
Decreased Expression and Uncoupling of Endothelial Nitric Oxide Synthase in the Cerebral Cortex of Rats with Thioacetamide-Induced Acute Liver Failure


Abstract
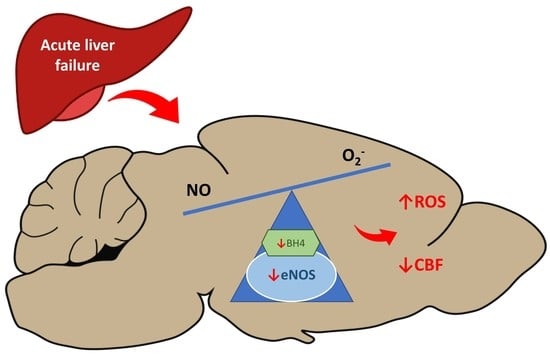
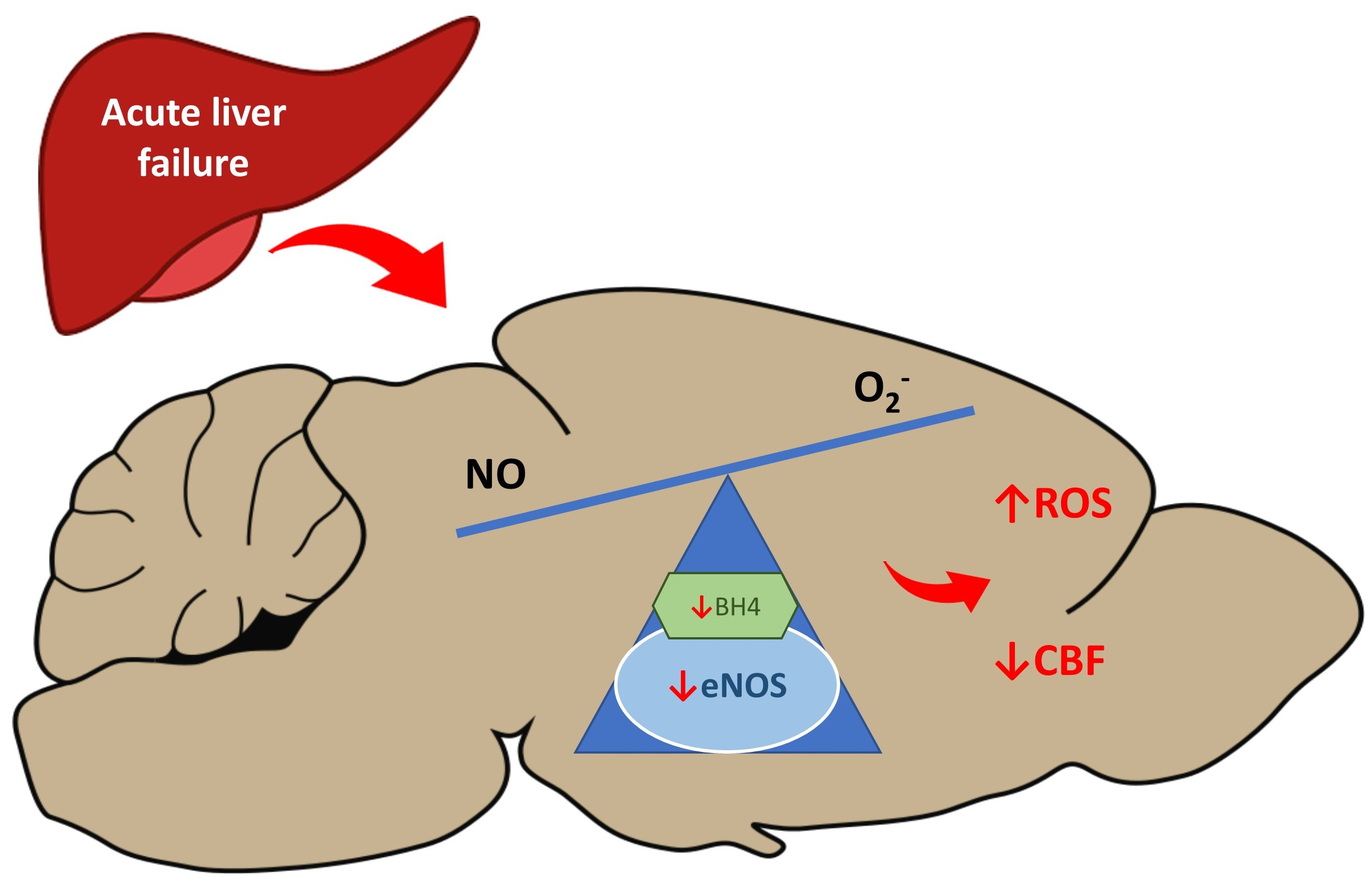
:
1. Introduction
2. Results
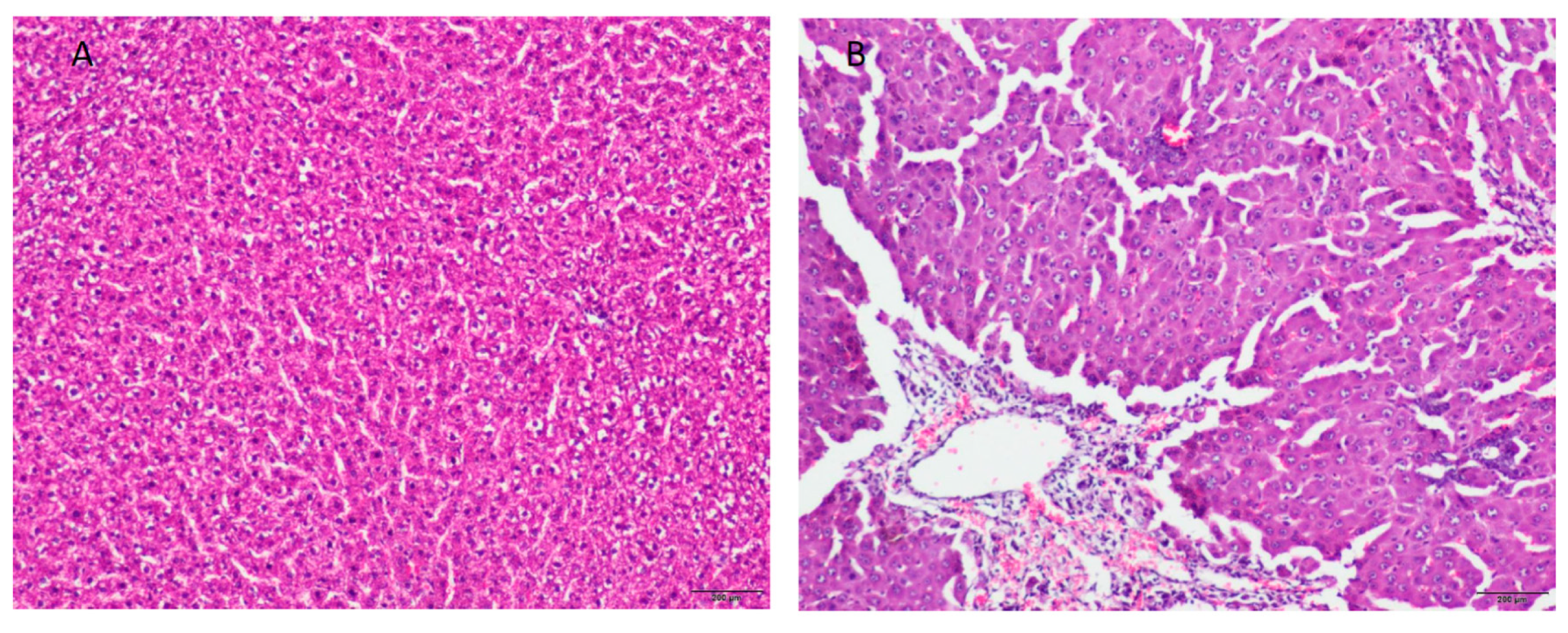
2.1. Rat Model of Acute Liver Failure
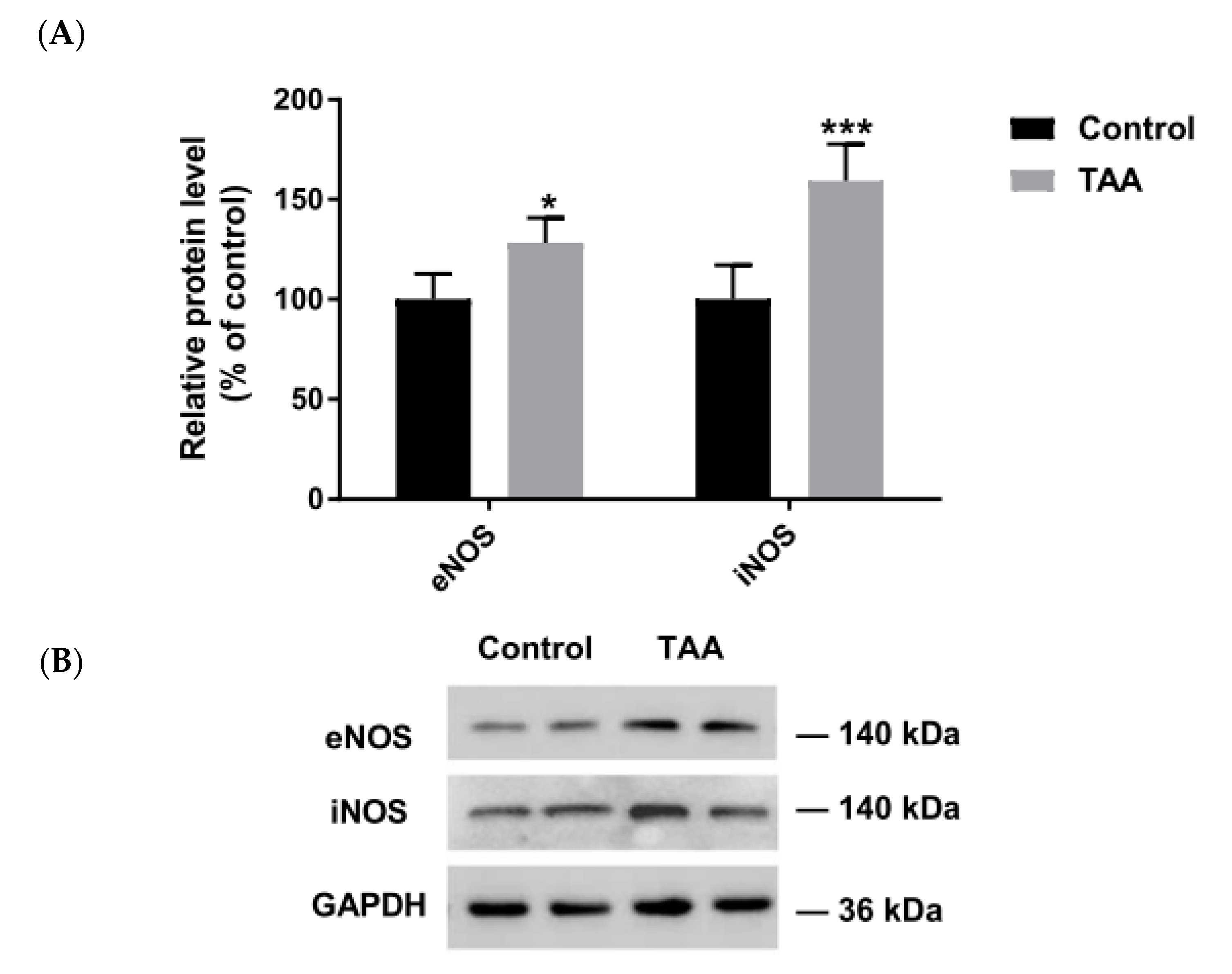
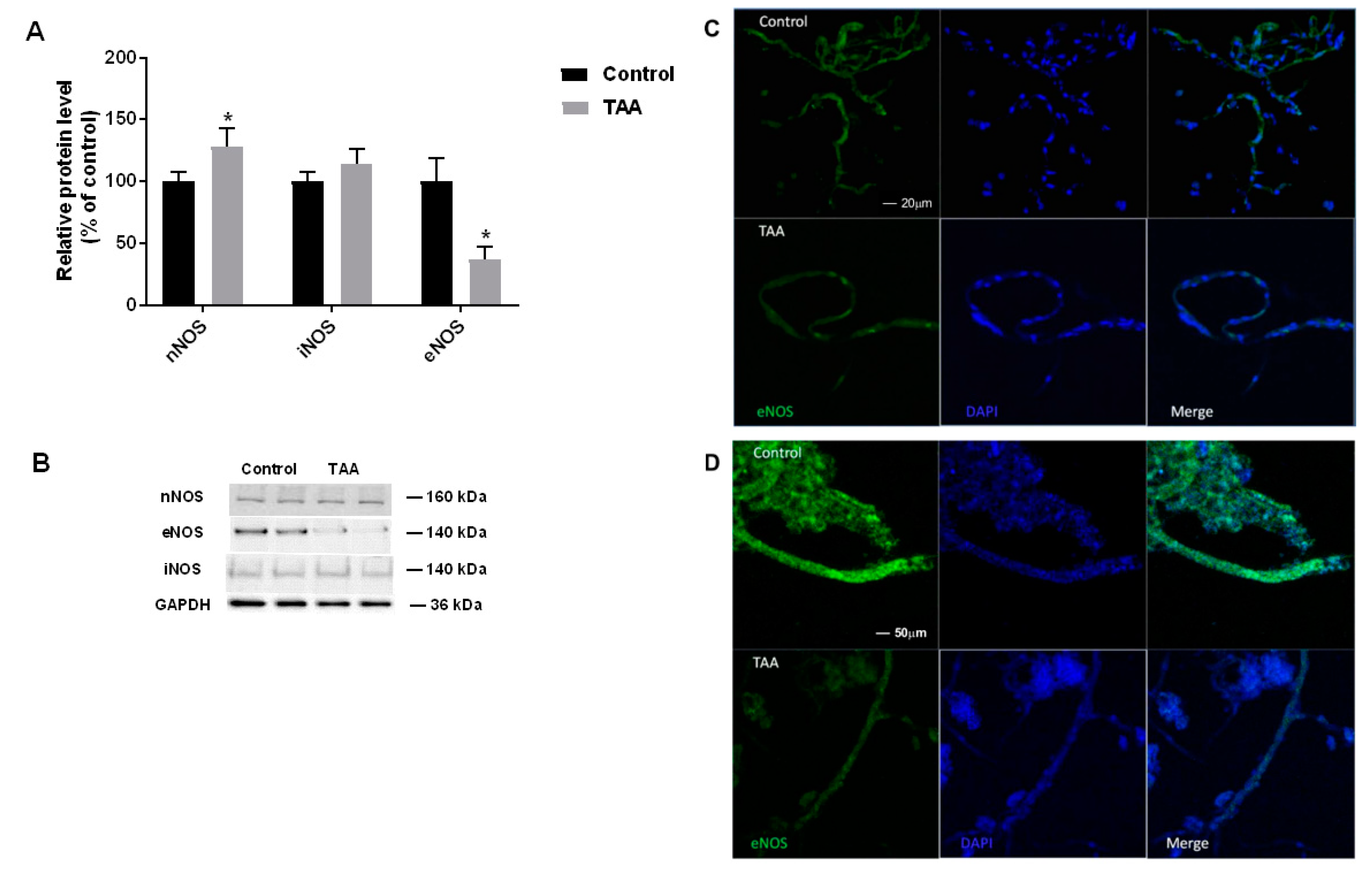
2.2. The Expression of NOS Isoforms in the Liver and Prefrontal Cortex
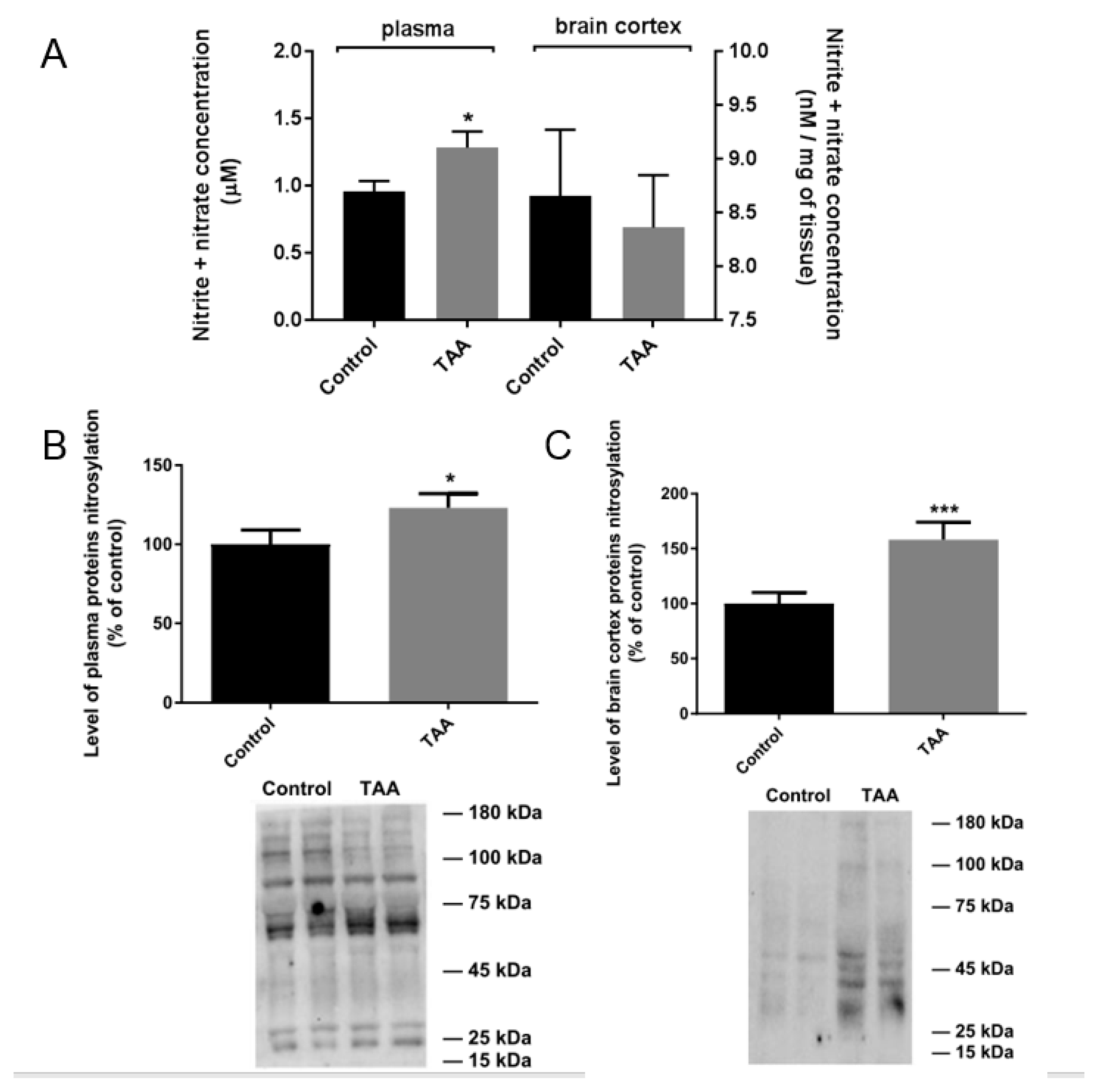
2.3. NOx Measurement and Nitration of Plasma and Prefrontal Cortex Proteins in TAA Rats
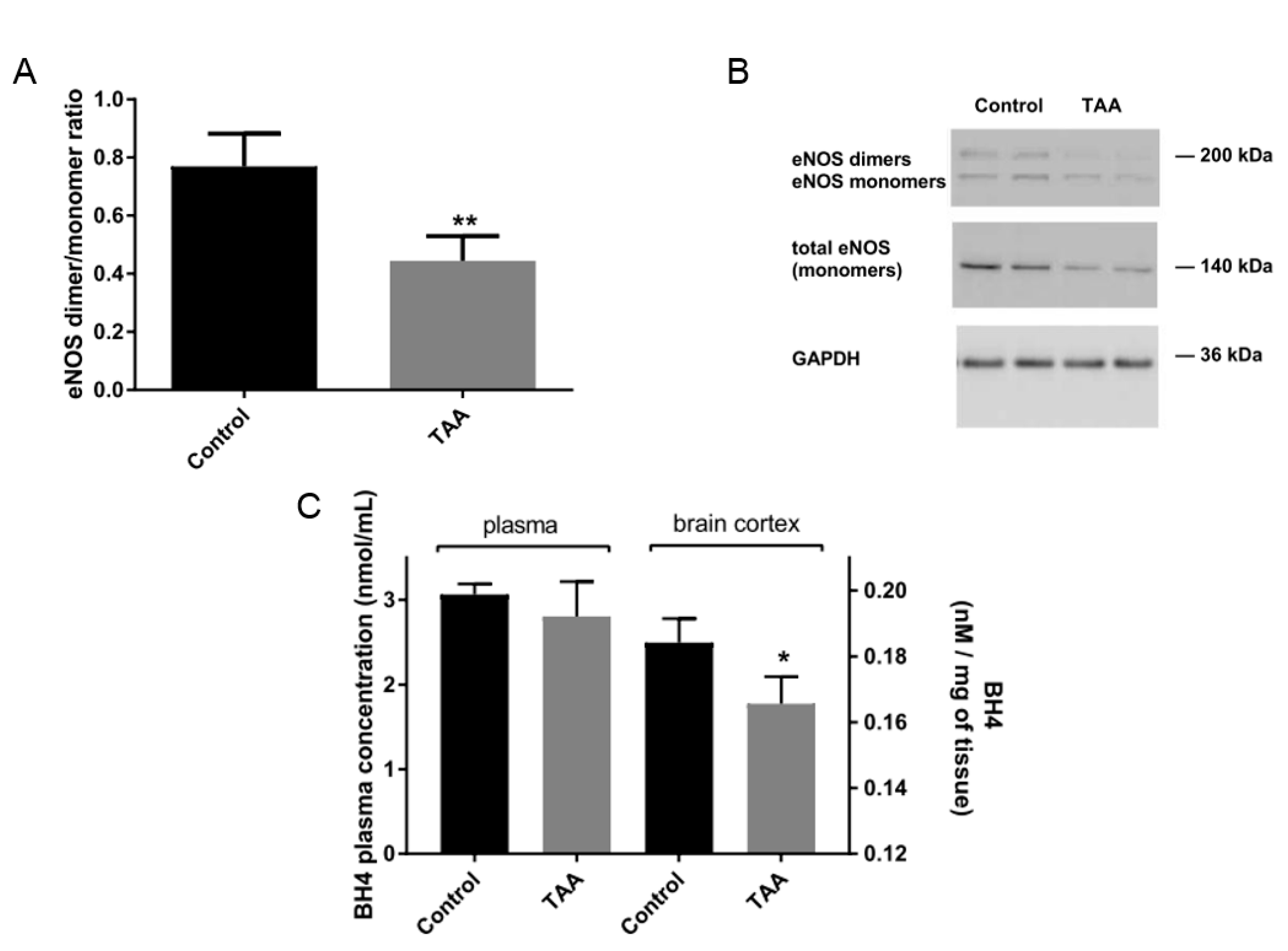
2.4. eNOS Dimer/Monomer Ratio Status and BH4 Level in the Brain Prefrontal Cortex
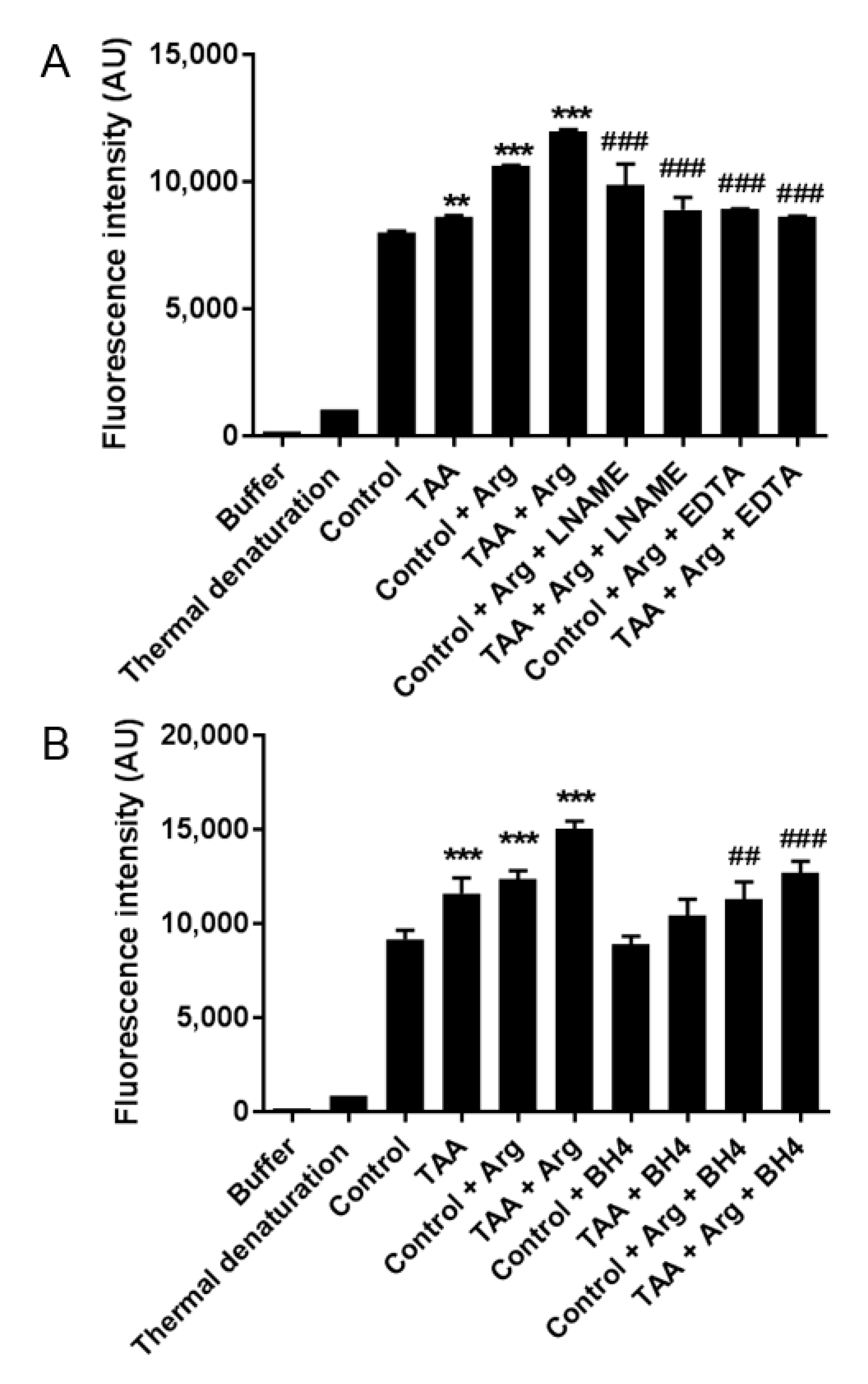
2.5. The Relative Contribution of NOS Isoforms to ROS Production in the Prefrontal Cortex
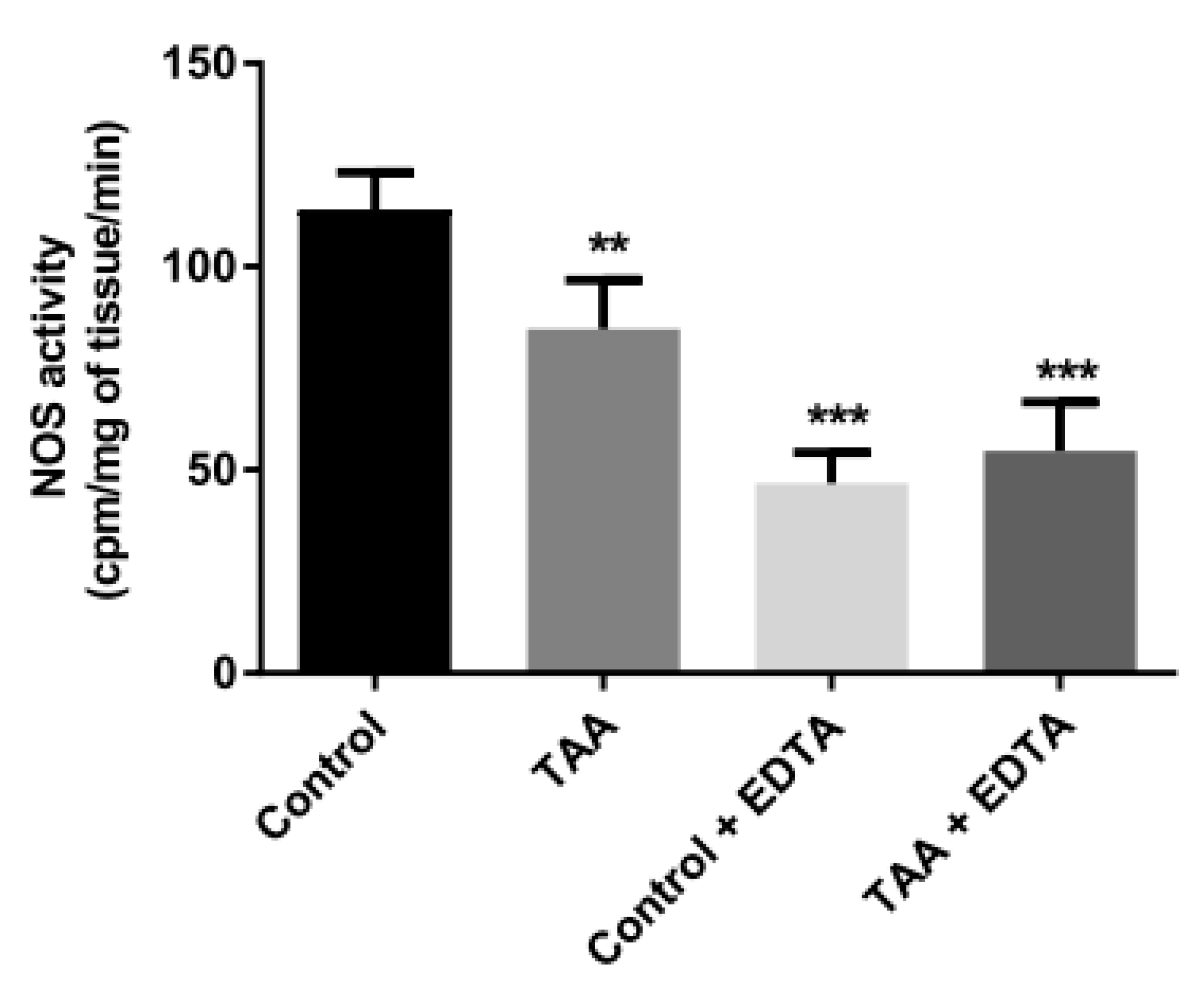
2.6. Total NOS Activity
2.7. CBF in the Prefrontal Cortex of TAA Rats
3. Discussion
4. Materials and Methods
4.1. Study Design, Acute Liver Failure Model and Biochemical Analysis
4.2. Protein Isolation, Western Blot, and eNOS Dimerization Analysis
4.3. The Isolation of a Capillary Fraction
4.4. Confocal Microscopy Visualization of eNOS
4.5. NOS Activity Measurement
4.6. Measurement of Nitrite and Nitrate
4.7. BH4 Measurement
4.8. Reactive Oxygen Species Measurement
4.9. Cerebral Blood Flow Measurement
4.10. Statistical Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| ALF | Acute liver failure |
| BBB | Blood–brain barrier |
| BH4 | Tetrahydrobiopterin |
| CBF | Cerebral blood flow |
| eNOS | Endothelial nitric oxide synthase |
| HE | Hepatic encephalopathy |
| NO | Nitric oxide |
| nNOS | Neuronal nitric oxide synthase |
| ONS | Oxidative nitrosative stress |
| ROS | Reactive oxygen species |
| TAA | Thioacetamide |
| TCA | Tricarboxylic acid |
References
- Prakash, R.; Mullen, K.D. Mechanisms, diagnosis and management of hepatic encephalopathy. Nat. Rev. Gastroenterol. Hepatol. 2010, 7, 515–525. [Google Scholar] [CrossRef]
- Felipo, V. Hepatic encephalopathy: Effects of liver failure on brain function. Nat. Rev. Neurosci. 2013, 14, 851–858. [Google Scholar] [CrossRef] [PubMed]
- Liere, V.; Sandhu, G.; De Morrow, S. Recent advances in hepatic encephalopathy. F1000Research 2017, 6, 1637. [Google Scholar] [CrossRef] [Green Version]
- McPhail, M.J.; Bajaj, J.S.; Thomas, H.C.; Taylor-Robinson, S.D. Pathogenesis and diagnosis of hepatic encephalopathy. Expert Rev. Gastroenterol. Hepatol. 2010, 4, 365–378. [Google Scholar] [CrossRef]
- Gorg, B.; Schliess, F.; Haussinger, D. Osmotic and oxidative/nitrosative stress in ammonia toxicity and hepatic encephalopathy. Arch. Biochem. Biophys. 2013, 536, 158–163. [Google Scholar] [CrossRef]
- Gorg, B.; Karababa, A.; Schutz, E.; Paluschinski, M.; Schrimpf, A.; Shafigullina, A.; Castoldi, M.; Bidmon, H.J.; Haussinger, D. O-GlcNAcylation-dependent upregulation of HO1 triggers ammonia-induced oxidative stress and senescence in hepatic encephalopathy. J. Hepatol. 2019, 71, 930–941. [Google Scholar] [CrossRef]
