
Access to New Cytotoxic Triterpene and Steroidal Acid-TEMPO Conjugates by Ugi Multicomponent-Reactions †

 ,
,  , , and
, , and
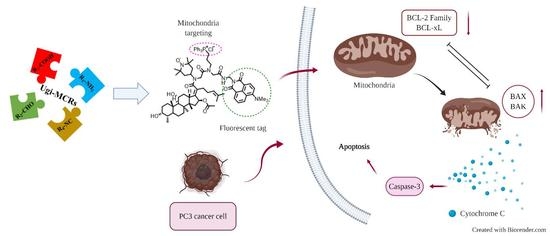
Abstract
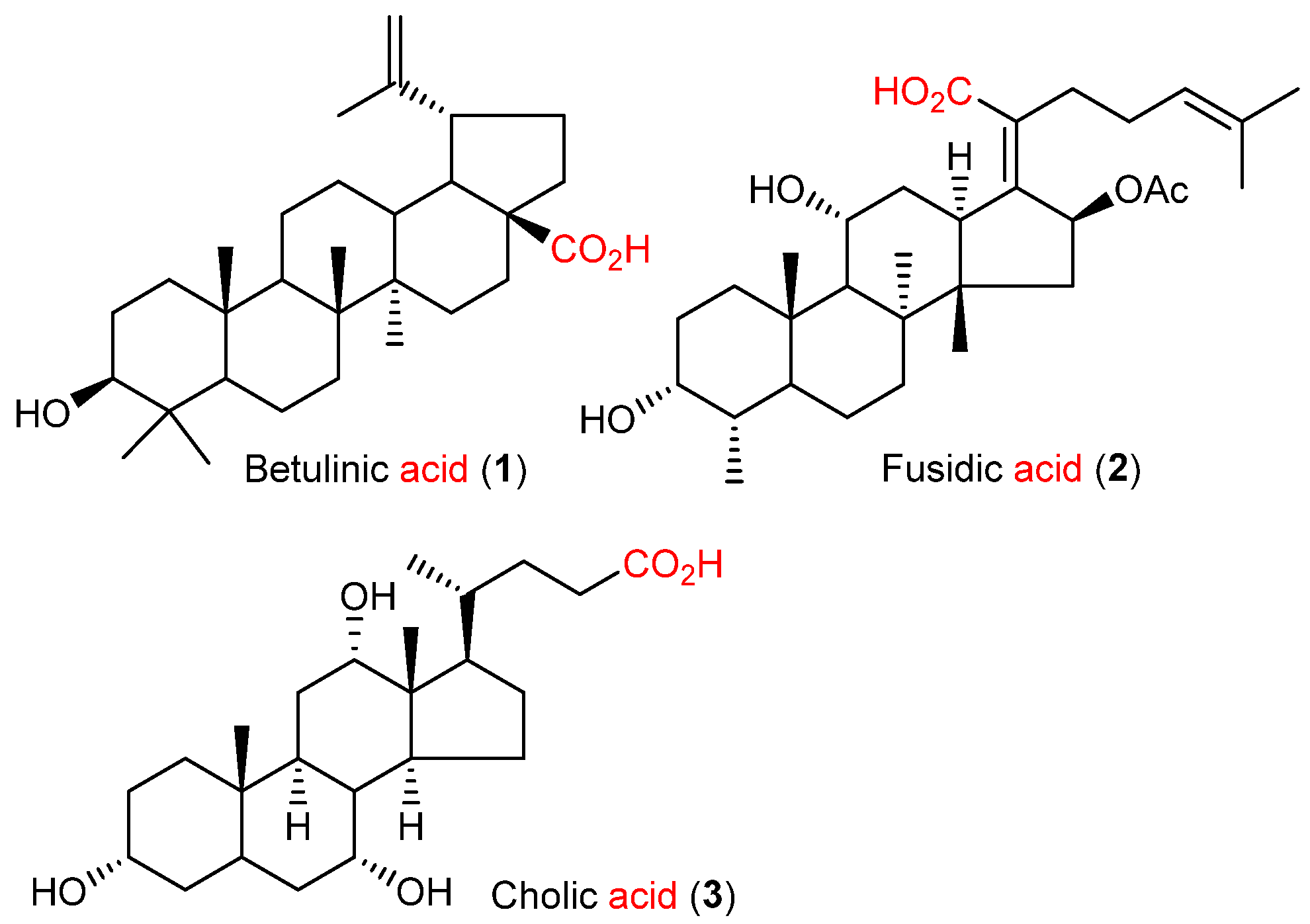
:
1. Introduction
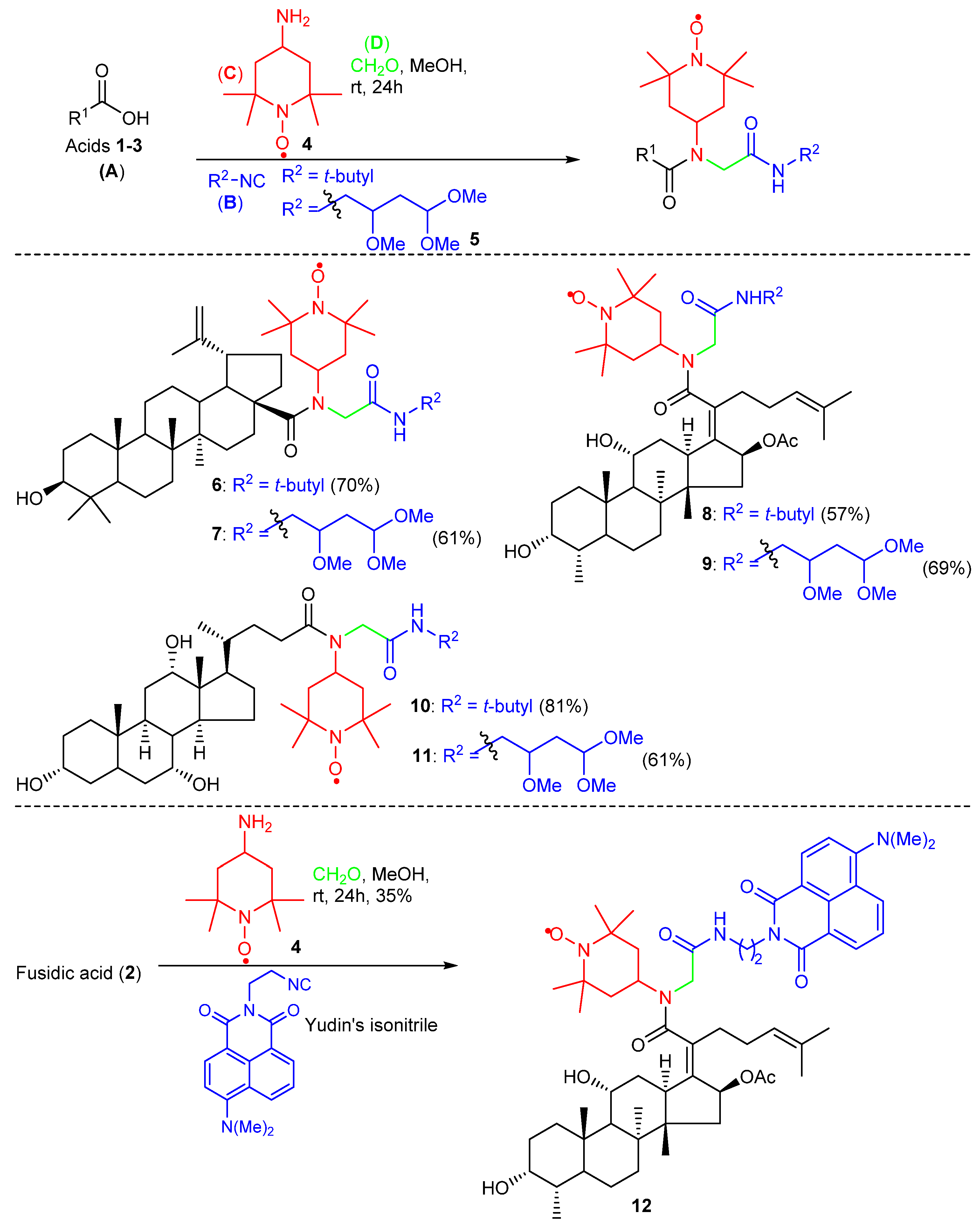
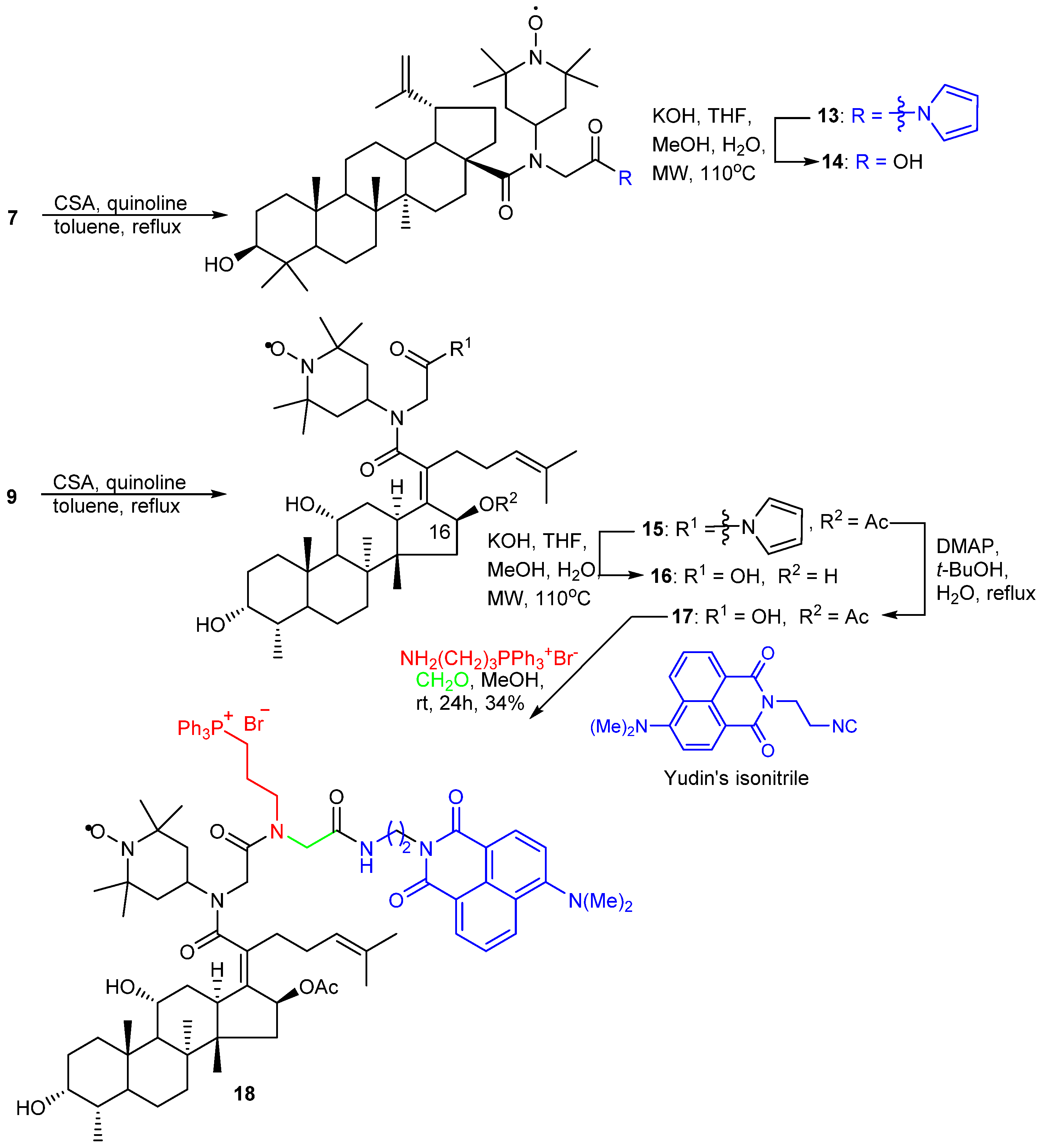
2. Results and Discussion
2.1. Chemistry
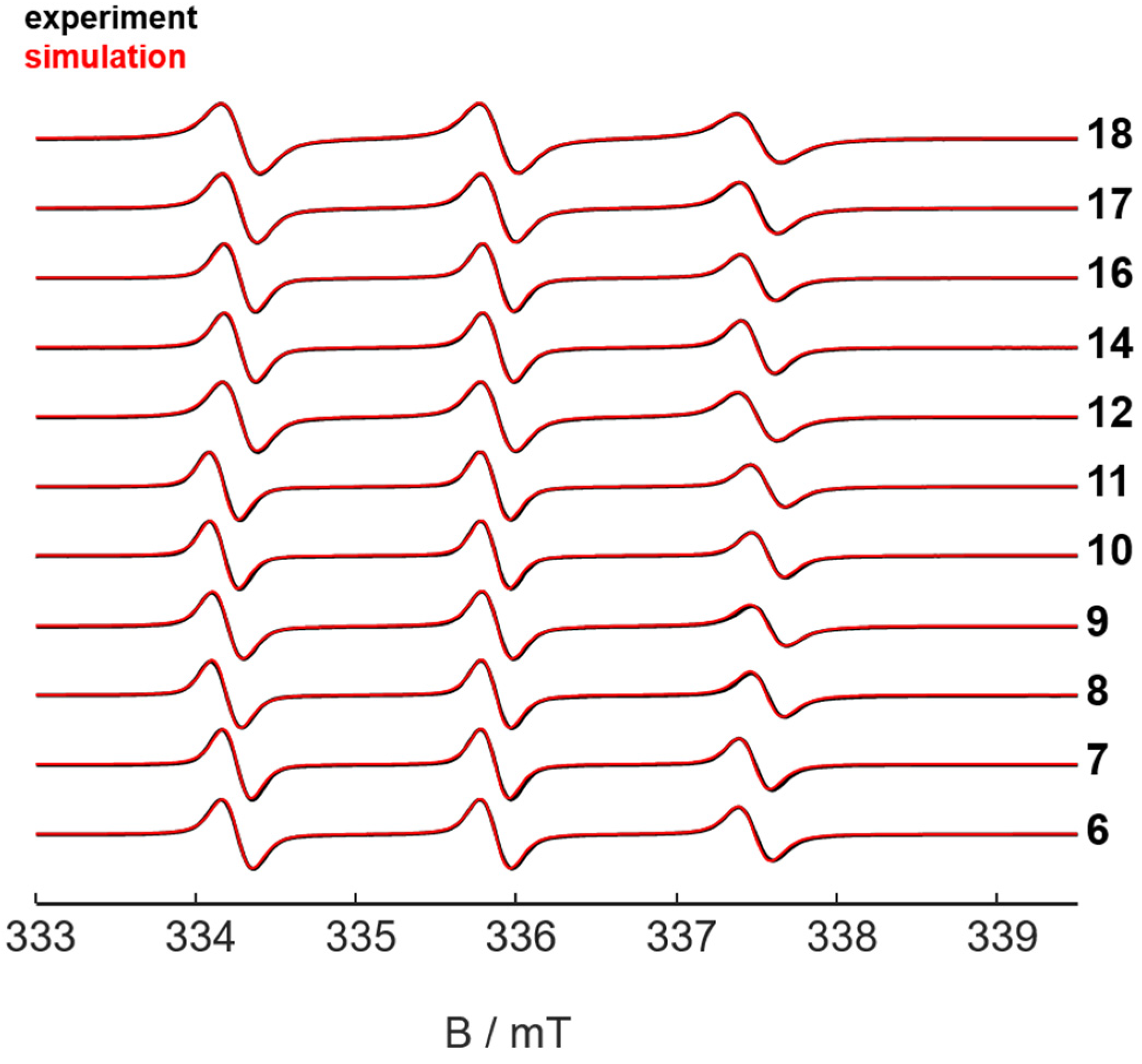
2.2. Characterization of the Nitroxide Conjugated Compounds by Electron Paramagnetic Resonance (EPR) Spectroscopy
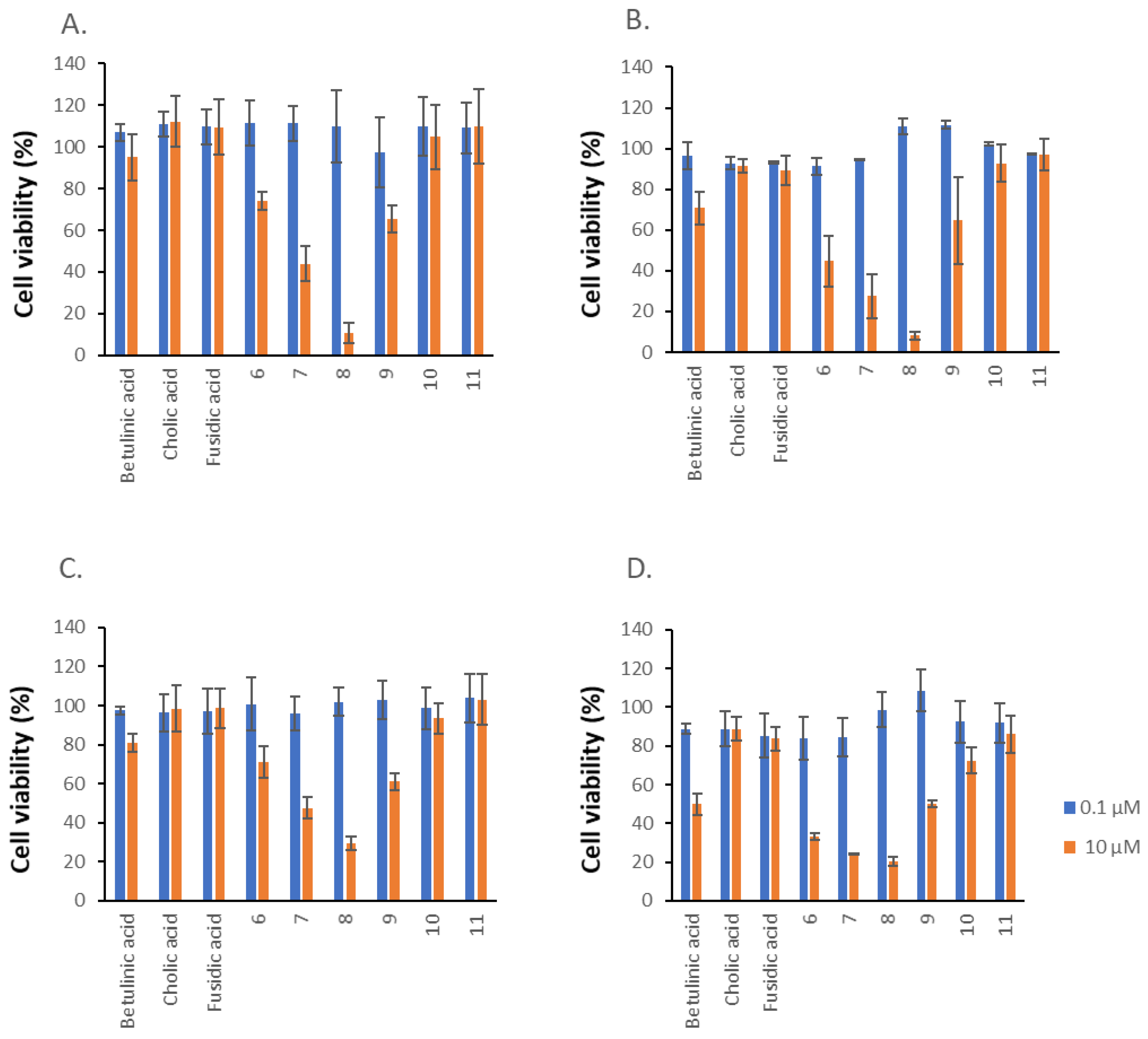
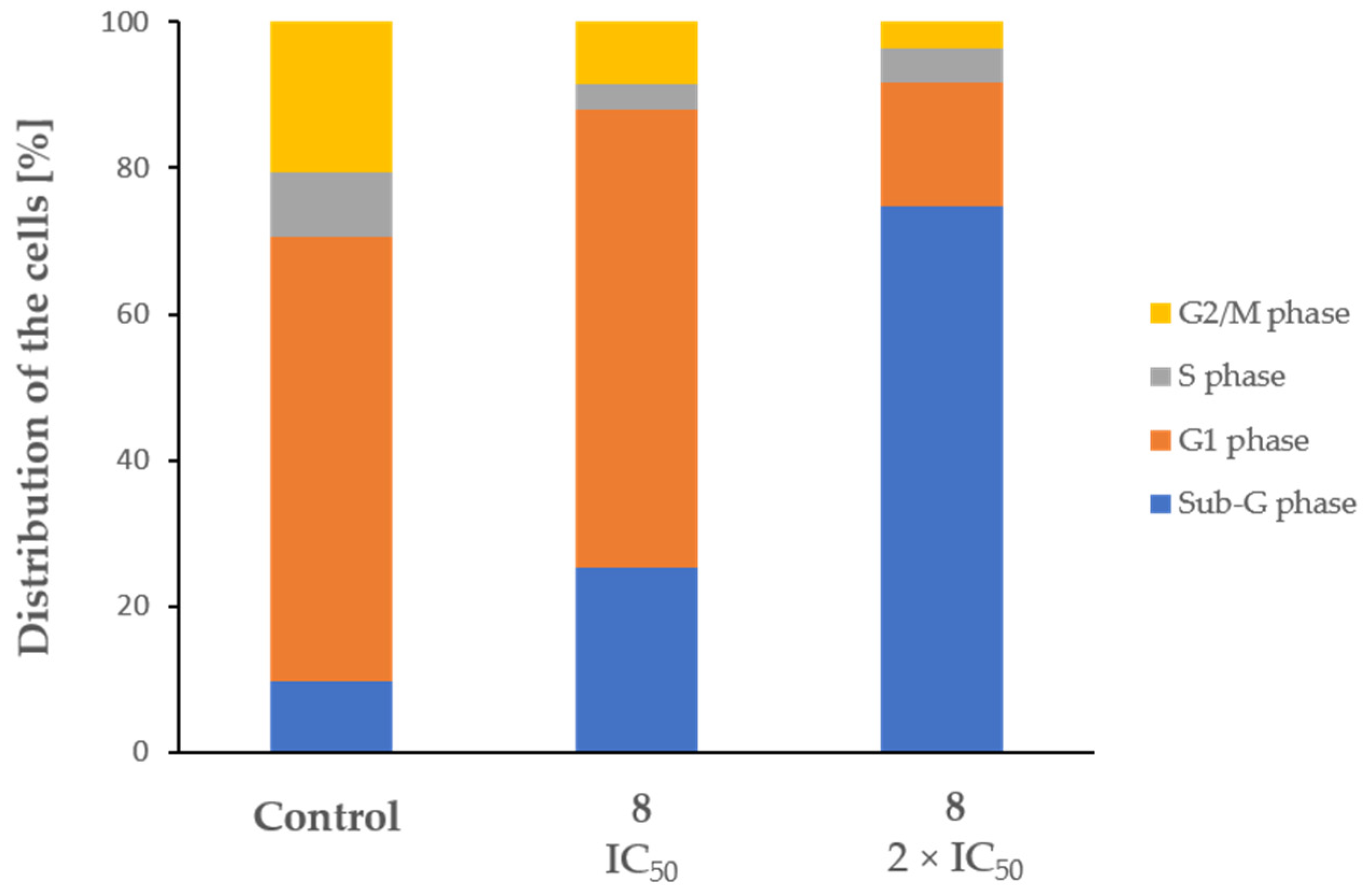
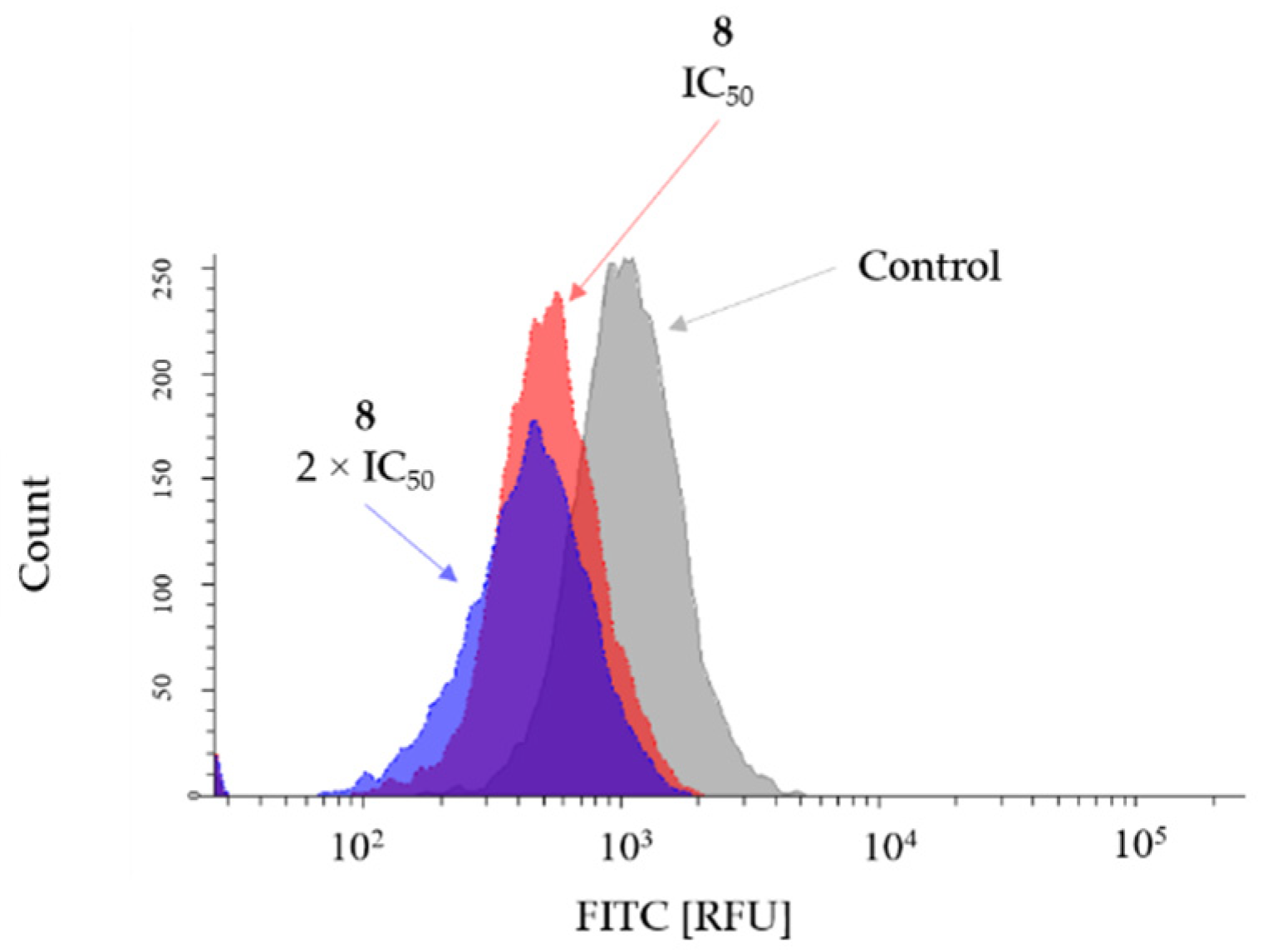
2.3. Cytotoxic Activity
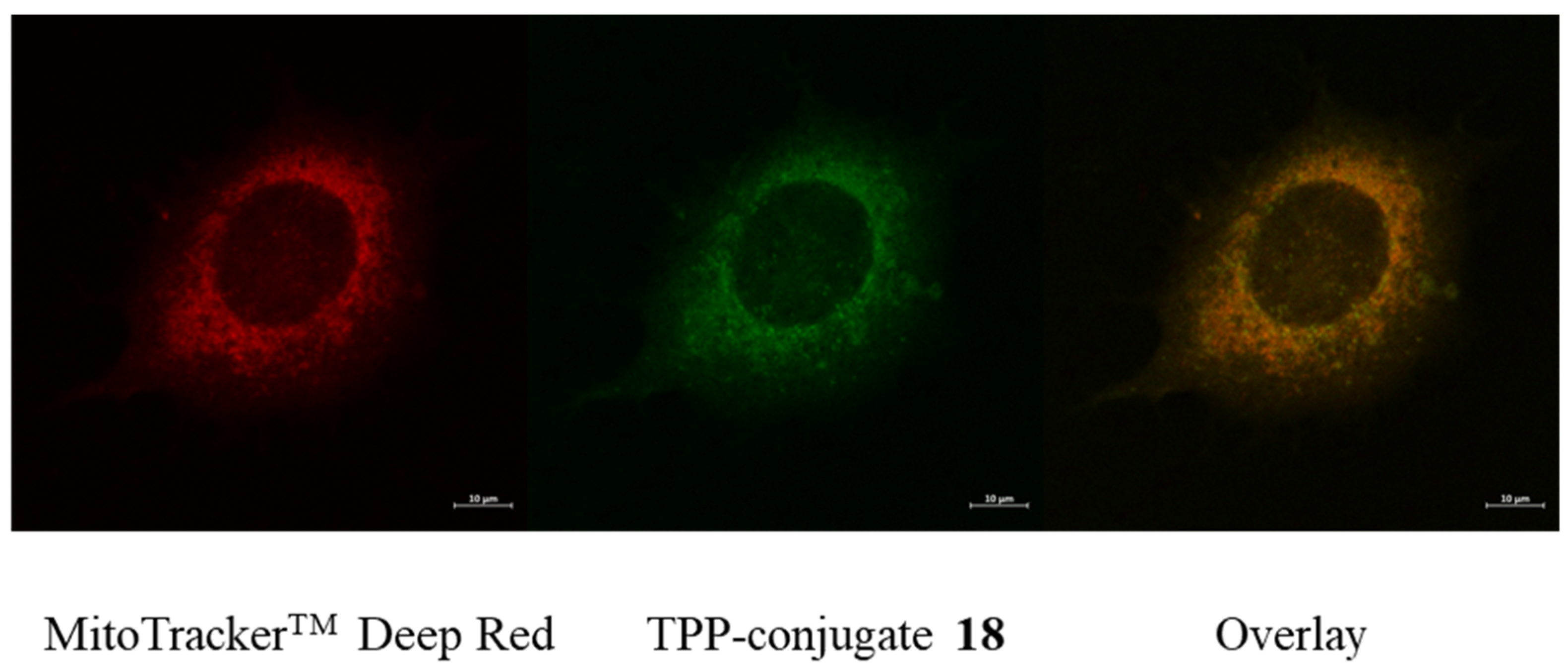
2.4. Fluorescent Imaging Study
3. Conclusions
4. Materials and Methods
4.1. Chemistry
4.1.1. Materials
4.1.2. General Procedure A for the Ugi-4CR
4.1.3. General Procedure B for the Conversion of Ugi Products 7 and 9 to Corresponding Spin-Labelled N-Acylpyrroles 13 and 15
4.1.4. General Procedure C for the Conversion of the N-Acylpyrroles 13 and 15 into Their Corresponding Carboxylic Acids 14 and 16
Method C1
Method C2
4.2. EPR Spectroscopy and Sample Preparation
4.3. Biology
4.3.1. Cell Lines and Cultivation
4.3.2. MTT and CV Assays
4.3.3. Apoptosis Analysis
4.3.4. Cell Cycle Analysis
4.3.5. Western Blot Analysis
4.3.6. Investigation of ROS Production
4.4. Microscopy
Fluorescent Microscopy
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Kumari, S.; Badana, A.K.; Murali Mohan, G.; Shailender, G.; Malla, R.R. Reactive Oxygen Species: A Key Constituent in Cancer Survival. Biomark. Insights 2018, 13, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Liou, G.-Y.; Storz, P. Reactive oxygen species in cancer. Free Radic. Res. 2010, 44, 479–496. [Google Scholar] [CrossRef] [Green Version]
