
Functional Characterization of Two Novel Mutations in SCN5A Associated with Brugada Syndrome Identified in Italian Patients

 , , , ,
, , , ,  , , , , ,
, , , , ,
Abstract
:1. Introduction
2. Results
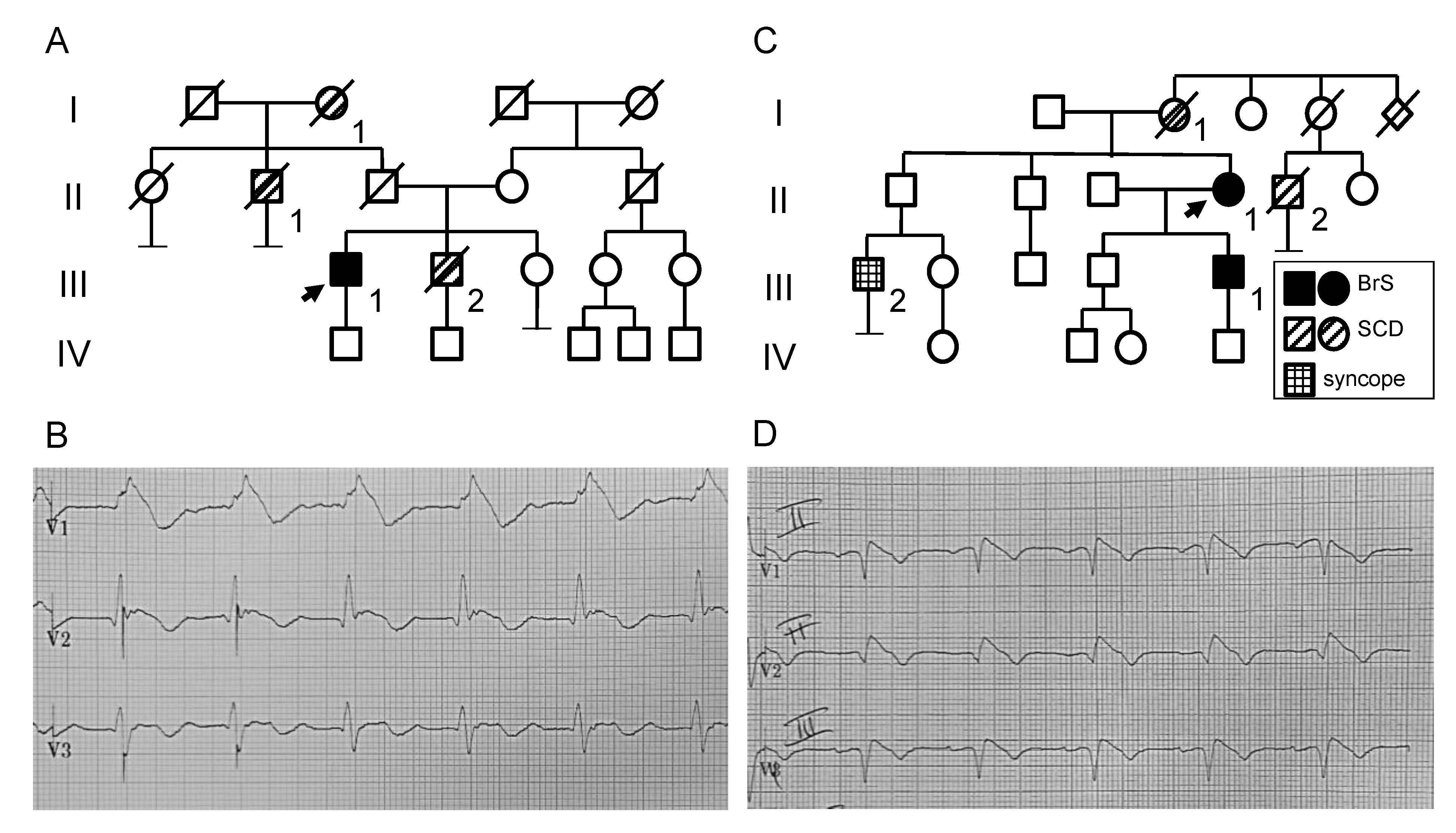
2.1. Clinical and Genetic Analysis
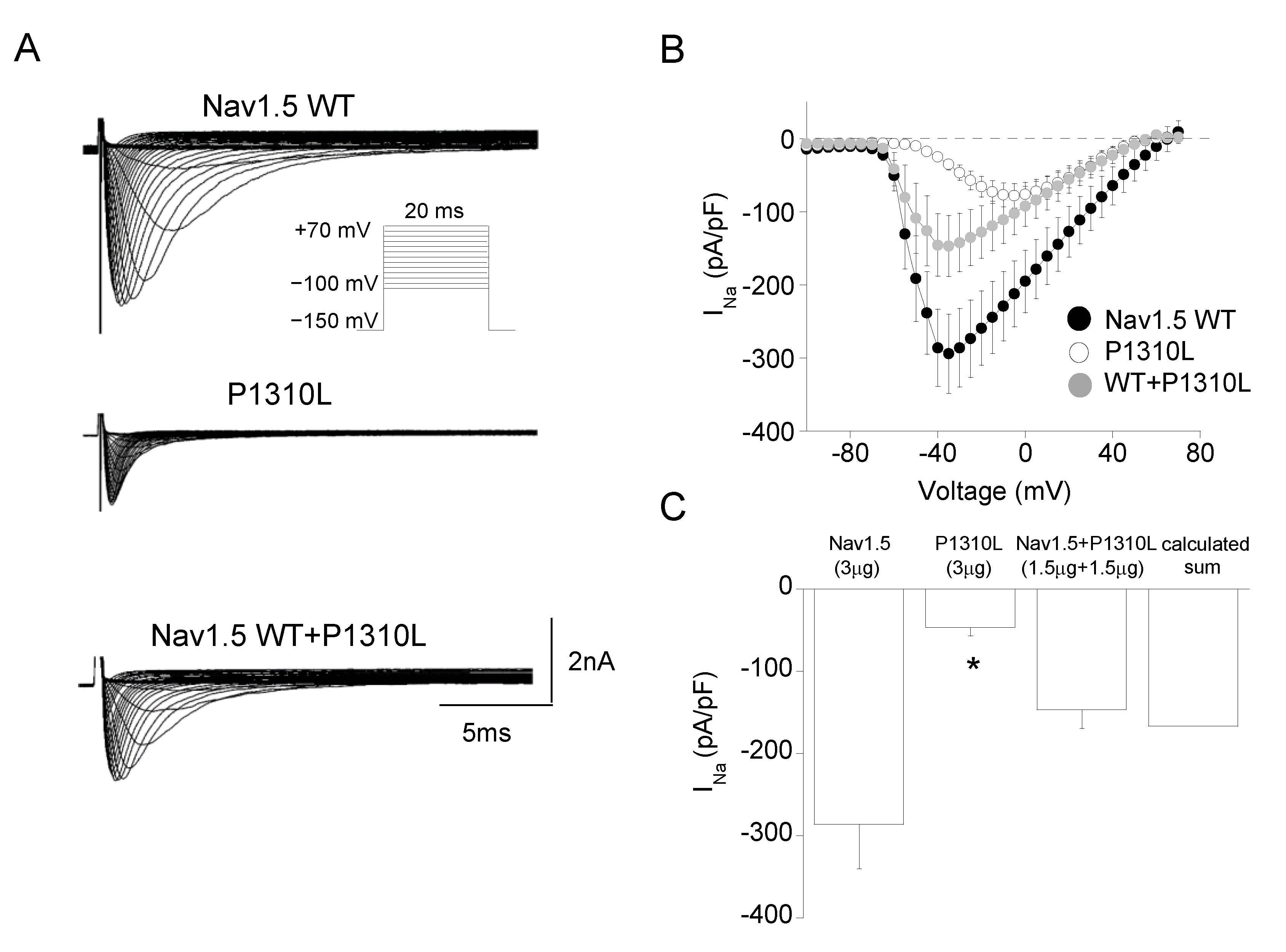
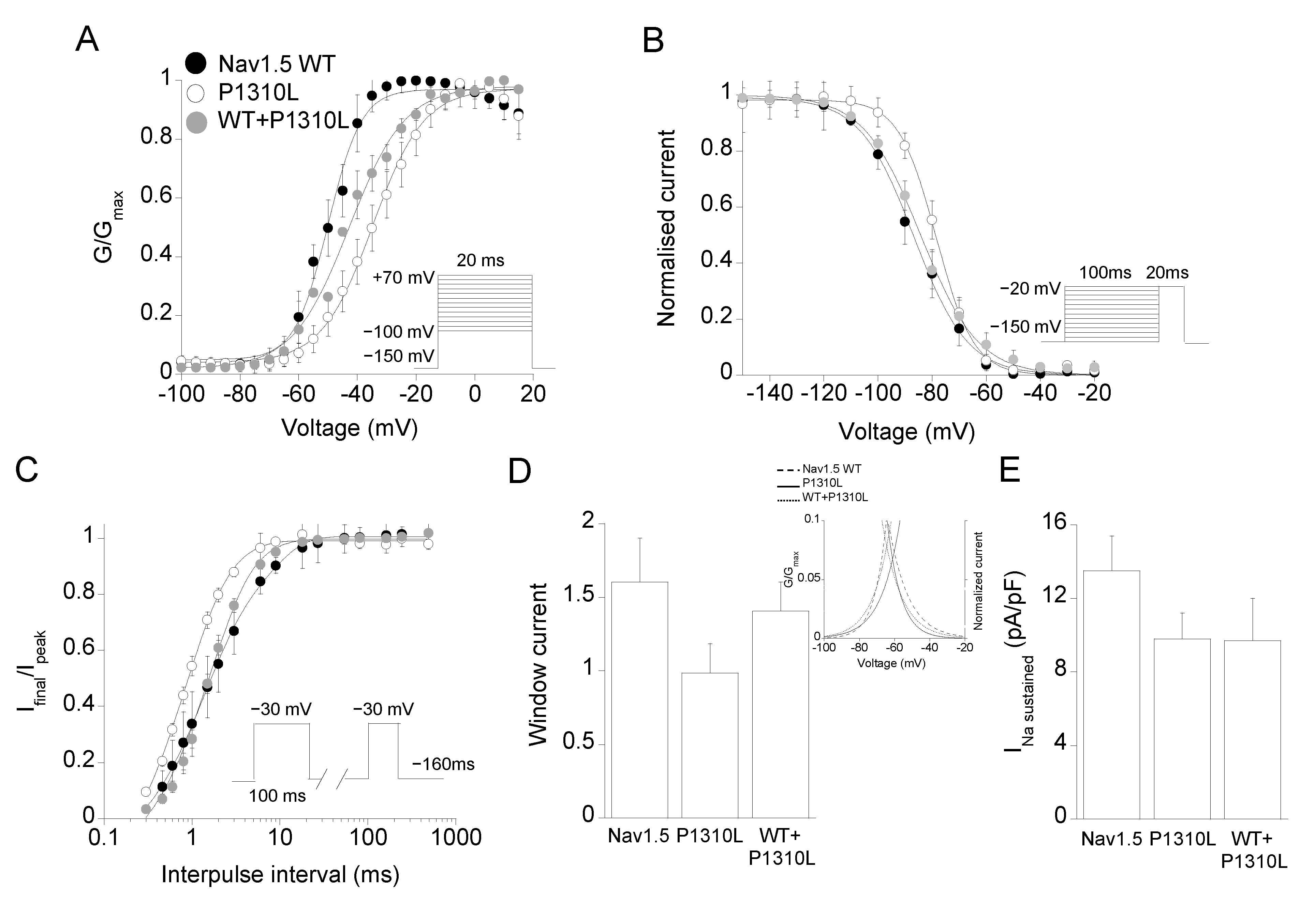
2.2. Functional Characterization of P1310L Mutant Channels
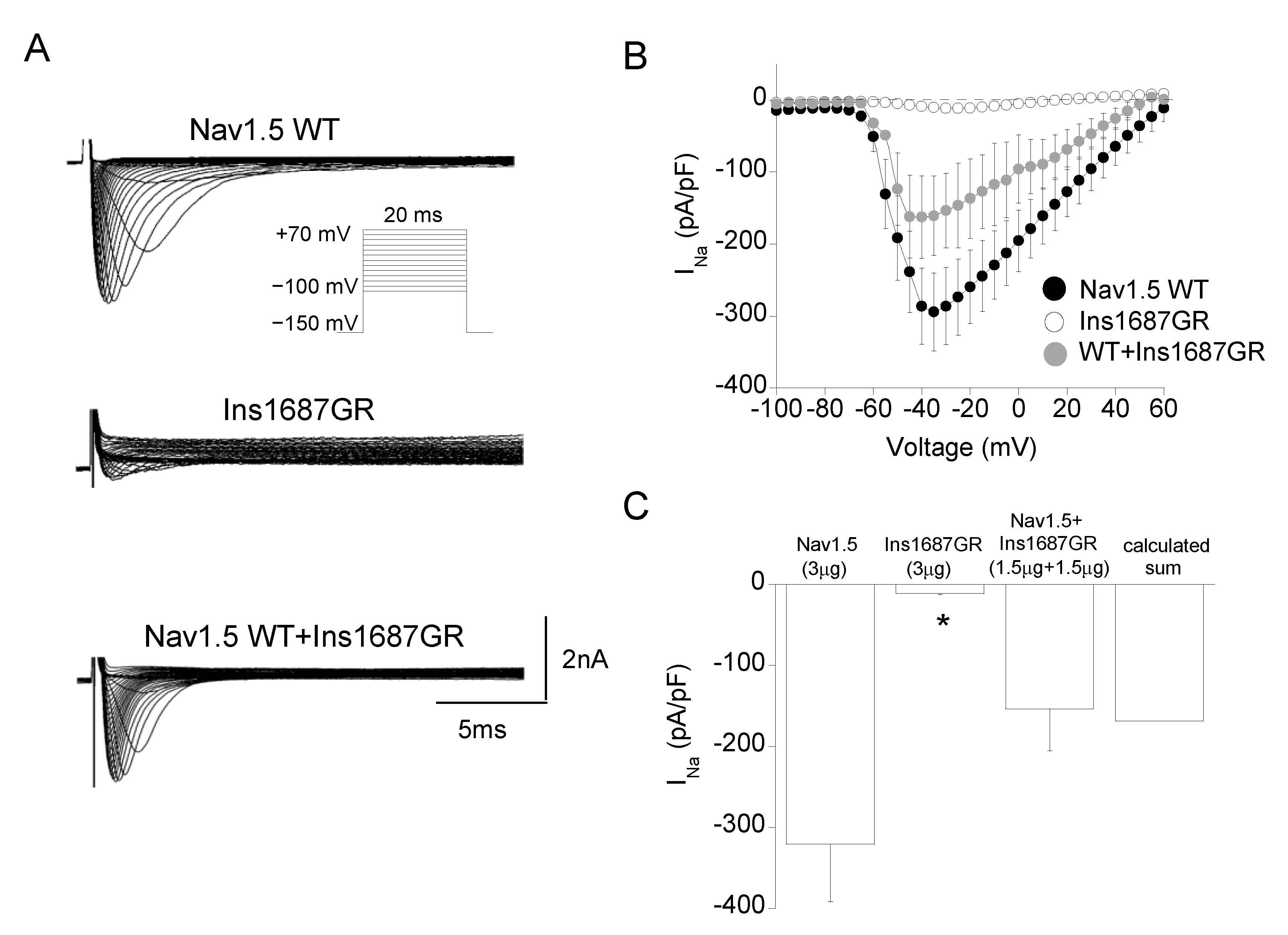
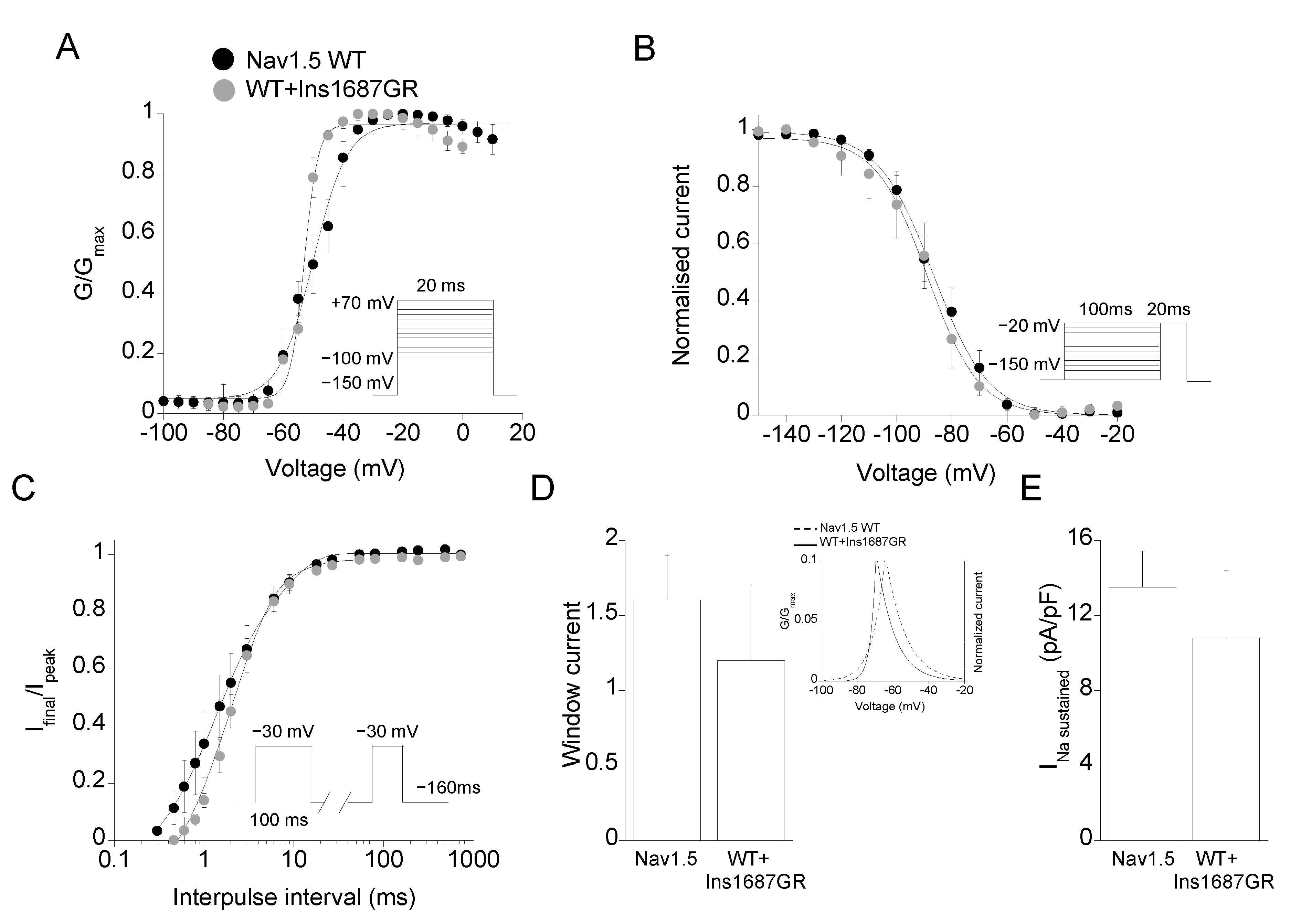
2.3. Functional Characterization of Ins1687GR Mutant Channels
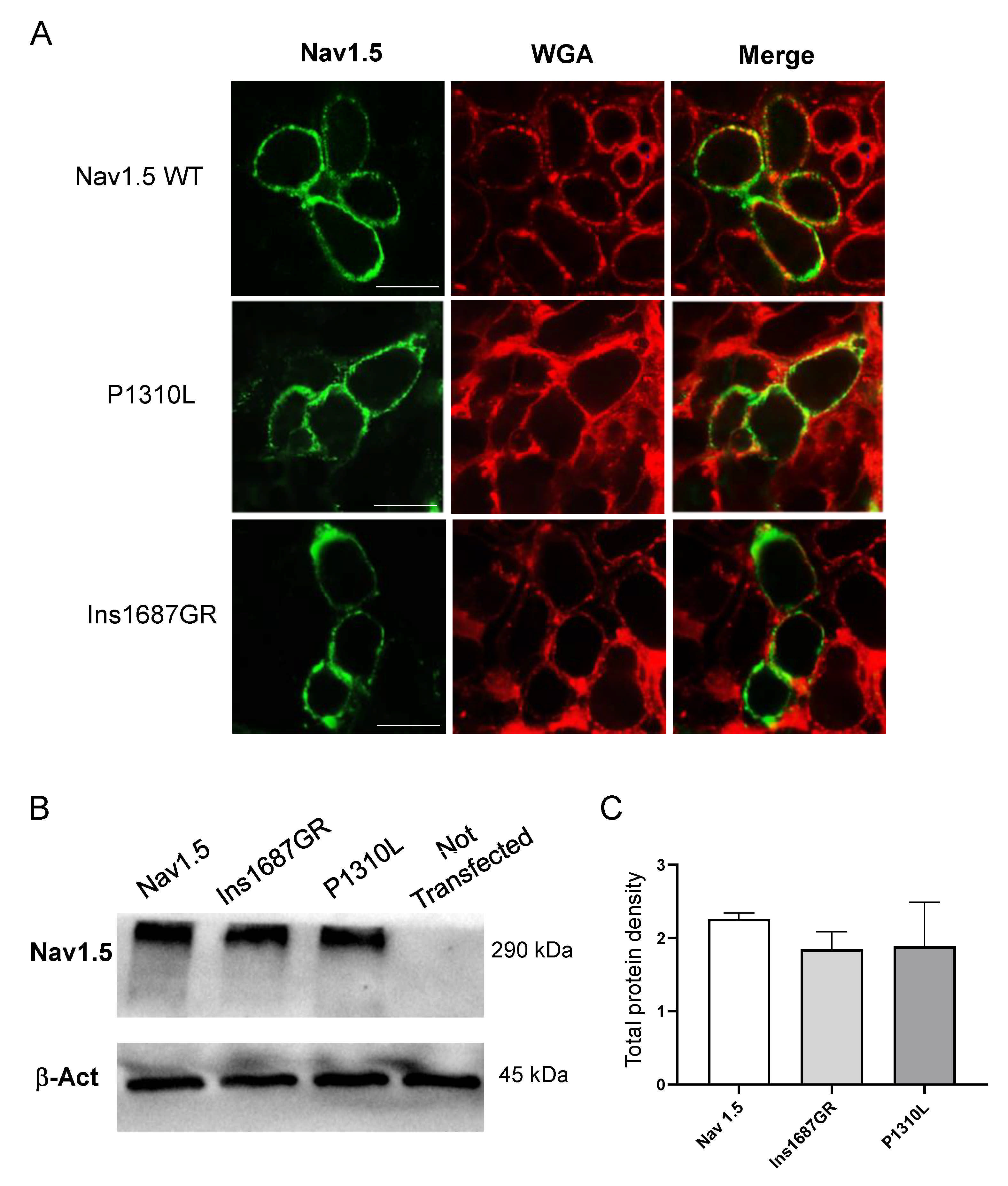
2.4. Cellular Localization of Nav1.5 WT, P1310L and Ins1687GR Mutant Channels
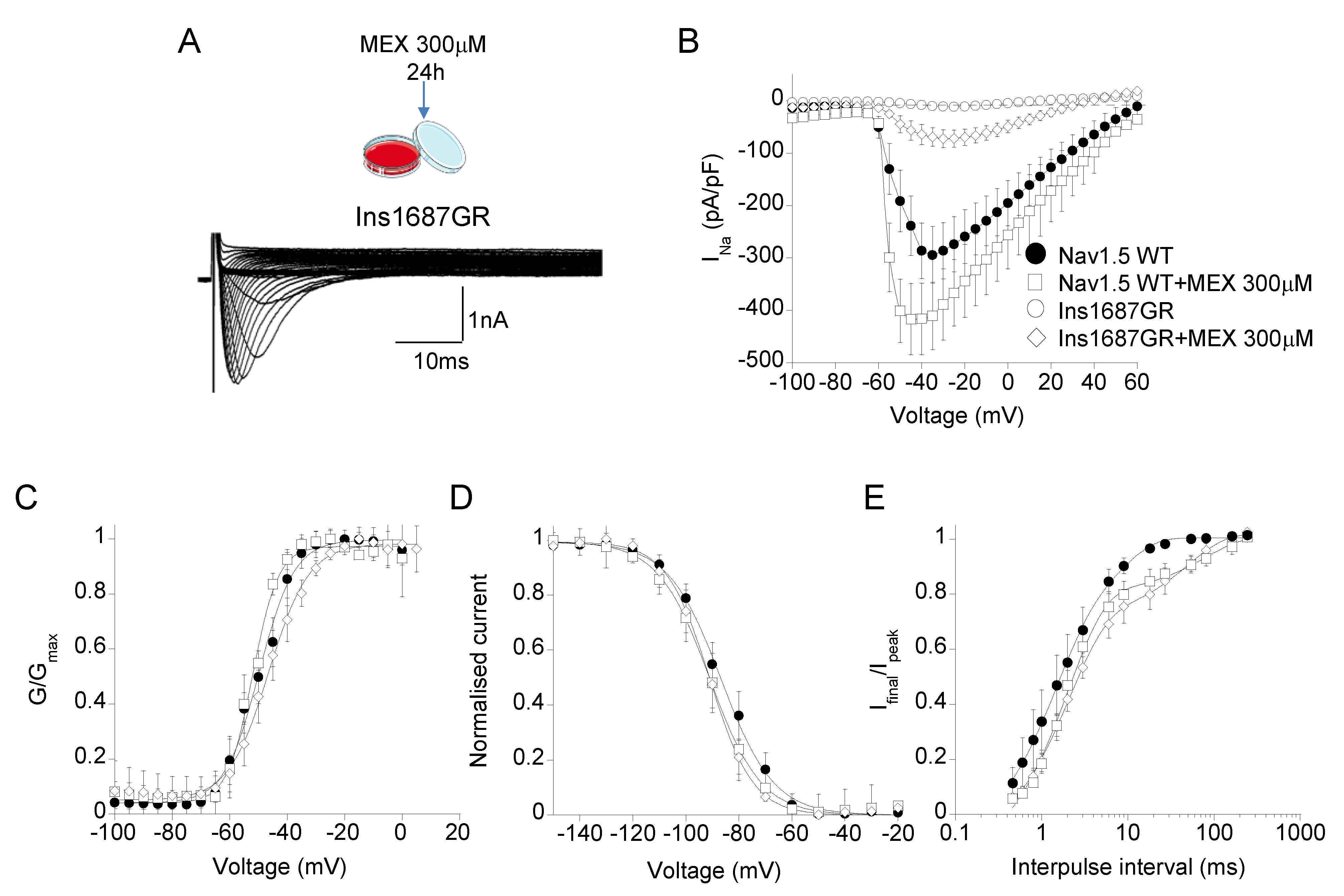
2.5. Effect of Mexiletine Incubation on Nav1.5 WT and Ins1687GR Mutant Channels
3. Discussion
3.1. Genotype-Phenotype Correlation
3.2. Pathophysiology
4. Materials and Methods
4.1. Clinical and Genetic Analysis
4.2. Mutagenesis and Nav1.5 Channel Expression
4.3. Electrophysiology
4.4. Homology Modeling
4.5. Immunocytochemistry
4.6. Western Blot Analysis
4.7. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Brugada, J.; Campuzano, O.; Arbelo, E.; Sarquella-Brugada, G.; Brugada, R. Present Status of Brugada Syndrome: JACC State-of-the-Art Review. J. Am. Coll. Cardiol. 2018, 72, 1046–1059. [Google Scholar] [CrossRef] [PubMed]
- Coppola, G.; Corrado, E.; Curnis, A.; Maglia, G.; Oriente, D.; Mignano, A.; Brugada, P. Update on Brugada Syndrome. Curr. Probl. Cardiol. 2019, 23, 100454. [Google Scholar] [CrossRef]
- Shi, S.; Barajas-Martinez, H.; Liu, T.; Sun, Y.; Yang, B.; Huang, C.; Hu, D. Prevalence of spontaneous Brugada ECG pattern recorded at standard intercostal leads: A meta-analysis. Int. J. Cardiol. 2018, 254, 151–156. [Google Scholar] [CrossRef]
- Garcia-Elias, A.; Benito, B. Ion Channel Disorders and Sudden Cardiac Death. Int. J. Mol. Sci. 2018, 19, 692. [Google Scholar] [CrossRef] [Green Version]
