
The Roles of the IGF Axis in the Regulation of the Metabolism: Interaction and Difference between Insulin Receptor Signaling and IGF-I Receptor Signaling


Abstract
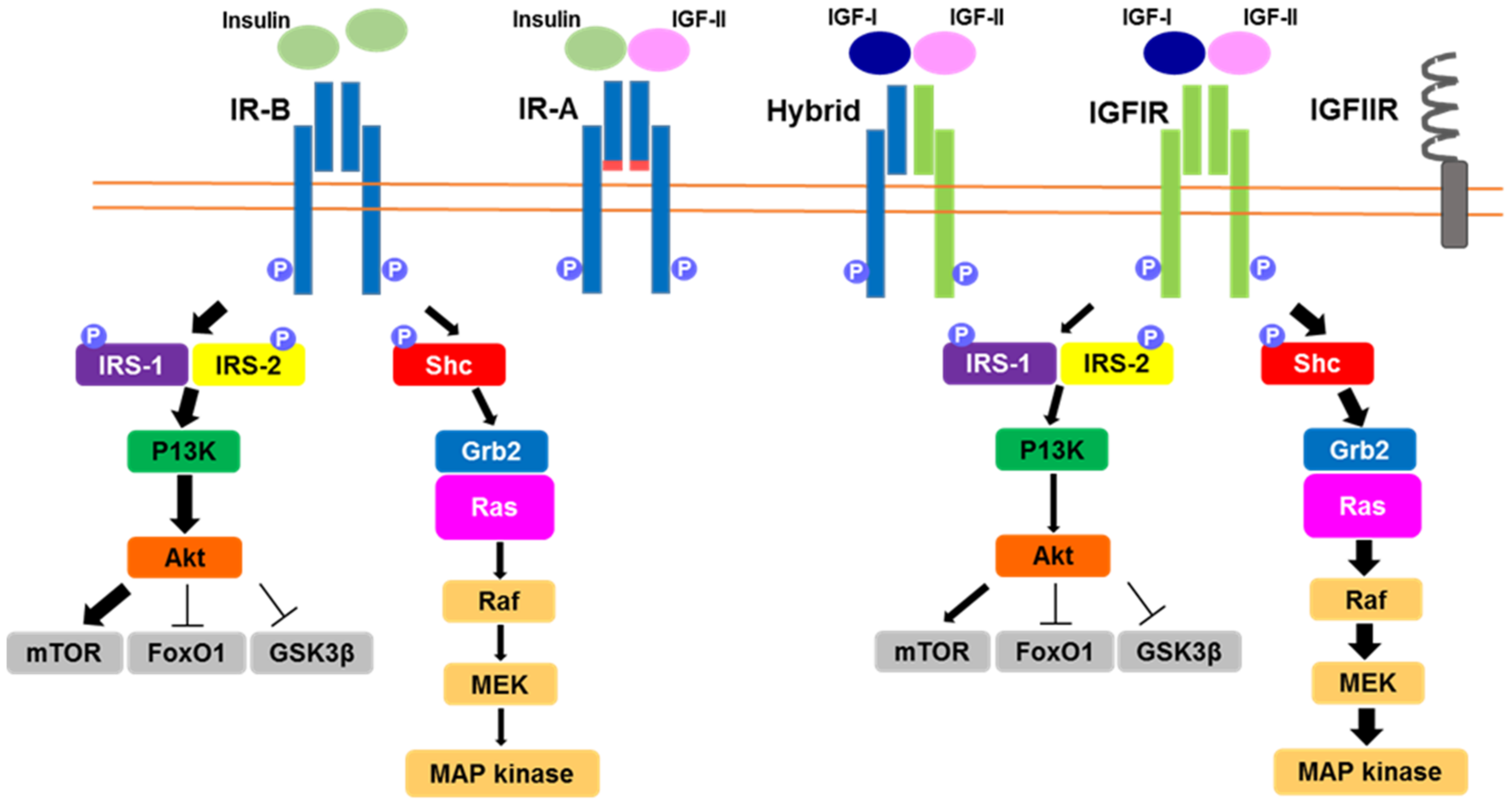
:1. Introduction
2. IGF-I and Insulin Signaling in Metabolic Organs
2.1. IGF-I and Insulin Signaling in the Liver
2.2. IGF-I and Insulin Signaling in Skeletal Muscle
2.3. IGF-I and Insulin Signaling in Adipose Tissue
2.4. IGF-I and Insulin Signaling in Pancreatic β-Cells
2.5. IGF-I and Insulin Signaling in the Brain
3. Metabolic Phenotype in OSI-906 Treated Mice
4. Perspective
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Sjögren, K.; Liu, J.L.; Blad, K.; Skrtic, S.; Vidal, O.; Wallenius, V.; LeRoith, D.; Törnell, J.; Isaksson, O.G.; Jansson, J.O.; et al. Liver-derived insulin-like growth factor I (IGF-I) is the principal source of IGF-I in blood but is not required for postnatal body growth in mice. Proc. Natl. Acad. Sci. USA 1999, 96, 7088–7092. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yakar, S.; Liu, J.L.; Stannard, B.; Butler, A.; Accili, D.; Sauer, B.; LeRoith, D. Normal growth and development in the absence of hepatic insulin-like growth factor I. Proc. Natl. Acad. Sci. USA 1999, 96, 7324–7329. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yakar, S.; Pennisi, P.; Kim, C.H.; Zhao, H.; Toyoshima, Y.; Gavrilova, O.; LeRoith, D. Studies involving the GH-IGF axis: Lessons from IGF-I and IGF-I receptor gene targeting mouse models. J. Endocrinol. Investig. 2005, 28 (Suppl. 5), 19–22. [Google Scholar]
- McRory, J.E.; Sherwood, N.M. Ancient divergence of insulin and insulin-like growth factor. DNA Cell Biol. 1997, 16, 939–949. [Google Scholar] [CrossRef] [PubMed]
- Pandini, G.; Frasca, F.; Mineo, R.; Sciacca, L.; Vigneri, R.; Belfiore, A. Insulin/insulin-like growth factor I hybrid receptors have different biological characteristics depending on the insulin receptor isoform involved. J. Biol. Chem. 2002, 277, 39684–39695. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hartog, H.; Wesseling, J.; Boezen, H.M.; van der Graaf, W.T. The insulin-like growth factor 1 receptor in cancer: Old focus, new future. Eur. J. Cancer 2007, 43, 1895–1904. [Google Scholar] [CrossRef] [PubMed]
- Vigneri, R.; Goldfine, I.D.; Frittitta, L. Insulin, insulin receptors, and cancer. J. Endocrinol. Investig. 2016, 39, 1365–1376. [Google Scholar] [CrossRef] [PubMed]
- De Meyts, P.; Whittaker, J. Structural biology of insulin and IGF1 receptors: Implications for drug design. Nat. Rev. Drug Discov. 2002, 1, 769–783. [Google Scholar] [CrossRef]
- Moller, D.E.; Yokota, A.; Caro, J.F.; Flier, J.S. Tissue-specific expression of two alternatively spliced insulin receptor mRNAs in man. Mol. Endocrinol. 1989, 3, 1263–1269. [Google Scholar] [CrossRef] [Green Version]