- Firuzi, O.; Miri, R.; Tavakkoli, M.; Saso, L. Antioxidant therapy: Current status and future prospects. Curr. Med. Chem. 2011, 18, 3871–3888. [Google Scholar] [CrossRef]
- Krasowska, A.; Piasecki, A.; Murzyn, A.; Sigler, K. Assaying the antioxidant and radical scavenging properties of aliphatic mono- and di-N-oxides in superoxide dismutase-deficient yeast and in a chemiluminescence test. Folia Microbiol. 2007, 52, 45–51. [Google Scholar] [CrossRef]
- Anderson, R.F.; Shinde, S.S.; Hay, M.P.; Denny, W.A. Potentiation of the cytotoxicity of the anticancer agent tirapazamine by benzotriazine N-oxides: the role of redox equilibria. J. Am. Chem. Soc. 2006, 128, 245–249. [Google Scholar] [CrossRef]
- Hordyjewska, A.; Ostapiuk, A.; Horecka, A.; Kurzepa, J. Betulin and betulinic acid: Triterpenoids derivatives with a powerful biological potential. Phytochem. Rev. 2019, 18, 929–951. [Google Scholar] [CrossRef] [Green Version]
- Bednarczyk-Cwynar, B. An overwiew on the chemistry and biochemistry of triterpenoids. Mini-Rev. Org. Chem. 2014, 11, 251–252. [Google Scholar] [CrossRef]
- Amiria, S.; Dastghaibb, S.; Ahmadic, M.; Mehrbodd, P.; Khademe, F.; Behroujb, H.; Aghanoorif, M.R.; Machajg, F.; Ghamsaric, M.; Rosikg, J.; et al. Betulin and its derivatives as novel compounds with different pharmacological effects. Biotechnol. Adv. 2020, 38, 107409. [Google Scholar] [CrossRef]
- Fulda, S.; Kroemer, G. Targeting mitochondrial apoptosis by betulinic acid in human cancers. Drug Discov. Today 2009, 14, 885–890. [Google Scholar] [CrossRef]
- Kumar, P.; Bhadauria, A.S.; Singh, A.K.; Saha, S. Betulinic acid as apoptosis activator: Molecular mechanisms, mathematical modeling and chemical modifications. Life Sci. 2018, 209, 24–33. [Google Scholar] [CrossRef]
