
Therapeutic Targeting of the Leukaemia Microenvironment
by
 , and
, and
Vincent Kuek
1,2,3,† ,
,
Anastasia M. Hughes
1,2,†,
Rishi S. Kotecha
1,2,4,5 and
Laurence C. Cheung
1,2,* 1
Leukaemia Translational Research Laboratory, Telethon Kids Cancer Centre, Telethon Kids Institute, Perth, WA 6009, Australia
2
Curtin Medical School, Curtin University, Perth, WA 6102, Australia
3
School of Biomedical Sciences, University of Western Australia, Perth, WA 6009, Australia
4
Department of Clinical Haematology, Oncology, Blood and Marrow Transplantation, Perth Children’s Hospital, Perth, WA 6009, Australia
5
School of Medicine, University of Western Australia, Perth, WA 6009, Australia
*
Author to whom correspondence should be addressed.
†
These authors contributed equally to this work.
Int. J. Mol. Sci. 2021, 22(13), 6888; https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22136888
Submission received: 31 May 2021
/
Revised: 18 June 2021
/
Accepted: 23 June 2021
/
Published: 26 June 2021
(This article belongs to the Special Issue Recent Advances of Leukemia Pathophysiology: From Molecular Basis to Targeted Therapies)
Abstract
:In recent decades, the conduct of uniform prospective clinical trials has led to improved remission rates and survival for patients with acute myeloid leukaemia and acute lymphoblastic leukaemia. However, high-risk patients continue to have inferior outcomes, where chemoresistance and relapse are common due to the survival mechanisms utilised by leukaemic cells. One such mechanism is through hijacking of the bone marrow microenvironment, where healthy haematopoietic machinery is transformed or remodelled into a hiding ground or “sanctuary” where leukaemic cells can escape chemotherapy-induced cytotoxicity. The bone marrow microenvironment, which consists of endosteal and vascular niches, can support leukaemogenesis through intercellular “crosstalk” with niche cells, including mesenchymal stem cells, endothelial cells, osteoblasts, and osteoclasts. Here, we summarise the regulatory mechanisms associated with leukaemia–bone marrow niche interaction and provide a comprehensive review of the key therapeutics that target CXCL12/CXCR4, Notch, Wnt/b-catenin, and hypoxia-related signalling pathways within the leukaemic niches and agents involved in remodelling of niche bone and vasculature. From a therapeutic perspective, targeting these cellular interactions is an exciting novel strategy for enhancing treatment efficacy, and further clinical application has significant potential to improve the outcome of patients with leukaemia.
1. Introduction
The bone marrow microenvironment (BMM) is a primary site for haematopoiesis and consists of unique bone marrow (BM) cells, which facilitate haematopoietic cell regeneration and renewal in specialised regions known as “niches”. These BM niches provide critical factors and signals that support the physiological maintenance of haematopoietic stem cells (HSCs) as well as the formation of lineage-committed cells in a highly regulated and controlled manner [1]. However, emerging evidence has pointed to their role in malignant transformation and, subsequently, pathogenesis of leukaemia. While genomic aberrations underpin leukaemic transformation, the BMM is being increasingly recognised as a key player in leukaemia progression, treatment resistance, and disease relapse. The ability of leukaemic stem or progenitor cells to interact with their neighbouring supportive cells can severely disrupt haematopoietic homeostasis, leading to the formation of a leukaemic-friendly niche or “safe haven” for malignant cells to escape chemotherapy [2]. Importantly, this can also compromise bone remodelling, a key process whereby osteoblasts and osteoclasts continuously remould and reshape the bone matrix in units known as bone remodelling compartments to maintain structural integrity [3]. Indeed, impaired bone remodelling can lead to the manifestation of secondary bone conditions commonly seen in patients with leukaemia, including osteolytic lesions, osteoporosis, and increased fracture risk [4,5]. Although the niche microenvironments of acute myeloid leukaemia (AML) and chronic myeloid leukaemia (CML) are well established, others such as acute lymphoblastic leukaemia (ALL) and chronic lymphocytic leukaemia (CLL) have not been thoroughly investigated, in part due to the lack of reliable animal models.
This review provides a current understanding of the BMM and the mechanisms involved in the maintenance of the leukaemic niche. In addition, we provide perspective on current BMM-targeted therapies for the treatment of myeloid and lymphoid malignancies and discuss the strengths and limitations associated with these treatment approaches. Knowledge gained from these studies will lead to improved treatment strategies and clinical outcomes for patients with high-risk leukaemia.
2. The Bone Marrow Niche
Studies conducted using various mouse models have delineated BM niches as endosteal or central, depending on their anatomical location within the BM structure [6]. The endosteal niche consists of osteolineage cells (e.g., osteoprogenitors, osteoblasts, osteoclasts) and mesenchymal stem cells (MSCs), which are localised on the inner bone lining surface and supported by the endosteal vessel network [7,8]. Insight into the modulation of HSCs by osteoblasts has been investigated using mouse models [9]. The mechanisms of osteoblast–HSC interaction have been validated as direct cell–cell contact (e.g., Notch1/Jagged1) and paracrine interactions via secretory factors (e.g., osteopontin and angiopoietin-1) to promote HSC maintenance, retention, and quiescence [9,10,11]. In addition, osteoblasts promote the maintenance of early lymphoid progenitors by producing C-X-C motif chemokine 12 (CXCL12), a prominent chemoattractant of haematopoietic cells [12]. This finding endorses the concept of osteoblasts and the overall endosteal niche support for haematopoiesis. In contrast, the central niche comprises vasculature and perivascular stromal cells, such as leptin receptor+ MSCs and CXCL12-abundant reticular cells [6]. These perivascular stromal cells, together with endothelial cells, are actively involved in HSC maintenance through local secretion of cytokines [13,14]. Importantly, these cells also regulate B lymphopoiesis through interleukin 7 production [15].
The dynamic nature of the BMM is attributable to a myriad of cytokines, chemokines, and adhesion molecules, which support crosstalk between haematopoietic and BM niche cells, resulting in healthy production and replenishment of myeloid and lymphoid cells. However, the presence of malignant cells outcompetes normal HSCs, leading to “hijacking” of the BM niche. Indeed, gradual changes of healthy BMM into a leukaemic niche or “sanctuary” is an important hallmark of leukaemogenesis, whereby leukaemic cells establish their own dynamic ecosystem and promote dormancy, survival, proliferation, and resistance to chemotherapy (Figure 1). The structure of the normal HSC niche [6,7,8], as well as cell-intrinsic/extrinsic niche factors that regulate clonal haematopoiesis and leukaemia progression [16,17], have been extensively reviewed. Thus, this review will primarily focus on BMM-targeted therapies.
3. Leukaemia Niche Regulation
3.1. Niche-Driven Malignant Transformation
Significant progress has been made over recent decades in understanding how genetic mutations or functional alterations of niche cells can promote leukaemogenesis. Deletion of certain genes (e.g., Rarg, Rb1, and Mib1) in non-haematopoietic cells is a major risk factor for the induction of myeloproliferative neoplasia (MPN)-like disease, a pre-leukaemic disorder, in several mouse models [6]. Deletion of Dicer1 in osteoprogenitors has been shown to result in myelodysplastic syndrome (MDS) with sporadic transformation to AML [18]. Apart from loss-of-function, constitutive activation of certain genes can also promote leukaemogenesis. For instance, osteoblast-associated activating mutation of β-catenin, present in 38% of patients with MDS or AML, can promote the development of leukaemia [19]. Likewise, activation of the parathyroid hormone receptor in osteoblasts attenuates BCR-ABL1-induced CML-like MPN and enhances KMT2A-MLLT3 oncogene-induced AML, providing further evidence that osteoblasts are capable of influencing haematological malignant transformation [20]. Endothelial cells have also been implicated in the development of MPN-like disorder through miR-155 microRNA-induced nuclear factor κB (NF-κB) activation and pro-inflammatory cytokine production [21]. The oncogenic role of MSCs has recently been further highlighted, where activating Ptpn11 mutations in MSCs and osteoprogenitors induce MPN through excessive production of chemokine ligand 3 (CCL3) [22]. This evidence points towards malignant transformation being dependent on the oncogenic tendencies of the surrounding cells, with such predisposition contributing to establishment of the BMM as a fertile ground for leukaemogenesis.
3.2. Leukaemic Remodelling of the Vasculature and Endosteal Niche
Leukaemic cells are also capable of remodelling the BMM into a cancer-supportive environment, known to facilitate tumour survival, resistance to therapy, and immune escape. AML cells co-expressing BCR-ABL1 and Nup98/HoxA9 fusion gene have been shown to disrupt bone homeostasis by inhibition of mature osteoblasts, likely via leukaemic cell-secreted CCL3 in vivo [23]. Similarly, KMT2A-MLLT3 AML cells impair HSC niche function and induce commitment of MSCs to differentiate into osteoprogenitor cells but not mature osteoblasts, leading to reduced bone mineralisation in vivo [24]. Furthermore, a study using a BCR-ABL driven CML/MPN mouse model showed that leukaemic myeloid cells remodelled the endosteal BM niche by promoting aberrant expansion of osteoblastic lineage cells [25]. These cells exhibited compromised HSC-supportive activity and favoured abnormal BM myelofibrosis, which is often associated with poor prognosis in CML [25]. Recently, T-ALL cells have also been implicated in inducing osteoblast apoptosis and impairing haematopoiesis [26] Further study revealed that the mechanism of T-ALL-mediated osteoblast suppression is via aberrant Notch activation, which plays a role in negatively regulating CXCL12 on osteoblasts [27].
Leukaemic cells are also known to cluster around BM blood vessels, where AML engraftment directly contributes to an altered vascular architecture, increased endothelial cells, and increased vascular permeability [28]. This finding converges on a model whereby the vascular niche, similar to the endosteal niche, is being remodelled by leukaemic cells to favour leukaemogenesis. Several studies have associated increased vascularity in leukaemic BM with the production of angiogenic factors and inflammatory cytokines. AML cells secrete vascular endothelial growth factor A (VEGF-A), which promotes both leukaemic cell proliferation and tumour-supportive angiogenesis [29]. Increased plasma VEGF levels in patients with AML has been associated with poor clinical outcome [30]. Activation of endothelial cells by VEGF-A can also contribute to increased leukaemic cell protection by the vascular niche, allowing the cells to escape from therapy-induced cytotoxicity [31]. Furthermore, AML engraftment can lead to the formation of a hypoxic niche microenvironment, alter the molecular signature of endothelial cells, and promote the production of hypoxia-responsive reactive oxygen species and nitric oxide (NO), a mediator of vascular permeability [28]. Importantly, NO-induced permeability and vessel leakiness are associated with a dysregulated blood supply and poor delivery of therapeutic agents to malignant target cells [28].
Remodelling of the vasculature by leukaemic cells is not strictly confined to the vascular sinusoids located within the central marrow, but also the endosteal vessels. Indeed, endosteal remodelling by AML can lead to the loss of blood vessels, correlating with progressive depletion of stromal cells, osteoblasts, and HSCs [32]. This striking phenotype indicates area-specific changes with differential remodelling of vessels in central and endosteal BM regions. Further characterisation of the mechanisms revealed that endosteal AML cells produced pro-inflammatory signals such as tumour necrosis factor and anti-angiogenic cytokines, which play a major role in local suppression and destruction of endosteal vasculature [32]. In addition, emerging evidence also suggests that leukaemic cell-secreted inflammatory mediators are pivotal in regulating the expression of endothelial adhesion molecules (e.g., E-selectin) and initiate leukaemic–endothelial interaction, which in turn modulates leukaemogenesis in a positive feedback loop mechanism [33,34]. Collectively, the mechanisms associated with BM vascular remodelling by leukaemic cells represent an important approach for therapeutic targeting in haematological malignancies.
Our recent understanding of the BMM in driving leukaemia manifestations and therapy resistance has significantly advanced due to investigations using a range of transgenic, syngeneic, and patient-derived xenograft leukaemia engraftment models. While most studies have investigated the microenvironment of myeloid malignancies, emerging evidence points to specificity of microenvironment remodelling for different types of leukaemia. For instance, endosteal vessels were found to be decreased in AML-burdened mice but were unaffected by T-ALL [26,32]. Furthermore, the manifestation of bone loss in pre-B ALL is different from AML: BCR-ABL1+ pre-B ALL mice have been shown to develop progressive but severe bone loss during leukaemogenesis caused by impaired osteogenesis, coupled with enhanced osteoclastogenesis and bone resorption via excessive production of RANKL by leukaemic cells [35]. Further characterisation of the pre-B ALL BMM using single-cell RNA sequencing revealed significant impairment of the haematopoietic stem and progenitor cells (HSPCs) compartment, intercellular communication changes in monocytes and HSPCs, and alteration of molecular signatures associated with pro-B and pre-B cell responses [36]. The different leukaemia-specific mechanisms of remodelling remain significant areas of interest for further investigation.
3.3. Adipocytic Niche in Leukaemia
Adipocytes are one of the key cellular components of the BMM. They occupy the majority of the BM space and play an important role in regulating normal haematopoiesis. Accumulating evidence also suggests that BM adipose tissues can impact on the survival and proliferation of leukaemic cells in AML and T-ALL [37,38]. Furthermore, adipocytes are also known to contribute to leukaemic chemoresistance by metabolising and inactivating drugs in the BM [39]. Thus, the concept of targeting adipocytes in the leukaemic BMM remains an attractive proposition due to their influence as a protective niche for malignant cells and has been extensively reviewed [40,41].
4. Targeting the Leukaemia Microenvironment: Mechanisms of Treatment Resistance and Therapeutic Strategies
Recognising that the tumour microenvironment contributes to treatment failure or success has led to a recent paradigm shift in cancer therapy. In leukaemia, targeting components or niche-associated signalling pathways of the disease microenvironment could present a novel means of enhancing therapeutic effectiveness, particularly in children with high-risk leukaemia, as dose-limiting toxicities of conventional chemotherapeutic agents have prevented further improvements in survival. As some BM niche factors involved in the survival mechanism of leukaemic cells are also major regulators of normal HSC mobilisation/trafficking [42], therapeutically targeting these factors to modulate the mobilisation of leukaemic cells could improve clinical outcomes. The therapeutic agents described and their mechanisms of action within the leukaemia BMM have been summarised in Table 1 and illustrated in Figure 2. The clinical trial outcomes for each therapeutic agent have been summarised in Table 2.
4.1. CXCL12/CXCR4 Signalling Pathway
The C-X-C motif chemokine 12/C-X-C chemokine receptor type 4 (CXCL12/CXCR4) axis has been highlighted as a key driver of chemoresistance in leukaemia. In AML cells, chemotherapy has been reported to upregulate CXCR4 expression, thereby conferring resistance to therapy-induced apoptosis when co-cultured with stroma cells [105]. Likewise, stromal CXCL12 is required for homing, retention, and repopulation of pre-B ALL cells in the BM [106]. Interestingly, direct contact of T-ALL cells with CXCL12-producing endothelial cells has been implicated in the formation of a vascular niche for T-ALL [50].
Numerous preclinical studies have demonstrated that CXCR4 inhibitors can improve treatment strategies for leukaemia. Plerixafor (AMD3100) is a potent, U.S. Food and Drug Administration (FDA)-approved bicyclam CXCR4 antagonist for the treatment of multiple myeloma and lymphoma. In vivo studies have shown that plerixafor can mobilise and sensitise leukaemic cells to the chemotoxicity of treatment by disrupting CXCL12/CXCR4 interactions between leukaemic cells and the stromal niche [43,44]. The safety and tolerability of plerixafor has been demonstrated in phase 1 trials for patients with relapsed/refractory acute leukaemia and in patients with AML undergoing allogeneic haematopoietic stem cell transplantation (HSCT) [46,47,48]. Other derivatives of plerixafor, such as AMD3465 and AMD11070, have also been reported to elicit anti-leukaemic activity. Treatment with AMD3465 in vivo led to the reduction of leukaemic burden in pre-B ALL and T-ALL, while AMD3465 was shown to antagonise stroma-induced migration of AML and enhance the susceptibility of leukaemic cells to chemotherapy [45,49,50]. Likewise, AMD11070 has been shown to enhance the therapeutic efficacy of chemotherapy in mice transplanted with pre-B ALL cells [51]. Whilst plerixafor is the only reported bicyclam CXCR4 inhibitor to have undergone clinical assessment, in vivo data suggest further clinical investigation of others is warranted.
In recent years, synthetically designed peptides have emerged as another group of promising agents owing to their high-affinity for CXCR4 binding and ability to mobilise leukaemic cells from their protective niche. BL-8040 has been shown to induce leukaemic cell apoptosis in both AML-bearing mice and patients [52,53]. Identification of another potent CXCR4 peptide antagonist, LY2510924, led to a phase 1 study that demonstrated safety and yielded a response signal in patients with relapsed/refractory AML, supporting further clinical investigation [54,55]. Other emerging CXCR4 peptide antagonists capable of mobilising leukaemic cells include E5 and POL6326, with the latter currently under clinical evaluation (NCT01413568) [57,58]. Interestingly, low anticoagulant 2-O, 3-O desulphated heparin (CX-01), an analog of heparin with reduced anticoagulation, has been suggested as a potential therapeutic agent due to its CXCL12/CXCR4-inhibitory properties [63]. Encouraging complete remission rates were seen when CX-01 was administered to patients with AML in combination with standard induction therapy, with disruption of CXCL12/CXCR4-mediated protection of leukaemic stem cells (LSCs) in the BM niche playing a potential role in treatment response [63].