- Campuzano, O.; Sarquella-Brugada, G.; Fernandez-Falgueras, A.; Cesar, S.; Coll, M.; Mates, J.; Arbelo, E.; Perez-Serra, A.; Del Olmo, B.; Jordá, P.; et al. Genetic interpretation and clinical translation of minor genes related to Brugada syndrome. Hum. Mutat. 2019, 40, 749–764. [Google Scholar] [CrossRef] [PubMed]
- Monasky, M.M.; Micaglio, E.; Locati, E.T.; Pappone, C. Evaluating the Use of Genetics in Brugada Syndrome Risk Stratification. Front. Cardiovasc. Med. 2021, 8, 652027. [Google Scholar] [CrossRef] [PubMed]
- Catterall, W.A.; Lenaeus, M.J.; Gamal El-Din, T.M. Structure and Pharmacology of Voltage-Gated Sodium and Calcium Channels. Ann. Rev. Pharmacol. Toxicol. 2020, 60, 133–154. [Google Scholar] [CrossRef] [Green Version]
- Kapplinger, J.D.; Tester, D.J.; Alders, M.; Benito, B.; Berthet, M.; Brugada, J.; Brugada, P.; Fressart, V.; Guerchicoff, A.; Harris-Kerr, C.; et al. An international compendium of mutations in the SCN5A-encoded cardiac sodium channel in patients referred for Brugada syndrome genetic testing. Heart Rhythm 2010, 7, 33–46. [Google Scholar] [CrossRef] [Green Version]
- Denham, N.C.; Pearman, C.M.; Ding, W.Y.; Waktare, J.; Gupta, D.; Snowdon, R.; Hall, M.; Cooper, R.; Modi, S.; Todd, D.; et al. Systematic re-evaluation of SCN5A variants associated with Brugada syndrome. J. Cardiovasc. Electrophysiol. 2019, 30, 118–127. [Google Scholar] [CrossRef] [Green Version]
- Wilde, A.A.M.; Amin, A.S. Clinical Spectrum of SCN5A Mutations: Long QT Syndrome, Brugada Syndrome, and Cardiomyopathy. JACC Clin. Electrophysiol. 2018, 4, 569–579. [Google Scholar] [CrossRef]
- Yamagata, K.; Horie, M.; Aiba, T.; Ogawa, S.; Aizawa, Y.; Ohe, T.; Yamagishi, M.; Makita, N.; Sakurada, H.; Tanaka, T.; et al. Genotype-Phenotype Correlation of SCN5A Mutation for the Clinical and Electrocardiographic Characteristics of Probands With Brugada Syndrome: A Japanese Multicenter Registry. Circulation 2017, 135, 2255–2270. [Google Scholar] [CrossRef]