- Csuk, R.; Barthel, A.; Schwarz, S.; Kommera, H.; Paschke, R. Synthesis and biological evaluation of antitumor-active γ-butyrolactone substituted betulin derivatives. Bioorganic Med. Chem. 2010, 18, 2549–2558. [Google Scholar] [CrossRef] [PubMed]
- Sommerwerk, S.; Heller, L.; Kerzig, C.; Kramell, A.E.; Csuk, R. Rhodamine B conjugates of triterpenoic acids are cytotoxic mitocans even at nanomolar concentrations. Eur. J. Med. Chem. 2017, 127, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Wiemann, J.; Heller, L.; Perl, V.; Kluge, R.; Ströhl, D.; Csuk, R. Betulinic acid derived hydroxamates and betulin derived carbamates are interesting scaffolds for the synthesis of novel cytotoxic compounds. Eur. J. Med. Chem. 2015, 106, 194–210. [Google Scholar] [CrossRef] [PubMed]
- Wolfram, R.K.; Fischer, L.; Kluge, R.; Ströhl, D.; Al-Harrasi, A.; Csuk, R. Homopiperazine-rhodamine B adducts of triterpenoic acids are strong mitocans. Eur. J. Med. Chem. 2018, 155, 869–879. [Google Scholar] [CrossRef] [PubMed]
- Wolfram, R.K.; Heller, L.; Csuk, R. Targeting mitochondria: Esters of rhodamine B with triterpenoids are mitocanic triggers of apoptosis. Eur. J. Med. Chem. 2018, 152, 21–30. [Google Scholar] [CrossRef]
- Antimonova, A.N.; Petrenko, N.I.; Shults, E.E.; Polienko, I.F.; Shakirov, M.M.; Irtegova, I.G.; Pokrovskii, M.A.; Sherman, K.M.; Grigor’ev, I.A. Synthetic transformations of higher terpenoids. XXX. Synthesis and cytotoxic activity of betulonic acid amides with a piperidine or pyrrolidine nitroxide moiety. Bioorganicheskaia khimiia 2013, 39, 206–211. [Google Scholar]