Another CXCL12/CXCR4-targeted therapeutic strategy involves the use of monoclonal antibodies. Ulocuplumab (BMS-936564/MDX-1338) is a first-in-class human IgG4 monoclonal anti-CXCR4 antibody that has therapeutic potential for several haematologic malignancies [59]. Preclinical studies have shown that ulocuplumab inhibits CXCL12-induced cell migration, induces apoptosis in leukaemic cells and exhibits anti-tumour effects when used as monotherapy in AML mouse xenograft models [59]. Moreover, clinical studies have revealed the ability of ulocuplumab to mobilise leukaemic cells into the circulation, rendering them more susceptible to chemotherapy-induced toxicity in patients with AML [60,61]. LY2624587, another humanised CXCR4 antagonistic antibody, was found to elicit dose-dependent apoptosis of CCRF-CEM T-ALL in vitro and tumour growth inhibition in vivo [62]. However, the clinical trial status of LY2624587 in leukaemia is currently unknown.
4.2. Notch Signalling Pathway
Notch signalling is another important mechanism that contributes to leukaemia progression. Abnormal Notch1 activating mutations are found in over 50% of T-ALL cases, and various studies have shown that stromal-induced maintenance and growth of human T-ALL cells, and BM engraftment of T-ALL, are initiated by Notch1 activation [107,108]. In addition, pre-B ALL samples collected from high-risk patients showed elevated expression of Notch3/4 and Jagged2, and that Notch 3 and 4 signalling pathways were responsible for a stromal-mediated anti-apoptotic effect on pre-B ALL cells [109,110]. Furthermore, the role of Notch signalling has also been recognised in the pathogenesis of AML. Bone marrow MSCs (BM-MSCs) isolated from patients with AML expressed higher levels of Notch1 and Jagged1 proteins compared to healthy donors and promoted the proliferation, survival, and chemoresistance of AML cells [111]. In the vascular niche, endothelial cells also interact with T-ALL cells in a Notch-mediated fashion, thus promoting tumour growth and survival [112].
The relevance of Notch signalling in the crosstalk between leukaemic cells and BM stromal/MSCs render this pathway as attractive therapeutic target. One such strategy is the use of monoclonal antibodies with targeted specificity against extracellular Notch receptors or ligands. Antibody-blocking Notch ligand DLL4 has been shown to impair tumour growth and enhance apoptosis in T-ALL, partly through the modulation of stromal cells [113]. While humanised anti-DLL4 antibodies such as demcizumab and enoticumab have undergone clinical trials for solid tumours, neither have been tested in haematological malignancies [114,115]. The gamma–secretase complex plays a pivotal role in the activation of Notch signalling. During receptor–ligand interaction, Notch receptors are cleaved by ADAM/TACE metalloproteinases, followed by another cleavage by the gamma–secretase complex to induce Notch activation [116]. Therefore, inhibiting this crucial step with gamma–secretase inhibitors (GSIs) is an appealing treatment approach for haematological malignancies. Treatment of pre-B ALL-bearing mice with GSI-XII resulted in enhanced chemo-sensitivity of ALL cells, leading to reduced BM leukaemic burden and prolonged survival [110]. Another GSI, MK-0752, was clinically evaluated in seven patients with T-ALL and one with AML [117]. While MK-0752 was poorly tolerated due to gastrointestinal toxicities caused by the “on-target” abrogation of Notch1/2 in the gut, such adverse effects could be overcome by combining the therapy with glucocorticoids and adopting an intermittent dosing regimen [118,119]. Indeed, minimal gastrointestinal toxicity of this treatment regimen was verified using PF-03084014, a GSI in clinical development [64]. PF-03084014, in combination with fludarabine, has been shown to exhibit synergistic anti-tumour effects against Notch1-mutated CLL cells in vitro, even in the presence of protective stromal cells [120]. Furthermore, PF-03084014 synergistically enhanced glucocorticoid-induced reduction of tumour burden in T-ALL-bearing mice [65]. A subsequent phase 1 trial has demonstrated promising anti-tumour activity in patients with T-ALL, thus supporting the potential use of PF-03084014 in the clinical setting [66]. Another promising GSI currently under clinical development is BMS-906024. In a phase 1 trial, 32% of patients with relapsed/refractory T-ALL showed at least 50% reduction in BM blasts, and treatment with BMS-906024 was generally well tolerated [68]. MRK-560 is a highly selective GSI that inhibits the presenilin-1 (PSEN1) subclass of gamma–secretase complexes, which has high expression in T-ALL. A recent study has demonstrated that MRK-560 treatment can impair leukaemogenesis and prolong survival in T-ALL bearing mice without inducing gastrointestinal toxicity, indicating a potential therapeutic advantage over other broad-spectrum GSIs [69].
4.3. Wnt/β-Catenin Signalling Pathway
Aberrant activation of Wnt/β-catenin signalling is a major inducer of survival and recurrence in many cancers, including leukaemia. A recent genomic analysis of relapsed childhood ALL has identified the Wnt/β-catenin pathway as one of the top dysregulated pathways [121]. Aberrant activation of Wnt/β-catenin signalling, either through methylation of Wnt antagonists and/or mutations in Wnt signalling regulatory proteins, is known to cause disease progression in AML [122]. While cell-intrinsic mechanisms of Wnt signalling are directly involved in malignant transformation, compelling evidence points to the BM niche as an extrinsic activator of Wnt signalling and leukaemogenesis. For instance, leukaemogenesis initiated by osteoblasts is modulated by Notch/Jagged1 interactions in HSCs, and Jagged1 expression in osteoblasts is stimulated by β-catenin activation through interaction with the transcription factor, FoxO1 [123]. BM stromal cells also contribute to the chemoprotective effect of leukaemic cells via the Wnt signalling pathway [70]. Intriguingly, Wnt signalling has also been implicated in the pro-survival regulation of AML cells by the BM vascular niche [124]. E-selectin, an endothelial-specific adhesion molecule and regulator of HSCs, has been reported to interact with AML cells and enhance leukaemic survival via Wnt activation [124]. In addition, the expression of BM E-selectin was found to be increased during AML development, which correlated with increased E-selectin-binding potential of AML blasts. This suggests that multiple niche-derived factors, including Wnt, could be involved in the complex interaction between leukaemic cells and niche resident cells to support leukaemia cell attachment and retention.
While there are currently no known FDA-approved Wnt-targeting agents, several investigational drugs have been assessed in preclinical and clinical studies. XAV939, a Wnt signalling antagonist, exhibited anti-leukaemic activity by inhibiting AML cell viability and attenuating MSC-induced chemoprotective effects of ALL cells in vitro [70,125]. In addition, XAV939 enhanced chemotherapy-induced apoptosis in ALL and improved overall survival of ALL-bearing mice [70]. PRI-724, a β-catenin inhibitor currently undergoing clinical evaluation (NCT01606579), has also demonstrated anti-leukaemic activity in AML and CML. In a blast crisis CML-bearing mouse model, combined treatment of PRI-724 and nilotinib led to impaired engraftment of CML cells, prolonged survival of tumour-bearing mice, and synergistically induced cell death in tyrosine kinase inhibitor-resistant leukaemia cells [71]. Similarly, PRI-724 has been shown to synergistically enhance the anti-tumour effect of FLT3 tyrosine kinase inhibitors in vitro and in vivo by abrogating the BMM-induced protection of FLT3-mutated AML cells [72]. BC2059, an anthraquinone oxime-analogue, has been shown to synergistically induce apoptosis in primary AML blasts and prolong survival of AML xenograft mice following administration with panobinostat [73]. CWP232291, a novel peptidomimetic that possess broad preclinical anti-cancer activities, has recently been tested in a phase 1 trial in patients with AML, with minimal/modest efficacy [74,126,127]. Future studies have been planned to explore the potential synergistic effects of CWP232291 with other chemotherapeutic agents [74]. SKLB-677, a recently developed dual inhibitor of FLT3 and Wnt/β-catenin signalling pathways, was found to suppress the viability of FLT3-driven AML cells and prolonged the survival of AML xenograft mice in a dose-dependent manner [75].
Collectively, preclinical and clinical studies to date have supported the potential for Wnt/β-catenin inhibitors to treat leukaemia, largely due to their cytotoxicity on leukaemia cells and their ability to modulate the leukaemic microenvironment. However, the challenges of targeting the Wnt pathway have been highlighted by a study that found variable response to Wnt inhibition in primary blasts from different patients with normal karyotype AML, indicating that certain subtypes of AML may be more susceptible to anti-Wnt therapy, necessitating further research in this area [128].
4.4. Cell–Cell Adhesion
Direct cell–cell contact via adhesion molecules is an indispensable strategy not only for physiological BM homeostasis, but also for the survival of leukaemic cells. For instance, the interaction between lymphocyte function-associated antigen 1 expressed on T-ALL cells and its ligand, intercellular adhesion molecule 1 expressed on BM-MSCs, promotes leukaemic cell survival, highlighting its critical role in the T-ALL niche [129]. Another important adhesion molecule, very late antigen-4 (VLA-4) (α4β1 integrin), is known to be highly expressed on leukaemic blasts from children with relapsed pre-B ALL and is associated with poor prognosis and overall survival [130]. VLA-4 plays an integral role in mediating leukaemia cell adhesion and chemoresistance by interacting with vascular cell adhesion molecule 1 (VCAM-1) expressed on BM MSCs [131]. Importantly, VLA-4/VCAM-1 is also involved in mediating the adhesion of AML to endothelial cells, suggesting a role within the BM vascular niche [31].
Recognition of the VLA-4/VCAM-1 interaction within the leukaemia–BMM niche has led to development of numerous pharmaceuticals targeting this specific pathway. Natalizumab is a clinically available, humanised monoclonal anti-VLA-4 with anti-tumour potential in AML and pre-B ALL [78,79]. Mechanistically, natalizumab promotes mobilisation and chemosensitivity of leukaemic cells by inhibiting VLA-4-mediated stromal adhesion [78,79]. However, its widespread therapeutic use has been limited due to the risk of multifocal leukoencephalopathy [132]. Small molecules, such as TBC3486, have shown therapeutic potential by competing with VCAM-1 for integrin α4 binding, thereby sensitising pre-B ALL cells to chemotherapy [80]. FNIII14 peptide, another potent inhibitor of VLA-4, successfully eradicated minimal residual disease in the BM of mice with AML when combined with cytarabine, achieving a remarkable 100% survival rate [81].
CD44 is recognised as another key cell surface adhesion molecule that promotes the leukaemia–BMM niche interaction and BM engraftment of leukaemic cells [108]. Using a mouse model xenografted with haematopoietic progenitors expressing oncogenic Notch1 to drive T-ALL development, treatment with anti-CD44 monoclonal antibody was shown to antagonise leukaemic cell activity and prolong survival by impairing the interaction between leukaemic cells and stromal niches [108]. In addition, several other studies have supported the therapeutic potential of CD44-targeted antibodies for the treatment of AML [76,133]. RG7356 (or ARH460-16-2), a humanised anti-CD44 monoclonal antibody, has been investigated in a phase 1 dose-escalation study for patients with relapsed/refractory AML and was found to be safe and well tolerated [77].
Another emerging adhesion-mediated therapeutic target is E-selectin, an important contributor to chemoresistance and disease relapse. Uproleselan (GMI-1271) is an endothelial niche modulator and a small molecule mimetic that targets E-selectin [34]. A recent study has shown that inhibition of the E-selectin-mediated pro-survival signalling pathway with uproleselan suppressed AML blast regeneration and enhanced survival of leukaemia-bearing mice harbouring the human KMT2A-MLLT3 oncogene via a synergistic effect with chemotherapy [34]. Encouragingly, an early phase study revealed that administration of uproleselan with chemotherapy led to high remission rates and promising survival outcomes in patients with relapsed/refractory AML [82]. Phase 2/3 clinical trials are currently underway to further evaluate the efficacy of uproselesan in AML (NCT03701308, NCT03616470). GMI-1359, a dual inhibitor of CXCR4/E-selectin, has been shown to efficiently mobilise leukaemic cells into the circulation and strongly extend survival of mice xenografted with FLT3-internal tandem duplication (FLT3-ITD) mutant AML cells either alone or in combination with the FLT3-ITD inhibitor sorafenib [83]. Strikingly, administration of GMI-1359 and sorafenib was also found to increase regeneration of normal BM haematopoietic cell populations [83]. As haematopoiesis is often perturbed during leukaemia development, a dual therapeutic strategy of eliminating leukaemic cells and restoring haematopoiesis constitutes an attractive strategy to improve clinical outcomes.
4.5. Bone Remodelling Signalling Pathways
Osteolytic lesions or pathological fractures are common features of dysregulated bone remodelling in certain cancers such as multiple myeloma and metastatic breast cancer, where the interactions between cancer cells and the BMM impair the homeostatic balance between bone formation and resorption, leading to bone loss and destruction. Despite manifestations of bone diseases in many patients with leukaemia, therapeutic clinical evaluation of bone anabolic agents is lacking. One promising candidate is zoledronic acid, an FDA-approved drug for the treatment of osteoporosis. Mechanistically, zoledronic acid directly inhibits farnesyl diphosphonate synthase in osteoclasts, thereby inhibiting resorption and survival [134]. Administration of zoledronic acid led to an improved pain profile in children with ALL who developed treatment-related osteonecrosis [135]. Furthermore, attenuating osteoclast-induced bone loss with zoledronic acid has been shown to reduce the leukaemia burden in the BM and extend survival in a syngeneic BCR-ABL1+ pre-B ALL mouse model [35]. These findings provide support for clinical investigation of zoledronic acid to restore the BMM in patients with leukaemia.
Studies investigating the use of bone anabolic agents as treatment applications for malignancies other than leukaemia have yielded positive outcomes, prompting further interest in their potential therapeutic efficacy for leukaemia. Interestingly, some of these agents have only been investigated in preclinical and clinical studies for their cytotoxicity against leukaemia. Everolimus, a mammalian target of the rapamycin (mTOR) inhibitor, is an inhibitor of osteoclast resorption and has been shown to prevent bone loss in ovariectomised mice [136]. The clinical efficacy of everolimus administration in combination with multi-agent chemotherapy has been evaluated in adult and paediatric relapsed/refractory ALL. Everolimus was shown to be safe and effective in T-ALL (50% response) when combined with chemotherapy [84]. Likewise, the combination of everolimus with four-drug reinduction therapy yielded favourable second complete remission rates and low levels of minimal residual disease following reinduction [85]. However, these studies did not assess bone parameters that could provide important validation regarding the effects of everolimus on leukaemia-induced bone loss. Another promising agent, cabozantinib, exhibits potent activity against multiple receptor tyrosine kinases that modulate tumour survival and metastasis [137]. Notably, cabozantinib is also capable of remodelling the BMM through inhibition of osteoclast differentiation and resorption, as well as modulating the RANKL/osteoprotegerin ratio in osteoblasts [138]. In a recent preclinical evaluation, cabozantinib was found to selectively induce cytotoxic effects in AML cells with FLT3-ITD in vivo [86]. Importantly, a dose-escalation study demonstrated that cabozantinib was well tolerated in patients with AML, thus establishing a precedent for larger efficacy trials in the future [87].
The ubiquitin–proteasome pathway also presents as an attractive target due to its role in the degradation of regulatory proteins, including cell cycle proteins, tumour suppressor proteins, and transcription factors associated with bone metabolism [139]. Bortezomib is an FDA-approved proteasome inhibitor that can normalise bone remodelling in multiple myeloma by exerting inhibitory effects on osteoclast activity and stimulatory effects on osteoblast differentiation/growth [140]. A preclinical study found that bortezomib exerted an anti-tumour effect on human T-ALL by inhibiting Notch1 target genes [88]. Similarly, bortezomib has been shown to potentiate volasertib-induced mitotic arrest in AML cells by inhibiting slow degradation of cyclin B and prolonged survival of volasertib-treated AML-bearing mice [89]. Several clinical studies have evaluated bortezomib in combination with four-drug induction therapy for relapsed ALL. Although these studies have shown an improvement in response rate, a survival advantage is yet to be elucidated [90]. The addition of bortezomib to standard chemotherapy has also failed to improve survival in children with AML [91]. A number of second-generation proteasome inhibitors have subsequently been developed. These include carfilzomib and ixazomib, both of which are capable of exerting bone anabolic activity via dual effects of promoting osteoblastic bone formation and suppressing osteoclastic resorption [141,142,143]. Carfilzomib has also been shown to induce leukaemia cell apoptosis and achieved modest anti-leukaemic activity in a phase 1 study in patients with AML [92,94]. Several early phase clinical trials examining the anti-leukaemic effect of carfilzomib are currently recruiting (NCT02303821 and NCT02512926) [95]. A recent pre-clinical study by our group demonstrated that cafilzomib treatment did not confer a survival advantage when combined with multi-agent induction chemotherapy of vincristine, dexamethasone, and L-asparaginase, although the impact of treatment on bone was not examined [93]. Likewise, an encouraging response rate was seen in a phase 1 clinical trial that evaluated the efficacy of ixazomib administered in combination with mitoxantrone, etoposide, and cytarabine [96]. However, due to limited in vivo assessment, it remains unclear whether proteasome inhibitors could reverse defective bone remodelling in the leukaemia BMM.
4.6. Hypoxia-Related Signalling Pathways
In the BMM, low oxygen distribution provides a hypoxic niche where leukaemic cells preferentially reside, contributing to chemoresistance [144]. Furthermore, activation of hypoxia-inducible factor 1α (HIF-1α) in leukaemic cells, either via hypoxia-induced stabilisation or in response to oncogenic stimuli, can facilitate angiogenesis and production of pro-survival factors that favour chemoresistance [145,146]. However, the option of therapeutically targeting HIF-1α remains controversial in light of recent findings that showed that the loss of HIF-1α could accelerate leukaemogenesis [147,148]. In this context, an emerging group of therapeutic candidates known as hypoxia activated prodrugs, which are inert in normoxia and active only in hypoxia, are being evaluated for treatment of leukaemia.