- Shawcross, D.L.; Sharifi, Y.; Canavan, J.B.; Yeoman, A.D.; Abeles, R.D.; Taylor, N.J.; Auzinger, G.; Bernal, W.; Wendon, J.A. Infection and systemic inflammation, not ammonia, are associated with Grade 3/4 hepatic encephalopathy, but not mortality in cirrhosis. J. Hepatol. 2011, 54, 640–649. [Google Scholar] [CrossRef]
- Reinehr, R.; Gorg, B.; Becker, S.; Qvartskhava, N.; Bidmon, H.J.; Selbach, O.; Haas, H.L.; Schliess, F.; Haussinger, D. Hypoosmotic swelling and ammonia increase oxidative stress by NADPH oxidase in cultured astrocytes and vital brain slices. Glia 2007, 55, 758–771. [Google Scholar] [CrossRef]
- Youn, J.Y.; Gao, L.; Cai, H. The p47phox- and NADPH oxidase organiser 1 (NOXO1)-dependent activation of NADPH oxidase 1 (NOX1) mediates endothelial nitric oxide synthase (eNOS) uncoupling and endothelial dysfunction in a streptozotocin-induced murine model of diabetes. Diabetologia 2012, 55, 2069–2079. [Google Scholar] [CrossRef] [Green Version]
- Li, Q.; Youn, J.-Y.; Cai, H. Mechanisms and consequences of endothelial nitric oxide synthase dysfunction in hypertension. J. Hypertens. 2015, 33, 1128–1136. [Google Scholar] [CrossRef] [Green Version]
- Palenzuela, L.; Oria, M.; Romero-Giménez, J.; Garcia-Lezana, T.; Chavarria, L.; Cordoba, J. Gene expression profiling of brain cortex microvessels may support brain vasodilation in acute liver failure rat models. Metab. Brain Dis. 2016, 31, 1405–1417. [Google Scholar] [CrossRef]
- Siragusa, M.; Fleming, I. The eNOS signalosome and its link to endothelial dysfunction. Pflügers Arch. Eur. J. Physiol. 2016, 468, 1125–1137. [Google Scholar] [CrossRef] [PubMed]
- Hong, W.K.; Shim, K.Y.; Baik, S.K.; Kim, M.Y.; Cho, M.Y.; Jang, Y.O.; Park, Y.S.; Han, J.; Kim, G.; Cho, Y.Z.; et al. Relationship between Tetrahydrobiopterin and Portal Hypertension in Patients with Chronic Liver Disease. J. Korean Med. Sci. 2014, 29, 392–399. [Google Scholar] [CrossRef] [Green Version]
- Balasubramaniyan, V.; Wright, G.; Sharma, V.; Davies, N.A.; Sharifi, Y.; Habtesion, A.; Mookerjee, R.P.; Jalan, R. Ammonia reduction with ornithine phenylacetate restores brain eNOS activity via the DDAH-ADMA pathway in bile duct-ligated cir-rhotic rats. Am. J. Physiol. Gastrointest. Liver Physiol. 2012, 302, G145–G152. [Google Scholar] [CrossRef]
- Matei, V.; Rodriguez-Vilarrupla, A.; Deulofeu, R.; Colomer, D.; Fernandez, M.; Bosch, J.; Garcia-Pagan, J.C. The eNOS cofactor tetrahydrobiopterin improves endothelial dysfunction in livers of rats with CCl4 cirrhosis. Hepatology 2006, 44, 44–52. [Google Scholar] [CrossRef]
- Hilgier, W.; Olson, J.E. Brain Ion and Amino Acid Contents During Edema Development in Hepatic Encephalopathy. J. Neurochem. 1994, 62, 197–204. [Google Scholar] [CrossRef] [PubMed]
- Schemitt, E.G.; Hartmann, R.M.; Colares, J.R.; Licks, F.; Salvi, J.O.; Marroni, C.A.; Marroni, N.P. Protective action of glutamine in rats with severe acute liver failure. World J. Hepatol. 2019, 11, 273–286. [Google Scholar] [CrossRef]
- Mohi-Ud-Din, R.; Mir, R.H.; Sawhney, G.; Dar, M.A.; Bhat, Z.A. Possible Pathways of Hepatotoxicity Caused by Chemical Agents. Curr. Drug Metab. 2019, 20, 867–879. [Google Scholar] [CrossRef]