- Meregalli, P.G.; Tan, H.L.; Probst, V.; Koopmann, T.T.; Tanck, M.W.; Bhuiyan, Z.A.; Sacher, F.; Kyndt, F.; Schott, J.J.; Albuisson, J.; et al. Type of SCN5A mutation determines clinical severity and degree of conduction slowing in loss-of-function sodium channelopathies. Heart Rhythm 2009, 6, 341–348. [Google Scholar] [CrossRef]
- Gando, I.; Morganstein, J.; Jana, K.; McDonald, T.V.; Tang, Y.; Coetzee, W.A. Infant sudden death: Mutations responsible for impaired Nav1.5 channel trafficking and function. Pacing Clin. Electrophysiol. 2017, 40, 703–712. [Google Scholar] [CrossRef]
- Amin, A.S.; Reckman, Y.J.; Arbelo, E.; Spanjaart, A.M.; Postema, P.G.; Tadros, R.; Tanck, M.W.; Van den Berg, M.P.; Wilde, A.A.M.; Tan, H.L. SCN5A mutation type and topology are associated with the risk of ventricular arrhythmia by sodium channel blockers. Int. J. Cardiol. 2018, 266, 128–132. [Google Scholar] [CrossRef] [Green Version]
- Antzelevitch, C.; Patocskai, B. Brugada Syndrome: Clinical, Genetic, Molecular, Cellular, and Ionic Aspects. Curr. Probl. Cardiol. 2016, 41, 7–57. [Google Scholar] [CrossRef] [Green Version]
- Pappone, C.; Monasky, M.M.; Ciconte, G. Epicardial ablation in genetic cardiomyopathies: A new frontier. Eur. Heart J. Suppl. 2019, 21, B61–B66. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schwartz, P.J.; Ackerman, M.J.; Wilde, A.A.M. Channelopathies as Causes of Sudden Cardiac Death. Card. Electrophysiol. Clin. 2017, 9, 537–549. [Google Scholar] [CrossRef]
- Walsh, R.; Wilde, A.A.M. SCN5A variants in Brugada syndrome: True, true false, or false true. J. Cardiovasc. Electrophysiol. 2019, 30, 128–131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gualandi, F.; Zaraket, F.; Malagù, M.; Parmeggiani, G.; Trabanelli, C.; Fini, S.; Dang, X.; Wei, X.; Fang, M.; Bertini, M.; et al. Mutation Load of Multiple Ion Channel Gene Mutations in Brugada Syndrome. Cardiology 2017, 137, 256–260. [Google Scholar] [CrossRef] [PubMed]
