
Molecular Inversion Probe-Based Sequencing of USH2A Exons and Splice Sites as a Cost-Effective Screening Tool in USH2 and arRP Cases

 , , , , , , , , , , , , , , , , , , and
, , , , , , , , , , , , , , , , , , and
Abstract
:1. Introduction
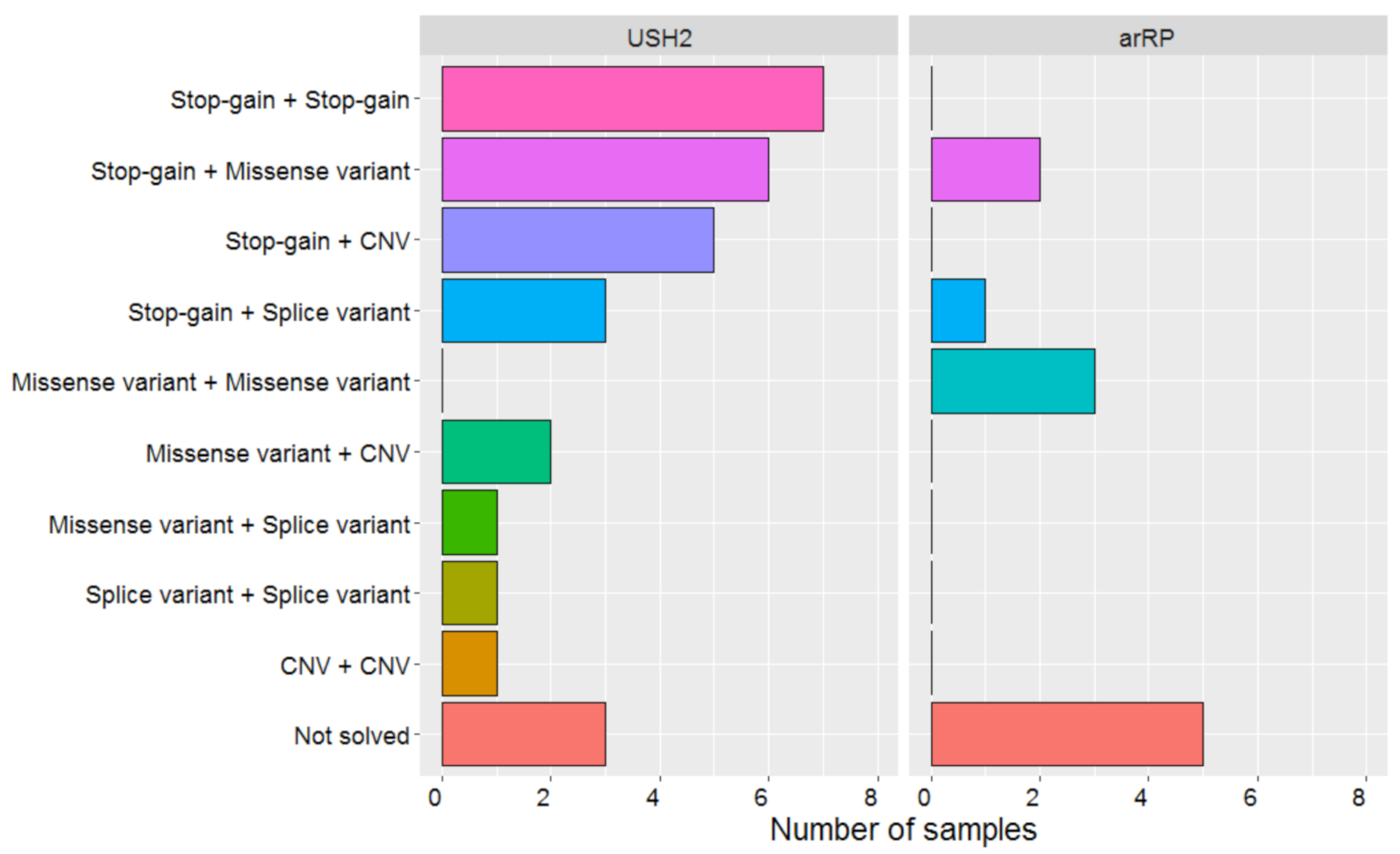
2. Results
2.1. One Hundred and Seven SNVs Were Identified, Including Four Novel Variants
2.2. Four Copy Number Variants Were Detected
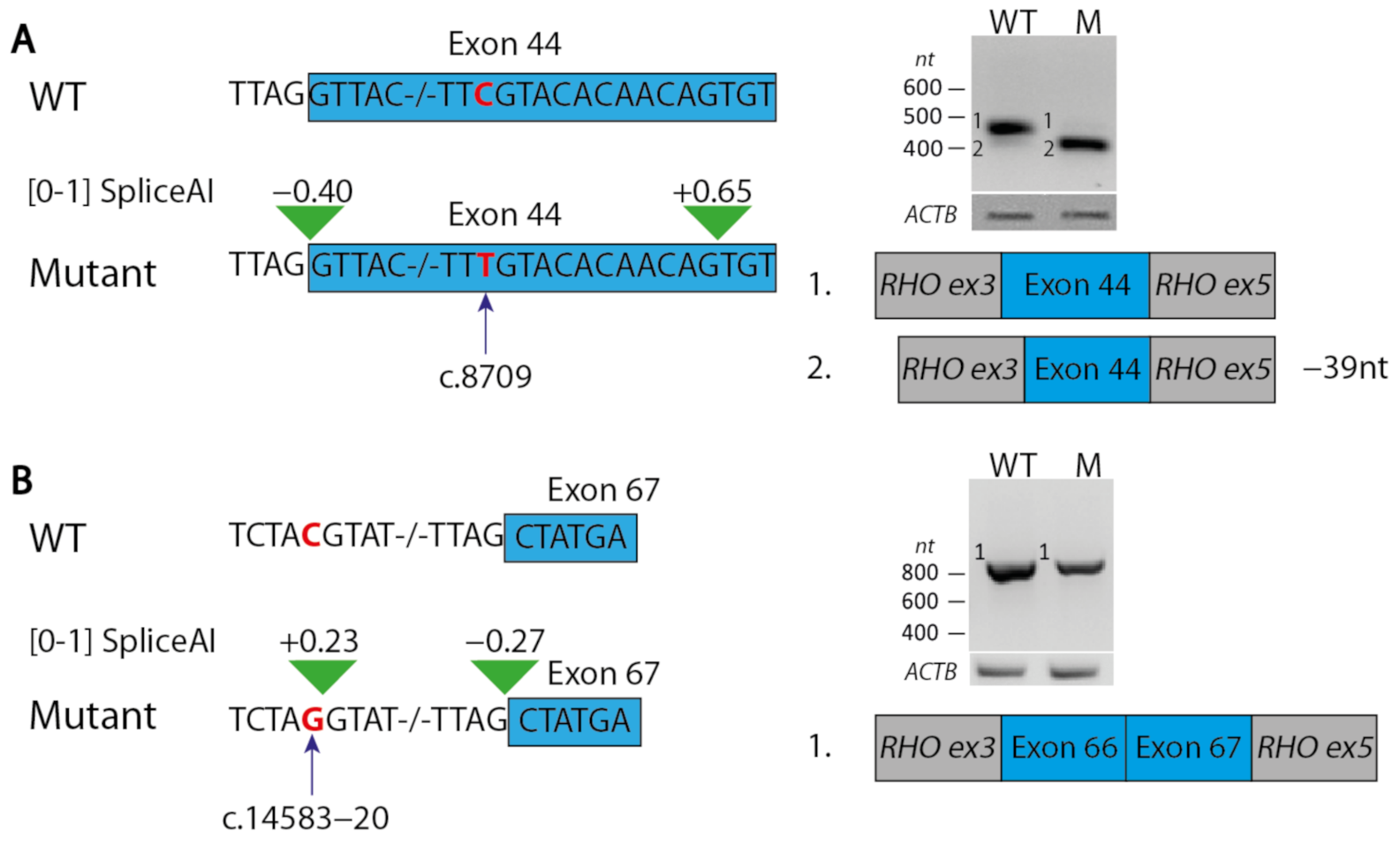
2.3. Two In-Frame Deletions Were Classified as Likely Pathogenic after Modelling
3. Discussion
4. Materials and Methods
4.1. Inclusion of Samples
4.2. MIP-Based Sequencing and Data Analysis
4.3. Prioritization and Classification of Variants
4.4. Minigene Splice Assays
4.5. Modelling the Effect of Two In-Frame Deletions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Daiger, S.R.; Greenberg, J.; Christoffels, A.; Hide, W. RetNet, the Retinal Information Network. Available online: https://sph.uth.edu/RetNet/ (accessed on 9 March 2021).