PR-104 is a phosphate ester prodrug that can be converted into alcohol PR-104A in vivo and act as a hypoxia-activated prodrug [149]. Mechanistically, PR-104A is selectively reduced to reactive cytotoxic nitrogen mustards (hydroxylamine PR-104H and amine PR-104M) upon encountering a hypoxic environment and induces cell cytotoxicity via DNA cross-linking [149]. PR-104 has been shown to induce hypoxia-selective growth inhibition of pre-B ALL cells in vitro, and the administration of PR-104 in AML and ALL mouse xenografts reduced leukaemic burden and prolonged survival [97]. Another reported mechanism of PR-104 involves hypoxia-independent activation by the enzyme aldo-keto reductase 1C3 (AKR1C3) [150]. Primary T-ALL cells have been found to be more sensitive to the cytotoxicity of PR-104 compared to pre-B ALL cells due to higher expression of AKR1C3 [98]. In a phase 1/2 clinical trial of relapsed/refractory ALL and AML, PR-104 was found to exert measurable clinical activity [99]. However, significant toxicity was also observed in some participants, likely due to the higher dose administered compared to the dose used in solid tumour trials. Further clinical evaluation of PR-104 should consider lower doses and tolerability in combination with chemotherapeutic agents. Another extensively examined hypoxia-activated prodrug is evofosfamide (TH-302), a 2-nitroimidazole prodrug of the cytotoxin bromo-isophosphoramide mustard, which has shown efficacy in a range of different tumour models [151]. The anti-tumour effect of evofosfamide has been demonstrated in primary leukaemia samples in vitro, as well as in vivo, where evofosfamide treatment of AML xenografts reduced disease burden and prolonged survival without overt haematological toxicity [100,101]. A phase 1 study revealed that evofosfamide therapy significantly suppressed hypoxia markers in the leukaemic BM; however, there was limited activity in heavily pre-treated patients with leukaemia, which may hamper further clinical investigation [102].
4.7. The Vasculature
Another treatment approach that has been extensively investigated is the use of anti-angiogenic agents that target neovascularisation within the leukaemic niche. However, clinical outcomes have largely been disappointing due to significant adverse effects and limited response rates in patients with leukaemia. For instance, VEGF receptor inhibitors such as vatalanib and sorafenib have not been reported to confer additional survival benefit for myeloid neoplasms in adult and elderly patients, even when used in conjunction with chemotherapy [152,153]. However, sorafenib’s potential benefit has been noted in FLT3/ITD+ AML patients, which may be related to its direct anti-leukaemic effect [154]. An alternative approach is the use of vascular disrupting agents, which can induce breakdown of the vascular architecture, leading to disruption of the vascular supply to tumour cells [155]. Combretastatin-A1-diphosphate (OXi4503/CA1P), a microtubule-destabilising agent, has been shown to induce vascular disruption, delay tumour growth, and increase survival in HL60 leukaemia-bearing mice [103]. In addition, a recently completed clinical study demonstrated that CA1P administered with cytarabine showed early clinical activity and was well tolerated by patients with relapsed/refractory AML [104]. CA1P has been granted Rare Paediatric Disease designation by the FDA for children with AML, paving the way for further future clinical investigation.
The therapeutic approach of targeting angiogenesis within the leukaemia microenvironment is further complicated by differential remodelling of the vasculature, depending on the niche location [32]. In AML, leukaemic infiltration results in a loss of endosteal vessels. A vessel-poor microenvironment or abnormal vessels incapable of supplying oxygen or nutrients could hinder the efficiency of drug delivery and thus promote chemoresistance. Therefore, a niche-specific strategy is needed to overcome the non-specificity of targeting the BM vasculature. In this context, agents that can rescue endosteal vessels prior to chemotherapy could be highly beneficial for patients. For instance, deferoxamine has been shown to promote HSC homing and endosteal vessel formation in AML-bearing mice, which could potentially confer additional treatment benefits [32]. Thus, clinical studies are needed to investigate the potential of incorporating an endosteal antagonist into conventional chemotherapeutic regimens to improve survival outcomes.
4.8. Emerging Therapies: Immunotherapies and Mitochondrial-Targeted Therapies
In recent years, the immune cell microenvironment of haematologic malignancies has been increasingly recognised as an important target for immunotherapies. In particular, tumour-associated macrophages (TAMs) are capable of being reprogrammed by tumour cells into an immunosuppressive and pro-tumourigenic phenotype, thus contributing to subdued immune responses and promoting disease progression [156]. Immune-based therapies that target macrophages in haematologic malignancies have been extensively reviewed [157]. In particular, CD47 (a surface protein that is expressed by leukaemic cells) has emerged as a key target for immunotherapy due to its role in innate immune evasion by inhibiting macrophage phagocytosis [158], with a recent review reporting current clinical trials involving the use of CD47 antagonists in haematologic malignancies [156]. The feasibility and effectiveness of other emerging immune-based therapies, such as genetically-engineered T-cells expressing a chimeric antigen receptor (CAR T-cell therapy), has been demonstrated in clinical trials, inspiring new and innovative approaches to management of leukaemia [159].
Finally, intercellular mitochondrial transfer through tunneling nanotubes and extracellular vesicles is regarded as a new and exciting crosstalk mechanism between BMM cells and leukaemic cells. Functional exchange of mitochondria between donor BM-MSCs and recipient leukaemic cells contribute to enhanced leukaemic cell metabolism, increased proliferative capacity, drug resistance, and disease relapse [160,161]. Therefore, targeting molecular components involved in mitochondrial transfer, such as CD38, could present a novel avenue for elimination of minimal residual disease in treatment for leukaemia [161]. Other promising mitochondrial-targeted approaches have been extensively reviewed elsewhere [162].
5. Open Questions and Future Perspectives
In recent years, extensive use of preclinical models has provided valuable insights into the pathophysiology of leukaemia. Through research, it is now clear that leukaemia is no longer viewed solely as a spontaneous cancer induced by genetic defects, but rather a disease highly dependent on both intrinsic and extrinsic mechanisms exerted by BM niche cells. Conversely, the BMM can also be remodelled into an oncogenic sanctuary by leukaemic cells to provide significant survival advantage and therapy resistance. Crosstalk between the leukaemic cells and BMM are highly dependent on cell–cell interactions and pathways, including CXCL12/CXCR4, VLA-4/VCAM-1, Notch, and Wnt, amongst others. As these mechanisms are often associated with haematopoietic niche regulation, leukaemia-induced changes in the BMM will compromise healthy haematopoiesis and blood cell turnover, ultimately leading to clinical manifestations of the disease.
Due to the indispensable role of the BMM in leukaemogenesis, targeting the sheltered leukaemia niche presents a feasible strategy to improve treatment outcomes. However, this approach has its own share of challenges, and effective therapeutic targeting has often remained elusive. In many cases, significant toxicities and limited efficacies exerted by the therapeutic agents in clinical trials urgently need to be addressed. For instance, a recent phase 3 clinical trial demonstrated that bortezomib increased toxicity but failed to improve overall survival in a large cohort of children with AML [91]. Similarly, administering PR-104 in patients with relapsed/refractory AML and ALL resulted in significant adverse events, such as myelosuppression, febrile neutropenia, infection, and enterocolitis [99]. Intriguingly, preclinical survival data supported the potential use of bortezomib for treatment of AML [89], and a similar survival advantage of PR-104 was also demonstrated in preclinical models of ALL [97,98]. It is important to note that due to practical limitations on the size of preclinical studies, treatment-related adverse events that are rare in patients may not be easily identified in animals despite similar or even higher dose levels/duration of drug administration [163]. In addition, early phase clinical trials often enrol heavily pre-treated patients with relapsed/refractory disease, as opposed to the young treatment-naive animals used in preclinical settings. Thus, increasing preclinical sample sizes and/or developing new patient-derived xenograft models that recapitulate patients with relapsed/refractory leukaemia could improve predictions of adverse events in humans.
While phase 1 clinical studies are primarily designed to explore safety and tolerability and are not powered for efficacy, outcomes from these studies often provide an indication as to whether certain leukaemia types respond better than others. For instance, it was found that patients with T-ALL appeared to be better responders compared to those with pre-B ALL (50% vs. 39%) when treated with everolimus in combination with HyperCVAD chemotherapy [84]. Of note, the patients with T-ALL in this study were also heavily pre-treated with a median of four prior therapies compared to those with pre-B ALL who had received a median of one prior therapy. While these findings ought to be interpreted with caution as the sample size was small, it was found that everolimus could increase the levels of PTEN, a major negative regulator of PI3K/Akt/mTOR signalling, in patients with T-ALL who achieved a response [84]. In contrast, patients with B-ALL had elevated PTEN levels but did not respond. This interesting finding indicates that the mTOR/PTEN pathway may play a more central role in T-ALL leukaemogenesis, and therefore further investigation into targeting this pathway for T-ALL is warranted. Others also point to certain genetic features and molecular markers as reliable predictors of therapeutic responses. For example, correlative studies have found that patients with increased levels of IFI30 and RORα were associated with improved response to ixazomib [96]. Furthermore, E-selectin ligand expression in LSCs correlated with improved response and survival following uproleselan treatment in relapsed/refractory AML [82]. This suggests that gene expression/transcriptome profiling may be useful for predicting which patients are likely to benefit from or exhibit resistance to treatments. As such, future clinical trials that investigate novel agents targeting the BMM should implement such approaches to help identify biomarkers predictive of response.
Recently, cell mobilising agents have been intensely investigated due to their ability to enhance chemotherapy effectiveness by sensitising the leukaemic cells from their protective niche. In particular, agents that target the CXCL12/CXCR4 and cell–cell contact pathways are at the forefront of clinical trials. Remarkably, agents such as plerixafor can also enhance the haematologic recovery post-myeloablative allogeneic HSCT [164]. Other agents such as GMI-1359 have been shown to improve haematopoiesis during treatment [83]. Indeed, haematopoietic restoration is a vital clinical outcome during treatment and for recovery of marrow function post-treatment, and clinical use of drugs that can simultaneously potentiate anti-leukaemic activity and haematopoiesis is lacking. Therefore, such promising drug candidates should be prioritised for evaluation in clinical trials in combination with conventional therapy.
Another treatment strategy that remains significantly under-investigated is the use of bone anabolic agents as a therapeutic approach to reverse leukaemia-induced osteoporosis in patients. While some agents tested in clinical trials for leukaemia are known to be modulators of osteoblasts and osteoclasts, none of the trials evaluated the bone-normalising potential of these agents. A proof of concept study demonstrated that inhibiting osteoclast resorption in pre-B ALL mice with zoledronic acid could achieve a dual clinical outcome: normalising bone loss and prolonging survival [35]. In this study, zoledronic acid did not directly affect survival of the pre-B ALL cells, and whether it is the osteoclasts that directly contribute to survival remain to be explored [35]. As zoledronic acid’s safety profile in children has been demonstrated, future clinical trials could evaluate its efficacy as an adjuvant therapy in conjunction with conventional chemotherapy in childhood ALL. Other RANKL antagonists, such as the monoclonal antibody denosumab, have also displayed a good safety profile and may present an alternative therapeutic option [165]. Interestingly, a leukaemia-induced reduction in osteoblast numbers contributed to bone loss in pre-B ALL-bearing mice [35]. The mechanisms by which the leukaemic cells impair osteoblasts, either through suppression of osteoblastogenesis and/or induction of osteoblast apoptosis, are hitherto unclear and remain to be verified. It is plausible to postulate that increasing osteoblastic bone formation through therapeutic means may hinder leukaemogenesis and restore healthy haematopoiesis.
Advances in modern treatments have led to significant improvements in the prognosis of patients with leukaemia. Nevertheless, high-risk subgroups, including hypodiploid pre-B ALL, infant KMT2A-rearranged ALL, relapsed T-ALL, and AML, continue to have inferior outcomes. While older children, adolescents, and young adults are better able to tolerate intensified therapy, applying the same approach to infants and older adults is challenging. Over the years, infant-specific collaborative trials have made tremendous advances in optimising treatment protocols; however, despite this progress, infants with KMT2A-rearranged ALL continue to demonstrate inferior outcomes [166]. For older adults with AML, survival outcomes remain poor. Thus, we are approaching a therapeutic plateau and novel agents are desperately required to improve clinical outcome. Hence, the potential synergistic benefits of BMM-targeting agents with existing therapy should be explored more broadly, particularly in high-risk leukaemias that are difficult to treat. In this regard, well-characterised patient-derived xenograft models of high-risk leukaemia should be used for the evaluation of novel therapies due to their ability to mimic human disease and consideration as the “gold standard” models for preclinical testing. Future clinical trials should focus on integrating innovative therapies, identifying optimal dosing regimens and predictive biomarkers to maximise trial success.
6. Summary
The development of novel niche-targeted therapeutic agents currently has a cellular and pathway-specific focus, incorporating crosstalk disruption and/or direct cellular cytotoxicity as the main anti-leukaemic mechanisms. However, the strategy of blocking signalling pathways often carries the risk of counterproductive and unpredictable adverse events. Indeed, these limiting factors often act as roadblocks for clinical development of an investigational agent on its path towards FDA-approval. Therefore, it is important to examine the efficacy of these agents with conventionally used chemotherapeutic agents in clinical trials, deciphering the optimal doses and drug combinations to eradicate mobilised leukaemic cells without increasing toxicity. Finally, novel crosstalk mechanisms are constantly being discovered, such as leukaemic cell-derived exosomes and tunnelling nanotubes, thus opening up new and exciting avenues for therapeutic targeting. Future studies, combining aspects of both basic and clinical translational research, will facilitate better understanding of the pathological mechanisms within the leukaemia microenvironment, ultimately leading to the development of improved therapeutic approaches and outcomes for patients with leukaemia.
Author Contributions
V.K. and A.M.H. wrote the manuscript. R.S.K. and L.C.C. supervised manuscript preparation and revised the manuscript. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by the Children’s Leukaemia and Cancer Research Foundation (CLCRF) Perth, Western Australia, and by grant 1184963 awarded through the 2019 Priority-driven Collaborative Cancer Research Scheme and co-funded by Cancer Australia, Cure Cancer, and Leukaemia Foundation of Australia. RS Kotecha is supported by a Fellowship from the National Health and Medical Research Council of Australia (NHMRC APP1142627).
Acknowledgments
Figures were created with BioRender.com.
Conflicts of Interest
The authors declare that they have no conflict of interest.