- Sekine, A.; Fukuwatari, T. Acute liver failure increases kynurenic acid production in rat brain via changes in tryptophan metabolism in the periphery. Neurosci. Lett. 2019, 701, 14–19. [Google Scholar] [CrossRef] [PubMed]
- Rahman, T.M.; Hodgson, H.J.F. Animal models of acute hepatic failure. Int. J. Exp. Pathol. 2000, 81, 145–157. [Google Scholar] [CrossRef]
- Butterworth, R.F.; Norenberg, M.D.; Felipo, V.; Ferenci, P.; Albrecht, J.; Blei, A.T. Experimental models of hepatic encephalopathy: ISHEN guidelines. Liver Int. 2009, 29, 783–788. [Google Scholar] [CrossRef]
- Huang, H.-C.; Wang, S.-S.; Chan, C.-Y.; Chen, Y.-C.; Lee, F.-Y.; Chang, F.-Y.; Chu, C.-J.; Lin, H.-C.; Lu, R.-H.; Lee, S.-D. Role of Hepatic Nitric Oxide Synthases in Rats with Thioacetamide-induced Acute Liver Failure and Encephalopathy. J. Chin. Med. Assoc. 2007, 70, 16–23. [Google Scholar] [CrossRef] [Green Version]
- Jiang, W.; Desjardins, P.; Butterworth, R.F. Direct evidence for central proinflammatory mechanisms in rats with experimental acute liver failure: Protective effect of hypothermia. J. Cereb. Blood Flow Metab. Off. J. Int. Soc. Cereb. Blood Flow Metab. 2009, 29, 944–952. [Google Scholar] [CrossRef] [Green Version]
- Pacher, P.; Beckman, J.S.; Liaudet, L. Nitric Oxide and Peroxynitrite in Health and Disease. Physiol. Rev. 2007, 87, 315–424. [Google Scholar] [CrossRef] [Green Version]
- Shishehbor, M.H.; Aviles, R.J.; Brennan, M.-L.; Fu, X.; Goormastic, M.; Pearce, G.L.; Gokce, N.; Keaney, J.J.F.; Penn, M.S.; Sprecher, D.L.; et al. Association of Nitrotyrosine Levels with Cardiovascular Disease and Modulation by Statin Therapy. JAMA 2003, 289, 1675–1680. [Google Scholar] [CrossRef] [Green Version]
- Winyard, P.G.; Ryan, B.; Eggleton, P.; Nissim, A.; Taylor, E.; Faro, M.L.L.; Burkholz, T.; Szabó-Taylor, K.E.; Fox, B.; Viner, N.; et al. Measurement and meaning of markers of reactive species of oxygen, nitrogen and sulfur in healthy human subjects and patients with inflammatory joint disease. Biochem. Soc. Trans. 2011, 39, 1226–1232. [Google Scholar] [CrossRef]
- Montoliu, C.; Cauli, O.; Urios, A.; El Mlili, N.; Serra, M.A.; Giner-Duran, R.; Gonzalez-Lopez, O.; del Olmo, J.A.; Wassel, A.; Rodrigo, J.M.; et al. 3-nitro-tyrosine as a peripheral biomarker of minimal hepatic encephalopathy in patients with liver cirrhosis. Am. J. Gastroenterol. 2011, 106, 1629–1637. [Google Scholar] [CrossRef]
- Katakam, P.V.; Snipes, J.A.; Steed, M.M.; Busija, D.W. Insulin-induced generation of reactive oxygen species and uncou-pling of nitric oxide synthase underlie the cerebrovascular insulin resistance in obese rats. J. Cereb. Blood Flow Metab. Off. J. Int. Soc. Cereb. Blood Flow Metab. 2012, 32, 792–804. [Google Scholar] [CrossRef]
- Ponnuswamy, P.; Ostermeier, E.; Schröttle, A.; Chen, J.; Huang, P.L.; Ertl, G.; Nieswandt, B.; Kuhlencordt, P.J. Oxidative Stress and Compartment of Gene Expression Determine Proatherosclerotic Effects of Inducible Nitric Oxide Synthase. Am. J. Pathol. 2009, 174, 2400–2410. [Google Scholar] [CrossRef] [Green Version]