- Cerrone, M.; Remme, C.A.; Tadros, R.; Bezzina, C.R.; Delmar, M. Beyond the One Gene-One Disease Paradigm: Complex Genetics and Pleiotropy in Inheritable Cardiac Disorders. Circulation 2019, 140, 595–610. [Google Scholar] [CrossRef]
- Schwartz, P.J.; Crotti, L.; George, A.L., Jr. Modifier genes for sudden cardiac death. Eur. Heart J. 2018, 39, 3925–3931. [Google Scholar] [CrossRef]
- Chen, C.; Tan, Z.; Zhu, W.; Fu, L.; Kong, Q.; Xiong, Q.; Yu, J.; Hong, K. Brugada syndrome with SCN5A mutations exhibits more pronounced electrophysiological defects and more severe prognosis: A meta-analysis. Clin. Genet. 2020, 97, 198–208. [Google Scholar] [CrossRef] [Green Version]
- Núñez, L.; Barana, A.; Amorós, I.; de la Fuente, M.G.; Dolz-Gaitón, P.; Gómez, R.; Rodríguez-García, I.; Mosquera, I.; Monserrat, L.; Delpón, E.; et al. p.D1690N Nav1.5 rescues p.G1748D mutation gating defects in a compound heterozygous Brugada syndrome patient. Heart Rhythm 2013, 10, 264–272. [Google Scholar] [CrossRef]
- Jiang, D.; Shi, H.; Tonggu, L.; Gamal El-Din, T.M.; Lenaeus, M.J.; Zhao, Y.; Yoshioka, C.; Zheng, N.; Catterall, W.A. Structure of the Cardiac Sodium Channel. Cell 2020, 180, 122–134. [Google Scholar] [CrossRef]
- Wang, H.G.; Zhu, W.; Kanter, R.J.; Silva, J.R.; Honeywell, C.; Gow, R.M.; Pitt, G.S. A novel NaV1.5 voltage sensor mutation associated with severe atrial and ventricular arrhythmias. J. Mol. Cell. Cardiol. 2016, 92, 52–62. [Google Scholar] [CrossRef] [Green Version]
- Clatot, J.; Hoshi, M.; Wan, X.; Liu, H.; Jain, A.; Shinlapawittayatorn, K.; Marionneau, C.; Ficker, E.; Ha, T.; Deschênes, I. Voltage-gated sodium channels assemble and gate as dimers. Nat. Commun. 2017, 8, 2077. [Google Scholar] [CrossRef] [Green Version]
- Clatot, J.; Zheng, Y.; Girardeau, A.; Liu, H.; Laurita, K.R.; Marionneau, C.; Deschênes, I. Mutant voltage-gated Na+ channels can exert a dominant negative effect through coupled gating. Am. J. Physiol. Heart Circ. Physiol. 2018, 315, H1250–H1257. [Google Scholar] [CrossRef] [PubMed]