- Belfiore, A.; Frasca, F.; Pandini, G.; Sciacca, L.; Vigneri, R. Insulin receptor isoforms and insulin receptor/insulin-like growth factor receptor hybrids in physiology and disease. Endocr. Rev. 2009, 30, 586–623. [Google Scholar] [CrossRef] [Green Version]
- Belfiore, A.; Malaguarnera, R.; Vella, V.; Lawrence, M.C.; Sciacca, L.; Frasca, F.; Morrione, A.; Vigneri, R. Insulin Receptor Isoforms in Physiology and Disease: An Updated View. Endocr. Rev. 2017, 38, 379–431. [Google Scholar] [CrossRef]
- Pullen, R.A.; Lindsay, D.G.; Wood, S.P.; Tickle, I.J.; Blundell, T.L.; Wollmer, A.; Krail, G.; Brandenburg, D.; Zahn, H.; Gliemann, J.; et al. Receptor-binding region of insulin. Nature 1976, 259, 369–373. [Google Scholar] [CrossRef]
- Shirakawa, J.; Okuyama, T.; Yoshida, E.; Shimizu, M.; Horigome, Y.; Tuno, T.; Hayasaka, M.; Abe, S.; Fuse, M.; Togashi, Y.; et al. Effects of the antitumor drug OSI-906, a dual inhibitor of IGF-1 receptor and insulin receptor, on the glycemic control, β-cell functions, and β-cell proliferation in male mice. Endocrinology 2014, 155, 2102–2111. [Google Scholar] [CrossRef] [Green Version]
- Tajima, K.; Shirakawa, J.; Togashi, Y.; Yamazaki, S.; Okuyama, T.; Kyohara, M.; Konishi, H.; Terauchi, Y. Metabolic recovery of lipodystrophy, liver steatosis, and pancreatic β cell proliferation after the withdrawal of OSI-906. Sci. Rep. 2017, 7, 4119. [Google Scholar]
- Shirakawa, J.; Tajima, K.; Okuyama, T.; Kyohara, M.; Togashi, Y.; De Jesus, D.F.; Basile, G.; Kin, T.; Shapiro, A.M.J.; Kulkarni, R.N.; et al. Luseogliflozin increases beta cell proliferation through humoral factors that activate an insulin receptor- and IGF-1 receptor-independent pathway. Diabetologia 2020, 63, 577–587. [Google Scholar] [CrossRef]
- Okuyama, T.; Shirakawa, J.; Tajima, K.; Ino, Y.; Vethe, H.; Togashi, Y.; Kyohara, M.; Inoue, R.; Miyashita, D.; Li, J.; et al. Linagliptin Ameliorates Hepatic Steatosis via Non-Canonical Mechanisms in Mice Treated with a Dual Inhibitor of Insulin Receptor and IGF-1 Receptor. Int. J. Mol. Sci. 2020, 21, 7815. [Google Scholar] [CrossRef]
- Satake, S.; Moore, M.C.; Igawa, K.; Converse, M.; Farmer, B.; Neal, D.W.; Cherrington, A.D. Direct and indirect effects of insulin on glucose uptake and storage by the liver. Diabetes 2002, 51, 1663–1671. [Google Scholar] [CrossRef] [Green Version]
- Yakar, S.; Liu, J.L.; Fernandez, A.M.; Wu, Y.; Schally, A.V.; Frystyk, J.; Chernausek, S.D.; Mejia, W.; Le Roith, D. Liver-specific igf-1 gene deletion leads to muscle insulin insensitivity. Diabetes 2001, 50, 1110–1118. [Google Scholar] [CrossRef] [Green Version]