- Popov, S.A.; Shpatov, A.V.; Grigor’ev, I.A. Synthesis of substituted esters of ursolic, betulonic, and betulinic acids containing the nitroxyl radical 4-Amino-2,2,6,6-tetramethylpiperidine-1-oxyl. Chem. Nat. Compd. 2015, 51, 87–90. [Google Scholar] [CrossRef]
- Zhao, M.; Gödecke, T.; Gunn, J.; Duan, J.A.; Che, C.T. Protostane and fusidane triterpenes. Molecules 2013, 18, 4054–4080. [Google Scholar] [CrossRef] [PubMed]
- Zykova, T.; Zhu, F.; Wang, L.; Li, H.; Lim, D.Y.; Yao, K.; Roh, E.; Yoon, S.P.; Kim, H.G.; Bae, K.B.; et al. Targeting PRPK function blocks colon cancer metastasis. Mol. Cancer Ther. 2018, 17, 1101–1113. [Google Scholar] [CrossRef] [Green Version]
- Ni, J.; Guo, M.; Cao, Y.; Lei, L.; Liu, K.; Wang, B.; Lu, F.; Zhai, R.; Gao, X.; Yan, C.; et al. Discovery, synthesis of novel fusidic acid derivatives possessed amino-terminal groups at the 3-hydroxyl position with anticancer activity. Eur. J. Med. Chem. 2019, 162, 122–131. [Google Scholar] [CrossRef]
- Magnuson, B.A.; Carr, I.; Bird, R.P. Ability of aberrant crypt foci characteristics to predict colonic tumor incidence in rats fed cholic acid. Cancer Res. 1993, 53, 4499–4504. [Google Scholar]
- Peterlik, M. Role of bile acid secretion in human colorectal cancer. Wien Med. Wochenschr. 2008, 158, 539–541. [Google Scholar] [CrossRef]
- Govdi, A.I.; Sokolova, N.V.; Sorokina, I.V.; Baev, D.S.; Tolstikova, T.G.; Mamatyuk, V.I.; Fadeev, D.S.; Vasilevsky, S.F.; Nenajdenko, V.G. Synthesis of new betulinic acid–peptide conjugates and in vivo and in silico studies of the influence of peptide moieties on the triterpenoid core activity. Med. Chem. Commun. 2015, 6, 230–238. [Google Scholar] [CrossRef]