- Perea-Romero, I.; The ESRETNET Study Group; Gordo, G.; Iancu, I.F.; Del Pozo-Valero, M.; Almoguera, B.; Blanco-Kelly, F.; Carreño, E.; Jimenez-Rolando, B.; Lopez-Rodriguez, R.; et al. Genetic landscape of 6089 inherited retinal dystrophies affected cases in Spain and their therapeutic and extended epidemiological implications. Sci. Rep. 2021, 11, 1–13. [Google Scholar] [CrossRef]
- Stone, E.M.; Andorf, J.L.; Whitmore, S.S.; DeLuca, A.P.; Giacalone, J.C.; Streb, L.M.; Braun, T.A.; Mullins, R.F.; Scheetz, T.E.; Sheffield, V.C.; et al. Clinically Focused Molecular Investigation of 1000 Consecutive Families with Inherited Retinal Disease. Ophthalmology 2017, 124, 1314–1331. [Google Scholar] [CrossRef] [PubMed]
- Jouret, G.; Poirsier, C.; Spodenkiewicz, M.; Jaquin, C.; Gouy, E.; Arndt, C.; Labrousse, M.; Gaillard, D.; Doco-Fenzy, M.; Lebre, A.-S. Genetics of Usher Syndrome: New Insights from a Meta-analysis. Otol. Neurotol. 2019, 40, 121–129. [Google Scholar] [CrossRef] [PubMed]
- McGee, T.L.; Seyedahmadi, B.J.; Sweeney, M.O.; Dryja, T.P.; Berson, E.L. Novel mutations in the long isoform of the USH2A gene in patients with Usher syndrome type II or non-syndromic retinitis pigmentosa. J. Med. Genet. 2010, 47, 499–506. [Google Scholar] [CrossRef]
- Toualbi, L.; Toms, M.; Moosajee, M. USH2A-retinopathy: From genetics to therapeutics. Exp. Eye Res. 2020, 201, 108330. [Google Scholar] [CrossRef]
- Boughman, J.A.; Conneally, P.M.; Nance, W.E. Population genetic studies of retinitis pigmentosa. Am. J. Hum. Genet. 1980, 32, 223–235. [Google Scholar]
- Puech, B.; Kostrubiec, B.; Hache, J.C.; François, P. Epidemiology and prevalence of hereditary retinal dystrophies in the Northern France. J. Fr. Ophtalmol. 1991, 14, 153–164. [Google Scholar]
- Rosenberg, T.; Haim, M.; Hauch, A.-M.; Parving, A. The prevalence of Usher syndrome and other retinal dystrophy-hearing impairment associations. Clin. Genet. 2008, 51, 314–321. [Google Scholar] [CrossRef]
- Spandau, U.H.; Rohrschneider, K. Prevalence and geographical distribution of Usher syndrome in Germany. Graefes Arch. Clin. Exp. Ophthalmol. 2002, 240, 495–498. [Google Scholar] [CrossRef]
- Van Wijk, E.; Pennings, R.J.; te Brinke, H.; Claassen, A.; Yntema, H.G.; Hoefsloot, L.H.; Cremers, F.P.; Cremers, C.W.; Kremer, H. Identification of 51 Novel Exons of the Usher Syndrome Type 2A (USH2A) Gene That Encode Multiple Conserved Functional Domains and That Are Mutated in Patients with Usher Syndrome Type II. Am. J. Hum. Genet. 2004, 74, 738–744. [Google Scholar] [CrossRef] [Green Version]
- Adato, A.; Lefèvre, G.; Delprat, B.; Michel, V.; Michalski, N.; Chardenoux, S.; Weil, D.; El-Amraoui, A.; Petit, C. Usherin, the defective protein in Usher syndrome type IIA, is likely to be a component of interstereocilia ankle links in the inner ear sensory cells. Hum. Mol. Genet. 2005, 14, 3921–3932. [Google Scholar] [CrossRef] [PubMed]
- Eudy, J.D.; Weston, M.D.; Yao, S.; Hoover, D.M.; Rehm, H.L.; Ma-Edmonds, M.; Yan, D.; Ahmad, I.; Cheng, J.J.; Ayuso, C.; et al. Mutation of a Gene Encoding a Protein with Extracellular Matrix Motifs in Usher Syndrome Type IIa. Science 1998, 280, 1753–1757. [Google Scholar] [CrossRef] [PubMed]
