Circulation 2008, 118, 1021–1033. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pesant, M.; Sueur, S.; Dutartre, P.; Tallandier, M.; Grimaldi, P.A.; Rochette, L.; Connat, J.-L. Peroxisome proliferator-activated receptor δ (PPARδ) activation protects H9c2 cardiomyoblasts from oxidative stress-induced apoptosis. Cardiovasc. Res. 2006, 69, 440–449. [Google Scholar] [CrossRef]
- Liu, J.; Wang, P.; Luo, J.; Huang, Y.; He, L.; Yang, H.; Li, Q.; Wu, S.; Zhelyabovska, O.; Yang, Q. Peroxisome proliferator-activated receptor β/δ activation in adult hearts facilitates mitochondrial function and cardiac performance under pressure-overload condition. Hypertension 2011, 57, 223–230. [Google Scholar] [CrossRef] [Green Version]
- Villarroya, F.; Iglesias, R.; Giralt, M. PPARs in the Control of Uncoupling Proteins Gene Expression. PPAR Res. 2006, 2007, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Anedda, A.; López-Bernardo, E.; Acosta-Iborra, B.; Suleiman, M.S.; Landázuri, M.O.; Cadenas, S. The transcription factor Nrf2 promotes survival by enhancing the expression of uncoupling protein 3 under conditions of oxidative stress. Free. Radic. Biol. Med. 2013, 61, 395–407. [Google Scholar] [CrossRef] [PubMed]
- Wagner, K.-D.; Wagner, N. Peroxisome proliferator-activated receptor beta/delta (PPARβ/δ) acts as regulator of metabolism linked to multiple cellular functions. Pharmacol. Ther. 2010, 125, 423–435. [Google Scholar] [CrossRef]
- Csala, M.; Kardon, T.; Legeza, B.; Lizák, B.; Mandl, J.; Margittai, É.; Puskás, F.; Száraz, P.; Szelényi, P.; Bánhegyi, G. On the role of 4-hydroxynonenal in health and disease. Biochim. Biophys. Acta (BBA) Mol. Basis Dis. 2015, 1852, 826–838. [Google Scholar] [CrossRef] [Green Version]
- Münzel, T.; Daiber, A. The potential of aldehyde dehydrogenase 2 as a therapeutic target in cardiovascular disease. Expert Opin. Ther. Targets 2018, 22, 217–231. [Google Scholar] [CrossRef]
- Chen, C.-H.; Ferreira, J.C.B.; Mochly-Rosen, D. ALDH2 and Cardiovascular Disease. Aldehyde Dehydrogenases 2019, 1193, 53–67. [Google Scholar] [CrossRef]
- Ma, H.; Guo, R.; Yu, L.; Zhang, Y.; Ren, J. Aldehyde dehydrogenase 2 (ALDH2) rescues myocardial ischaemia/reperfusion injury: Role of autophagy paradox and toxic aldehyde. Eur. Heart J. 2010, 32, 1025–1038. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hua, Y.; Chen, H.; Zhao, X.; Liu, M.; Jin, W.; Yan, W.; Wu, Y.; Tan, Z.; Fan, H.; Wu, Y.; et al. Alda-1, an aldehyde dehydrogenase-2 agonist, improves long-term survival in rats with chronic heart failure following myocardial infarction. Mol. Med. Rep. 2018, 18, 3159–3166. [Google Scholar] [CrossRef]
- Xia, G.; Fan, F.; Liu, M.; Wang, S.; Wu, J.; Shen, C.; Han, S.; Wang, C.; Jia, J.; Zou, Y.; et al. Aldehyde dehydrogenase 2 deficiency blunts compensatory cardiac hypertrophy through modulating Akt phosphorylation early after transverse aorta constriction in mice. Biochim. Biophys. Acta (BBA) Mol. Basis Dis. 2016, 1862, 1587–1593. [Google Scholar] [CrossRef] [PubMed]
- Ji, W.; Wei, S.; Hao, P.; Xing, J.; Yuan, Q.; Wang, J.; Xu, F.; Chen, Y. Aldehyde Dehydrogenase 2 Has Cardioprotective Effects on Myocardial Ischaemia/Reperfusion Injury via Suppressing Mitophagy. Front. Pharmacol. 2016, 7, 101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, B.; Wang, J.; Li, M.; Yuan, Q.; Xue, M.; Xu, F.; Chen, Y. Inhibition of ALDH2 by O-GlcNAcylation contributes to the hyperglycemic exacerbation of myocardial ischemia/reperfusion injury. Oncotarget 2016, 8, 19413–19426. [Google Scholar] [CrossRef]
- Doorn, J.A.; Hurley, T.; Petersen, D.R. Inhibition of Human Mitochondrial Aldehyde Dehydrogenase by 4-Hydroxynon-2-enal and 4-Oxonon-2-enal. Chem. Res. Toxicol. 2006, 19, 102–110. [Google Scholar] [CrossRef]