- Govdi, A.I.; Vasilevsky, S.F.; Sokolova, N.V.; Sorokina, I.V.; Tolstikova, T.G.; Nenajdenko, V.G. Betulonic acid–peptide conjugates: Synthesis and evaluation of anti-inflammatory activity. Mendeleev Commun. 2013, 23, 260–261. [Google Scholar] [CrossRef]
- Sultani, H.N.; Haeri, H.H.; Hinderberger, D.; Westermann, B. Spin-labelled diketopiperazines and peptide–peptoid chimera by Ugi-multi-component-reactions. Org. Biomol. Chem. 2016, 14, 11336–11341. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neves Filho, R.A.W.; Stark, S.; Morejon, M.C.; Westermann, B.; Wessjohann, L.A. 4-Isocyanopermethylbutane-1,1,3-triol (IPB): A convertible isonitrile for multicomponent reactions. Tetrahedron Lett. 2012, 53, 5360–5363. [Google Scholar] [CrossRef]
- Rotstein, B.H.; Mourtada, R.; Kelley, S.O.; Yudin, A.K. Solvatochromic reagents for multicomponent reactions and their utility in the development of cell-permeable macrocyclic peptide vectors. Chem. Eur. J. 2011, 17, 12257–12261. [Google Scholar] [CrossRef] [PubMed]
- Stoll, S.; Schweiger, A. EasySpin, a comprehensive software package for spectral simulation and analysis in EPR. J. Magn. Reson. 2006, 178, 42–55. [Google Scholar] [CrossRef] [PubMed]
- Ondar, M.A.; Grinberg, O.Y.; Dubinskii, A.A.; Lebedev, Y.S. Study of the effect of the medium on the magnetic-resonance parameters of nitroxyl radicals by high-resolution EPR spectroscopy. Sov. J. Chem. Phys. 1985, 3, 781–792. [Google Scholar]
- Snipes, W.; Cupp, J.; Cohn, G.; Keith, A. Electron spinal resonance analysis of the nitroxide spin label 2,2,6,6-tetramethylpipidone-N-oxyl (Tempone) in single crystals of the reduced tempone matrix. Biophys. J. 1974, 14, 20–32. [Google Scholar] [CrossRef] [Green Version]
- Kawamura, T.; Matsunami, S.; Yonezawa, T. Solvent effects on the g-value of di-t-butyl nitric oxide. Bull. Chem. Soc. Jpn. 1967, 40, 1111–1115. [Google Scholar] [CrossRef] [Green Version]