- Michael, M.D.; Kulkarni, R.N.; Postic, C.; Previs, S.F.; Shulman, G.I.; Magnuson, M.A.; Kahn, C.R. Loss of insulin signaling in hepatocytes leads to severe insulin resistance and progressive hepatic dysfunction. Mol. Cell 2000, 6, 87–97. [Google Scholar] [CrossRef]
- Meek, S.E.; Persson, M.; Ford, G.C.; Nair, K.S. Differential regulation of amino acid exchange and protein dynamics across splanchnic and skeletal muscle beds by insulin in healthy human subjects. Diabetes 1998, 47, 1824–1835. [Google Scholar] [CrossRef]
- Chow, L.S.; Albright, R.C.; Bigelow, M.L.; Toffolo, G.; Cobelli, C.; Nair, K.S. Mechanism of insulin’s anabolic effect on muscle: Measurements of muscle protein synthesis and breakdown using aminoacyl-tRNA and other surrogate measures. Am. J. Physiol. Endocrinol. Metab. 2006, 291, E729–E736. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, T.; Delafontaine, P. Mechanisms of IGF-1-Mediated Regulation of Skeletal Muscle Hypertrophy and Atrophy. Cells 2020, 9, 1970. [Google Scholar] [CrossRef] [PubMed]
- O’Neill, B.T.; Lee, K.Y.; Klaus, K.; Softic, S.; Krumpoch, M.T.; Fentz, J.; Stanford, K.I.; Robinson, M.M.; Cai, W.; Kleinridders, A.; et al. Insulin and IGF-1 receptors regulate FoxO-mediated signaling in muscle proteostasis. J. Clin. Investig. 2016, 126, 3433–3446. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laustsen, P.G.; Russell, S.J.; Cui, L.; Entingh-Pearsall, A.; Holzenberger, M.; Liao, R.; Kahn, C.R. Essential role of insulin and insulin-like growth factor 1 receptor signaling in cardiac development and function. Mol. Cell. Biol. 2007, 27, 1649–1664. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Neill, B.T.; Lauritzen, H.P.; Hirshman, M.F.; Smyth, G.; Goodyear, L.J.; Kahn, C.R. Differential Role of Insulin/IGF-1 Receptor Signaling in Muscle Growth and Glucose Homeostasis. Cell Rep. 2015, 11, 1220–1235. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miniou, P.; Tiziano, D.; Frugier, T.; Roblot, N.; Le Meur, M.; Melki, J. Gene targeting restricted to mouse striated muscle lineage. Nucleic Acids Res. 1999, 27, e27–e30. [Google Scholar] [CrossRef] [Green Version]
- Mavalli, M.D.; DiGirolamo, D.J.; Fan, Y.; Riddle, R.C.; Campbell, K.S.; van Groen, T.; Frank, S.J.; Sperling, M.A.; Esser, K.A.; Bamman, M.M.; et al. Distinct growth hormone receptor signaling modes regulate skeletal muscle development and insulin sensitivity in mice. J. Clin. Investig. 2010, 120, 4007–4020. [Google Scholar] [CrossRef] [Green Version]
- Anderson, C.M.; Hu, J.; Barnes, R.M.; Heidt, A.B.; Cornelissen, I.; Black, B.L. Myocyte enhancer factor 2C function in skeletal muscle is required for normal growth and glucose metabolism in mice. Skelet. Muscle 2015, 5, 7. [Google Scholar] [CrossRef] [Green Version]