References
- Morrison, S.J.; Scadden, D.T. The bone marrow niche for haematopoietic stem cells. Nature 2014, 505, 327–334. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chiarini, F.; Lonetti, A.; Evangelisti, C.; Buontempo, F.; Orsini, E.; Evangelisti, C.; Cappellini, A.; Neri, L.M.; McCubrey, J.; Martelli, A.M. Advances in understanding the acute lymphoblastic leukemia bone marrow microenvironment: From biology to therapeutic targeting. Biochim. Biophys. Acta Bioenerg. 2016, 1863, 449–463. [Google Scholar] [CrossRef] [PubMed]
- Kular, J.; Tickner, J.; Chim, S.M.; Xu, J. An overview of the regulation of bone remodelling at the cellular level. Clin. Biochem. 2012, 45, 863–873. [Google Scholar] [CrossRef] [PubMed]
- Mostoufi-Moab, S.; Halton, J. Bone Morbidity in Childhood Leukemia: Epidemiology, Mechanisms, Diagnosis, and Treatment. Curr. Osteoporos. Rep. 2014, 12, 300–312. [Google Scholar] [CrossRef] [Green Version]
- Wilson, C.L.; Ness, K.K. Bone Mineral Density Deficits and Fractures in Survivors of Childhood Cancer. Curr. Osteoporos. Rep. 2013, 11, 329–337. [Google Scholar] [CrossRef] [Green Version]
- Méndez-Ferrer, S.; Bonnet, D.; Steensma, D.P.; Hasserjian, R.P.; Ghobrial, I.M.; Gribben, J.G.; Andreeff, M.; Krause, D.S. Bone marrow niches in haematological malignancies. Nat. Rev. Cancer 2020, 20, 285–298. [Google Scholar] [CrossRef]
- Pinho, S.; Frenette, P.S. Haematopoietic stem cell activity and interactions with the niche. Nat. Rev. Mol. Cell Biol. 2019, 20, 303–320. [Google Scholar] [CrossRef]
- Wang, A.; Zhong, H. Roles of the bone marrow niche in hematopoiesis, leukemogenesis, and chemotherapy resistance in acute myeloid leukemia. Hematology 2018, 23, 729–739. [Google Scholar] [CrossRef] [Green Version]
- Calvi, L.M.; Adams, G.B.; Weibrecht, K.W.; Weber, J.; Olson, D.P.; Knight, M.C.; Martin, R.P.; Schipani, E.; Divieti, P.; Bringhurst, F.R.; et al. Osteoblastic cells regulate the haematopoietic stem cell niche. Nature 2003, 425, 841–846. [Google Scholar] [CrossRef]
- Arai, F.; Hirao, A.; Ohmura, M.; Sato, H.; Matsuoka, S.; Takubo, K.; Ito, K.; Koh, G.Y.; Suda, T. Tie2/Angiopoietin-1 Signaling Regulates Hematopoietic Stem Cell Quiescence in the Bone Marrow Niche. Cell 2004, 118, 149–161. [Google Scholar] [CrossRef] [Green Version]
- Nilsson, S.K.; Johnston, H.M.; Whitty, G.A.; Williams, B.; Webb, R.J.; Denhardt, D.T.; Bertoncello, I.; Bendall, L.J.; Simmons, P.J.; Haylock, D.N. Osteopontin, a key component of the hematopoietic stem cell niche and regulator of primitive hematopoietic progenitor cells. Blood 2005, 106, 1232–1239. [Google Scholar] [CrossRef]
- Ding, L.; Morrison, S.J. Haematopoietic stem cells and early lymphoid progenitors occupy distinct bone marrow niches. Nature 2013, 495, 231–235. [Google Scholar] [CrossRef]
- Greenbaum, A.; Hsu, Y.-M.S.; Day, R.B.; Schuettpelz, L.G.; Christopher, M.J.; Borgerding, J.N.; Nagasawa, T.; Link, D.C. CXCL12 in early mesenchymal progenitors is required for haematopoietic stem-cell maintenance. Nature 2013, 495, 227–230. [Google Scholar] [CrossRef] [Green Version]
- Ding, L.; Saunders, T.; Enikolopov, G.; Morrison, S.J. Endothelial and perivascular cells maintain haematopoietic stem cells. Nature 2012, 481, 457–462. [Google Scholar] [CrossRef] [Green Version]
- Gomes, A.C.; Hara, T.; Lim, V.Y.; Herndler-Brandstetter, D.; Nevius, E.; Sugiyama, T.; Tani-Ichi, S.; Schlenner, S.; Richie, E.; Rodewald, H.R.; et al. Hematopoietic Stem Cell Niches Produce Lineage-Instructive Signals to Control Multipotent Progenitor Differentiation. Immunity 2016, 45, 1219–1231. [Google Scholar] [CrossRef] [Green Version]
- Park, S.J.; Bejar, R. Clonal Hematopoiesis in Aging. Curr. Stem Cell Rep. 2018, 4, 209–219. [Google Scholar] [CrossRef]
- King, K.Y.; Huang, Y.; Nakada, D.; Goodell, M.A. Environmental influences on clonal hematopoiesis. Exp. Hematol. 2020, 83, 66–73. [Google Scholar] [CrossRef] [Green Version]
- Raaijmakers, M.H.G.P.; Mukherjee, S.; Guo, S.; Zhang, S.; Kobayashi, T.; Schoonmaker, J.A.; Ebert, B.L.; Al-Shahrour, F.; Hasserjian, R.P.; Scadden, E.O.; et al. Bone progenitor dysfunction induces myelodysplasia and secondary leukaemia. Nature 2010, 464, 852–857. [Google Scholar] [CrossRef] [Green Version]
- Kode, A.; Manavalan, J.S.; Mosialou, I.; Bhagat, G.; Rathinam, C.V.; Luo, N.; Khiabanian, H.; Lee, A.; Murty, V.V.; Friedman, R.; et al. Leukaemogenesis induced by an activating beta-catenin mutation in osteoblasts. Nature 2014, 506, 240–244. [Google Scholar] [CrossRef] [Green Version]
- Krause, D.S.; Fulzele, K.; Catic, A.; Sun, C.C.; Dombkowski, D.; Hurley, M.P.; Lezeau, S.; Attar, E.; Wu, J.Y.; Lin, H.Y.; et al. Differential regulation of myeloid leukemias by the bone marrow microenvironment. Nat. Med. 2013, 19, 1513–1517. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Zhang, H.; Rodriguez, S.; Cao, L.; Parish, J.; Mumaw, C.; Zollman, A.; Kamoka, M.M.; Mu, J.; Chen, D.Z.; et al. Notch-dependent repression of miR-155 in the bone marrow niche regulates hematopoiesis in an NF-kappaB-dependent manner. Cell Stem Cell 2014, 15, 51–65. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dong, L.; Yu, W.-M.; Zheng, H.; Loh, M.L.; Bunting, S.T.; Pauly, M.; Huang, G.; Zhou, M.; Broxmeyer, H.E.; Scadden, D.T.; et al. Leukaemogenic effects of Ptpn11 activating mutations in the stem cell microenvironment. Nature 2016, 539, 304–308. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frisch, B.J.; Ashton, J.M.; Xing, L.; Becker, M.W.; Jordan, C.T.; Calvi, L.M. Functional inhibition of osteoblastic cells in an in vivo mouse model of myeloid leukemia. Blood 2012, 119, 540–550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hanoun, M.; Zhang, D.; Mizoguchi, T.; Pinho, S.; Pierce, H.; Kunisaki, Y.; Lacombe, J.; Armstrong, S.A.; Dührsen, U.; Frenette, P.S. Acute Myelogenous Leukemia-Induced Sympathetic Neuropathy Promotes Malignancy in an Altered Hematopoietic Stem Cell Niche. Cell Stem Cell 2014, 15, 365–375. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schepers, K.; Pietras, E.M.; Reynaud, D.; Flach, J.; Binnewies, M.; Garg, T.; Wagers, A.J.; Hsiao, E.C.; Passegué, E. Myeloproliferative Neoplasia Remodels the Endosteal Bone Marrow Niche into a Self-Reinforcing Leukemic Niche. Cell Stem Cell 2013, 13, 285–299. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hawkins, E.D.; Duarte, D.; Akinduro, O.; Khorshed, R.A.; Passaro, D.; Nowicka, M.; Straszkowski, L.; Scott, M.K.; Rothery, S.; Ruivo, N.; et al. T-cell acute leukaemia exhibits dynamic interactions with bone marrow microenvironments. Nature 2016, 538, 518–522. [Google Scholar] [CrossRef] [Green Version]
- Wang, W.; Zimmerman, G.; Huang, X.; Yu, S.; Myers, J.; Wang, Y.; Moreton, S.; Nthale, J.; Awadallah, A.; Beck, R.; et al. Aberrant Notch Signaling in the Bone Marrow Microenvironment of Acute Lymphoid Leukemia Suppresses Osteoblast-Mediated Support of Hematopoietic Niche Function. Cancer Res. 2016, 76, 1641–1652. [Google Scholar] [CrossRef] [Green Version]
- Passaro, D.; di Tullio, A.; Abarrategi, A.; Rouault-Pierre, K.; Foster, K.; Ariza-McNaughton, L.; Montaner, B.; Chakravarty, P.; Bhaw, L.; Diana, G.; et al. Increased Vascular Permeability in the Bone Marrow Microenvironment Contributes to Disease Progression and Drug Response in Acute Myeloid Leukemia. Cancer Cell 2017, 32, 324–341.e6. [Google Scholar] [CrossRef] [Green Version]
- Kampen, K.R.; Ter Elst, A.; de Bont, E.S.J.M. Vascular endothelial growth factor signaling in acute myeloid leukemia. Cell. Mol. Life Sci. 2013, 70, 1307–1317. [Google Scholar] [CrossRef] [Green Version]
- Aguayo, A.; Kantarjian, H.M.; Estey, E.H.; Giles, F.J.; Verstovsek, S.; Manshouri, T.; Gidel, C.; O’Brien, S.; Keating, M.J.; Albitar, M. Plasma vascular endothelial growth factor levels have prognostic significance in patients with acute myeloid leukemia but not in patients with myelodysplastic syndromes. Cancer 2002, 95, 1923–1930. [Google Scholar] [CrossRef]
- Poulos, M.G.; Gars, E.J.; Gutkin, M.C.; Kloss, C.C.; Ginsberg, M.; Scandura, J.; Rafii, S.; Butler, J.M. Activation of the vascular niche supports leukemic progression and resistance to chemotherapy. Exp. Hematol. 2014, 42, 976–986.e3. [Google Scholar] [CrossRef] [Green Version]
- Duarte, D.; Hawkins, E.; Akinduro, O.; Ang, H.; de Filippo, K.; Kong, I.Y.; Haltalli, M.; Ruivo, N.; Straszkowski, L.; Vervoort, S.J.; et al. Inhibition of Endosteal Vascular Niche Remodeling Rescues Hematopoietic Stem Cell Loss in AML. Cell Stem Cell 2018, 22, 64–77.e6. [Google Scholar] [CrossRef] [Green Version]
- Pezeshkian, B.; Donnelly, C.; Tamburo, K.; Geddes, T.; Madlambayan, G.J. Leukemia Mediated Endothelial Cell Activation Modulates Leukemia Cell Susceptibility to Chemotherapy through a Positive Feedback Loop Mechanism. PLoS ONE 2013, 8, e60823. [Google Scholar] [CrossRef] [Green Version]
- Barbier, V.; Erbani, J.; Fiveash, C.; Davies, J.M.; Tay, J.; Tallack, M.R.; Lowe, J.; Magnani, J.L.; Pattabiraman, D.R.; Perkins, A.; et al. Endothelial E-selectin inhibition improves acute myeloid leukaemia therapy by disrupting vascular niche-mediated chemoresistance. Nat. Commun. 2020, 11, 1–15. [Google Scholar] [CrossRef]
- Cheung, L.C.; Tickner, J.; Hughes, A.M.; Skut, P.; Howlett, M.; Foley, B.; Oommen, J.; Wells, J.E.; He, B.; Singh, S.; et al. New therapeutic opportunities from dissecting the pre-B leukemia bone marrow microenvironment. Leukemia 2018, 32, 2326–2338. [Google Scholar] [CrossRef]
- Anderson, D.; Skut, P.; Hughes, A.M.; Ferrari, E.; Tickner, J.; Xu, J.; Mullin, B.H.; Tang, D.; Malinge, S.; Kees, U.R.; et al. The bone marrow microenvironment of pre-B acute lymphoblastic leukemia at single-cell resolution. Sci. Rep. 2020, 10, 1–14. [Google Scholar] [CrossRef]
- Cahu, X.; Calvo, J.; Poglio, S.; Prade, N.; Colsch, B.; Arcangeli, M.-L.; Leblanc, T.; Petit, A.; Baleydier, F.; Baruchel, A.; et al. Bone marrow sites differently imprint dormancy and chemoresistance to T-cell acute lymphoblastic leukemia. Blood Adv. 2017, 1, 1760–1772. [Google Scholar] [CrossRef]
- Shafat, M.S.; Oellerich, T.; Mohr, S.; Robinson, S.; Edwards, D.; Marlein, C.R.; Piddock, R.E.; Fenech, M.; Zaitseva, L.; Abdul-Aziz, A.; et al. Leukemic blasts program bone marrow adipocytes to generate a protumoral microenvironment. Blood 2017, 129, 1320–1332. [Google Scholar] [CrossRef]
- Sheng, X.; Parmentier, J.-H.; Tucci, J.; Pei, H.; Cortez-Toledo, O.; Dieli-Conwright, C.M.; Oberley, M.J.; Neely, M.; Orgel, E.; Louie, S.G.; et al. Adipocytes Sequester and Metabolize the Chemotherapeutic Daunorubicin. Mol. Cancer Res. 2017, 15, 1704–1713. [Google Scholar] [CrossRef] [Green Version]
- Zinngrebe, J.; Debatin, K.-M.; Fischer-Posovszky, P. Adipocytes in hematopoiesis and acute leukemia: Friends, enemies, or innocent bystanders? Leukemia 2020, 34, 2305–2316. [Google Scholar] [CrossRef]
- Samimi, A.; Ghanavat, M.; Shahrabi, S.; Azizidoost, S.; Saki, N. Role of bone marrow adipocytes in leukemia and chemotherapy challenges. Cell. Mol. Life Sci. 2019, 76, 2489–2497. [Google Scholar] [CrossRef] [PubMed]
- Wasnik, S.; Chen, W.; Ahmed, A.S.I.; Zhang, X.-B.; Tang, X.; Baylink, D.J. Chapter 5—HSC Niche: Regulation of Mobilization and Homing. In Biology and Engineering of Stem Cell Niches; Vishwakarma, A., Karp, J.M., Eds.; Academic Press: Boston, MA, USA, 2017; pp. 63–73. [Google Scholar]
- Welschinger, R.; Liedtke, F.; Basnett, J.; Pena, A.D.; Juarez, J.G.; Bradstock, K.F.; Bendall, L.J. Plerixafor (AMD3100) induces prolonged mobilization of acute lymphoblastic leukemia cells and increases the proportion of cycling cells in the blood in mice. Exp. Hematol. 2013, 41, 293–302.e1. [Google Scholar] [CrossRef] [PubMed]
- Sison, E.A.R.; Rau, R.; McIntyre, E.; Li, L.; Small, D.; Brown, P. MLL-rearranged acute lymphoblastic leukaemia stem cell interactions with bone marrow stroma promote survival and therapeutic resistance that can be overcome with CXCR4 antagonism. Br. J. Haematol. 2013, 160, 785–797. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Juárez, J.; Pena, A.D.; Baraz, R.; Hewson, J.; Khoo, M.; Cisterne, A.; Fricker, S.; Fujii, N.; Bradstock, K.F.; Bendall, L.J. CXCR4 antagonists mobilize childhood acute lymphoblastic leukemia cells into the peripheral blood and inhibit engraftment. Leukemia 2007, 21, 1249–1257. [Google Scholar] [CrossRef] [Green Version]
- Cooper, T.M.; Sison, E.A.R.; Baker, S.; Li, L.; Ahmed, A.; Trippett, T.; Gore, L.; Macy, M.E.; Narendran, A.; August, K.; et al. A phase 1 study of the CXCR4 antagonist plerixafor in combination with high-dose cytarabine and etoposide in children with relapsed or refractory acute leukemias or myelodysplastic syndrome: A Pediatric Oncology Experimental Therapeutics Investigators’ Consortium Study (POE 10-03). Pediatr. Blood Cancer 2017, 64, e26414. [Google Scholar] [CrossRef]
- Michelis, F.V.; Hedley, D.W.; Malhotra, S.; Chow, S.; Loach, D.; Gupta, V.; Kim, D.D.; Kuruvilla, J.; Lipton, J.H.; Viswabandya, A.; et al. Mobilization of Leukemic Cells Using Plerixafor as Part of a Myeloablative Preparative Regimen for Patients with Acute Myelogenous Leukemia Undergoing Allografting: Assessment of Safety and Tolerability. Biol. Blood Marrow Transplant. 2019, 25, 1158–1163. [Google Scholar] [CrossRef]