- Roda, A.; Minutello, A.; Angellotti, M.A.; Fini, A. Bile acid structure-activity relationship: Evaluation of bile acid lipophilicity using 1-octanol/water partition coefficient and reverse phase HPLC. J. Lipid Res. 1990, 31, 1433–1443. [Google Scholar] [CrossRef]
- Fulda, S. Betulinic acid is a natural product with a range of biological effects. Int. J. Mol. Sci. 2008, 9, 1096–1107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reyes, F.J.; Centelles, J.J.; Lupiáñez, J.A.; Cascante, M. (2α,3β)-2,3-Dihydroxyolean-12-en-28-oic acid, a new natural triterpene from Olea europea, induces caspase dependent apoptosis selectively in colon adenocarcinoma cells. FEBS Lett. 2006, 580, 6302–6310. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suy, S.; Mitchell, J.B.; Samuni, A.; Mueller, S.; Kasid, U. Nitroxide tempo, a small molecule, induces apoptosis in prostate carcinoma cells and suppresses tumor growth in athymic mice. Cancer 2005, 103, 1302–1313. [Google Scholar] [CrossRef]
- Nie, X.; Li, C.; Hu, S.; Xue, F.; Kang, Y.J.; Zhang, W. An appropriate loading control for western blot analysis in animal models of myocardial ischemic infarction. Biochem. Biophys. Rep. 2017, 12, 108–113. [Google Scholar] [CrossRef]
- Oropesa-Ávila, M.; Fernández-Vega, A.; de La Mata, M.; Maraver, J.G.; Cordero, M.D.; Cotán, D.; Calero, C.P.; Paz, M.V.; Pavón, A.D.; Sánchez, M.A.; et al. Apoptotic microtubules delimit an active caspase free area in the cellular cortex during the execution phase of apoptosis. Cell Death Dis. 2013, 4, e527. [Google Scholar] [CrossRef] [Green Version]