- Chidlow, J.H.; Sessa, W.C. Caveolae, caveolins, and cavins: Complex control of cellular signalling and inflammation. Cardiovasc. Res. 2010, 86, 219–225. [Google Scholar] [CrossRef] [Green Version]
- Hernández, R.; Martínez-Lara, E.; del Moral, M.L.; Blanco, S.; Cañuelo, A.; Siles, E.; Esteban, F.J.; Pedrosa, J.A.; Peinado, M.A. Upregulation of endothelial nitric oxide synthase maintains nitric oxide production in the cerebellum of thioacetamide cirrhotic rats. Neuroscience 2004, 126, 879–887. [Google Scholar] [CrossRef]
- Hermenegildo, C.; Monfort, P.; Felipo, V. Activation of N-methyl-D-aspartate receptors in rat brainin vivo following acute ammonia intoxication: Characterization byin vivo brain microdialysis. Hepatology 2000, 31, 709–715. [Google Scholar] [CrossRef]
- Schneider, F.; Lutun, P.; Boudjema, K.; Wolf, P.; Tempe, J.D. In vivo evidence of enhanced guanylyl cyclase activation dur-ing the hyperdynamic circulation of acute liver failure. Hepatology 1994, 19, 38–44. [Google Scholar] [CrossRef] [PubMed]
- Hilgier, W.; Anderzhanova, E.; Oja, S.S.; Saransaari, P.; Albrecht, J. Taurine reduces ammonia- and N-methyl-d-aspartate-induced accumulation of cyclic GMP and hydroxyl radicals in microdialysates of the rat striatum. Eur. J. Pharmacol. 2003, 468, 21–25. [Google Scholar] [CrossRef]
- Llansola, M.; Rodrigo, R.; Monfort, P.; Montoliu, C.; Kosenko, E.; Cauli, O.; Piedrafita, B.; El Mlili, N.; Felipo, V. NMDA receptors in hyperammonemia and hepatic encephalopathy. Metab. Brain Dis. 2007, 22, 321–335. [Google Scholar] [CrossRef] [PubMed]
- Rodrigo, R.; Felipo, V. Brain regional alterations in the modulation of the glutamate-nitric oxide-cGMP pathway in liver cirrhosis. Role of hyperammonemia and cell types involved. Neurochem. Int. 2006, 48, 472–477. [Google Scholar] [CrossRef]
- Corbalán, R.; Chatauret, N.; Behrends, S.; Butterworth, R.F.; Felipo, V. Region selective alterations of soluble guanylate cyclase content and modulation in brain of cirrhotic patients. Hepatology 2002, 36, 1155–1162. [Google Scholar] [CrossRef] [PubMed]
- Nishida, K.; Harrison, D.G.; Navas, J.P.; Fisher, A.A.; Dockery, S.P.; Uematsu, M.; Nerem, R.M.; Alexander, R.W.; Murphy, T.J. Molecular cloning and characterization of the constitutive bovine aortic endothelial cell nitric oxide synthase. J. Clin. Investig. 1992, 90, 2092–2096. [Google Scholar] [CrossRef] [Green Version]
- Bouloumie, A.; Schini-Kerth, V.B.; Busse, R. Vascular endothelial growth factor up-regulates nitric oxide synthase expression in endothelial cells. Cardiovasc. Res. 1999, 41, 773–780. [Google Scholar] [CrossRef] [Green Version]
- Saura, M.; Zaragoza, C.; Cao, W.; Bao, C.; Rodriguez-Puyol, M.; Rodriguez-Puyol, D.; Lowenstein, C.J. Smad2 mediates transforming growth factor-beta induction of endothelial nitric oxide synthase expression. Circ. Res. 2002, 91, 806–813. [Google Scholar] [CrossRef] [Green Version]
- Searles, C.D. Transcriptional and posttranscriptional regulation of endothelial nitric oxide synthase expression. Am. J. Physiol.-Cell Physiol. 2006, 291, C803–C816. [Google Scholar] [CrossRef] [Green Version]
- Neumann, P.; Gertzberg, N.; Johnson, A. TNF-α induces a decrease in eNOS promoter activity. Am. J. Physiol. Cell. Mol. Physiol. 2004, 286, L452–L459. [Google Scholar] [CrossRef]