- Cremers, F.P.M.; Kimberling, W.J.; Külm, M.; De Brouwer, A.P.; Van Wijk, E.; Brinke, H.T.; Cremers, C.W.R.J.; Hoefsloot, L.H.; Banfi, S.; Simonelli, F.; et al. Development of a genotyping microarray for Usher syndrome. J. Med. Genet. 2006, 44, 153–160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dulla, K.; Slijkerman, R.; van Diepen, H.C.; Albert, S.; Dona, M.; Beumer, W.; Turunen, J.J.; Chan, H.L.; Schulkens, I.A.; Vorthoren, L.; et al. Antisense oligonucleotide-based treatment of retinitis pigmentosa caused by USH2A exon 13 mutations. Mol. Ther. 2021. [Google Scholar] [CrossRef]
- Vaché, C.; Besnard, T.; le Berre, P.; García-García, G.; Baux, D.; Larrieu, L.; Abadie, C.; Blanchet, C.; Bolz, H.J.; Millan, J.; et al. Usher syndrome type 2 caused by activation of an USH2A pseudoexon: Implications for diagnosis and therapy. Hum. Mutat. 2012, 33, 104–108. [Google Scholar] [CrossRef]
- Slijkerman, R.W.; Vaché, C.; Dona, M.; García-García, G.; Claustres, M.; Hetterschijt, L.; A Peters, T.; Hartel, B.P.; Pennings, R.J.; Millan, J.M.; et al. Antisense Oligonucleotide-based Splice Correction for USH2A-associated Retinal Degeneration Caused by a Frequent Deep-intronic Mutation. Mol. Ther. Nucleic Acids 2016, 5, e381. [Google Scholar] [CrossRef] [Green Version]
- Liquori, A.; Vaché, C.; Baux, D.; Blanchet, C.; Hamel, C.P.; Malcolm, S.; Koenig, M.; Claustres, M.; Roux, A.-F. Whole USH2A Gene Sequencing Identifies Several New Deep Intronic Mutations. Hum. Mutat. 2016, 37, 184–193. [Google Scholar] [CrossRef]
- Baux, D.; Vaché, C.; Blanchet, C.; Willems, M.; Baudoin, C.; Moclyn, M.; Faugère, V.; Touraine, R.; Isidor, B.; Dupin-Deguine, D.; et al. Combined genetic approaches yield a 48% diagnostic rate in a large cohort of French hearing-impaired patients. Sci. Rep. 2017, 7, 16783. [Google Scholar] [CrossRef] [Green Version]
- Hardenbol, P.; Baner, J.; Jain, M.; Nilsson, M.; Namsaraev, E.A.; Karlin-Neumann, G.A.; Fakhrai-Rad, H.; Ronaghi, M.; Willis, T.D.; Landegren, U.; et al. Multiplexed genotyping with sequence-tagged molecular inversion probes. Nat. Biotechnol. 2003, 21, 673–678. [Google Scholar] [CrossRef]
- O’Roak, B.J.; Vives, L.; Fu, W.; Egertson, J.D.; Stanaway, I.B.; Phelps, I.G.; Carvill, G.; Kumar, A.; Lee, C.; Ankenman, K.; et al. Multiplex Targeted Sequencing Identifies Recurrently Mutated Genes in Autism Spectrum Disorders. Science 2012, 338, 1619–1622. [Google Scholar] [CrossRef] [Green Version]
- Le Guédard-Méreuze, S.; Vaché, C.; Baux, D.; Faugère, V.; Larrieu, L.; Abadie, C.; Janecke, A.; Claustres, M.; Roux, A.-F.; Tuffery-Giraud, S. Ex vivo splicing assays of mutations at noncanonical positions of splice sites in USHER genes. Hum. Mutat. 2010, 31, 347–355. [Google Scholar] [CrossRef]
- Vaché, C.; Besnard, T.; Blanchet, C.; Baux, D.; Larrieu, L.; Faugère, V.; Mondain, M.; Hamel, C.; Malcolm, S.; Claustres, M.; et al. Nasal epithelial cells are a reliable source to study splicing variants in Usher syndrome. Hum. Mutat. 2010, 31, 734–741. [Google Scholar] [CrossRef] [Green Version]
- Baux, D.; Blanchet, C.; Hamel, C.; Meunier, I.; Larrieu, L.; Faugère, V.; Vaché, C.; Castorina, P.; Puech, B.; Bonneau, D.; et al. Enrichment of LOVD-USHbases with 152 USH2A Genotypes Defines an Extensive Mutational Spectrum and Highlights Missense Hotspots. Hum. Mutat. 2014, 35, 1179–1186. [Google Scholar] [CrossRef]