- Xu, J.; Zeng, F.; Wu, H.; Wu, S. A mitochondrial-targeting and NO-based anticancer nanosystem with enhanced photo-controllability and low dark-toxicity. J. Mater. Chem. B 2015, 3, 4904–4912. [Google Scholar] [CrossRef]
- Lee, T.D.; Keana, J.F.W. In situ reduction of nitroxide spin labels with phenylhydrazine in deuteriochloroform solution. Convenient method for obtaining structural information on nitroxides using nuclear magnetic resonance spectroscopy. J. Org. Chem. 1975, 40, 3145–3147. [Google Scholar] [CrossRef]
- Li, Y.; Lei, X.; Li, X.; Lawler, R.G.; Murata, Y.; Komatsu, K.; Turro, N.J. Indirect 1H NMR characterization of H2@C60 nitroxide derivatives and their nuclear spin relaxation. Chem. Commun. 2011, 47, 12527–12529. [Google Scholar] [CrossRef]
- Sladowski, D.; Steer, S.J.; Clothier, R.H.; Balls, M. An improved MTT assay. J. Immunol. Methods 1993, 157, 203–207. [Google Scholar] [CrossRef]
- Kaluđerović, G.N.; Krajnović, T.; Momcilovic, M.; Stosic-Grujicic, S.; Mijatović, S.; Maksimović-Ivanić, D.; Hey-Hawkins, E. Ruthenium(II) p-cymene complex bearing 2,2′-dipyridylamine targets caspase 3 deficient MCF-7 breast cancer cells without disruption of antitumor immune response. J. Inorg. Biochem. 2015, 153, 315–321. [Google Scholar] [CrossRef] [PubMed]
- Krajnović, T.; Kaluđerović, G.N.; Wessjohann, L.A.; Mijatović, S.; Maksimović-Ivanić, D. Versatile antitumor potential of isoxanthohumol: Enhancement of paclitaxel activity in vivo. Pharmacol. Res. 2016, 105, 62–73. [Google Scholar] [CrossRef] [PubMed]
- Maksimovic-Ivanic, D.; Mijatovic, S.; Harhaji, L.; Miljkovic, D.; Dabideen, D.; Fan Cheng, K.; Mangano, K.; Malaponte, G.; Al-Abed, Y.; Libra, M.; et al. Anticancer properties of the novel nitric oxide-donating compound (S,R)-3-phenyl-4,5-dihydro-5-isoxazole acetic acid-nitric oxide in vitro and in vivo. Mol. Cancer Ther. 2008, 7, 510–520. [Google Scholar] [CrossRef] [PubMed] [Green Version]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | PC3 | HT29 | ||