- Valdivia, C.R.; Tester, D.J.; Rok, B.A.; Porter, C.J.; Munger, T.M.; Jahangir, A.; Makielski, J.C.; Ackerman, M.J. A Trafficking Defective, Brugada Syndrome-Causing SCN5A Mutation Rescued by Drugs. Cardiovasc. Res. 2004, 62, 53–62. [Google Scholar] [CrossRef]
- Tan, B.H.; Valdivia, C.R.; Song, C.; Makielski, J.C. Partial expression defect for the SCN5A missense mutation G1406R depends on splice variant background Q1077 and rescue by mexiletine. Am. J. Physiol. Heart Circ. Physiol. 2006, 291, H1822–H1828. [Google Scholar] [CrossRef] [Green Version]
- Ackerman, M.J.; Splawski, I.; Makielski, J.C.; Tester, D.J.; Will, M.L.; Timothy, K.W.; Keating, M.T.; Jones, G.; Chadha, M.; Burrow, C.R.; et al. Spectrum and prevalence of cardiac sodium channel variants among black, white, Asian, and Hispanic individuals: Implications for arrhythmogenic susceptibility and Brugada/long QT syndrome genetic testing. Heart Rhythm 2004, 1, 600–607. [Google Scholar] [CrossRef]
- Minier, M.; Probst, V.; Berthome, P.; Tixier, R.; Briand, J.; Geoffroy, O.; Clementy, N.; Mansourati, J.; Jesel, L.; Dupuis, J.M.; et al. Age at diagnosis of Brugada syndrome: Influence on clinical characteristics and risk of arrhythmia. Heart Rhythm 2020, 17, 743–749. [Google Scholar] [CrossRef] [PubMed]
- Ciconte, G.; Monasky, M.M.; Santinelli, V.; Micaglio, E.; Vicedomini, G.; Anastasia, L.; Negro, G.; Borrelli, V.; Giannelli, L.; Santini, F.; et al. Brugada syndrome genetics is associated with phenotype severity. Eur. Heart J. 2021, 42, 1082–1090. [Google Scholar] [CrossRef] [PubMed]
- Tadros, R.; Ton, A.T.; Fiset, C.; Nattel, S. Sex differences in cardiac electrophysiology and clinical arrhythmias: Epidemiology, therapeutics, and mechanisms. Can. J. Cardiol. 2014, 30, 783–792. [Google Scholar] [CrossRef]
