
Iron Chelator Induces Apoptosis in Osteosarcoma Cells by Disrupting Intracellular Iron Homeostasis and Activating the MAPK Pathway


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
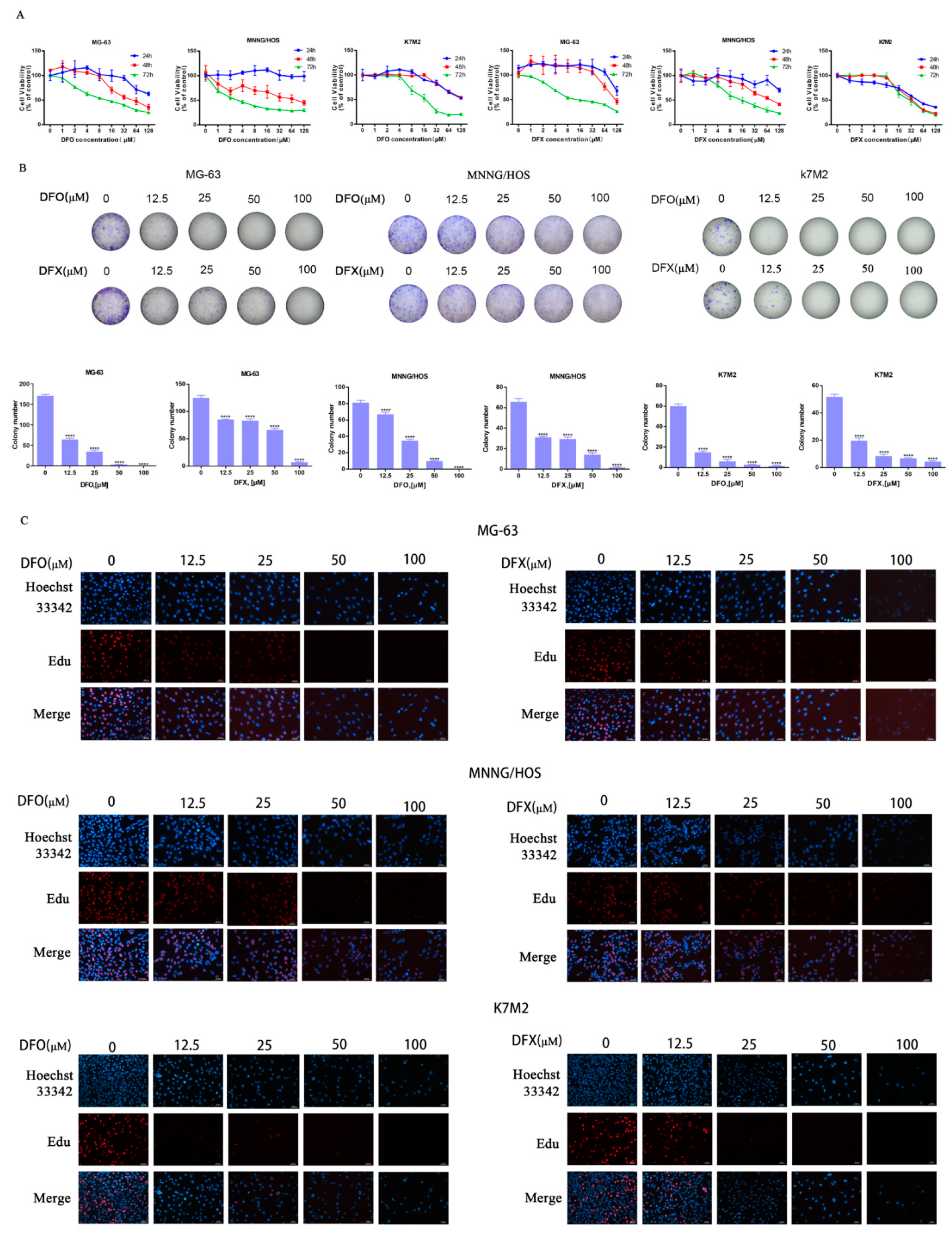
2.1. Iron Chelators DFO and DFX Inhibited Viability of Osteosarcoma Cells and Proliferation In Vitro
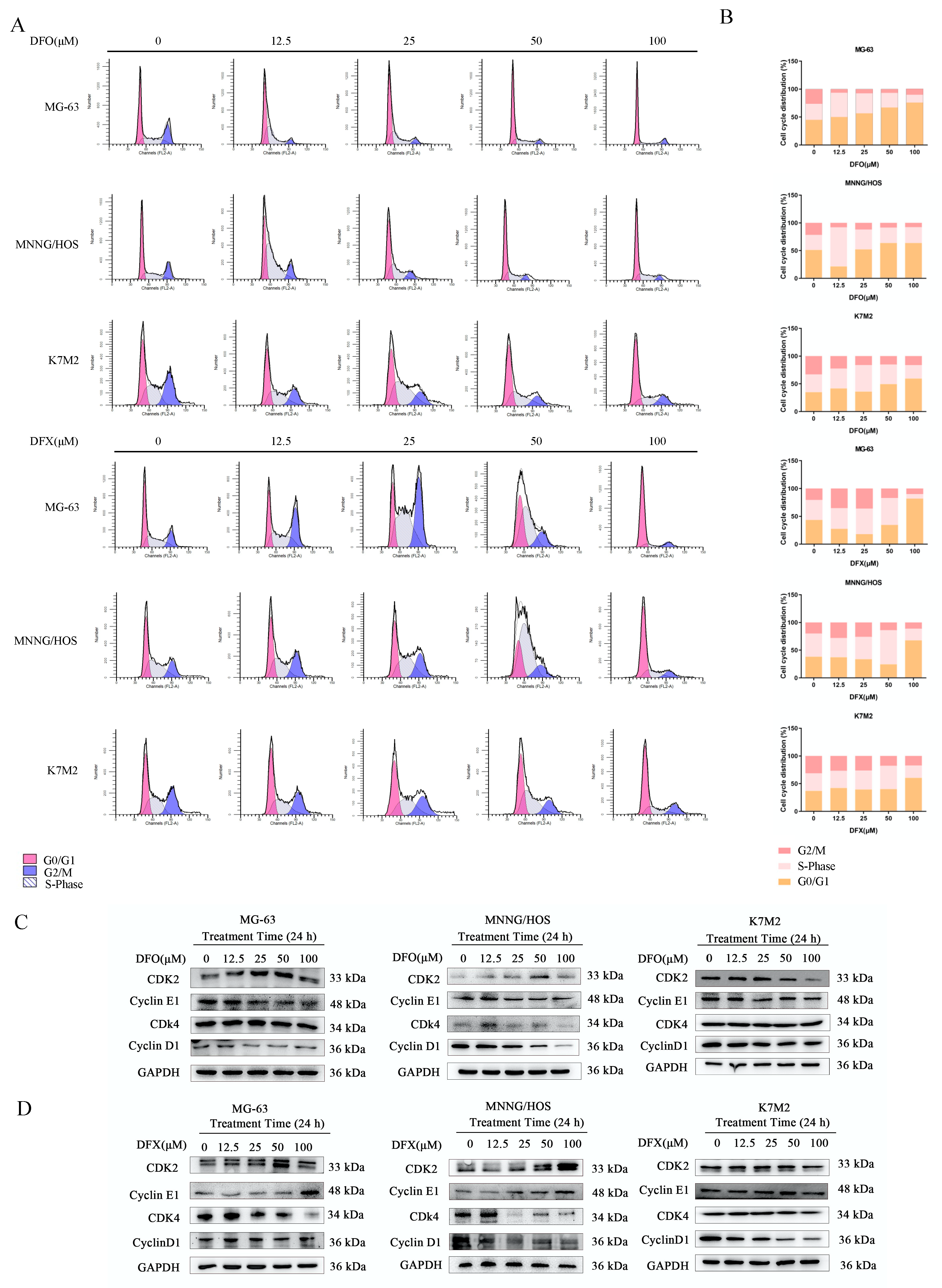
2.2. Iron Chelators Induced Cell-Cycle Arrest in Osteosarcoma Cells
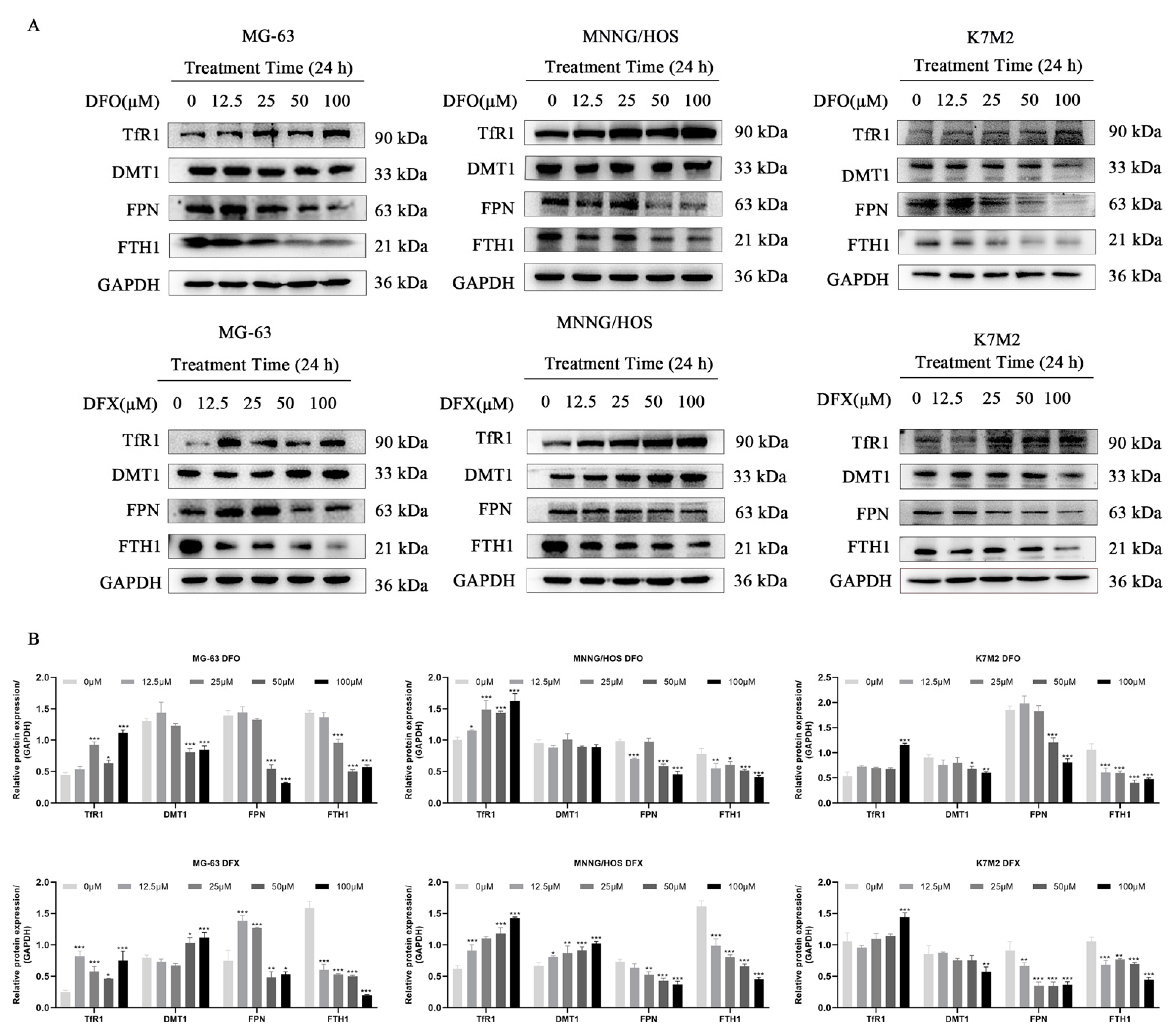
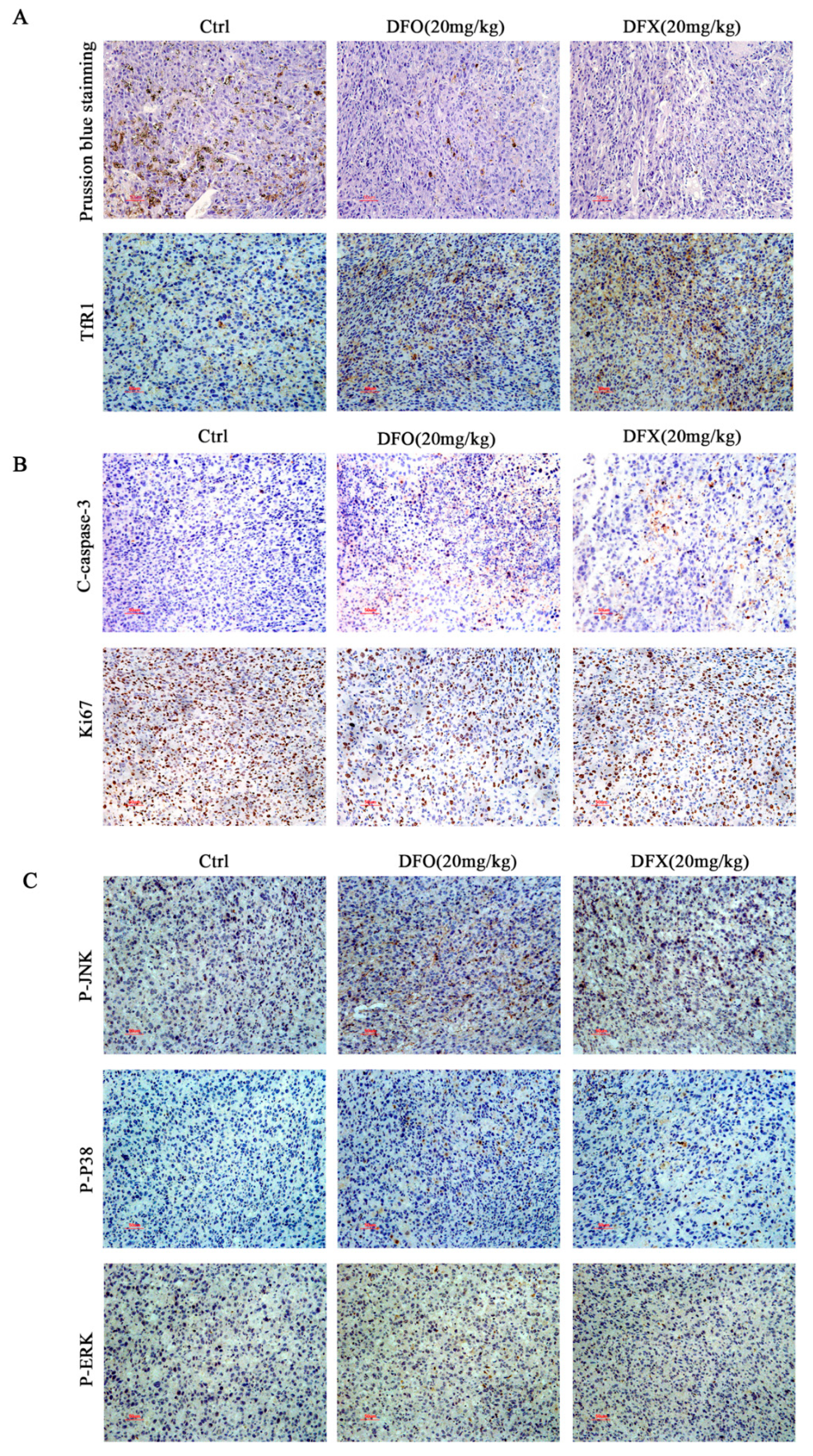
2.3. Iron Chelators Altered Iron Metabolism in Osteosarcoma Cells
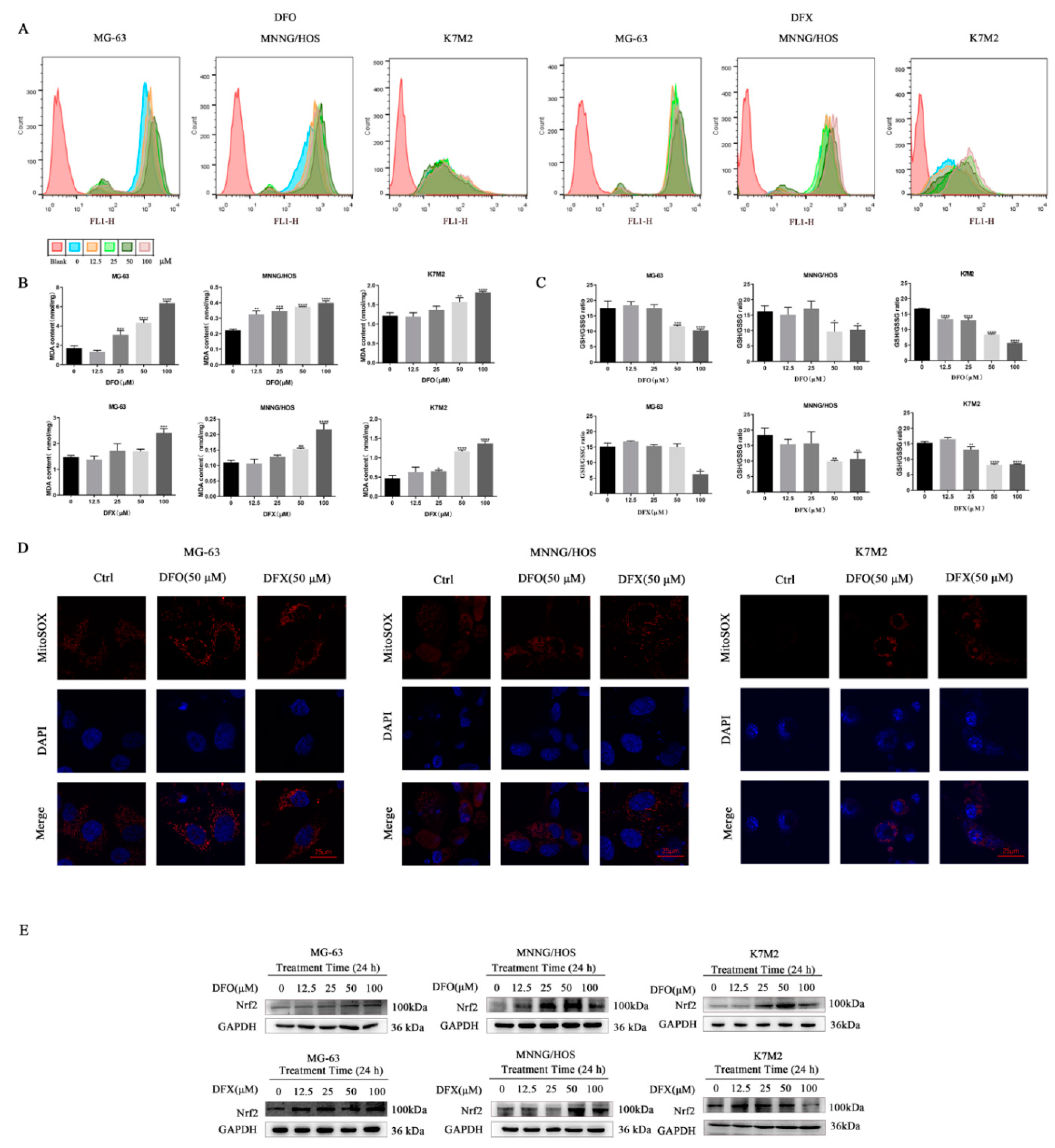
2.4. Iron Chelators Induced Oxidative Stress in Osteosarcoma Cells
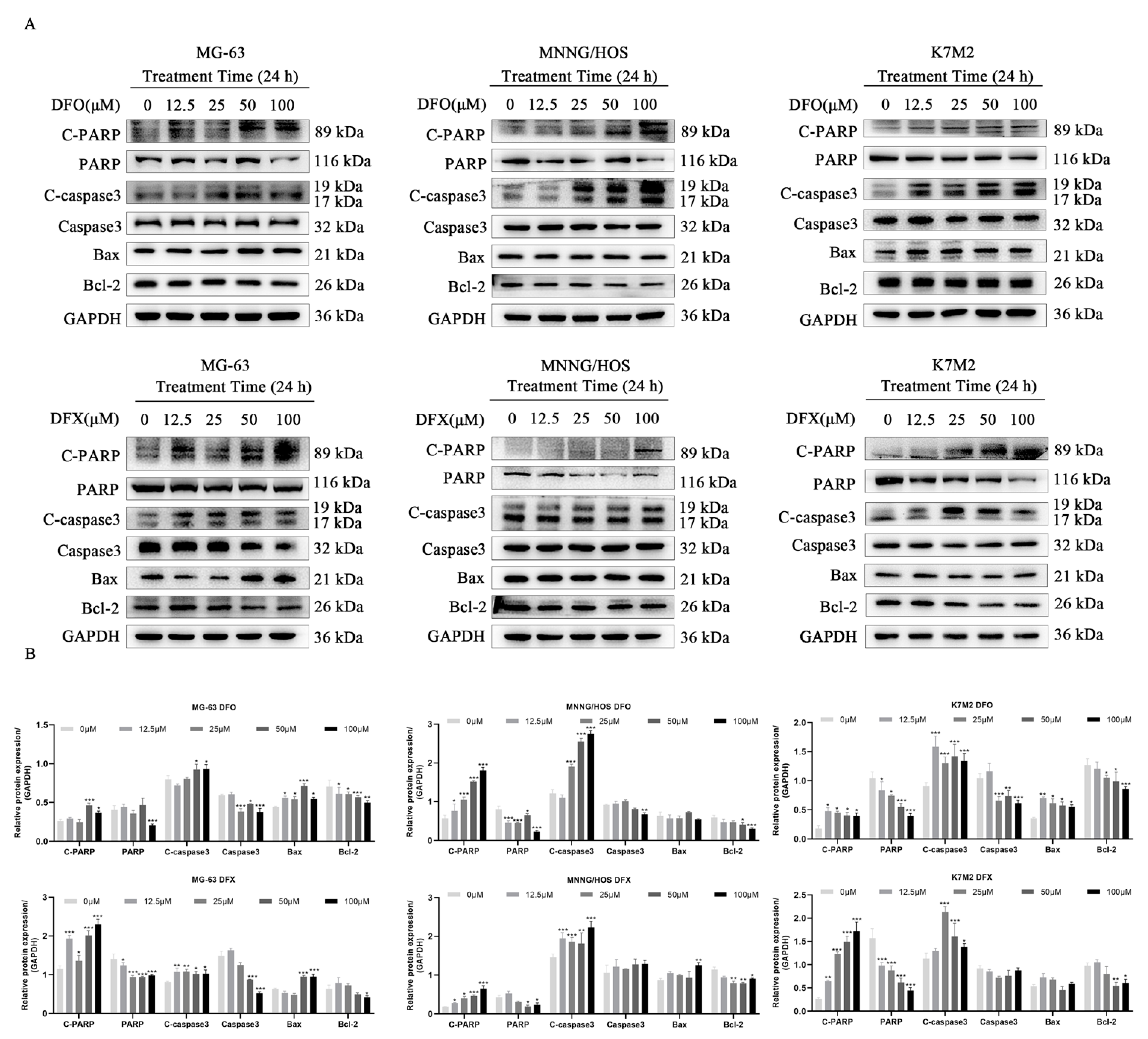
2.5. Iron Chelators Induced Apoptosis in Osteosarcoma Cells