- Baux, D.; Larrieu, L.; Blanchet, C.; Hamel, C.; Ben Salah, S.; Vielle, A.; Gilbert-Dussardier, B.; Holder, M.; Calvas, P.; Philip, N.; et al. Molecular and in silico analyses of the full-length isoform of usherin identify new pathogenic alleles in Usher type II patients. Hum. Mutat. 2007, 28, 781–789. [Google Scholar] [CrossRef]
- Dad, S.; Rendtorff, N.D.; Kann, E.; Albrechtsen, A.; Mehrjouy, M.M.; Bak, M.; Tommerup, N.; Tranebjærg, L.; Rosenberg, T.; Jensen, H.; et al. Partial USH2A deletions contribute to Usher syndrome in Denmark. Eur. J. Hum. Genet. 2015, 23, 1646–1651. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zampaglione, E.; Kinde, B.; Ms, E.M.P.; Bs, D.N.-G.; Ma, C.F.; Jamshidi, F.; Ms, S.N.; Bs, J.A.M.; Finn, C.; Schlegel, D.; et al. Copy-number variation contributes 9% of pathogenicity in the inherited retinal degenerations. Genet. Med. 2020, 22, 1079–1087. [Google Scholar] [CrossRef] [Green Version]
- Schultz, J.; Milpetz, F.; Bork, P.; Ponting, C.P. SMART, a simple modular architecture research tool: Identification of signaling domains. Proc. Natl. Acad. Sci. USA 1998, 95, 5857–5864. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Albert, S.; Garanto, A.; Sangermano, R.; Khan, M.; Bax, N.; Hoyng, C.B.; Zernant, J.; Lee, W.; Allikmets, R.; Collin, R.W.; et al. Identification and Rescue of Splice Defects Caused by Two Neighboring Deep-Intronic ABCA4 Mutations Underlying Stargardt Disease. Am. J. Hum. Genet. 2018, 102, 517–527. [Google Scholar] [CrossRef] [Green Version]
- Bonnet, C.; Riahi, Z.; Chantot-Bastaraud, S.; Smagghe, L.; Letexier, M.; Marcaillou, C.; Lefèvre, G.M.; Hardelin, J.-P.; El-Amraoui, A.; Singh-Estivalet, A.; et al. An innovative strategy for the molecular diagnosis of Usher syndrome identifies causal biallelic mutations in 93% of European patients. Eur. J. Hum. Genet. 2016, 24, 1730–1738. [Google Scholar] [CrossRef] [Green Version]
- Collins, R.L.; Team, G.A.D.P.; Brand, H.; Karczewski, K.J.; Zhao, X.; Alföldi, J.; Francioli, L.C.; Khera, A.V.; Lowther, C.; Gauthier, L.D.; et al. A structural variation reference for medical and population genetics. Nat. Cell Biol. 2020, 581, 444–451. [Google Scholar] [CrossRef]
- Fairley, S.; Lowy-Gallego, E.; Perry, E.; Flicek, P. The International Genome Sample Resource (IGSR) collection of open human genomic variation resources. Nucleic Acids Res. 2020, 48, D941–D947. [Google Scholar] [CrossRef]
- Lenarduzzi, S.; Morgan, A.; Faletra, F.; Cappellani, S.; Morgutti, M.; Mezzavilla, M.; Peruzzi, A.; Ghiselli, S.; Ambrosetti, U.; Graziano, C.; et al. Next generation sequencing study in a cohort of Italian patients with syndromic hearing loss. Hear. Res. 2019, 381, 107769. [Google Scholar] [CrossRef]
- Dreyer, B.; Brox, V.; Tranebjærg, L.; Rosenberg, T.; Sadeghi, A.M.; Möller, C.; Nilssen, Ø. Spectrum of USH2A mutations in Scandinavian patients with Usher syndrome type II. Hum. Mutat. 2008, 29, 451. [Google Scholar] [CrossRef]
- Karczewski, K.J.; Francioli, L.C.; Tiao, G.; Cummings, B.B.; Alföldi, J.; Wang, Q.; Collins, R.L.; Laricchia, K.M.; Ganna, A.; Birnbaum, D.P.; et al. Variation across 141,456 human exomes and genomes reveals the spectrum of loss-of-function intolerance across human protein-coding genes. bioRxiv 2019, 531210. [Google Scholar] [CrossRef]