- Brüning, J.C.; Michael, M.D.; Winnay, J.N.; Hayashi, T.; Hörsch, D.; Accili, D.; Goodyear, L.J.; Kahn, C.R. A muscle-specific insulin receptor knockout exhibits features of the metabolic syndrome of NIDDM without altering glucose tolerance. Mol. Cell 1998, 2, 559–569. [Google Scholar] [CrossRef]
- Wang, G.; Yu, Y.; Cai, W.; Batista, T.M.; Suk, S.; Noh, H.L.; Hirshman, M.; Nigro, P.; Li, M.E.; Softic, S.; et al. Muscle-Specific Insulin Receptor Overexpression Protects Mice from Diet-Induced Glucose Intolerance but Leads to Postreceptor Insulin Resistance. Diabetes 2020, 69, 2294–2309. [Google Scholar] [CrossRef]
- Blüher, M.; Michael, M.D.; Peroni, O.D.; Ueki, K.; Carter, N.; Kahn, B.B.; Kahn, C.R. Adipose tissue selective insulin receptor knockout protects against obesity and obesity-related glucose intolerance. Dev. Cell 2002, 3, 25–38. [Google Scholar] [CrossRef] [Green Version]
- Boucher, J.; Mori, M.A.; Lee, K.Y.; Smyth, G.; Liew, C.W.; Macotela, Y.; Rourk, M.; Bluher, M.; Russell, S.J.; Kahn, C.R. Impaired thermogenesis and adipose tissue development in mice with fat-specific disruption of insulin and IGF-1 signalling. Nat. Commun. 2012, 3, 902. [Google Scholar] [CrossRef]
- Klöting, N.; Koch, L.; Wunderlich, T.; Kern, M.; Ruschke, K.; Krone, W.; Brüning, J.C.; Blüher, M. Autocrine IGF-1 action in adipocytes controls systemic IGF-1 concentrations and growth. Diabetes 2008, 57, 2074–2082. [Google Scholar] [CrossRef] [Green Version]
- Boucher, J.; Softic, S.; El Ouaamari, A.; Krumpoch, M.T.; Kleinridders, A.; Kulkarni, R.N.; O’Neill, B.T.; Kahn, C.R. Differential Roles of Insulin and IGF-1 Receptors in Adipose Tissue Development and Function. Diabetes 2016, 65, 2201–2213. [Google Scholar] [CrossRef] [Green Version]
- Kulkarni, R.N.; Holzenberger, M.; Shih, D.Q.; Ozcan, U.; Stoffel, M.; Magnuson, M.A.; Kahn, C.R. Beta-cell-specific deletion of the Igf1 receptor leads to hyperinsulinemia and glucose intolerance but does not alter beta-cell mass. Nat. Genet. 2002, 31, 111–115. [Google Scholar] [CrossRef]
- Xuan, S.; Kitamura, T.; Nakae, J.; Politi, K.; Kido, Y.; Fisher, P.E.; Morroni, M.; Cinti, S.; White, M.F.; Herrera, P.L.; et al. Defective insulin secretion in pancreatic beta cells lacking type 1 IGF receptor. J. Clin. Investig. 2002, 110, 1011–1019. [Google Scholar] [CrossRef]
- Cornu, M.; Yang, J.Y.; Jaccard, E.; Poussin, C.; Widmann, C.; Thorens, B. Glucagon-like peptide-1 protects beta-cells against apoptosis by increasing the activity of an IGF-2/IGF-1 receptor autocrine loop. Diabetes 2009, 58, 1816–1825. [Google Scholar] [CrossRef] [Green Version]
- Nica, A.C.; Ongen, H.; Irminger, J.C.; Bosco, D.; Berney, T.; Antonarakis, S.E.; Halban, P.A.; Dermitzakis, E.T. Cell-type, allelic, and genetic signatures in the human pancreatic beta cell transcriptome. Genome Res. 2013, 23, 1554–1562. [Google Scholar] [CrossRef] [Green Version]