- Lizotte, E.; Junttila, M.J.; Dube, M.P.; Hong, K.; Benito, B.; De Zutter, M.; Henkens, S.; Sarkozy, A.; Huikuri, H.V.; Towbin, J.; et al. Genetic modulation of brugada syndrome by a common polymorphism. J. Cardiovasc. Electrophysiol. 2009, 20, 1137–1141. [Google Scholar] [CrossRef]
- Matsumura, H.; Nakano, Y.; Ochi, H.; Onohara, Y.; Sairaku, A.; Tokuyama, T.; Tomomori, S.; Motoda, C.; Amioka, M.; Hironobe, N.; et al. H558R, a common SCN5A polymorphism, modifies the clinical phenotype of Brugada syndrome by modulating DNA methylation of SCN5A promoters. J. Biomed. Sci. 2017, 24, 91. [Google Scholar] [CrossRef]
- Rook, M.B.; Evers, M.M.; Vos, M.A.; Bierhuizen, M.F. Biology of cardiac sodium channel Nav1.5 expression. Cardiovasc. Res. 2012, 93, 12–23. [Google Scholar] [CrossRef] [Green Version]
- Abriel, H.; Rougier, J.S.; Jalife, J. Ion channel macromolecular complexes in cardiomyocytes: Roles in sudden cardiac death. Circ. Res. 2015, 116, 1971–1988. [Google Scholar] [CrossRef] [Green Version]
- Sieira, J.; Dendramis, G.; Brugada, P. Pathogenesis and management of Brugada syndrome. Nat. Rev. Cardiol. 2016, 13, 744–756. [Google Scholar] [CrossRef]
- Corrado, D.; Migliore, F.; Zorzi, A. Brugada Syndrome: In Search of a Cause. J. Am. Coll. Cardiol. 2018, 72, 2758–2760. [Google Scholar] [CrossRef]
- Rivaud, M.R.; Delmar, M.; Remme, C.A. Heritable Arrhythmia Syndromes Associated With Abnormal Cardiac Sodium Channel Function: Ionic and Non-Ionic Mechanisms. Cardiovasc. Res. 2020, 116, 1557–1570. [Google Scholar] [CrossRef] [Green Version]
- Pappone, C.; Monasky, M.M.; Micaglio, E.; Ciconte, G. Right ventricular electromechanical abnormalities in Brugada syndrome: Is this a cardiomyopathy? Eur. Heart J. Suppl. 2020, 22, E101–E104. [Google Scholar] [CrossRef] [PubMed]