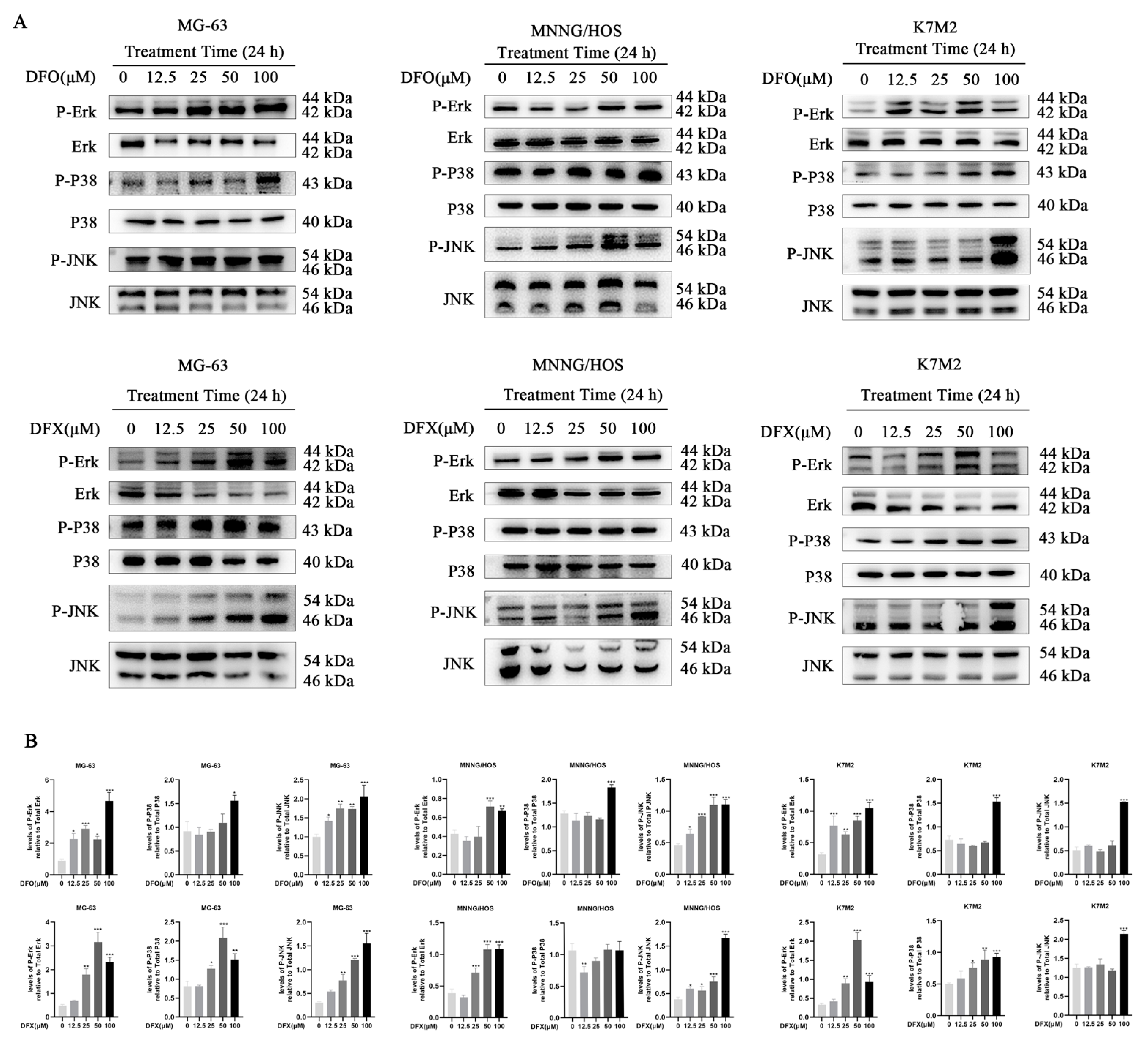
2.6. DFO and DFX Induced Apoptosis by Activating the Ros-Mediated Mapk Pathway in Osteosarcoma Cells
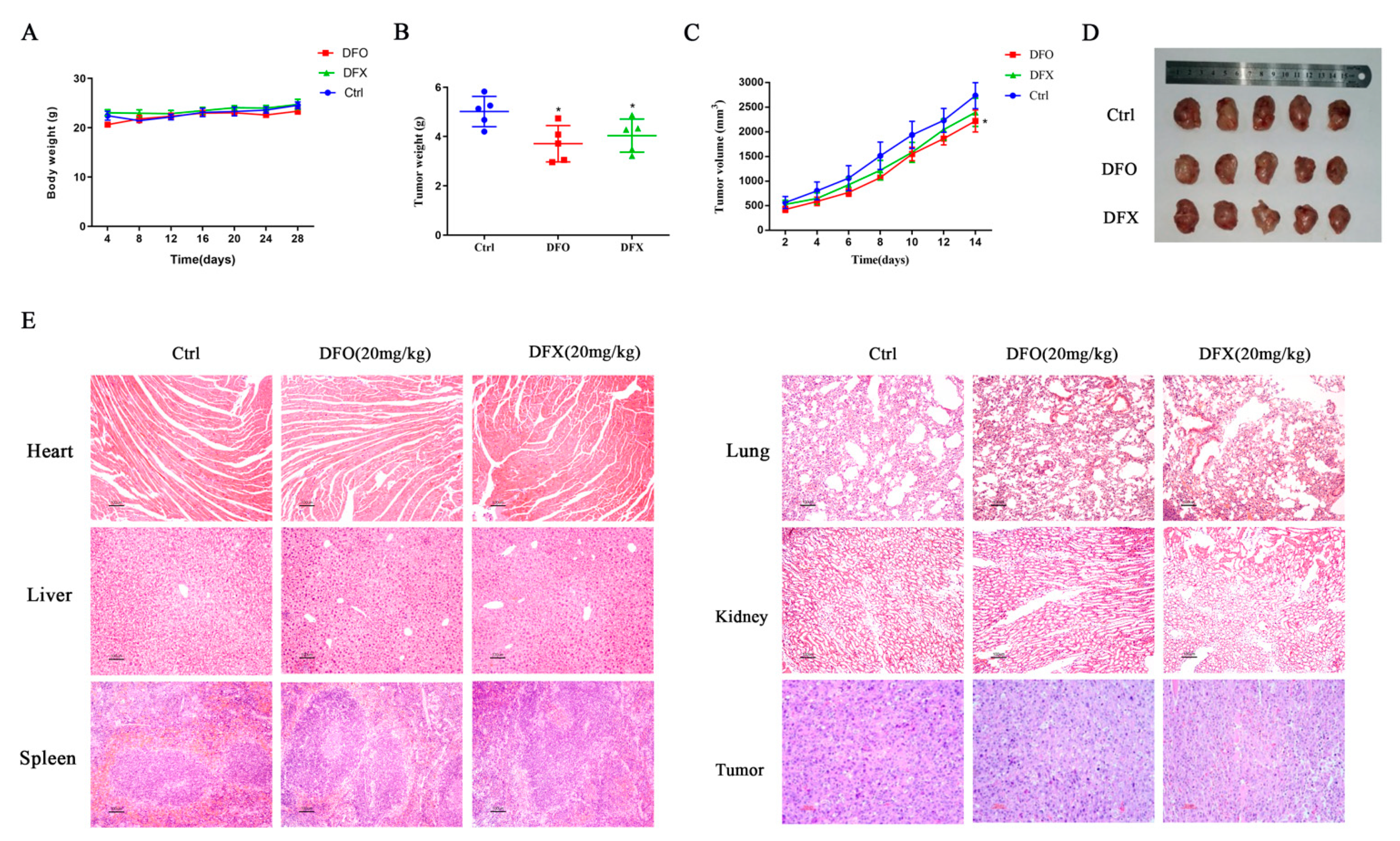
2.7. Iron Chelators Exerted Anti-Tumor Effects in Mice with Osteosarcoma Tumor Allografts
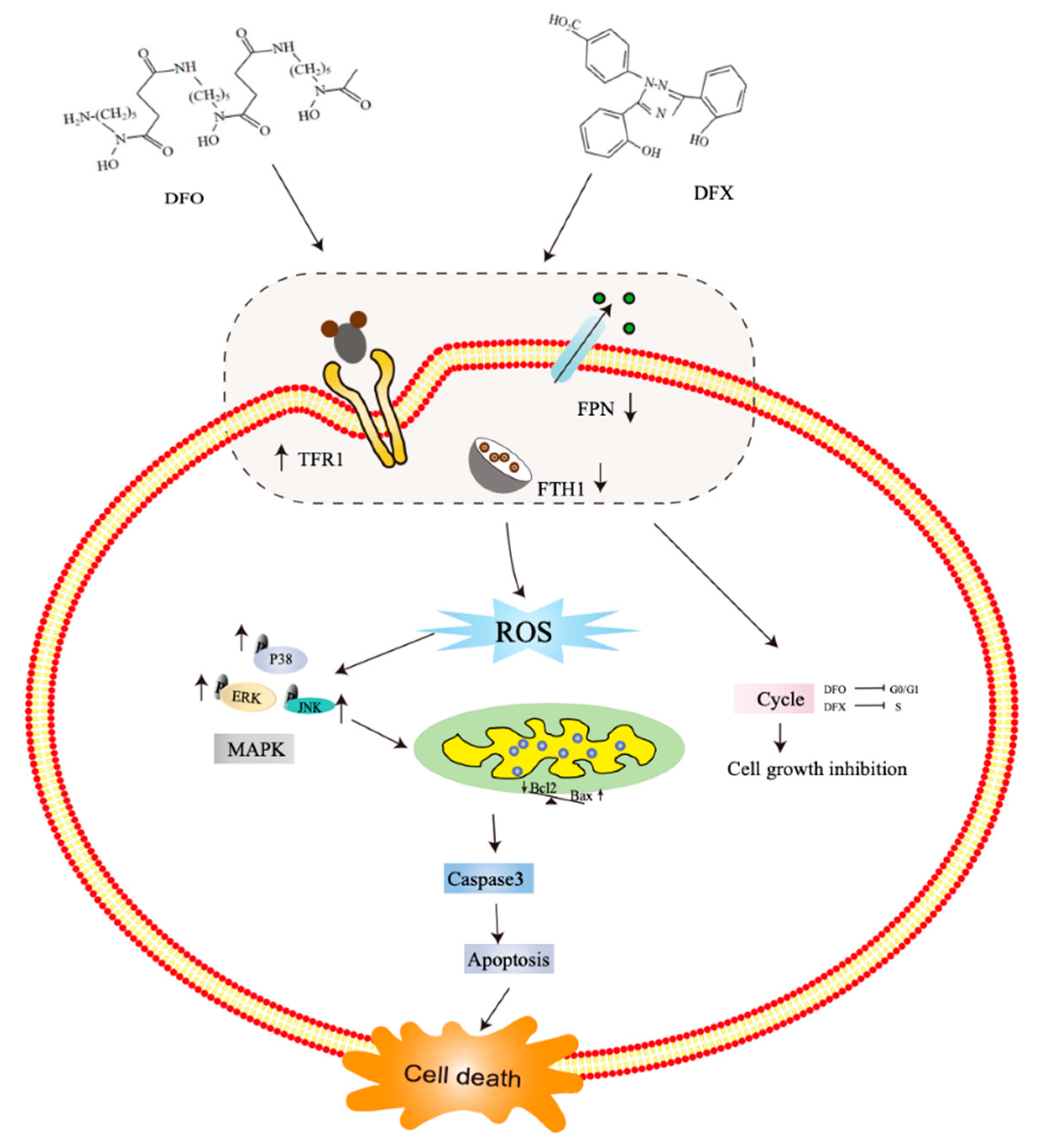
3. Discussion
4. Materials and Methods
4.1. Cell Culture and Chemicals
4.2. Cell Viability Assay
4.3. Colony Formation Assay
4.4. Cell Cycle Analysis
4.5. Measurement of Cytosolic ROS
4.6. Assessment of Mitochondrial Superoxide Production
4.7. Measurement of Malondialdehyde
4.8. Measurement of GSH/GSSG
4.9. Western Blot Analysis
4.10. Orthotopic Transplantation Tumors of k7M2 in Balb/C Mice
4.11. Histological Analysis
4.12. Immunohistochemistry Analysis