- Modi, H.; Jacovetti, C.; Tarussio, D.; Metref, S.; Madsen, O.D.; Zhang, F.P.; Rantakari, P.; Poutanen, M.; Nef, S.; Gorman, T.; et al. Autocrine Action of IGF2 Regulates Adult β-Cell Mass and Function. Diabetes 2015, 64, 4148–4157. [Google Scholar] [CrossRef] [Green Version]
- Sandovici, I.; Hammerle, C.M.; Virtue, S.; Vivas-Garcia, Y.; Izquierdo-Lahuerta, A.; Ozanne, S.E.; Vidal-Puig, A.; Medina-Gómez, G.; Constância, M. Autocrine IGF2 programmes β-cell plasticity under conditions of increased metabolic demand. Sci. Rep. 2021, 11, 7717. [Google Scholar] [CrossRef]
- Estil les, E.; Téllez, N.; Escoriza, J.; Montanya, E. Increased β-cell replication and β-cell mass regeneration in syngeneically transplanted rat islets overexpressing insulin-like growth factor II. Cell Transplant. 2012, 21, 2119–2129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Petrik, J.; Pell, J.M.; Arany, E.; McDonald, T.J.; Dean, W.L.; Reik, W.; Hill, D.J. Overexpression of insulin-like growth factor-II in transgenic mice is associated with pancreatic islet cell hyperplasia. Endocrinology 1999, 140, 2353–2363. [Google Scholar] [CrossRef] [PubMed]
- Devedjian, J.C.; George, M.; Casellas, A.; Pujol, A.; Visa, J.; Pelegrín, M.; Gros, L.; Bosch, F. Transgenic mice overexpressing insulin-like growth factor-II in beta cells develop type 2 diabetes. J. Clin. Investig. 2000, 105, 731–740. [Google Scholar] [CrossRef] [Green Version]
- Lu, Y.; Herrera, P.L.; Guo, Y.; Sun, D.; Tang, Z.; LeRoith, D.; Liu, J.L. Pancreatic-specific inactivation of IGF-I gene causes enlarged pancreatic islets and significant resistance to diabetes. Diabetes 2004, 53, 3131–3141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, J.; Liu, K.C.; Schulz, N.; Karampelias, C.; Charbord, J.; Hilding, A.; Rautio, L.; Bertolino, P.; Östenson, C.G.; Brismar, K.; et al. IGFBP1 increases β-cell regeneration by promoting α- to β-cell transdifferentiation. EMBO J. 2016, 35, 2026–2044. [Google Scholar] [CrossRef] [PubMed]
- Palau, N.; Rebuffat, S.A.; Altirriba, J.; Piquer, S.; Hanzu, F.A.; Gomis, R.; Barbera, A. Role of IGFBP-3 in the regulation of β-cell mass during obesity: Adipose tissue/β-cell cross talk. Endocrinology 2012, 153, 177–187. [Google Scholar] [CrossRef] [PubMed]
- Kulkarni, R.N.; Brüning, J.C.; Winnay, J.N.; Postic, C.; Magnuson, M.A.; Kahn, C.R. Tissue-specific knockout of the insulin receptor in pancreatic beta cells creates an insulin secretory defect similar to that in type 2 diabetes. Cell 1999, 96, 329–339. [Google Scholar] [CrossRef] [Green Version]