- Lu, J.L.; Schmiege, L.M., III; Kuo, L.; Liao, J.C. Downregulation of endothelial constitutive nitric oxide synthase expres-sion by lipopolysaccharide. Biochem. Biophys. Res. Commun. 1996, 225, 1–5. [Google Scholar] [CrossRef]
- McQuillan, L.P.; Leung, G.K.; Marsden, P.A.; Kostyk, S.K.; Kourembanas, S. Hypoxia inhibits expression of eNOS via transcriptional and posttranscriptional mechanisms. Am. J. Physiol. Circ. Physiol. 1994, 267, H1921–H1927. [Google Scholar] [CrossRef]
- Pedersen, C.L.; Faggiano, S.; Helbo, S.; Gesser, H.; Fago, A. Roles of nitric oxide, nitrite and myoglobin on myocardial efficiency in trout (Oncorhynchus mykiss) and goldfish (Carassius auratus): Implications for hypoxia tolerance. J. Exp. Biol. 2010, 213, 2755–2762. [Google Scholar] [CrossRef] [Green Version]
- Bonauer, A.; Carmona, G.; Iwasaki, M.; Mione, M.; Koyanagi, M.; Fischer, A.; Burchfield, J.; Fox, H.; Doebele, C.; Ohtani, K.; et al. MicroRNA-92a controls angiogenesis and functional recovery of ischemic tissues in mice. Science 2009, 324, 1710–1713. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qin, B.; Shu, Y.; Long, L.; Li, H.; Men, X.; Feng, L.; Yang, H.; Lu, Z. MicroRNA-142-3p Induces Atherosclerosis-Associated Endothelial Cell Apoptosis by Directly Targeting Rictor. Cell. Physiol. Biochem. 2018, 47, 1589–1603. [Google Scholar] [CrossRef]
- Miao, Y.; Ajami, N.E.; Huang, T.-S.; Lin, F.-M.; Lou, C.-H.; Wang, Y.-T.; Li, S.; Kang, J.; Munkacsi, H.; Maurya, M.R.; et al. Enhancer-associated long non-coding RNA LEENE regulates endothelial nitric oxide synthase and endothelial function. Nat. Commun. 2018, 9, 1–13. [Google Scholar] [CrossRef]
- Garcia, V.; Sessa, W.C. Endothelial NOS: Perspective and recent developments. Br. J. Pharmacol. 2019, 176, 189–196. [Google Scholar] [CrossRef] [Green Version]
- Bauer, P.M.; Fulton, D.; Boo, Y.C.; Sorescu, G.P.; Kemp, B.E.; Jo, H.; Sessa, W.C. Compensatory phosphorylation and protein-protein interactions revealed by loss of function and gain of function mutants of multiple serine phosphorylation sites in endothelial nitric-oxide synthase. J. Biol. Chem. 2003, 278, 14841–14849. [Google Scholar] [CrossRef] [Green Version]
- Mount, P.F.; Kemp, B.E.; Power, D.A. Regulation of endothelial and myocardial NO synthesis by multi-site eNOS phos-phorylation. J. Mol. Cell. Cardiol. 2007, 4, 271–279. [Google Scholar] [CrossRef] [PubMed]
- Karbach, S.; Wenzel, P.; Waisman, A.; Munzel, T.; Daiber, A. eNOS uncoupling in cardiovascular diseases-the role of oxida-tive stress and inflammation. Curr. Pharm. Des. 2014, 20, 3579–3594. [Google Scholar] [CrossRef] [PubMed]
- Milewski, K.; Hilgier, W.; Albrecht, J.; Zielinska, M. The dimethylarginine (ADMA)/nitric oxide pathway in the brain and periphery of rats with thioacetamide-induced acute liver failure: Modulation by histidine. Neurochem. Int. 2015, 88, 26–31. [Google Scholar] [CrossRef] [PubMed]