- Borthakur, G.; Zeng, Z.; Cortes, J.E.; Chen, H.; Huang, X.; Konopleva, M.; Ravandi, F.; Kadia, T.; Patel, K.P.; Daver, N.; et al. Phase 1 study of combinatorial sorafenib, G-CSF, and plerixafor treatment in relapsed/refractory, FLT3-ITD-mutated acute myelogenous leukemia patients. Am. J. Hematol. 2020, 95, 1296–1303. [Google Scholar] [CrossRef]
- Zeng, Z.; Shi, Y.X.; Samudio, I.J.; Wang, R.-Y.; Ling, X.; Frolova, O.; Levis, M.; Rubin, J.B.; Negrin, R.R.; Estey, E.H.; et al. Targeting the leukemia microenvironment by CXCR4 inhibition overcomes resistance to kinase inhibitors and chemotherapy in AML. Blood 2009, 113, 6215–6224. [Google Scholar] [CrossRef] [Green Version]
- Pitt, L.A.; Tikhonova, A.N.; Hu, H.; Trimarchi, T.; King, B.; Gong, Y.; Sanchez-Martin, M.; Tsirigos, A.; Littman, D.R.; Ferrando, A.A.; et al. CXCL12-Producing Vascular Endothelial Niches Control Acute T Cell Leukemia Maintenance. Cancer Cell 2015, 27, 755–768. [Google Scholar] [CrossRef] [Green Version]
- Parameswaran, R.; Yu, M.; Lim, M.; Groffen, J.; Heisterkamp, N. Combination of drug therapy in acute lymphoblastic leukemia with a CXCR4 antagonist. Leukemia 2011, 25, 1314–1323. [Google Scholar] [CrossRef]
- Abraham, M.; Klein, S.; Bulvik, B.; Wald, H.; Weiss, I.D.; Olam, D.; Weiss, L.; Beider, K.; Eizenberg, O.; Wald, O.; et al. The CXCR4 inhibitor BL-8040 induces the apoptosis of AML blasts by downregulating ERK, BCL-2, MCL-1 and cyclin-D1 via altered miR-15a/16-1 expression. Leukemia 2017, 31, 2336–2346. [Google Scholar] [CrossRef]
- Borthakur, G.; Tallman, M.S.; Ofran, Y.; Foran, J.M.; Uy, G.L.; D iPersio, J.F.; Nagler, A.; Rowe, J.M.; Showel, M.M.; Altman, J.; et al. The Selective Anti Leukemic Effect of BL-8040, a Peptidic CXCR4 Antagonist, Is Mediated by Induction of Leukemic Blast Mobilization, Differentiation and Apoptosis: Results of Correlative Studies from a Ph2a Trial in Acute Myeloid Leukemia. Blood 2016, 128, 2745. [Google Scholar] [CrossRef]
- Cho, B.-S.; Zeng, Z.; Mu, H.; Wang, Z.; Konoplev, S.; McQueen, T.; Protopopova, M.; Cortes, J.; Marszalek, J.R.; Peng, S.-B.; et al. Antileukemia activity of the novel peptidic CXCR4 antagonist LY2510924 as monotherapy and in combination with chemotherapy. Blood 2015, 126, 222–232. [Google Scholar] [CrossRef]
- Boddu, P.; Borthakur, G.; Koneru, M.; Huang, X.; Naqvi, K.; Wierda, W.; Bose, P.; Jabbour, E.; Estrov, Z.; Burger, J.; et al. Initial Report of a Phase I Study of LY2510924, Idarubicin, and Cytarabine in Relapsed/Refractory Acute Myeloid Leukemia. Front. Oncol. 2018, 8, 369. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Guo, H.; Yang, Y.; Meng, J.; Liu, J.; Wang, C.; Xu, H. A designed peptide targeting CXCR4 displays anti-acute myelocytic leukemia activity in vitro and in vivo. Sci. Rep. 2015, 4, 6610. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Guo, H.; Duan, H.; Yang, Y.; Meng, J.; Liu, J.; Wang, C.; Xu, H. Improving chemotherapeutic efficiency in acute myeloid leukemia treatments by chemically synthesized peptide interfering with CXCR4/CXCL12 axis. Sci. Rep. 2015, 5, 16228. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Zeng, Z.; Shi, Y.; Jacamo, R.; Ludin, C.; Dembowsky, K.; Frenette, P.S.; Konopleva, M.; Andreeff, M. Targeting CXCR4, SDF1 and Beta-Adrenergic Receptors in the AML Microenvironment by Novel Antagonist POL6326, G-CSF and Isoproterenol. Blood 2010, 116, 2179. [Google Scholar] [CrossRef]
- Kuhne, M.R.; Mulvey, T.; Belanger, B.; Chen, S.; Pan, C.; Chong, C.; Cao, F.; Niekro, W.; Kempe, T.; Henning, K.A.; et al. BMS-936564/MDX-1338: A Fully Human Anti-CXCR4 Antibody Induces Apoptosis In Vitro and Shows Antitumor Activity In Vivo in Hematologic Malignancies. Clin. Cancer Res. 2013, 19, 357–366. [Google Scholar] [CrossRef] [Green Version]
- Chien, S.; Beyerle, L.E.; Wood, B.L.; Estey, E.H.; Appelbaum, F.R.; Cardarelli, P.P.M.; Sabbatini, P.; Shelat, M.-P.S.; Cohen, L.; Becker, P.S. Mobilization of Blasts and Leukemia Stem Cells by Anti-CXCR4 Antibody BMS-936564 (MDX 1338) in Patients with Relapsed/Refractory Acute Myeloid Leukemia. Blood 2013, 122, 3882. [Google Scholar] [CrossRef]
- Becker, P.S.; Foran, J.M.; Altman, J.K.; Yacoub, M.A.; Castro, J.E.; Sabbatini, P.; Dilea, C.; Wade, M.; Xing, G.; Gutierrez, A.; et al. Targeting the CXCR4 Pathway: Safety, Tolerability and Clinical Activity of Ulocuplumab (BMS-936564), an Anti-CXCR4 Antibody, in Relapsed/Refractory Acute Myeloid Leukemia. Blood 2014, 124, 386. [Google Scholar] [CrossRef]
- Peng, S.-B.; Zhang, X.; Paul, D.; Kays, L.M.; Ye, M.; Vaillancourt, P.; Dowless, M.; Stancato, L.F.; Stewart, J.; Uhlik, M.T.; et al. Inhibition of CXCR4 by LY2624587, a Fully Humanized Anti-CXCR4 Antibody Induces Apoptosis of Hematologic Malignancies. PLoS ONE 2016, 11, e0150585. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kovacsovics, T.J.; Mims, A.; Salama, M.E.; Pantin, J.; Rao, N.; Kosak, K.M.; Ahorukomeye, P.; Glenn, M.J.; Deininger, M.W.N.; Boucher, K.M.; et al. Combination of the low anticoagulant heparin CX-01 with chemotherapy for the treatment of acute myeloid leukemia. Blood Adv. 2018, 2, 381–389. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wei, P.; Walls, M.; Qiu, M.; Ding, R.; Denlinger, R.H.; Wong, A.; Tsaparikos, K.; Jani, J.P.; Hosea, N.; Sands, M.; et al. Evaluation of selective gamma-secretase inhibitor PF-03084014 for its antitumor efficacy and gastrointestinal safety to guide optimal clinical trial design. Mol. Cancer. Ther. 2010, 9, 1618–1628. [Google Scholar] [CrossRef] [Green Version]
- Samon, J.B.; Castillo-Martin, M.; Hadler, M.; Ambesi-Impiobato, A.; Paietta, E.; Racevskis, J.; Wiernik, P.H.; Rowe, J.M.; Jakubczak, J.; Randolph, S.; et al. Preclinical analysis of the gamma-secretase inhibitor PF-03084014 in combination with glucocorticoids in T-cell acute lymphoblastic leukemia. Mol. Cancer. Ther. 2012, 11, 1565–1575. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Papayannidis, C.; De Angelo, D.J.; Stock, W.; Huang, B.; Shaik, M.N.; Cesari, R.; Zheng, X.; Reynolds, J.M.; English, P.A.; Ozeck, M.; et al. A Phase 1 study of the novel gamma-secretase inhibitor PF-03084014 in patients with T-cell acute lymphoblastic leukemia and T-cell lymphoblastic lymphoma. Blood Cancer J. 2015, 5, e350. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gavai, A.V.; Quesnelle, C.A.; Norris, D.J.; Han, W.-C.; Gill, P.S.; Shan, W.; Balog, A.; Chen, K.; Tebben, A.J.; Rampulla, R.; et al. Discovery of Clinical Candidate BMS-906024: A Potent Pan-Notch Inhibitor for the Treatment of Leukemia and Solid Tumors. ACS Med. Chem. Lett. 2015, 6, 523–527. [Google Scholar] [CrossRef] [PubMed]
- Zweidler-McKay, M.P.A.; de Angelo, M.D.J.; Douer, D.; Dombret, H.; Ottmann, O.G.; Vey, N.; Thomas, D.A.; Zhu, M.L.; Huang, F.; Bajaj, G.; et al. The Safety and Activity of BMS-906024, a Gamma Secretase Inhibitor (GSI) with Anti-Notch Activity, in Patients with Relapsed T-Cell Acute Lymphoblastic Leukemia (T-ALL): Initial Results of a Phase 1 Trial. Blood 2014, 124, 968. [Google Scholar] [CrossRef]
- Habets, R.A.; de Bock, C.E.; Serneels, L.; Lodewijckx, I.; Verbeke, D.; Nittner, D.; Narlawar, R.; Demeyer, S.; Dooley, J.; Liston, A.; et al. Safe targeting of T cell acute lymphoblastic leukemia by pathology-specific NOTCH inhibition. Sci. Transl. Med. 2019, 11, eaau6246. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.; Mallampati, S.; Sun, B.; Zhang, J.; Kim, S.-B.; Lee, J.-S.; Gong, Y.; Cai, Z.; Sun, X. Wnt pathway contributes to the protection by bone marrow stromal cells of acute lymphoblastic leukemia cells and is a potential therapeutic target. Cancer Lett. 2013, 333, 9–17. [Google Scholar] [CrossRef] [Green Version]
- Zhou, H.; Mak, P.Y.; Mu, H.; Mak, D.H.; Zeng, Z.; Cortes, J.; Liu, Q.; Andreeff, M.; Carter, B.Z. Combined inhibition of beta-catenin and Bcr-Abl synergistically targets tyrosine kinase inhibitor-resistant blast crisis chronic myeloid leukemia blasts and progenitors in vitro and in vivo. Leukemia 2017, 31, 2065–2074. [Google Scholar] [CrossRef]
- Jiang, X.; Mak, P.Y.; Mu, H.; Tao, W.; Mak, D.H.; Kornblau, S.; Zhang, Q.; Ruvolo, P.; Burks, J.K.; Zhang, W.; et al. Disruption of Wnt/beta-Catenin Exerts Antileukemia Activity and Synergizes with FLT3 Inhibition in FLT3-Mutant Acute Myeloid Leukemia. Clin. Cancer Res. 2018, 24, 2417–2429. [Google Scholar] [CrossRef] [Green Version]
- Fiskus, W.; Sharma, S.; Saha, S.; Shah, B.; Devaraj, S.G.; Sun, B.; Horrigan, S.; Leveque, C.; Zu, Y.; Iyer, S.; et al. Pre-clinical efficacy of combined therapy with novel beta-catenin antagonist BC2059 and histone deacetylase inhibitor against AML cells. Leukemia 2015, 29, 1267–1278. [Google Scholar] [CrossRef]
- Lee, J.-H.; Faderl, S.; Pagel, J.M.; Jung, C.W.; Yoon, S.-S.; Pardanani, A.D.; Becker, P.S.; Lee, H.; Choi, J.; Lee, K.; et al. Phase 1 study of CWP232291 in patients with relapsed or refractory acute myeloid leukemia and myelodysplastic syndrome. Blood Adv. 2020, 4, 2032–2043. [Google Scholar] [CrossRef]
- Ma, S.; Yang, L.-L.; Niu, T.; Cheng, C.; Zhong, L.; Zheng, M.-W.; Xiong, Y.; Li, L.-L.; Xiang, R.; Chen, L.-J.; et al. SKLB-677, an FLT3 and Wnt/β-catenin signaling inhibitor, displays potent activity in models of FLT3-driven AML. Sci. Rep. 2015, 5, 15646. [Google Scholar] [CrossRef]
- Da Cruz, L.; McConkey, F.; Feng, N.; Pereira, D.; Hahn, S.; Rubinstein, D.; Young, D. Anti-CD44 antibody, ARH460-16-2, binds to human AML CD34+CD38− cancer stem cells and demonstrates anti-tumor activity in an AML xenograft model. Cancer Res. 2008, 68 (Suppl. 9), 3976. [Google Scholar]
- Vey, N.; Delaunay, J.; Martinelli, G.; Fiedler, W.; Raffoux, E.; Prebet, T.; Gomez-Roca, C.; Papayannidis, C.; Kebenko, M.; Paschka, P.; et al. Phase I clinical study of RG7356, an anti-CD44 humanized antibody, in patients with acute myeloid leukemia. Oncotarget 2016, 7, 32532–32542. [Google Scholar] [CrossRef] [Green Version]
- Hsieh, Y.-T.; Jiang, E.; Pham, J.; Kim, H.-N.; Abdel-Azim, H.; Khazal, S.; Bug, G.; Spohn, P.G.; Bonig, H.; Kim, Y.-M. VLA4 Blockade in Acute Myeloid Leukemia. Blood 2013, 122, 3944. [Google Scholar] [CrossRef]
- Hsieh, Y.-T.; Gang, E.J.; Geng, H.; Park, E.; Huantes, S.; Chudziak, R.; Dauber, K.; Schaefer, P.; Scharman, C.; Shimada, H.; et al. Integrin alpha4 blockade sensitizes drug resistant pre-B acute lymphoblastic leukemia to chemotherapy. Blood 2013, 121, 1814–1818. [Google Scholar] [CrossRef] [Green Version]
- Hsieh, Y.T.; Gang, E.J.; Shishido, S.N.; Kim, H.N.; Pham, J.; Khazal, S.; Osborne, A.; Esguerra, Z.A.; Kwok, E.; Jang, J.; et al. Effects of the small-molecule inhibitor of integrin α4, TBC3486, on pre-B-ALL cells. Leukemia 2014, 28, 2101–2104. [Google Scholar] [CrossRef] [Green Version]
- Matsunaga, T.; Fukai, F.; Miura, S.; Nakane, Y.; Owaki, T.; Kodama, H.; Tanaka, M.; Nagaya, T.; Takimoto, R.; Takayama, T.; et al. Combination therapy of an anticancer drug with the FNIII14 peptide of fibronectin effectively overcomes cell adhesion-mediated drug resistance of acute myelogenous leukemia. Leukemia 2007, 22, 353–360. [Google Scholar] [CrossRef] [Green Version]
- DeAngelo, D.J.; Jonas, B.A.; Liesveld, J.L.; Bixby, D.L.; Advani, A.S.; Marlton, P.; O’Dwyer, M.E.; Fogler, W.E.; Wolfgang, C.D.; Magnani, J.L.; et al. Uproleselan (GMI-1271), an E-Selectin Antagonist, Improves the Efficacy and Safety of Chemotherapy in Relapsed/Refractory (R/R) and Newly Diagnosed Older Patients with Acute Myeloid Leukemia: Final, Correlative, and Subgroup Analyses. Blood 2018, 132 (Suppl. 1), 331. [Google Scholar] [CrossRef]
- Zhang, W.; Ly, C.; Zhang, Q.; Mu, H.; Battula, V.L.; Patel, N.; Schober, W.; Han, X.; Fogler, W.E.; Magnani, J.L.; et al. Dual E-Selectin/CXCR4 Antagonist GMI-1359 Exerts Efficient Anti-Leukemia Effects in a FLT3 ITD Mutated Acute Myeloid Leukemia Patient-Derived Xenograft Murine Model. Blood 2016, 128, 3519. [Google Scholar] [CrossRef]
- Daver, N.; Boumber, Y.; Kantarjian, H.; Ravandi, F.; Cortes, J.; Rytting, M.E.; Kawedia, J.D.; Basnett, J.; Culotta, K.S.; Zeng, Z.; et al. A Phase I/II Study of the mTOR Inhibitor Everolimus in Combination with HyperCVAD Chemotherapy in Patients with Relapsed/Refractory Acute Lymphoblastic Leukemia. Clin. Cancer Res. 2015, 21, 2704–2714. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Place, A.E.; Pikman, Y.; Stevenson, K.E.; Harris, M.H.; Pauly, M.; Sulis, M.-L.; Hijiya, N.; Gore, L.; Cooper, T.M.; Loh, M.L.; et al. Phase I trial of the mTOR inhibitor everolimus in combination with multi-agent chemotherapy in relapsed childhood acute lymphoblastic leukemia. Pediatr. Blood Cancer 2018, 65, e27062. [Google Scholar] [CrossRef]
- Lu, J.-W.; Wang, A.-N.; Liao, H.-A.; Chen, C.-Y.; Hou, H.-A.; Hu, C.-Y.; Tien, H.-F.; Ou, D.-L.; Lin, L.-I. Cabozantinib is selectively cytotoxic in acute myeloid leukemia cells with FLT3-internal tandem duplication (FLT3-ITD). Cancer Lett. 2016, 376, 218–225. [Google Scholar] [CrossRef]
- Fathi, A.T.; Blonquist, T.M.; Hernandez, D.; Amrein, P.C.; Ballen, K.K.; McMasters, M.; Avigan, D.E.; Joyce, R.; Logan, E.K.; Hobbs, G.; et al. Cabozantinib is well tolerated in acute myeloid leukemia and effectively inhibits the resistance-conferring FLT3/tyrosine kinase domain/F691 mutation. Cancer 2018, 124, 306–314. [Google Scholar] [CrossRef] [Green Version]