- Okada, T.; Liew, C.W.; Hu, J.; Hinault, C.; Michael, M.D.; Krtzfeldt, J.; Yin, C.; Holzenberger, M.; Stoffel, M.; Kulkarni, R.N. Insulin receptors in beta-cells are critical for islet compensatory growth response to insulin resistance. Proc. Natl. Acad. Sci. USA 2007, 104, 8977–8982. [Google Scholar] [CrossRef] [Green Version]
- Otani, K.; Kulkarni, R.N.; Baldwin, A.C.; Krutzfeldt, J.; Ueki, K.; Stoffel, M.; Kahn, C.R.; Polonsky, K.S. Reduced beta-cell mass and altered glucose sensing impair insulin-secretory function in betaIRKO mice. Am. J. Physiol. Endocrinol. Metab. 2004, 286, E41–E49. [Google Scholar] [CrossRef]
- Shirakawa, J.; Fernandez, M.; Takatani, T.; El Ouaamari, A.; Jungtrakoon, P.; Okawa, E.R.; Zhang, W.; Yi, P.; Doria, A.; Kulkarni, R.N. Insulin Signaling Regulates the FoxM1/PLK1/CENP-A Pathway to Promote Adaptive Pancreatic β Cell Proliferation. Cell Metab. 2017, 25, 868–882.e5. [Google Scholar] [CrossRef] [Green Version]
- Ueki, K.; Okada, T.; Hu, J.; Liew, C.W.; Assmann, A.; Dahlgren, G.M.; Peters, J.L.; Shackman, J.G.; Zhang, M.; Artner, I.; et al. Total insulin and IGF-I resistance in pancreatic beta cells causes overt diabetes. Nat. Genet. 2006, 38, 583–588. [Google Scholar] [CrossRef]
- Ansarullah; Jain, C.; Far, F.F.; Homberg, S.; Wißmiller, K.; von Hahn, F.G.; Raducanu, A.; Schirge, S.; Sterr, M.; Bilekova, S.; et al. Inceptor counteracts insulin signalling in β-cells to control glycaemia. Nature 2021, 590, 326–331. [Google Scholar] [CrossRef]
- Folli, F.; Okada, T.; Perego, C.; Gunton, J.; Liew, C.W.; Akiyama, M.; D’Amico, A.; La Rosa, S.; Placidi, C.; Lupi, R.; et al. Altered insulin receptor signalling and β-cell cycle dynamics in type 2 diabetes mellitus. PLoS ONE 2011, 6, e28050. [Google Scholar] [CrossRef] [Green Version]
- Kubota, N.; Terauchi, Y.; Tobe, K.; Yano, W.; Suzuki, R.; Ueki, K.; Takamoto, I.; Satoh, H.; Maki, T.; Kubota, T.; et al. Insulin receptor substrate 2 plays a crucial role in beta cells and the hypothalamus. J. Clin. Investig. 2004, 114, 917–927. [Google Scholar] [CrossRef] [Green Version]
- Hashimoto, N.; Kido, Y.; Uchida, T.; Asahara, S.; Shigeyama, Y.; Matsuda, T.; Takeda, A.; Tsuchihashi, D.; Nishizawa, A.; Ogawa, W.; et al. Ablation of PDK1 in pancreatic beta cells induces diabetes as a result of loss of beta cell mass. Nat. Genet. 2006, 38, 589–593. [Google Scholar] [CrossRef]
- Brüning, J.C.; Gautam, D.; Burks, D.J.; Gillette, J.; Schubert, M.; Orban, P.C.; Klein, R.; Krone, W.; Müller-Wieland, D.; Kahn, C.R. Role of brain insulin receptor in control of body weight and reproduction. Science 2000, 289, 2122–2125. [Google Scholar] [CrossRef]
- Kappeler, L.; De Magalhaes Filho, C.; Dupont, J.; Leneuve, P.; Cervera, P.; Périn, L.; Loudes, C.; Blaise, A.; Klein, R.; Epelbaum, J.; et al. Brain IGF-1 receptors control mammalian growth and lifespan through a neuroendocrine mechanism. PLoS Biol. 2008, 6, e254. [Google Scholar] [CrossRef]