- Milewski, K.; Hilgier, W.; Fresko, I.; Polowy, R.; Podsiadlowska, A.; Zolocinska, E.; Grymanowska, A.W.; Filipkowski, R.K.; Albrecht, J.; Zielinska, M. Carnosine Reduces Oxidative Stress and Reverses Attenuation of Righting and Postural Re-flexes in Rats with Thioacetamide-Induced Liver Failure. Neurochem. Res. 2016, 41, 376–384. [Google Scholar] [CrossRef] [Green Version]
- Sydow, K.; Münzel, T. ADMA and oxidative stress. Atheroscler. Suppl. 2003, 4, 41–51. [Google Scholar] [CrossRef]
- Czarnecka, A.; Milewski, K.; Jaźwiec, R.; Zielińska, M. Intracerebral Administration of S-Adenosylhomocysteine or S-Adenosylmethionine Attenuates the Increases in the Cortical Extracellular Levels of Dimethylarginines Without Affecting cGMP Level in Rats with Acute Liver Failure. Neurotox. Res. 2016, 31, 99–108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Forstermann, U.; Munzel, T. Endothelial nitric oxide synthase in vascular disease: From marvel to menace. Circulation 2006, 113, 1708–1714. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosenkranz-Weiss, P.; Sessa, W.C.; Milstien, S.; Kaufman, S.; Watson, C.A.; Pober, J.S. Regulation of nitric oxide synthesis by proinflammatory cytokines in human umbilical vein endothelial cells. Elevations in tetrahydrobiopterin levels enhance endothelial nitric oxide synthase specific activity. J. Clin. Investig. 1994, 93, 2236–2243. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Werner, E.R.; Blau, N.; Thony, B. Tetrahydrobiopterin: Biochemistry and pathophysiology. Biochem. J. 2011, 438, 397–414. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vásquez-Vivar, J. Tetrahydrobiopterin, superoxide, and vascular dysfunction. Free Radic. Biol. Med. 2009, 47, 1108–1119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ullah, R.; Jo, M.H.; Riaz, M.; Alam, S.I.; Saeed, K.; Ali, W.; Rehman, I.U.; Ikram, M.; Kim, M.O. Glycine, the smallest amino acid, confers neuroprotection against d-galactose-induced neurodegeneration and memory impairment by regulating c-Jun N-terminal kinase in the mouse brain. J. Neuroinflamm. 2020, 17, 303. [Google Scholar] [CrossRef]
- Capeillere-Blandin, C.; Mathieu, D.; Mansuy, D. Reduction of ferric haemoproteins by tetrahydropterins: A kinetic study. Biochem. J. 2005, 392, 583–587. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yagita, Y.; Kitagawa, K.; Oyama, N.; Yukami, T.; Watanabe, A.; Sasaki, T.; Mochizuki, H. Functional deterioration of endo-thelial nitric oxide synthase after focal cerebral ischemia. J. Cereb. Blood Flow Metab. Off. J. Int. Soc. Cereb. Blood Flow Metab. 2013, 33, 1532–1539. [Google Scholar] [CrossRef] [PubMed]
- Ferrer-Sueta, G.; Campolo, N.; Trujillo, M.; Bartesaghi, S.; Carballal, S.; Romero, N.; Alvarez, B.; Radi, R. Biochemistry of Peroxynitrite and Protein Tyrosine Nitration. Chem. Rev. 2018, 118, 1338–1408. [Google Scholar] [CrossRef]
- Lang, J.; Marechal, A.; Couture, M.; Santolini, J. Reaction Intermediates and Molecular Mechanism of Peroxynitrite Activation by NO Synthases. Biophys. J. 2016, 111, 2099–2109. [Google Scholar] [CrossRef] [Green Version]
- Schliess, F.; Görg, B.; Fischer, R.; Desjardins, P.; Bidmon, H.J.; Herrmann, A.; Butterworth, R.F.; Zilles, K.; Häussinger, D. Ammonia induces MK-801-sensitive nitration and phosphorylation of protein tyrosine residues in rat astrocytes. FASEB J. 2002, 16, 739–741. [Google Scholar] [CrossRef] [PubMed]