- Huang, C.; Hu, X.; Wang, L.; Lü, S.; Cheng, H.; Song, X.; Wang, J. Bortezomib suppresses the growth of leukemia cells with Notch1 overexpression in vivo and in vitro. Cancer Chemother. Pharmacol. 2012, 70, 801–809. [Google Scholar] [CrossRef]
- Schnerch, D.; Schüler, J.; Follo, M.; Felthaus, J.; Wider, D.; Klingner, K.; Greil, C.; Duyster, J.; Engelhardt, M.; Wäsch, R. Proteasome inhibition enhances the efficacy of volasertib-induced mitotic arrest in AML in vitro and prolongs survival in vivo. Oncotarget 2017, 8, 21153–21166. [Google Scholar] [CrossRef] [Green Version]
- Messinger, Y.H.; Bostrom, B.C. Bortezomib-based four-drug induction does induce a response in advanced relapsed ALL but cure remains elusive. Pediatr. Blood Cancer 2019, 67, e28115. [Google Scholar] [CrossRef]
- Aplenc, R.; Meshinchi, S.; Sung, L.; Alonzo, T.; Choi, J.; Fisher, B.; Gerbing, R.; Hirsch, B.; Horton, T.; Kahwash, S.; et al. Bortezomib with standard chemotherapy for children with acute myeloid leukemia does not improve treatment outcomes: A report from the Children’s Oncology Group. Haematologica 2020, 105, 1879–1886. [Google Scholar] [CrossRef]
- Liu, C.Y.; Hsieh, F.S.; Chu, P.Y.; Tsai, W.C.; Huang, C.T.; Yu, Y.B.; Huang, T.T.; Ko, P.S.; Hung, M.H.; Wang, W.L.; et al. Carfilzomib induces leukaemia cell apoptosis via inhibiting ELK1/KIAA1524 (Elk-1/CIP2A) and activating PP2A not related to proteasome inhibition. Br. J. Haematol. 2017, 177, 726–740. [Google Scholar] [CrossRef] [Green Version]
- Cheung, L.C.; de Kraa, R.; Oommen, J.; Chua, G.-A.; Singh, S.; Hughes, A.M.; Ferrari, E.; Ford, J.; Chiu, S.K.; Stam, R.W.; et al. Preclinical Evaluation of Carfilzomib for Infant KMT2A-Rearranged Acute Lymphoblastic Leukemia. Front. Oncol. 2021, 11. [Google Scholar] [CrossRef]
- Wartman, L.D.; Fiala, M.A.; Fletcher, T.; Hawkins, E.R.; Cashen, A.; Di Persio, J.F.; Jacoby, M.A.; Stockerl-Goldstein, K.E.; Pusic, I.; Uy, G.L.; et al. A phase I study of carfilzomib for relapsed or refractory acute myeloid and acute lymphoblastic leukemia. Leuk. Lymphoma 2015, 57, 728–730. [Google Scholar] [CrossRef] [Green Version]
- Burke, M.J.; Ziegler, D.S.; Sirvent, F.J.B.; Attarbaschi, A.; Gore, L.; Locatelli, F.; O’Brien, M.M.; Pauly, M.; Obreja, M.; Morris, C.L.; et al. Phase 1b Study of Carfilzomib in Combination with Induction Chemotherapy in Children with Relapsed or Refractory Acute Lymphoblastic Leukemia (ALL). Blood 2019, 134 (Suppl. 1), 3873. [Google Scholar] [CrossRef]
- Advani, A.S.; Cooper, B.; Visconte, V.; Elson, P.; Chan, R.; Carew, J.; Wei, W.; Mukherjee, S.; Gerds, A.; Carraway, H.; et al. A Phase I/II Trial of MEC (Mitoxantrone, Etoposide, Cytarabine) in Combination with Ixazomib for Relapsed Refractory Acute Myeloid Leukemia. Clin. Cancer Res. 2019, 25, 4231–4237. [Google Scholar] [CrossRef] [Green Version]
- Benito, J.; Shi, Y.; Szymanska, B.; Carol, H.; Boehm, I.; Lu, H.; Konoplev, S.; Fang, W.; Zweidler-McKay, P.A.; Campana, D.; et al. Pronounced Hypoxia in Models of Murine and Human Leukemia: High Efficacy of Hypoxia-Activated Prodrug PR-104. PLoS ONE 2011, 6, e23108. [Google Scholar] [CrossRef]
- Moradi Manesh, D.; El-Hoss, J.; Evans, K.; Richmond, J.; Toscan, C.E.; Bracken, L.S.; Hedrick, A.; Sutton, R.; Marshall, G.M.; Wilson, W.R.; et al. AKR1C3 is a biomarker of sensitivity to PR-104 in preclinical models of T-cell acute lymphoblastic leukemia. Blood 2015, 126, 1193–1202. [Google Scholar] [CrossRef] [Green Version]
- Konopleva, M.; Thall, P.F.; Yi, C.A.; Borthakur, G.; Coveler, A.; Bueso-Ramos, C.; Benito, J.; Konoplev, S.; Gu, Y.; Ravandi, F.; et al. Phase I/II study of the hypoxia-activated prodrug PR104 in refractory/relapsed acute myeloid leukemia and acute lymphoblastic leukemia. Haematologica 2015, 100, 927–934. [Google Scholar] [CrossRef] [Green Version]
- Portwood, S.; Lal, D.; Hsu, Y.-C.; Vargas, R.; Johnson, M.K.; Wetzler, M.; Hart, C.; Wang, E.S. Activity of the Hypoxia-Activated Prodrug, TH-302, in Preclinical Human Acute Myeloid Leukemia Models. Clin. Cancer Res. 2013, 19, 6506–6519. [Google Scholar] [CrossRef] [Green Version]
- Benito, J.; Ramirez, M.S.; Millward, N.Z.; Velez, J.; Harutyunyan, K.G.; Lu, H.; Shi, Y.-X.; Matre, P.; Jacamo, R.; Ma, H.; et al. Hypoxia-Activated Prodrug TH-302 Targets Hypoxic Bone Marrow Niches in Preclinical Leukemia Models. Clin. Cancer Res. 2016, 22, 1687–1698. [Google Scholar] [CrossRef] [Green Version]
- Badar, T.; Handisides, D.R.; Benito, J.M.; Richie, M.A.; Borthakur, G.; Jabbour, E.; Harutyunyan, K.; Konoplev, S.; Faderl, S.; Kroll, S.; et al. Phase I study of evofosfamide, an investigational hypoxia-activated prodrug, in patients with advanced leukemia. Am. J. Hematol. 2016, 91, 800–805. [Google Scholar] [CrossRef] [Green Version]
- Benezra, M.; Phillips, E.L.; Tilki, D.; Ding, B.-S.; Butler, J.M.; Dobrenkov, K.; Siim, B.G.; Chaplin, D.D.; Rafii, S.; Rabbany, S.Y.; et al. Serial monitoring of human systemic and xenograft models of leukemia using a novel vascular disrupting agent. Leukemia 2012, 26, 1771–1778. [Google Scholar] [CrossRef] [Green Version]
- Uckun, F.M.; Cogle, C.R.; Lin, T.L.; Qazi, S.; Trieu, V.N.; Schiller, G.; Watts, J.M. A Phase 1B Clinical Study of Combretastatin A1 Diphosphate (OXi4503) and Cytarabine (ARA-C) in Combination (OXA) for Patients with Relapsed or Refractory Acute Myeloid Leukemia. Cancers 2019, 12, 74. [Google Scholar] [CrossRef] [Green Version]
- Sison, E.A.R.; McIntyre, E.; Magoon, D.; Brown, P. Dynamic Chemotherapy-Induced Upregulation of CXCR4 Expression: A Mechanism of Therapeutic Resistance in Pediatric AML. Mol. Cancer Res. 2013, 11, 1004–1016. [Google Scholar] [CrossRef] [Green Version]
- Spiegel, A.; Kollet, O.; Peled, A.; Abel, L.; Nagler, A.; Bielorai, B.; Rechavi, G.; Vormoor, J.; Lapidot, T. Unique SDF-1–induced activation of human precursor-B ALL cells as a result of altered CXCR4 expression and signaling. Blood 2004, 103, 2900–2907. [Google Scholar] [CrossRef]
- Armstrong, F.; de la Grange, P.B.; Gerby, B.; Rouyez, M.-C.; Calvo, J.; Fontenay, M.; Boissel, N.; Dombret, H.; Baruchel, A.; Landman-Parker, J.; et al. NOTCH is a key regulator of human T-cell acute leukemia initiating cell activity. Blood 2009, 113, 1730–1740. [Google Scholar] [CrossRef] [PubMed]
- García-Peydró, M.; Fuentes, P.; Mosquera, M.; García-León, M.J.; Alcain, J.; Rodríguez, A.; de Miguel, P.G.; Menéndez, P.; Weijer, K.; Spits, H.; et al. The NOTCH1/CD44 axis drives pathogenesis in a T cell acute lymphoblastic leukemia model. J. Clin. Investig. 2018, 128, 2802–2818. [Google Scholar] [CrossRef] [PubMed]
- Nwabo Kamdje, A.H.; Mosna, F.; Bifari, F.; Lisi, V.; Bassi, G.; Malpeli, G.; Ricciardi, M.; Perbellini, O.; Scupoli, M.T.; Pizzolo, G.; et al. Notch-3 and Notch-4 signaling rescue from apoptosis human B-ALL cells in contact with human bone marrow-derived mesenchymal stromal cells. Blood 2011, 118, 380–389. [Google Scholar] [CrossRef]
- Kamga, P.T.; dal Collo, G.; Midolo, M.; Adamo, A.; Delfino, P.; Mercuri, A.; Cesaro, S.; Mimiola, E.; Bonifacio, M.; Andreini, A.; et al. Inhibition of Notch Signaling Enhances Chemosensitivity in B-cell Precursor Acute Lymphoblastic Leukemia. Cancer Res. 2019, 79, 639–649. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kamga, P.T.; Bassi, G.; Cassaro, A.; Midolo, M.; di Trapani, M.; Gatti, A.; Carusone, R.; Resci, F.; Perbellini, O.; Gottardi, M.; et al. Notch signalling drives bone marrow stromal cell-mediated chemoresistance in acute myeloid leukemia. Oncotarget 2016, 7, 21713–21727. [Google Scholar] [CrossRef] [Green Version]
- Indraccolo, S.; Minuzzo, S.; Masiero, M.; Pusceddu, I.; Persano, L.; Moserle, L.; Reboldi, A.; Favaro, E.; Mecarozzi, M.; di Mario, G.; et al. Cross-talk between Tumor and Endothelial Cells Involving the Notch3-Dll4 Interaction Marks Escape from Tumor Dormancy. Cancer Res. 2009, 69, 1314–1323. [Google Scholar] [CrossRef] [Green Version]
- Minuzzo, S.; Agnusdei, V.; Pusceddu, I.; Pinazza, M.; Moserle, L.; Masiero, M.; Rossi, E.; Crescenzi, M.; Hoey, T.; Ponzoni, M.; et al. DLL4 regulates NOTCH signaling and growth of T acute lymphoblastic leukemia cells in NOD/SCID mice. Carcinogenesis 2015, 36, 115–121. [Google Scholar] [CrossRef] [Green Version]
- Smith, D.C.; Eisenberg, P.D.; Manikhas, G.; Chugh, R.; Gubens, M.A.; Stagg, R.J.; Kapoun, A.M.; Xu, L.; Dupont, J.; Sikic, B. A Phase I Dose Escalation and Expansion Study of the Anticancer Stem Cell Agent Demcizumab (Anti-DLL4) in Patients with Previously Treated Solid Tumors. Clin. Cancer Res. 2014, 20, 6295–6303. [Google Scholar] [CrossRef] [Green Version]
- Chiorean, E.G.; Lorusso, P.M.; Strother, R.M.; Diamond, J.R.; Younger, A.; Messersmith, W.A.; Adriaens, L.; Liu, L.; Kao, R.J.; DiCioccio, A.T.; et al. A Phase I First-in-Human Study of Enoticumab (REGN421), a Fully Human Delta-like Ligand 4 (Dll4) Monoclonal Antibody in Patients with Advanced Solid Tumors. Clin. Cancer Res. 2015, 21, 2695–2703. [Google Scholar] [CrossRef] [Green Version]
- Tejada, F.N.H.; Silva, J.R.G.; Zweidler-McKay, P.A. The challenge of targeting notch in hematologic malignancies. Front. Pediatr. 2014, 2, 54. [Google Scholar]
- DeAngelo, D.J.; Stone, R.M.; Silverman, L.B.; Stock, W.; Attar, E.C.; Fearen, I.; Dallob, A.; Matthews, C.; Stone, J.; Freedman, S.J.; et al. A phase I clinical trial of the notch inhibitor MK-0752 in patients with T-cell acute lymphoblastic leukemia/lymphoma (T-ALL) and other leukemias. J. Clin. Oncol. 2006, 24, 6585. [Google Scholar] [CrossRef]
- Cullion, K.; Draheim, K.M.; Hermance, N.; Tammam, J.; Sharma, V.M.; Ware, C.; Nikov, G.; Krishnamoorthy, V.; Majumder, P.K.; Kelliher, M.A. Targeting the Notch1 and mTOR pathways in a mouse T-ALL model. Blood 2009, 113, 6172–6181. [Google Scholar] [CrossRef] [Green Version]
- Real, P.J.; Tosello, V.; Palomero, T.; Castillo, M.; Hernando, E.; de Stanchina, E.; Sulis, M.L.; Barnes, K.; Sawai, C.; Homminga, I.; et al. Gamma-secretase inhibitors reverse glucocorticoid resistance in T cell acute lymphoblastic leukemia. Nat. Med. 2009, 15, 50–58. [Google Scholar] [CrossRef] [Green Version]
- López-Guerra, M.; Xargay-Torrent, S.; Rosich, L.; Montraveta, A.; Roldán, J.; Matas-Céspedes, A.; Villamor, N.; Aymerich, M.; López-Otín, C.; Pérez-Galán, P.; et al. The gamma-secretase inhibitor PF-03084014 combined with fludarabine antagonizes migration, invasion and angiogenesis in NOTCH1-mutated CLL cells. Leukemia 2015, 29, 96–106. [Google Scholar] [CrossRef]
- Hogan, L.E.; Meyer, J.A.; Yang, J.; Wang, J.; Wong, N.; Yang, W.; Condos, G.; Hunger, S.P.; Raetz, E.; Saffery, R.; et al. Integrated genomic analysis of relapsed childhood acute lymphoblastic leukemia reveals therapeutic strategies. Blood 2011, 118, 5218–5226. [Google Scholar] [CrossRef] [Green Version]
- Gruszka, A.M.; Valli, D.; Alcalay, M. Wnt Signalling in Acute Myeloid Leukaemia. Cells 2019, 8, 1403. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kode, A.; Mosialou, I.; Manavalan, S.J.; Rathinam, C.V.; Friedman, R.A.; Teruya-Feldstein, J.; Bhagat, G.; Berman, E.; Kousteni, S. FoxO1-dependent induction of acute myeloid leukemia by osteoblasts in mice. Leukemia 2016, 30, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chien, S.; Haq, S.U.; Pawlus, M.; Moon, R.T.; Estey, E.H.; Appelbaum, F.R.; Othus, M.; Magnani, J.L.; Becker, P.S. Adhesion of Acute Myeloid Leukemia Blasts To E-Selectin In The Vascular Niche Enhances Their Survival By Mechanisms Such As Wnt Activation. Blood 2013, 122, 61. [Google Scholar] [CrossRef]
- Yang, S.; Gu, Y.; Wang, G.; Hu, Q.; Chen, S.; Wang, Y.; Zhao, M. HMGA2 regulates acute myeloid leukemia progression and sensitivity to daunorubicin via Wnt/beta-catenin signaling. Int. J. Mol. Med. 2019, 44, 427–436. [Google Scholar] [PubMed] [Green Version]
- Pak, S.; Park, S.; Kim, Y.; Park, J.H.; Park, C.H.; Lee, K.J.; Kim, C.S.; Ahn, H. The small molecule WNT/beta-catenin inhibitor CWP232291 blocks the growth of castration-resistant prostate cancer by activating the endoplasmic reticulum stress pathway. J. Exp. Clin. Cancer Res. 2019, 38, 342. [Google Scholar]
- Cha, J.Y.; Jung, J.-E.; Lee, K.-H.; Briaud, I.; Tenzin, F.; Jung, H.K.; Pyon, Y.; Lee, N.; Chung, J.U.; Lee, J.H.; et al. Anti-Tumor Activity of Novel Small Molecule Wnt Signaling Inhibitor, CWP232291, In Multiple Myeloma. Blood 2010, 116, 3038. [Google Scholar] [CrossRef]
- Griffiths, E.A.; Golding, M.C.; Srivastava, P.; Povinelli, B.J.; James, S.R.; Ford, L.A.; Wetzler, M.; Wang, E.S.; Nemeth, M.J. Pharmacological targeting of beta-catenin in normal karyotype acute myeloid leukemia blasts. Haematologica 2015, 100, e49–e52. [Google Scholar] [CrossRef] [Green Version]
- Winter, S.S.; Sweatman, J.J.; Lawrence, M.B.; Rhoades, T.H.; Hart, A.L.; Larson, R.S. Enhanced T-lineage acute lymphoblastic leukaemia cell survival on bone marrow stroma requires involvement of LFA-1 and ICAM-1. Br. J. Haematol. 2001, 115, 862–871. [Google Scholar] [CrossRef]
- Shalapour, S.; Hof, J.; Kirschner-Schwabe, R.; Bastian, L.; Eckert, C.; Prada, J.; Henze, G.; von Stackelberg, A.; Seeger, K. High VLA-4 expression is associated with adverse outcome and distinct gene expression changes in childhood B-cell precursor acute lymphoblastic leukemia at first relapse. Haematologica 2011, 96, 1627–1635. [Google Scholar] [CrossRef]