4.13. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ottaviani, G.; Jaffe, N. The Epidemiology of Osteosarcoma. Cancer Treat. Res. 2009, 152, 3–13. [Google Scholar]
- Miller, B.J.; Cram, P.; Lynch, C.F.; Buckwalter, J.A. Risk factors for metastatic disease at presentation with osteosarcoma: An analysis of the SEER database. J. Bone Joint Surg. Am. 2013, 95. [Google Scholar] [CrossRef] [Green Version]
- Tsay, J.; Yang, Z.; Ross, F.P.; Cunningham-Rundles, S.; Lin, H.; Coleman, R.; Mayer-Kuckuk, P.; Doty, S.B.; Grady, R.W.; Giardina, P.J.; et al. Bone loss caused by iron overload in a murine model: Importance of oxidative stress. Blood 2010, 116, 2582–2589. [Google Scholar] [CrossRef]
- Harrison, P.; Neilson, J.R.; Marwah, S.S.; Madden, L.; Bareford, D.; Milligan, D.W. Role of non-transferrin bound iron in iron overload and liver dysfunction in long term survivors of acute leukaemia and bone marrow transplantation. J. Clin. Pathol. 1996, 49, 853–856. [Google Scholar] [CrossRef] [Green Version]
- Torti, S.V.; Torti, F.M. Iron and cancer: More ore to be mined. Nat. Rev. Cancer 2013, 13, 342–355. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raza, M.; Chakraborty, S.; Choudhury, M.; Ghosh, P.C.; Nag, A. Cellular iron homeostasis and therapeutic implications of iron chelators in cancer. Curr. Pharm. Biotechnol. 2014, 15, 1125–1140. [Google Scholar] [CrossRef] [PubMed]
- Bystrom, L.M.; Rivella, S. Cancer cells with irons in the fire. Free. Radic. Biol. Med. 2015, 79, 337–342. [Google Scholar] [CrossRef] [Green Version]