|---|---|---|---|---|
| CV | MTT | CV | MTT | |
| 1 | 24.64 ± 1.78 | 25.43 ± 4.35 | 24.97 ± 0.57 | 19.02 ± 2.26 |
| 6 | 13.69 ± 0.80 | 7.43 ± 0.72 | 13.16 ± 0.97 | 8.98 ± 0.43 |
| 7 | 10.59 ± 0.85 | 10.54 ± 0.91 | 13.82 ± 0.29 | 11.87 ± 0.94 |
| 8 | 7.44 ± 0.80 | 6.00 ± 1.09 | 8.10 ± 0.43 | 7.41 ± 0.56 |
| 9 | 15.26 ± 1.01 | 13.85 ± 2.04 | 6.98 ± 0.25 | 12.94 ± 1.03 |
| 18 | 9.27 ± 0.73 | 6.19 ± 0.20 | 16.30 ± 0.87 | 12.23 ± 0.67 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sultani, H.N.; Morgan, I.; Hussain, H.; Roos, A.H.; Haeri, H.H.; Kaluđerović, G.N.; Hinderberger, D.; Westermann, B. Access to New Cytotoxic Triterpene and Steroidal Acid-TEMPO Conjugates by Ugi Multicomponent-Reactions. Int. J. Mol. Sci. 2021, 22, 7125. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22137125
Sultani HN, Morgan I, Hussain H, Roos AH, Haeri HH, Kaluđerović GN, Hinderberger D, Westermann B. Access to New Cytotoxic Triterpene and Steroidal Acid-TEMPO Conjugates by Ugi Multicomponent-Reactions. International Journal of Molecular Sciences. 2021; 22(13):7125. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22137125
Chicago/Turabian StyleSultani, Haider N., Ibrahim Morgan, Hidayat Hussain, Andreas H. Roos, Haleh H. Haeri, Goran N. Kaluđerović, Dariush Hinderberger, and Bernhard Westermann. 2021. "Access to New Cytotoxic Triterpene and Steroidal Acid-TEMPO Conjugates by Ugi Multicomponent-Reactions" International Journal of Molecular Sciences 22, no. 13: 7125. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22137125