- Roede, J.R.; Jones, D.P. Reactive species and mitochondrial dysfunction: Mechanistic significance of 4-hydroxynonenal. Environ. Mol. Mutagen. 2010, 51, 380–390. [Google Scholar] [CrossRef] [Green Version]
- Zhang, R.; Liu, B.; Fan, X.; Wang, W.; Xu, T.; Wei, S.; Zheng, W.; Yuan, Q.; Gao, L.; Yin, X.; et al. Aldehyde Dehydrogenase 2 Protects Against Post-Cardiac Arrest Myocardial Dysfunction Through a Novel Mechanism of Suppressing Mitochondrial Reactive Oxygen Species Production. Front. Pharmacol. 2020, 11, 373. [Google Scholar] [CrossRef]
- Shearer, B.G.; Steger, D.J.; Way, J.M.; Stanley, T.B.; Lobe, D.C.; Grillot, D.A.; Iannone, M.A.; Lazar, M.A.; Willson, T.M.; Billin, A.N. Identification and Characterization of a Selective Peroxisome Proliferator-Activated Receptor β/δ (NR1C2) Antagonist. Mol. Endocrinol. 2008, 22, 523–529. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ravingerová, T.; Matejíková, J.; Neckář, J.; Andelová, E.; Kolář, F. Differential role of PI3K/Akt pathway in the infarct size limitation and antiarrhythmic protection in the rat heart. Mol. Cell. Biochem. 2007, 297, 111–120. [Google Scholar] [CrossRef] [PubMed]
- Buss, H.; Chan, T.P.; Sluis, K.B.; Domigan, N.M.; Winterbourn, C.C. Protein Carbonyl Measurement by a Sensitive ELISA Method. Free. Radic. Biol. Med. 1997, 23, 361–366. [Google Scholar] [CrossRef]
- Eigentler, A.; Draxl, A.; Wiethüchter, A.; Kuznetsov, A.V.; Lassing, B.; Gnaiger, E. Laboratory protocol: Citrate synthase, Mi-tochondrial marker enzyme. Mitochondr Physiol Netw. 2012, 1704, 1–11. [Google Scholar]
- Lamprecht, W.; Trautschold, I. ATP-determination with hexokinase and glucose-6-phosphate dehydrogenase. In Methods of Enzymatic Analysis; Bergmeyer, H.U., Ed.; Academic: New York, NY, USA, 1974; pp. 2101–2110. [Google Scholar]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene Name | Forward Primer | Reverse Primer |
|---|---|---|
| Pparβ/δ | GAGGGGTGCAAGGGCTTCTT | CACTTGTTGCGGTTCTTCTTCTG |
| Angptl4 | GACTTTTCCAGATCCAGCCT | TGATTGAAGTCCACAGAGCC |
| Mcad | CTTCGAGTTGACGGAGCAG | TTGATGAGAGGGAACGGGT |
| Sod2 | GGAGTCCAAGGTTCAGGCT | AGGTAGTAAGCGTGCTCCCA |
| Catalase | CCTGTGAACTGTCCCTACCG | ACCCAGTCCCATGCTCTCTC |
| Ucp3 | ACAAAGGATTCATGCCCTCC | GATTCCCGCAGTACCTGGAC |
| Aldh2 | CAGGTAGCCGAAGGGAACA | GCCAATCGGTACAACAGCC |
| Ppargc1a | AGAGTCACCAAATGACCCCA | GAGTTAGGCCTGCAGTTCCA |
| Idh2 | GGACAGTCACCCGCCATTAC | ACATTGCTGAGGCCATGGAT |
| β-Actin | GCCCTGAGGCACTCTTCCA | CGGATGTCCACGTCACACTTC |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Papatheodorou, I.; Galatou, E.; Panagiotidis, G.-D.; Ravingerová, T.; Lazou, A. Cardioprotective Effects of PPARβ/δ Activation against Ischemia/Reperfusion Injury in Rat Heart Are Associated with ALDH2 Upregulation, Amelioration of Oxidative Stress and Preservation of Mitochondrial Energy Production. Int. J. Mol. Sci. 2021, 22, 6399. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22126399
Papatheodorou I, Galatou E, Panagiotidis G-D, Ravingerová T, Lazou A. Cardioprotective Effects of PPARβ/δ Activation against Ischemia/Reperfusion Injury in Rat Heart Are Associated with ALDH2 Upregulation, Amelioration of Oxidative Stress and Preservation of Mitochondrial Energy Production. International Journal of Molecular Sciences. 2021; 22(12):6399. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22126399
Chicago/Turabian StylePapatheodorou, Ioanna, Eleftheria Galatou, Georgios-Dimitrios Panagiotidis, Táňa Ravingerová, and Antigone Lazou. 2021. "Cardioprotective Effects of PPARβ/δ Activation against Ischemia/Reperfusion Injury in Rat Heart Are Associated with ALDH2 Upregulation, Amelioration of Oxidative Stress and Preservation of Mitochondrial Energy Production" International Journal of Molecular Sciences 22, no. 12: 6399. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22126399