- Lv, H.; Zhen, C.; Liu, J.; Shang, P. beta-Phenethyl Isothiocyanate Induces Cell Death in Human Osteosarcoma through Altering Iron Metabolism, Disturbing the Redox Balance, and Activating the MAPK Signaling Pathway. Oxid. Med. Cell. Longev. 2020. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shen, Y.; Zhang, B.; Su, Y.; Badshah, S.A.; Wang, X.; Li, X.; Xue, Y.; Xie, L.; Wang, Z.; Yang, Z.; et al. Iron Promotes Dihydroartemisinin Cytotoxicity via ROS Production and Blockade of Autophagic Flux via Lysosomal Damage in Osteosarcoma. Front. Pharmacol. 2020, 11. [Google Scholar] [CrossRef]
- Zhang, J.; Hu, W.; Ding, C.; Yao, G.; Zhao, H.; Wu, S. Deferoxamine inhibits iron-uptake stimulated osteoclast differentiation by suppressing electron transport chain and MAPKs signaling. Toxicol. Lett. 2019, 313, 50–59. [Google Scholar] [CrossRef]
- Xue, Y.; Yang, J.; Luo, J.; Ren, L.; Shen, Y.; Dong, D.; Fang, Y.; Hu, L.; Liu, M.; Liao, Z.; et al. Disorder of Iron Metabolism Inhibits the Recovery of Unloading-Induced Bone Loss in Hypomagnetic Field. J. Bone Miner. Res. 2019, 35, 1163–1173. [Google Scholar] [CrossRef]
- Tam, T.F.; Leung-Toung, R.; Li, W.; Wang, Y.; Karimian, K.; Spino, M. Iron Chelator Research: Past, Present, and Future. Curr. Med. Chem. 2003, 10, 983–995. [Google Scholar] [CrossRef]
- Agarwal, M. Deferasirox: Oral, once daily iron chelator—An expert opinion. Indian J. Pediatr. 2010, 77, 185–191. [Google Scholar] [CrossRef]
- Crisponi, G.; Nurchi, V.M.; Lachowicz, J.I. Iron Chelation for Iron Overload in Thalassemia. Met. Ions Life Sci. 2019, 19. [Google Scholar] [CrossRef]