- Grantham, R. Amino Acid Difference Formula to Help Explain Protein Evolution. Science 1974, 185, 862–864. [Google Scholar] [CrossRef]
- Kircher, M.; Witten, D.M.; Jain, P.; O‘Roak, B.J.; Cooper, G.M.; Shendure, J. A general framework for estimating the relative pathogenicity of human genetic variants. Nat. Genet. 2014, 46, 310–315. [Google Scholar] [CrossRef] [Green Version]
- Pollard, K.S.; Hubisz, M.J.; Rosenbloom, K.R.; Siepel, A. Detection of nonneutral substitution rates on mammalian phylogenies. Genome Res. 2009, 20, 110–121. [Google Scholar] [CrossRef] [Green Version]
- Ng, P.C.; Henikoff, S. Predicting Deleterious Amino Acid Substitutions. Genome Res. 2001, 11, 863–874. [Google Scholar] [CrossRef] [Green Version]
- Schwarz, J.M.; Cooper, D.N.; Schuelke, M.; Seelow, D. MutationTaster2: Mutation prediction for the deep-sequencing age. Nat. Methods 2014, 11, 361–362. [Google Scholar] [CrossRef]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [Green Version]
- McKenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A.; Garimella, K.; Altshuler, D.; Gabriel, S.; Daly, M.; et al. The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010, 20, 1297–1303. [Google Scholar] [CrossRef] [Green Version]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–423. [Google Scholar] [CrossRef]
- Jaganathan, K.; Panagiotopoulou, S.K.; McRae, J.F.; Darbandi, S.F.; Knowles, D.; Li, Y.I.; Kosmicki, J.A.; Arbelaez, J.; Cui, W.; Schwartz, G.B.; et al. Predicting Splicing from Primary Sequence with Deep Learning. Cell 2019, 176, 535–548e24. [Google Scholar] [CrossRef] [Green Version]
- Khan, M.; Msc, S.S.C.; Msc, M.D.P.-V.; Whelan, L.; Runhart, E.H.; Mishra, K.; Bults, F.; AlSwaiti, Y.; AlTalbishi, A.; De Baere, E.; et al. Resolving the dark matter of ABCA4 for 1054 Stargardt disease probands through integrated genomics and transcriptomics. Genet. Med. 2020, 22, 1235–1246. [Google Scholar] [CrossRef]
- Sangermano, R.; Bax, N.; Bauwens, M.; Born, L.I.V.D.; De Baere, E.; Garanto, A.; Collin, R.W.; Goercharn-Ramlal, A.S.; Dijk, A.H.D.E.-V.; Rohrschneider, K.; et al. Photoreceptor Progenitor mRNA Analysis Reveals Exon Skipping Resulting from the ABCA4 c5461-10T→C Mutation in Stargardt Disease. Ophthalmology 2016, 123, 1375–1385. [Google Scholar] [CrossRef]
- Krieger, E.; Koraimann, G.; Vriend, G. Increasing the precision of comparative models with YASARA NOVA—A self-parameterizing force field. Proteins Struct. Funct. Bioinform. 2002, 47, 393–402. [Google Scholar] [CrossRef]
- Vriend, G. WHAT IF: A molecular modeling and drug design program. J. Mol. Graph. 1990, 8, 52–56. [Google Scholar] [CrossRef]
- Porebski, B.T.; Nickson, A.A.; Hoke, D.E.; Hunter, M.R.; Zhu, L.; McGowan, S.; Webb, G.I.; Buckle, A.M. Structural and dynamic properties that govern the stability of an engineered fibronectin type III domain. Protein Eng. Des. Sel. 2015, 28, 67–78. [Google Scholar] [CrossRef] [Green Version]
- Xu, K.; Tzvetkova-Robev, D.; Xu, Y.; Goldgur, Y.; Chan, Y.-P.; Himanen, J.P.; Nikolov, D.B. Insights into Eph receptor tyrosine kinase activation from crystal structures of the EphA4 ectodomain and its complex with ephrin-A5. Proc. Natl. Acad. Sci. USA 2013, 110, 14634–14639. [Google Scholar] [CrossRef] [Green Version]