- Mulvihill, M.J.; Cooke, A.; Rosenfeld-Franklin, M.; Buck, E.; Foreman, K.; Landfair, D.; O’Connor, M.; Pirritt, C.; Sun, Y.; Yao, Y.; et al. Discovery of OSI-906: A selective and orally efficacious dual inhibitor of the IGF-1 receptor and insulin receptor. Future Med. Chem. 2009, 1, 1153–1171. [Google Scholar] [CrossRef]
- Cioce, M.; Pulito, C.; Strano, S.; Blandino, G.; Fazio, V.M. Metformin: Metabolic Rewiring Faces Tumor Heterogeneity. Cells 2020, 9, 2439. [Google Scholar] [CrossRef]
- Holzenberger, M.; Dupont, J.; Ducos, B.; Leneuve, P.; Géloën, A.; Even, P.C.; Cervera, P.; Le Bouc, Y. IGF-1 receptor regulates lifespan and resistance to oxidative stress in mice. Nature 2003, 421, 182–187. [Google Scholar] [CrossRef]
- Vellai, T.; Takacs-Vellai, K.; Zhang, Y.; Kovacs, A.L.; Orosz, L.; Müller, F. Genetics: Influence of TOR kinase on lifespan in C. elegans. Nature 2003, 426, 620. [Google Scholar] [CrossRef] [PubMed]
- Kapahi, P.; Zid, B.M.; Harper, T.; Koslover, D.; Sapin, V.; Benzer, S. Regulation of lifespan in Drosophila by modulation of genes in the TOR signaling pathway. Curr. Biol. 2004, 14, 885–890. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spadaro, O.; Goldberg, E.L.; Camell, C.D.; Youm, Y.H.; Kopchick, J.J.; Nguyen, K.Y.; Bartke, A.; Sun, L.Y.; Dixit, V.D. Growth Hormone Receptor Deficiency Protects against Age-Related NLRP3 Inflammasome Activation and Immune Senescence. Cell Rep. 2016, 14, 1571–1580. [Google Scholar] [CrossRef] [Green Version]
- Suh, Y.; Atzmon, G.; Cho, M.O.; Hwang, D.; Liu, B.; Leahy, D.J.; Barzilai, N.; Cohen, P. Functionally significant insulin-like growth factor I receptor mutations in centenarians. Proc. Natl. Acad. Sci. USA 2008, 105, 3438–3442. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Heemst, D.; Beekman, M.; Mooijaart, S.P.; Heijmans, B.T.; Brandt, B.W.; Zwaan, B.J.; Slagboom, P.E.; Westendorp, R.G. Reduced insulin/IGF-1 signalling and human longevity. Aging Cell 2005, 4, 79–85. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Tissue | Promoter-Driven Cre | IGF-IRKO | IRKO | DKO |
|---|---|---|---|---|
| Liver | Albumin | - | severe insulin resistance | - |
| (Hepatocyte) | overt severe diabetes | |||
| increase in β-cell mass | ||||
| liver dysfunction (age-related) | ||||
| reduced serum triglycerides and free fatty acids | ||||
| Muscle | Creatine kinase | normal body weight | normal body weight | reduced body weight |
| normal glucose tolerance | normal glucose tolerance | normal glucose tolerance | ||
| normal cardiac performance | impaired cardiac performance | developed heart failure | ||