- Jacamo, R.; Chen, Y.; Wang, Z.; Ma, W.; Zhang, M.; Spaeth, E.L.; Wang, Y.; Battula, V.L.; Mak, P.Y.; Schallmoser, K.; et al. Reciprocal leukemia-stroma VCAM-1/VLA-4-dependent activation of NF-kappaB mediates chemoresistance. Blood 2014, 123, 2691–2702. [Google Scholar] [CrossRef]
- Ho, P.-R.; Koendgen, H.; Campbell, N.; Haddock, B.; Richman, S.; Chang, I. Risk of natalizumab-associated progressive multifocal leukoencephalopathy in patients with multiple sclerosis: A retrospective analysis of data from four clinical studies. Lancet Neurol. 2017, 16, 925–933. [Google Scholar] [CrossRef]
- Gadhoum, S.; Madhoun, N.; AbuElela, A.; Merzaban, J.S. Anti-CD44 antibodies inhibit both mTORC1 and mTORC2: A new rationale supporting CD44-induced AML differentiation therapy. Leukemia 2016, 30, 2397–2401. [Google Scholar] [CrossRef] [Green Version]
- Russell, R.G.G.; Watts, N.B.; Ebetino, F.H.; Rogers, M. Mechanisms of action of bisphosphonates: Similarities and differences and their potential influence on clinical efficacy. Osteoporos. Int. 2008, 19, 733–759. [Google Scholar] [CrossRef]
- Padhye, B.; dalla-Pozza, L.; Little, D.; Munns, C. Incidence and outcome of osteonecrosis in children and adolescents after intensive therapy for acute lymphoblastic leukemia (ALL). Cancer Med. 2016, 5, 960–967. [Google Scholar] [CrossRef] [Green Version]
- Kneissel, M.; Luong-Nguyen, N.-H.; Baptist, M.; Cortesi, R.; Zumstein-Mecker, S.; Kossida, S.; O’Reilly, T.; Lane, H.; Susa, M. Everolimus suppresses cancellous bone loss, bone resorption, and cathepsin K expression by osteoclasts. Bone 2004, 35, 1144–1156. [Google Scholar] [CrossRef]
- Yakes, F.M.; Chen, J.; Tan, J.; Yamaguchi, K.; Shi, Y.; Yu, P.; Qian, F.; Chu, F.; Bentzien, F.; Cancilla, B.; et al. Cabozantinib (XL184), a Novel MET and VEGFR2 Inhibitor, Simultaneously Suppresses Metastasis, Angiogenesis, and Tumor Growth. Mol. Cancer Ther. 2011, 10, 2298–2308. [Google Scholar] [CrossRef] [Green Version]
- Fioramonti, M.; Santini, D.; Iuliani, M.; Ribelli, G.; Manca, P.; Papapietro, N.; Spiezia, F.; Vincenzi, B.; Denaro, V.; Russo, A.; et al. Cabozantinib targets bone microenvironment modulating human osteoclast and osteoblast functions. Oncotarget 2017, 8, 20113–20121. [Google Scholar] [CrossRef] [Green Version]
- Terpos, E.; Sezer, O.; Croucher, P.; Dimopoulos, M. Myeloma bone disease and proteasome inhibition therapies. Blood 2007, 110, 1098–1104. [Google Scholar] [CrossRef]
- Mohty, M.; Malard, F.; Mohty, B.; Savani, B.; Moreau, P.; Terpos, E. The effects of bortezomib on bone disease in patients with multiple myeloma. Cancer 2014, 120, 618–623. [Google Scholar] [CrossRef] [Green Version]
- Accardi, F.; Toscani, D.; Bolzoni, M.; Palma, A.B.D.; Aversa, F.; Giuliani, N. Mechanism of Action of Bortezomib and the New Proteasome Inhibitors on Myeloma Cells and the Bone Microenvironment: Impact on Myeloma-Induced Alterations of Bone Remodeling. BioMed Res. Int. 2015, 2015, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Hurchla, M.A.; Garcia, A.; Hornick, M.C.; Ocio, E.M.; Li, A.; Blanco, J.F.; Collins, L.; Kirk, C.J.; Piwnica-Worms, D.; Vij, R.; et al. The epoxyketone-based proteasome inhibitors carfilzomib and orally bioavailable oprozomib have anti-resorptive and bone-anabolic activity in addition to anti-myeloma effects. Leukemia 2012, 27, 430–440. [Google Scholar] [CrossRef] [Green Version]
- Garcia, A.; Quwaider, D.; Canavese, M.; Ocio, E.M.; Tian, Z.; Blanco, J.F.; Berger, A.J.; de Solórzano, C.O.; Hernández-Iglesias, T.; Martens, A.C.; et al. Preclinical Activity of the Oral Proteasome Inhibitor MLN9708 in Myeloma Bone Disease. Clin. Cancer Res. 2014, 20, 1542–1554. [Google Scholar] [CrossRef] [Green Version]
- Jensen, P.O.; Mortensen, B.T.; Hodgkiss, R.J.; Iversen, P.O.; Christensen, I.J.; Helledie, N.; Larsen, J.K. Increased cellular hypoxia and reduced proliferation of both normal and leukaemic cells during progression of acute myeloid leukaemia in rats. Cell Prolif. 2000, 33, 381–395. [Google Scholar] [CrossRef]
- Frolova, O.; Samudio, I.; Benito, J.M.; Jacamo, R.; Kornblau, S.M.; Markovic, A.; Schober, W.; Lu, H.; Qiu, Y.H.; Buglio, D.; et al. Regulation of HIF-1α signaling and chemoresistance in acute lymphocytic leukemia under hypoxic conditions of the bone marrow microenvironment. Cancer Biol. Ther. 2012, 13, 858–870. [Google Scholar] [CrossRef] [PubMed]
- Irigoyen, M.; Garcia-Ruiz, J.C.; Berra, E. The hypoxia signalling pathway in haematological malignancies. Oncotarget 2017, 8, 36832–36844. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Velasco-Hernandez, T.; Hyrenius-Wittsten, A.; Rehn, M.; Bryder, D.; Cammenga, J. HIF-1α can act as a tumor suppressor gene in murine acute myeloid leukemia. Blood 2014, 124, 3597–3607. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Velasco-Hernandez, T.; Tornero, D.; Cammenga, J. Loss of HIF-1α accelerates murine FLT-3ITD-induced myeloproliferative neoplasia. Leukemia 2015, 29, 2366–2374. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patterson, A.; Ferry, D.M.; Edmunds, S.; Gu, Y.; Singleton, R.S.; Patel, K.; Pullen, S.M.; Hicks, K.O.; Syddall, S.P.; Atwell, G.J.; et al. Mechanism of Action and Preclinical Antitumor Activity of the Novel Hypoxia-Activated DNA Cross-Linking Agent PR-104. Clin. Cancer Res. 2007, 13, 3922–3932. [Google Scholar] [CrossRef] [Green Version]
- Guise, C.P.; Abbattista, M.R.; Singleton, R.S.; Holford, S.D.; Connolly, J.; Dachs, G.U.; Fox, S.B.; Pollock, R.; Harvey, J.; Guilford, P.; et al. The Bioreductive Prodrug PR-104A Is Activated under Aerobic Conditions by Human Aldo-Keto Reductase 1C3. Cancer Res. 2010, 70, 1573–1584. [Google Scholar] [CrossRef] [Green Version]
- Sun, J.D.; Liu, Q.; Wang, J.; Ahluwalia, D.; Ferraro, D.; Wang, Y.; Duan, J.-X.; Ammons, W.S.; Curd, J.G.; Matteucci, M.D.; et al. Selective Tumor Hypoxia Targeting by Hypoxia-Activated Prodrug TH-302 Inhibits Tumor Growth in Preclinical Models of Cancer. Clin. Cancer Res. 2012, 18, 758–770. [Google Scholar] [CrossRef] [Green Version]
- Gupta, P.; Mulkey, F.; Hasserjian, R.P.; Sanford, B.L.; Vij, R.; Hurd, D.D.; Odenike, O.M.; Bloomfield, C.D.; Owzar, K.; Stone, R.M.; et al. A phase II study of the oral VEGF receptor tyrosine kinase inhibitor vatalanib (PTK787/ZK222584) in myelodysplastic syndrome: Cancer and Leukemia Group B study 10105 (Alliance). Investig. New Drugs 2013, 31, 1311–1320. [Google Scholar] [CrossRef] [Green Version]
- Serve, H.; Krug, U.; Wagner, R.; Sauerland, M.C.; Heinecke, A.; Brunnberg, U.; Schaich, M.; Ottmann, O.; Duyster, J.; Wandt, H.; et al. Sorafenib in Combination with Intensive Chemotherapy in Elderly Patients with Acute Myeloid Leukemia: Results from a Randomized, Placebo-Controlled Trial. J. Clin. Oncol. 2013, 31, 3110–3118. [Google Scholar] [CrossRef]
- Pollard, J.A.; Alonzo, T.A.; Brown, P.A.; Gerbing, R.B.; Fox, E.; Choi, J.K.; Fisher, D.B.T.; Hirsch, B.A.; Kahwash, S.; Levine, M.J.E.; et al. Sorafenib in Combination with Standard Chemotherapy for Children with High Allelic Ratio FLT3/ITD+ AML Improves Event-Free Survival and Reduces Relapse Risk: A Report from the Children’s Oncology Group Protocol AAML1031. Blood 2019, 134 (Suppl. 1), 292. [Google Scholar] [CrossRef]
- Madlambayan, G.J.; Meacham, A.M.; Hosaka, K.; Mir, S.; Jorgensen, M.; Scott, E.W.; Siemann, D.W.; Cogle, C.R. Leukemia regression by vascular disruption and antiangiogenic therapy. Blood 2010, 116, 1539–1547. [Google Scholar] [CrossRef] [Green Version]
- Petty, A.J.; Yang, Y. Tumor-Associated Macrophages in Hematologic Malignancies: New Insights and Targeted Therapies. Cells 2019, 8, 1526. [Google Scholar] [CrossRef] [Green Version]
- Duan, Z.; Luo, Y. Targeting macrophages in cancer immunotherapy. Signal Transduct. Target. Ther. 2021, 6, 1–21. [Google Scholar] [CrossRef]
- Feng, M.; Jiang, W.; Kim, B.Y.S.; Zhang, C.C.; Fu, Y.-X.; Weissman, I.L. Phagocytosis checkpoints as new targets for cancer immunotherapy. Nat. Rev. Cancer 2019, 19, 568–586. [Google Scholar] [CrossRef]
- Pehlivan, K.C.; Duncan, B.B.; Lee, D.W. CAR-T Cell Therapy for Acute Lymphoblastic Leukemia: Transforming the Treatment of Relapsed and Refractory Disease. Curr. Hematol. Malig. Rep. 2018, 13, 396–406. [Google Scholar] [CrossRef]
- Griessinger, E.; Moschoi, R.; Biondani, G.; Peyron, J.-F. Mitochondrial Transfer in the Leukemia Microenvironment. Trends Cancer 2017, 3, 828–839. [Google Scholar] [CrossRef]
- Sahinbegovic, H.; Jelinek, T.; Hrdinka, M.; Bago, J.R.; Turi, M.; Sevcikova, T.; Kurtovic-Kozaric, A.; Hajek, R.; Simicek, M. Intercellular Mitochondrial Transfer in the Tumor Microenvironment. Cancers 2020, 12, 1787. [Google Scholar] [CrossRef]
- Panuzzo, C.; Jovanovski, A.; Pergolizzi, B.; Pironi, L.; Stanga, S.; Fava, C.; Cilloni, D. Mitochondria: A Galaxy in the Hematopoietic and Leukemic Stem Cell Universe. Int. J. Mol. Sci. 2020, 21, 3928. [Google Scholar] [CrossRef] [PubMed]
- Polson, A.G.; Fuji, R.N. The successes and limitations of preclinical studies in predicting the pharmacodynamics and safety of cell-surface-targeted biological agents in patients. Br. J. Pharmacol. 2012, 166, 1600–1602. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Green, M.M.B.; Chao, N.; Chhabra, S.; Corbet, K.; Gasparetto, C.; Horwitz, A.; Li, Z.; Venkata, J.K.; Long, G.; Mims, A.; et al. Plerixafor (a CXCR4 antagonist) following myeloablative allogeneic hematopoietic stem cell transplantation enhances hematopoietic recovery. J. Hematol. Oncol. 2016, 9, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stopeck, A.T.; Lipton, A.; Body, J.-J.; Steger, G.G.; Tonkin, K.; de Boer, R.H.; Lichinitser, M.; Fujiwara, Y.; Yardley, D.A.; Viniegra, M.; et al. Denosumab Compared with Zoledronic Acid for the Treatment of Bone Metastases in Patients with Advanced Breast Cancer: A Randomized, Double-Blind Study. J. Clin. Oncol. 2010, 28, 5132–5139. [Google Scholar] [CrossRef] [Green Version]
- Kotecha, R.S.; Gottardo, N.G.; Kees, U.R.; Cole, C.H. The evolution of clinical trials for infant acute lymphoblastic leukemia. Blood Cancer J. 2014, 4, e200. [Google Scholar] [CrossRef] [Green Version]
Figure 1.
Leukaemogenesis and remodelling of the bone marrow niche by leukaemic cells. (A) Oncogenic transformation of haematopoietic cells into leukaemic cells can be initiated by cell-intrinsic genetic alterations in surrounding bone marrow niche cells. (B) During leukaemogenesis, malignant cells can hijack the mechanisms of homing used by healthy haematopoietic stem cells and directly compete with haematopoietic stem cells for endosteal adhesion, resulting in transformation of the healthy niche microenvironment into a tumour-supportive niche. Furthermore, leukaemic cells are capable of remodelling the (C) central vascular and (D) endosteal vascular niche into leukaemia-supportive microenvironments that favour leukaemia cell survival and expansion, with hostility towards physiological haematopoiesis. (E) Leukaemic cells can promote remodelling of bone compartments by directly influencing the differentiation and/or function of osteoblasts and osteoclasts, leading to severely impaired bone homeostasis.
Figure 1.
Leukaemogenesis and remodelling of the bone marrow niche by leukaemic cells. (A) Oncogenic transformation of haematopoietic cells into leukaemic cells can be initiated by cell-intrinsic genetic alterations in surrounding bone marrow niche cells. (B) During leukaemogenesis, malignant cells can hijack the mechanisms of homing used by healthy haematopoietic stem cells and directly compete with haematopoietic stem cells for endosteal adhesion, resulting in transformation of the healthy niche microenvironment into a tumour-supportive niche. Furthermore, leukaemic cells are capable of remodelling the (C) central vascular and (D) endosteal vascular niche into leukaemia-supportive microenvironments that favour leukaemia cell survival and expansion, with hostility towards physiological haematopoiesis. (E) Leukaemic cells can promote remodelling of bone compartments by directly influencing the differentiation and/or function of osteoblasts and osteoclasts, leading to severely impaired bone homeostasis.

Figure 2.
Regulatory and survival pathways targeted by therapeutic agents in the leukaemia bone marrow niche. Therapeutic agents can block interactions between leukaemic cells and niche cells by interfering with cell adhesion (e.g., VLA-4/VCAM-1, E-selectin, Notch/Jagged1) and paracrine regulation (e.g., CXCL12/CXCR4, Wnt/β-catenin). Furthermore, agents that are capable of normalising bone homeostasis (by targeting osteoblast and/or osteoclast activities), as well as targeting tumour-supportive vasculature and the hypoxic environment in the leukaemia niche, show therapeutic potential.
Figure 2.
Regulatory and survival pathways targeted by therapeutic agents in the leukaemia bone marrow niche. Therapeutic agents can block interactions between leukaemic cells and niche cells by interfering with cell adhesion (e.g., VLA-4/VCAM-1, E-selectin, Notch/Jagged1) and paracrine regulation (e.g., CXCL12/CXCR4, Wnt/β-catenin). Furthermore, agents that are capable of normalising bone homeostasis (by targeting osteoblast and/or osteoclast activities), as well as targeting tumour-supportive vasculature and the hypoxic environment in the leukaemia niche, show therapeutic potential.


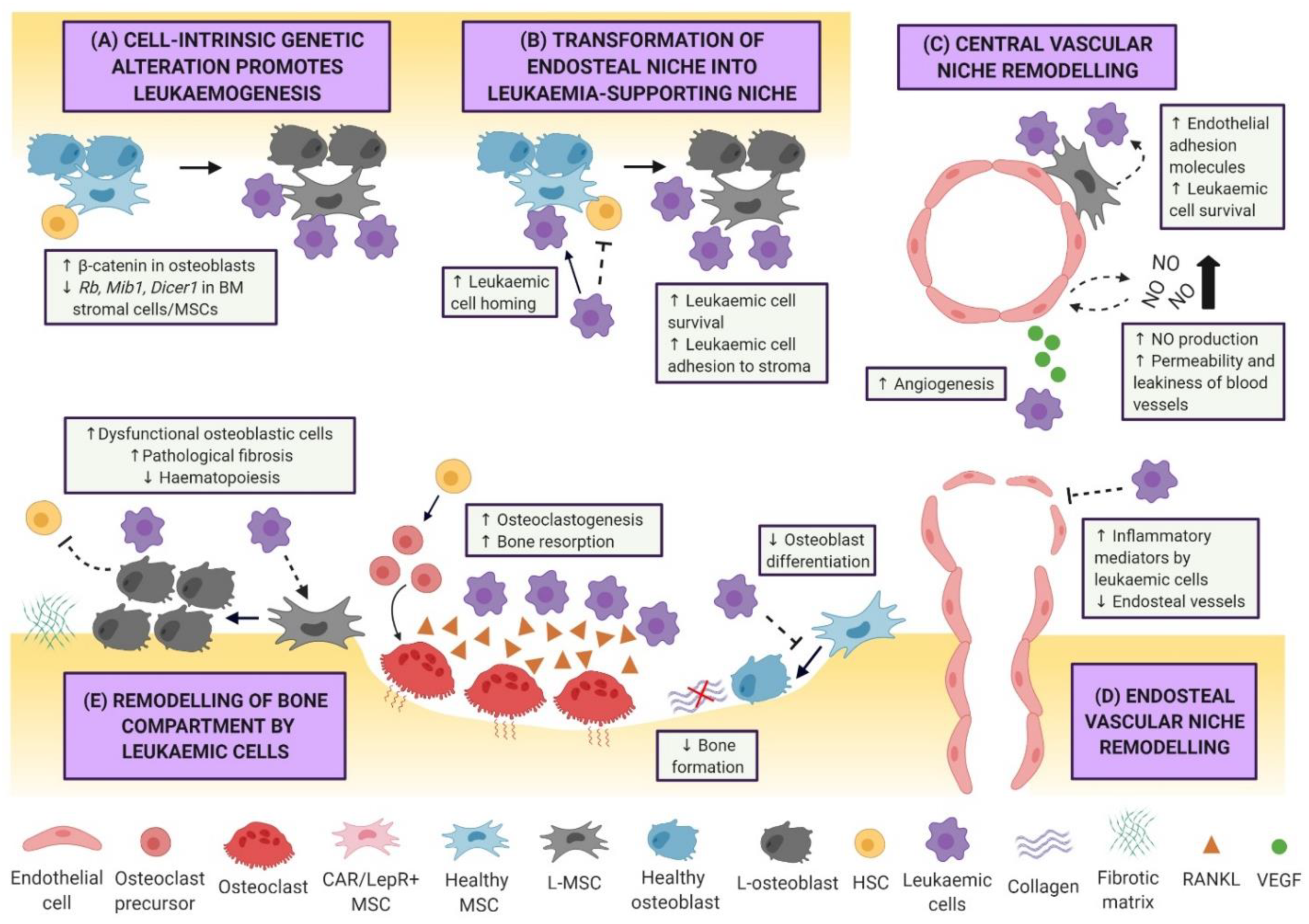{kind=link}
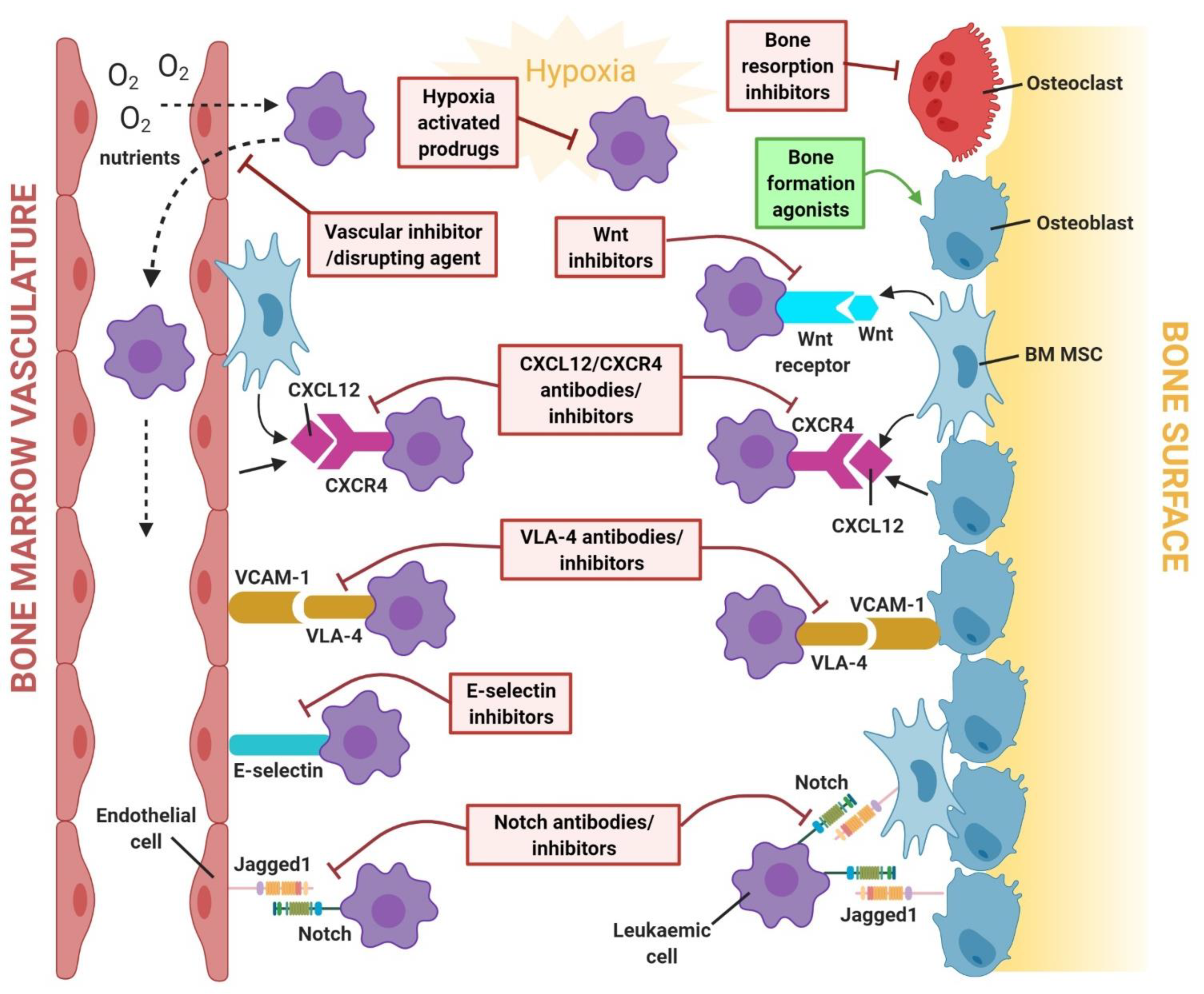{kind=link}
Table 1.