- Zielinska, M.; Milewski, K.; Skowronska, M.; Gajos, A.; Zieminska, E.; Beresewicz, A.; Albrecht, J. Induction of inducible nitric oxide synthase expression in ammonia-exposed cultured astrocytes is coupled to increased arginine transport by up-regulated y(+)LAT2 transporter. J. Neurochem. 2015, 135, 1272–1281. [Google Scholar] [CrossRef] [PubMed]
- Haussinger, D.; Gorg, B. Interaction of oxidative stress, astrocyte swelling and cerebral ammonia toxicity. Curr. Opin. Clin. Nutr. Metab. Care 2010, 13, 87–92. [Google Scholar] [CrossRef] [PubMed]
- Görg, B.; Qvartskhava, N.; Bidmon, H.-J.; Palomero-Gallagher, N.; Kircheis, G.; Zilles, K.; Häussinger, D. Oxidative stress markers in the brain of patients with cirrhosis and hepatic encephalopathy. Hepatology 2010, 52, 256–265. [Google Scholar] [CrossRef] [Green Version]
- El-Remessy, A.B.; Abou-Mohamed, G.; Caldwell, R.W.; Caldwell, R.B. High glucose-induced tyrosine nitration in endothelial cells: Role of eNOS uncoupling and aldose reductase activation. Investig. Ophthalmol. Vis. Sci. 2003, 44, 3135–3143. [Google Scholar] [CrossRef]
- Santhanam, A.V.R.; d’Uscio, L.V.; Smith, L.A.; Katusic, Z.S. Uncoupling of eNOS causes superoxide anion production and impairs NO signaling in the cerebral microvessels of hph-1 mice. J. Neurochem. 2012, 122, 1211–1218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bayir, H.; Kagan, V.E.; Clark, R.S.; Janesko-Feldman, K.; Rafikov, R.; Huang, Z.; Zhang, X.; Vagni, V.; Billiar, T.R.; Kochanek, P.M. Neuronal NOS-mediated nitration and inactivation of manganese superoxide dismutase in brain after experimental and human brain injury. J. Neurochem. 2007, 101, 168–181. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.; Chen, L.; Liu, C.; Qiu, P.; Wang, A.; Li, L.; Wang, H. Up-regulation of protein tyrosine nitration in methamphet-amine-induced neurotoxicity through DDAH/ADMA/NOS pathway. Neurochem. Int. 2013, 62, 1055–1064. [Google Scholar] [CrossRef] [PubMed]
- Cardounel, A.J.; Xia, Y.; Zweier, J.L. Endogenous methylarginines modulate superoxide as well as nitric oxide generation from neuronal nitric-oxide synthase: Differences in the effects of monomethyl- and dimethylarginines in the presence and absence of tetrahydrobiopterin. J. Biol. Chem. 2005, 280, 7540–7549. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Druhan, L.J.; Forbes, S.P.; Pope, A.J.; Chen, C.A.; Zweier, J.L.; Cardounel, A.J. Regulation of eNOS-derived superoxide by endogenous methylarginines. Biochemistry 2008, 47, 7256–7263. [Google Scholar] [CrossRef]
- Garry, P.S.; Ezra, M.; Rowland, M.J.; Westbrook, J.; Pattinson, K.T. The role of the nitric oxide pathway in brain injury and its treatment—From bench to bedside. Exp. Neurol. 2015, 263, 235–243. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Czarnecka, A.; Aleksandrowicz, M.; Jasinski, K.; Jazwiec, R.; Kalita, K.; Hilgier, W.; Zielinska, M. Cerebrovascular reactivity and cerebral perfusion of rats with acute liver failure: Role of L-glutamine and asymmetric dimethylarginine in L-arginine-induced response. J. Neurochem. 2018, 147, 692–704. [Google Scholar] [CrossRef]