- Imbrici, P.; Liantonio, A.; Camerino, G.M.; De Bellis, M.; Camerino, C.; Mele, A.; Giustino, A.; Pierno, S.; De Luca, A.; Tricarico, D.; et al. Therapeutic Approaches to Genetic Ion Channelopathies and Perspectives in Drug Discovery. Front. Pharmacol. 2016, 7, 121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Farinato, A.; Altamura, C.; Imbrici, P.; Maggi, L.; Bernasconi, P.; Mantegazza, R.; Pasquali, L.; Siciliano, G.; Lo Monaco, M.; Vial, C.; et al. Pharmacogenetics of myotonic hNav1.4 sodium channel variants situated near the fast inactivation gate. Pharmacol. Res. 2019, 141, 224–235. [Google Scholar] [CrossRef] [PubMed]
- Sali, A.; Blundell, T.L. Comparative protein modelling by satisfaction of spatial restraints. J. Mol. Biol. 1993, 234, 779–815. [Google Scholar] [CrossRef]
- Webb, B.; Sali, A. Comparative Protein Structure Modeling Using MODELLER. Curr. Protoc. Bioinform. 2016, 54, 5–6. [Google Scholar] [CrossRef] [Green Version]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Channel Type | Peak Current Density | Voltage Dependent Activation | Voltage Dependent Fast Inactivation | Time Constant of Inactivation | Recovery from Inactivation | Sustained Current Density | |||
|---|---|---|---|---|---|---|---|---|---|
| −30 mV, pA/pF | Vh, mV | k, mV | Vh, mV | k, mV | −30 mV, ms | τfast, ms (A1%) | τslow, ms (A2%) | −30 mV, 100 ms, pA/pF | |
| Nav1.5 WT | −286 ± 53 n = 11 | −49.6 ± 0.6 n = 6 | 6.0 ± 0.4 | −86.7 ± 0.5 n = 18 | 9.9 ± 0.4 | 0.86 ± 0.07 n = 14 | 1.2 ± 0.1 (87%) n = 12 | 6.7 ± 1.1 (13%) | 13.7 ± 1.9 n = 20 |
| P1310L | −46 ± 10 * n = 20 | −34.7 ± 0.6 * n = 19 | 8.7 ± 0.6 | −78.5 ± 0.4 * n = 15 | 7.0 ± 0.3 | 1.02 ± 0.09 n = 18 | 0.78 ± 0.2 * (78%) n = 8 | 2.4 ± 1.2 * (22%) | 9.8 ± 1.5 n = 16 |
| WT+ P1310L | −146 ± 23 n = 9 | −42.0 ± 0.5 n = 9 | 9.2 ± 0.8 | −84.0 ± 0.6 n = 15 | 10.0 ± 1.0 | 1.06 ± 0.17 n = 11 | 1.5 ± 0.6 (83%) n = 10 | 4.7 ± 1.2 (17%) | 9.7 ± 2.4 n = 13 |
| Ins1687GR | −11.1 ± 1.5 * n = 21 | / | / | / | / | / | / | / | / |
| WT+ Ins1687GR | −153 ± 52 n = 7 | −52.5 ± 0.4 n = 6 | 5.1 ± 0.3 | −88.0 ± 1.0 n = 6 | 9.3 ± 0.7 | 0.99 ± 0.12 n = 6 | 1.6 ± 0.4 (88%) n = 6 | 8.0 ± 3.1 (12%) | 10.8 ± 3.6 n = 6 |