Summary of therapeutic agents that target the leukaemic niche in the bone marrow microenvironment.
Table 1.
Summary of therapeutic agents that target the leukaemic niche in the bone marrow microenvironment.
| Target Pathways | Target Strategy | Agents | Mechanism of Action in the BMM | Leukaemia Type Investigated | Animal Studies | Clinical Studies |
|---|---|---|---|---|---|---|
| CXCL12/ CXCR4 signalling pathway | CXCR4 Inhibitor (bicyclams) | Plerixafor (AMD3100) | Induces mobilisation and chemosensitivity of leukaemic cells by targeting CXCL12/CXCR4 interactions. | AML, pre-B ALL | [43,44,45] | [46,47,48] FDA-A |
| AMD3465 | Induces mobilisation and chemosensitivity of leukaemic cells by targeting CXCL12/CXCR4 interactions. | AML, Pre-B ALL, T-ALL | [45,49,50] | N/R | ||
| AMD11070 | Induces mobilisation and chemosensitivity of leukaemic cells by targeting CXCL12/CXCR4 interactions. | Pre-B ALL | [51] | N/R | ||
| CXCR4 inhibitor (synthetic peptides) | BL-8040 (BKT140) | Induces mobilisation and chemosensitivity of leukaemic cells by targeting CXCL12/CXCR4 interactions. Induces apoptosis in leukaemic cells. | AML | [52] | [53] | |
| LY2510924 | Induces mobilisation and chemosensitivity of leukaemic cells by targeting CXCL12/CXCR4 interactions. Inhibits proliferation of leukaemic cells. | AML | [54] | [55] | ||
| E5 Peptide | Induces mobilisation and chemosensitivity of leukaemic cells by targeting CXCL12/CXCR4 interactions. Induces apoptosis in leukaemic cells. | AML | [56,57] | N/R | ||
| POL6326 | Induces mobilisation and chemosensitivity of leukaemic cells by targeting CXCL12/CXCR4 interactions. | AML | [58] | NCT01413568Phase I/II # | ||
| Anti-CXCR4 monoclonal antibody | Ulocuplumab | Induces apoptosis and blocks CXCL12-induced leukaemic cell migration. Induces mobilisation of leukaemic cells. | AML | [59] | [60,61] | |
| LY2624587 | Induces apoptosis and blocks CXCL12-induced leukaemic cell migration. | T-ALL | [62] | N/R | ||
| CXCL12 inhibitor | CX-01 | Inhibits binding of CXCL12 to immobilised heparin and enhances treatment efficacy. | AML | N/R | [63] | |
| Notch signalling pathway | Gamma-secretase inhibitor | PF-03084014 | Inhibits stromal-mediated chemoresistance and potentiates sensitivity of leukaemic cells to chemotherapy. Inhibits proliferation and induces apoptosis in leukaemic cells. | T-ALL | [64,65] | [66] |
| BMS-906024 | Inhibits growth and survival of leukaemic cells by targeting Notch signalling. | T-ALL | [67] | [68] | ||
| MRK-560 | Promote leukaemic cell cycle arrest by targeting PSEN1, a subclass of gamma-secretase complexes highly involved in activation of mutant Notch1. | T-ALL | [69] | N/R | ||
| Wnt/β-catenin signalling pathway | Wnt/β-catenin inhibitor | XAV939 | Attenuates BMM-induced protection of leukaemic cells by inhibiting Wnt signalling. Inhibits proliferation of leukaemic cells. | Pre-B ALL | [70] | N/R |
| PRI-724 | Attenuates BMM-induced protection of leukaemic cells by inhibiting Wnt signalling. Induces apoptosis in leukaemic cells. | AML, CML | [71,72] | NCT01606579Phase I/II # | ||
| BC2059 | Induces apoptosis of leukaemic cells by synergistically enhancing the effect of drug treatment. | AML | [73] | N/R | ||
| CWP232291 | Promotes endoplasmic reticulum stress activation, leading to degradation of β-catenin and apoptosis induction in leukaemic cells. | AML | N/R | [74] | ||
| Wnt/β-catenin/FLT3 inhibitor | SKLB-677 | Induces apoptosis in leukaemic cells. | AML | [75] | N/R | |
| Adhesion molecules signalling pathway | Anti-CD44 monoclonal antibody | RG7356/ ARH460-16-2 | Blocks leukaemia–stroma interaction by targeting CD44. | AML | [76] | [77] |
| Anti-α4β1/ VLA-4 monoclonal antibody | Natalizumab | Blocks leukaemia–stroma interaction by targeting VLA-4/VCAM-1, sensitising leukaemic cells to chemotherapy. | AML, Pre-B ALL | [78,79] | N/R FDA-A | |
| α4 inhibitor | TBC3486 | Blocks leukaemia–stroma interaction by targeting integrin α4, sensitising leukaemic cells to chemotherapy. | Pre-B ALL | [80] | N/R | |
| VLA-4 peptide antagonist | FNIII14 | Blocks cell adhesion by targeting VLA-4 to fibronectin interaction, sensitising leukaemic cells to chemotherapy. | AML | [81] | N/R | |
| E-selectin inhibitor | Uproleselan (GMI-1271) | Attenuates cell surface adhesion, regeneration and survival of leukaemic cells by antagonising E-selectin. Sensitises leukaemic cells to chemotherapy. | AML | [34] | [82] NCT03701308 NCT03616470 Phase II/III # | |
| Dual CXCR4/ E-selectin inhibitor | GMI-1359 | Promotes leukaemic cell mobilisation and restores normal haematopoiesis. | AML | [83] | N/R | |
| Bone remodelling signalling pathway | Bone resorption inhibitor | Zoledronic acid | Inhibits osteoclast resorption. | Pre-B ALL | [35] | N/R FDA-A |
| mTOR inhibitor | Everolimus | Inhibits osteoclast resorption. * | ALL | N/R | [84,85] FDA-A | |
| Receptor tyrosine kinase inhibitor | Cabozantinib | Inhibits osteoclast differentiation and resorption, modulates RANKL/osteoprotegerin in osteoblasts. * Induces apoptosis in leukaemic cells. | AML | [86] | [87] FDA-A | |
| Proteasome inhibitor | Bortezomib | Promotes osteoblast differentiation and suppress osteoclast activity. * Induces apoptosis in leukaemic cells. | AML, Pre-B ALL, T-ALL | [88,89] | [90,91] FDA-A | |
| Carfilzomib | Promotes osteoblast differentiation and suppress osteoclast activity. * Induces apoptosis in leukaemic cells. | AML, ALL | [92,93] | [94,95] NCT02303821 NCT02512926 Phase I FDA-A | ||
| Ixazomib | Promotes osteoblast differentiation and suppress osteoclast activity. * | AML | N/R | [96] FDA-A | ||
| Hypoxia-related signalling pathway | Hypoxia activated prodrug | PR-104 | Induces cytotoxicity in hypoxic leukaemic cells. | AML, Pre-B ALL, T-ALL | [97,98] | [99] |
| Evofosfamide (TH-302) | Induces cytotoxicity in hypoxic leukaemic cells. | AML, ALL | [100,101] | [102] | ||
| Vasculature-associated pathway | Vascular disrupting agent | CA1P (OXi4503) | Induces breakdown of vascular architecture. | AML | [103] | [104] |
N/R indicates not reported and status remains unclear. # ongoing clinical studies as described in the relevant reference or clinical trial number. * therapeutic mechanisms on bone remodelling confirmed using non-leukaemic animal models but remain to be confirmed in leukaemia animal models. FDA-A refers to agents clinically approved by the FDA for other disease treatments. ALL, acute lymphoblastic leukaemia; AML, acute myeloid leukaemia; BMM, bone marrow microenvironment.
Table 2.
Summary of clinical trial outcomes for therapeutic agents that target the leukaemic niche in the bone marrow microenvironment.
Table 2.
Summary of clinical trial outcomes for therapeutic agents that target the leukaemic niche in the bone marrow microenvironment.
| Agent | Studies/ Clinical Trial ID (Phases) | Treatment Design | Age Range (Median) | Disease Type (No. Patients Enrolled) | Ref | Outcomes |
|---|---|---|---|---|---|---|
| Plerixafor | NCT01319864 (Phase 1) | Dose escalation at four different dose levels (6, 9, 12, and 15 mg/m2/dose) as part of cytarabine and etoposide therapy. | AML 3–17 (13) ALL 12–21 (14) AML/ MDS 20 (20) | R/R AML (13) R/R ALL (5) R/R AML/MDS (1) | [46] |
|
| Plerixafor | NCT01141543 (Phase 1) | Dose escalation (240 μg/kg) as part of myeloablative conditioning regimen for patients undergoing allogeneic haematopoietic stem cell transplant. | 38–58 (49) | De novo AML (10) Secondary AML (2) | [47] |
|
| Plerixafor | NCT00943943 (Phase 1) | Plerixafor (240 μg/kg adjusted body weight, subcutaneously) combined with G-CSF and sorafenib dose-escalation. | 18–84 (58) | R/R AML with FLT3-ITD mutation (28) | [48] |
|
| BL-8040 | NCT01838395 (Phase 2a) | Once daily dose of BL-8040 (0.5–2 mg/kg) administered as monotherapy for days 1–2, followed by combination of BL-8040 with cytarabine. | (61) | R/R AML (45) | [53] |
|
| LY2510924 | NCT02652871 (Phase 1) | Dose escalation at two doses (10, 20 mg/day), administered as monotherapy daily for 7 days, followed by idarubicin and cytarabine combined with LY2510924. | 19–70 (55) | R/R AML (11) | [55] |
|
| Ulocuplumab | N/R (Phase 1) | Dose escalation at 0.3, 1, 3, and 10 mg/kg. Cohorts at 0.3 mg/kg received three weekly doses as monotherapy, followed by the same doses with MEC chemotherapy. Cohorts at 1, 3, and 10 mg/kg received 1 weekly dose as monotherapy, followed by the same combination regimen. | N/R | R/R AML (24) | [60] |
|
| Ulocuplumab | N/R (Phase 1) | Dose escalation (0.3, 1, 3, and 10 mg/kg) was given as a single infusion a week prior to MEC treatment, followed by 3 additional weekly doses per MEC cycle thereafter. For expansion, 10 mg/kg ulocuplumab + MEC regimen was used. | 21–79 (58) | R/R AML (73) | [61] |
|
| CX-01 | NCT02056782 (Phase 1) | For cytarabine/idarubicin induction, CX-01 (4 mg/kg) was given over 30 min after the first dose of idarubicin, followed by continuous infusion of 0.25 mg/kg per hour thereafter. For consolidation, CX-01 (4 mg/kg) was given over 30 min after the first dose of cytarabine, followed by continuous infusion of 0.25 mg/kg per hour thereafter. | 22–74 (54) | De novo AML (11) CMML-2 (1) | [63] |
|
| PF-03084014 | A8641014 (Phase 1) | PF-03084014 administered twice weekly (150 mg) in continuous cycles. | 18–43 (31) | R/R T-ALL (3) R/R T-LBL (5) | [66] |
|
| BMS-906024 | CA216002 (Phase 1) | BMS-906024 was given intravenously weekly at doses of 0.6, 4, and 6 mg. | 18–74 | R/R T-ALL/T-LBL (25) | [68] |
|
| CWP232291 | NCT01398462 (Phase 1) | Dose escalation at 4–334 mg/m2. Agent was administered intravenously daily for 7 days every 21 days. MTD was defined at 257 mg/m2. | 25–81 (64) | R/R AML (64) R/R MDS (5) | [74] |
|
| RG7356/ ARH460-16-2 | NCT01641250 (Phase 1) | RG7356 was administered intravenously at dosages ≤2400 mg every other week, or ≤1200 mg weekly or twice weekly. | 20–82 (69) | R/R AML (37) TN AML (7) | [77] |
|
| Uproleselan (GMI-1271) | N/R (Phase 1/2) | Dose escalation at 5–20 mg/kg in combination with MEC in patients with R/R AML. In phase 2, patients were given uproleselan with chemotherapy. RP2D was 10 mg/kg. | R/R patients 26–84 (59) TN patients 60–79 (67) | R/R AML (66) TN AML (25) | [82] |
|
| Everolimus | NCT00968253 (Phase 1/2) | Everolimus was given continuously at 5 or 10 mg/day with HyperCVAD treatment. MTD was defined at 5 mg/day. | 11–64 (25) | R/R Pre-B ALL (13) R/R T-ALL (10) MPAL (1) | [84] |
|
| Everolimus | NCT01523977 (Phase 1b) | Dose escalation at 2, 3, and 5 mg/m2/day for 32 days, co-administered with multi-agent reinduction chemotherapy. | 2.4–22.8 (11) | Relapsed B-ALL (21) Relapsed T-ALL (1) | [85] |
|
| Cabozantinib | NCT01961765 (Phase 1) | Dose escalation at 40, 60, and 80 mg daily in 28-day cycles. MTD was defined at 40 mg daily. | 27–85 (68) | R/R AML (16) Newly diagnosed AML (2) | [87] |
|
| Bortezomib | NCT01371981 (Phase 3) | Bortezomib (1.3 mg/m2) was incorporated into chemotherapy. Patients were randomly assigned to either standard AML therapy or standard therapy with bortezomib. | 0–29.5 (9.2) | De novo AML (1231) | [91] |
|
| Carfilzomib | NCT01137747 (Phase 1) | Dose escalation at 36, 45, and 56 mg/m2, administered as a 30 min infusion on a 28-day cycle. | 32–78 (70) | R/R AML (17) R/R ALL (1) | [94] |
|
| Carfilzomib | NCT02303821 (Phase 1b) | Dose escalation at 27–56 mg/m2 for patients treated with VXLD. Patients received one 4-week cycle of induction chemotherapy with VXLD, plus carfilzomib administered intravenously. | 1–19 (11) | R/R B-ALL (7) R/R T-ALL (8) | [95] |
|
| Ixazomib | NCT02070458 (Phase 1/2) | Dose escalation at 1, 2, and 3 mg in combination with MEC treatment. MTD was defined at 1 mg. | 31–70 (58) | R/R AML (30) | [96] |
|
| PR-104 | NCT01037556 (Phase 1/2) | Patients received PR-104 at doses ranging from 1.1–4 g/m2. For dose expansion, patients were treated at 3 or 4 g/m2. | 20–79 (62) | R/R AML (40) R/R ALL (10) | [99] |
|
| Evofosfamide (TH-302) | NCT01149915 (Phase 1) | Evifosfamide administered daily as a 30–60 min infusion (120–550 mg/m2; MTD 460 mg/m2), or as a continuous intravenous infusion over 120 h (MTD 330 mg/m2), on days 1–5 of 21-day cycles. | 23–76 (58) | R/R AML (39) R/R ALL (9) CML (1) | [102] |
|
| CA1P (OXi4503) | NCT02576301 (Phase 1B) | CA1P administered at doses ranging from 3.75 to 12.2 mg/m2 in combination with cytarabine. MTD defined at 9.76 mg/m2. | 26–78 (61) | R/R AML (27) R/R MDS (2) | [104] |
|
Abbreviations: AE, adverse event/effect; AML, acute myeloid leukaemia; ALL, acute lymphoblastic leukaemia; CMML-2, chronic myelomonocytic leukaemia-2; CR, complete remission; CRi, CR with incomplete haematologic recovery; CRp, CR with incomplete platelet recovery; CR2, second complete remission; MEC, mitoxantrone, etoposide, cytarabine; MDS, myelodysplastic syndrome; MLFS, morphological leukaemia-free state; MPAL, mixed phenotype acute leukaemia; MRD, minimal residual disease; MTD, maximum tolerated dose; N/R, not reported; ORR, overall response rate; PR, partial remission; RP2D, recommended phase 2 dose; R/R, relapsed/refractory; SD; stable disease; T-LBL, T-cell lymphoblastic lymphoma; TN, treatment-naïve; VXLD, vincristine, dexamethasone, PEG-asparaginase, daunorubicin. * indicates ORR was defined differently according to each individual study.
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Kuek, V.; Hughes, A.M.; Kotecha, R.S.; Cheung, L.C. Therapeutic Targeting of the Leukaemia Microenvironment. Int. J. Mol. Sci. 2021, 22, 6888. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22136888
AMA Style
Kuek V, Hughes AM, Kotecha RS, Cheung LC. Therapeutic Targeting of the Leukaemia Microenvironment. International Journal of Molecular Sciences. 2021; 22(13):6888. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22136888
Chicago/Turabian StyleKuek, Vincent, Anastasia M. Hughes, Rishi S. Kotecha, and Laurence C. Cheung. 2021. "Therapeutic Targeting of the Leukaemia Microenvironment" International Journal of Molecular Sciences 22, no. 13: 6888. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22136888
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.