- Yamagata, A.; Yoshida, T.; Sato, Y.; Goto-Ito, S.; Uemura, T.; Maeda, A.; Shiroshima, T.; Iwasawa-Okamoto, S.; Mori, H.; Mishina, M.; et al. Mechanisms of splicing-dependent trans-synaptic adhesion by PTPδ–IL1RAPL1/IL-1RAcP for synaptic differentiation. Nat. Commun. 2015, 6, 6926. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vilstrup, J.; Simonsen, A.; Birkefeldt, T.; Strandbygård, D.; Lyngsø, J.; Pedersen, J.S.; Thirup, S. Crystal and solution structures of fragments of the human leucocyte common antigen-related protein. Acta Crystallogr. Sect. D Struct. Biol. 2020, 76, 406–417. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Variant (NM_206933.2) | Protein Effect | Exon | PhyloP (≥2.7) | CADD_ PHRED (≥15) | Grantham (≥80) | SIFT | Mutation Taster | Domain | ACMG Classification |
|---|---|---|---|---|---|---|---|---|---|
| c.9388T>G | p.(Trp3130Gly) | 48 | 5.06 | 25.5 | 184 | Deleterious | Disease causing | Fibronectin type-III domain 18 | Likely Pathogenic |
| c.11683G>T | p.(Gly3895*) | 60 | 5.89 | 55 | - | - | - | Fibronectin type-III domain 23 | Pathogenic |
| c.14303A>C | p.(Tyr4768Ser) | 65 | 4.17 | 26.1 | 144 | Deleterious | Disease causing | Fibronectin type-III domain 32 | Likely Pathogenic |
| c.15286del | p.(Glu5096Lysfs*6) | 70 | - | 41 | - | - | - | - | Pathogenic |
| Variant (NM_206933.2) | Protein Effect | Exons | Domains (Partially) Deleted | Number of Alleles | Reference |
|---|---|---|---|---|---|
| c.1644+10004_1972−12164del | p.(Cys549_Gln657del) | 10-11 | Laminin EGF-like domains 1-3 | 1 | [24] |
| c.4627+25435_4987+660del | p.(Gly1543_Pro1662del) | 22-24 | Laminin G domain 1 | 1 | [25,26] |
| c.7121−8313_11048−962delins12 | p.(Val2374_Gly3683del) | 38-56 | Fibronectin type-III domains 10-21 | 6 | [27] |
| c.8559-?_8681+?del | p.(Tyr2854_Arg2894del) | 43 | Fibronectin type-III domain 15 | 1 | This study |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Reurink, J.; Dockery, A.; Oziębło, D.; Farrar, G.J.; Ołdak, M.; ten Brink, J.B.; Bergen, A.A.; Rinne, T.; Yntema, H.G.; Pennings, R.J.E.; et al. Molecular Inversion Probe-Based Sequencing of USH2A Exons and Splice Sites as a Cost-Effective Screening Tool in USH2 and arRP Cases. Int. J. Mol. Sci. 2021, 22, 6419. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22126419
Reurink J, Dockery A, Oziębło D, Farrar GJ, Ołdak M, ten Brink JB, Bergen AA, Rinne T, Yntema HG, Pennings RJE, et al. Molecular Inversion Probe-Based Sequencing of USH2A Exons and Splice Sites as a Cost-Effective Screening Tool in USH2 and arRP Cases. International Journal of Molecular Sciences. 2021; 22(12):6419. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22126419
Chicago/Turabian StyleReurink, Janine, Adrian Dockery, Dominika Oziębło, G. Jane Farrar, Monika Ołdak, Jacoline B. ten Brink, Arthur A. Bergen, Tuula Rinne, Helger G. Yntema, Ronald J. E. Pennings, and et al. 2021. "Molecular Inversion Probe-Based Sequencing of USH2A Exons and Splice Sites as a Cost-Effective Screening Tool in USH2 and arRP Cases" International Journal of Molecular Sciences 22, no. 12: 6419. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22126419