- Pluta, R.; Albrecht, J. Changes in arterial and cerebral venous blood gases, cerebral blood flow and cerebral oxygen con-sumption at different stages of thioacetamide-induced hepatogenic encephalopathy in rat. Resuscitation 1986, 14, 135–139. [Google Scholar] [PubMed]
- Montécot, C.; Borredon, J.; Seylaz, J.; Pinard, E. Nitric Oxide of Neuronal Origin is Involved in Cerebral Blood Flow Increase during Seizures Induced by Kainate. Br. J. Pharmacol. 1997, 17, 94–99. [Google Scholar] [CrossRef] [Green Version]
- Gotoh, J.; Kuang, T.-Y.; Nakao, Y.; Cohen, D.M.; Melzer, P.; Itoh, Y.; Pak, H.; Pettigrew, K.; Sokoloff, L. Regional differences in mechanisms of cerebral circulatory response to neuronal activation. Am. J. Physiol. Heart Circ. Physiol. 2001, 280, H821–H829. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hagioka, S.; Takeda, Y.; Zhang, S.; Sato, T.; Morita, K. Effects of 7-nitroindazole and N-nitro-l-arginine methyl ester on changes in cerebral blood flow and nitric oxide production preceding development of hyperbaric oxygen-induced seizures in rats. Neurosci. Lett. 2005, 382, 206–210. [Google Scholar] [CrossRef] [PubMed]
- Bjerring, P.N.; Gluud, L.L.; Larsen, F.S. Cerebral Blood Flow and Metabolism in Hepatic Encephalopathy—A Meta-Analysis. J. Clin. Exp. Hepatol. 2018, 8, 286–293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shah, V.; Webster, S.; Gottstein, J.; Blei, A.T. Reduction of cerebral perfusion precedes rise of intracranial pressure in rats with ischemic fulminant liver failure. Hepatology 1993, 17, 1117–1122. [Google Scholar] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Control | TAA | |
|---|---|---|
| AST (GOT) (U/L) | 142.3 ± 11.7 | 977.12 ± 124.38 *** |
| ALT (GPT) (U/L) | 54.62 ± 2.03 | 325.41 ± 43.84 *** |
| GGTP (U/L) | 2.65 ± 0.13 | 4.23 ± 0.33 ** |
| NH3 (µmol/L) | 64.54 ± 5.29 | 132.12 ± 18.64 ** |
| Initial body weight (g) | 208.5 ± 7.23 | 217.4 ± 8.1 |
| Final body weight (g) | 224.8 ± 9.47 | 196.1 ± 14.65 |
| Body weight diff. (g) | 16.3 | −21.3 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Milewski, K.; Czarnecka, A.M.; Albrecht, J.; Zielińska, M. Decreased Expression and Uncoupling of Endothelial Nitric Oxide Synthase in the Cerebral Cortex of Rats with Thioacetamide-Induced Acute Liver Failure. Int. J. Mol. Sci. 2021, 22, 6662. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22136662
Milewski K, Czarnecka AM, Albrecht J, Zielińska M. Decreased Expression and Uncoupling of Endothelial Nitric Oxide Synthase in the Cerebral Cortex of Rats with Thioacetamide-Induced Acute Liver Failure. International Journal of Molecular Sciences. 2021; 22(13):6662. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22136662
Chicago/Turabian StyleMilewski, Krzysztof, Anna Maria Czarnecka, Jan Albrecht, and Magdalena Zielińska. 2021. "Decreased Expression and Uncoupling of Endothelial Nitric Oxide Synthase in the Cerebral Cortex of Rats with Thioacetamide-Induced Acute Liver Failure" International Journal of Molecular Sciences 22, no. 13: 6662. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22136662