| elevated serum triglycerides and free fatty acids | ||||
| ACTA1 | normal body weight | normal body weight | severe muscle atrophy | |
| no change in muscle mass | no change in muscle mass | normal glucose tolerance | ||
| normal glucose tolerance | normal glucose tolerance | |||
| Mef2c | normal body weight | - | - | |
| normal serum glucose and triglycerides | ||||
| Adipose tissue | Adiponectin | reduced WAT and BAT mass | reduced body weight | reduced body weight |
| decreased lipogenic gene expression | reduced WAT and increased BAT mass | reduced WAT and BAT mass | ||
| lower plasma leptin and adiponectin level | lower plasma leptin and adiponectin level | lower plasma leptin and adiponectin level | ||
| normal glucose tolerance | overt diabetes | overt diabetes | ||
| normal insulin tolerance | severe insulin resistance | severe insulin resistance | ||
| ectopic lipid accumulation | ectopic lipid accumulation | |||
| dyslipidemia | dyslipidemia | |||
| pancreatic islet hyperplasia | pancreatic islet hyperplasia | |||
| cold intolerance | severe cold intolerance | |||
| increased basal energy expenditure | ||||
| Pancreatic β-cell | Rat insulin 2 promoter | no change in β-cell mass and β-cell proliferation | reduced β-cell mass and β-cell proliferation | markedly reduced β-cell mass |
| impaired glucose- and arginine-induced insulin secretion and impaired glucose tolerance | impaired glucose-stimulated insulin secretion and impaired glucose tolerance | overt diabetes | ||
| Brain | Rat nestin promoter | reduced brain size | normal brain size | |
| growth retardation | normal growth | |||
| behavioral changes | mild obesity | |||
| insulin resistance |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Okuyama, T.; Kyohara, M.; Terauchi, Y.; Shirakawa, J. The Roles of the IGF Axis in the Regulation of the Metabolism: Interaction and Difference between Insulin Receptor Signaling and IGF-I Receptor Signaling. Int. J. Mol. Sci. 2021, 22, 6817. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22136817
Okuyama T, Kyohara M, Terauchi Y, Shirakawa J. The Roles of the IGF Axis in the Regulation of the Metabolism: Interaction and Difference between Insulin Receptor Signaling and IGF-I Receptor Signaling. International Journal of Molecular Sciences. 2021; 22(13):6817. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22136817
Chicago/Turabian StyleOkuyama, Tomoko, Mayu Kyohara, Yasuo Terauchi, and Jun Shirakawa. 2021. "The Roles of the IGF Axis in the Regulation of the Metabolism: Interaction and Difference between Insulin Receptor Signaling and IGF-I Receptor Signaling" International Journal of Molecular Sciences 22, no. 13: 6817. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22136817