| Channel Type + Mexiletine 300 µM | Current Density | Voltage Dependent Activation | Voltage Dependent Inactivation | Time Constant of Inactivation | Recovery from Inactivation | |||
|---|---|---|---|---|---|---|---|---|
| −30 mV, pA/pF | Vh, mV | k, mV | Vh, mV | k, mV | −30 mV, ms | τfast, ms (A1%) | τslow, ms (A2%) | |
| Nav1.5 WT | −388 ± 78 n = 6 | −51.9 ± 0.4 n = 6 | 4.4 ± 0.3 | −91.0 ± 0.5 n = 6 | 9.7 ± 0.4 | 1.17 ± 0.13 n = 6 | 2.1 ± 0.1 (83%) n = 6 | 22 ± 4 (17%) |
| Ins1687GR | −72 ± 16 * n = 8 | −49.7 ± 0.5 n = 6 | 6.0 ± 0.5 | −91.0 ± 0.4 n = 7 | 8.2 ± 0.4 | 1.20 ± 0.20 n = 6 | 1.99 ± 0.1 (73%) n = 6 | 20 ± 4 (27%) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Balla, C.; Conte, E.; Selvatici, R.; Marsano, R.M.; Gerbino, A.; Farnè, M.; Blunck, R.; Vitali, F.; Armaroli, A.; Brieda, A.; et al. Functional Characterization of Two Novel Mutations in SCN5A Associated with Brugada Syndrome Identified in Italian Patients. Int. J. Mol. Sci. 2021, 22, 6513. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22126513
Balla C, Conte E, Selvatici R, Marsano RM, Gerbino A, Farnè M, Blunck R, Vitali F, Armaroli A, Brieda A, et al. Functional Characterization of Two Novel Mutations in SCN5A Associated with Brugada Syndrome Identified in Italian Patients. International Journal of Molecular Sciences. 2021; 22(12):6513. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22126513
Chicago/Turabian StyleBalla, Cristina, Elena Conte, Rita Selvatici, Renè Massimiliano Marsano, Andrea Gerbino, Marianna Farnè, Rikard Blunck, Francesco Vitali, Annarita Armaroli, Alessandro Brieda, and et al. 2021. "Functional Characterization of Two Novel Mutations in SCN5A Associated with Brugada Syndrome Identified in Italian Patients" International Journal of Molecular Sciences 22, no. 12: 6513. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22126513