- Kim, J.-L.; Kang, H.-N.; Kang, M.H.; Yoo, Y.A.; Kim, J.S.; Choi, C.W. The Oral Iron Chelator Deferasirox Induces Apoptosis in Myeloid Leukemia Cells by Targeting Caspase. Acta Haematol. 2011, 126, 241–245. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.G.; Kim, J.-L.; Park, J.; Lee, S.; Park, S.J.; Kim, J.S.; Choi, C.W. Effects of oral iron chelator deferasirox on human malignant lymphoma cells. Korean J. Hematol. 2012, 47, 194–201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lui, G.; Obeidy, P.; Ford, S.J.; Tselepis, C.; Sharp, D.M.; Jansson, P.; Kalinowski, D.S.; Kovacevic, Z.; Lovejoy, D.B.; Richardson, D. The Iron Chelator, Deferasirox, as a Novel Strategy for Cancer Treatment: Oral Activity Against Human Lung Tumor Xenografts and Molecular Mechanism of Action. Mol. Pharmacol. 2012, 83, 179–190. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ford, S.J.; Obeidy, P.; Lovejoy, D.B.; Bedford, M.; Nichols, L.; Chadwick, C.; Tucker, O.; Lui, G.Y.; Kalinowski, D.S.; Jansson, P.J.; et al. Deferasirox (ICL670A) effectively inhibits oesophageal cancer growth in vitro and in vivo. Br. J. Pharmacol. 2013, 168, 1316–1328. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buss, J.L.; Greene, B.T.; Turner, J.; Torti, F.M.; Torti, S.V. Iron chelators in cancer chemotherapy. Curr. Top. Med. Chem. 2004, 4, 1623–1635. [Google Scholar] [CrossRef]
- Kim, J.L.; Lee, D.-H.; Na, Y.J.; Kim, B.R.; Jeong, Y.A.; Lee, S.I.; Kang, S.; Joung, S.Y.; Lee, S.-Y.; Oh, S.C.; et al. Iron chelator-induced apoptosis via the ER stress pathway in gastric cancer cells. Tumor Biol. 2016, 37, 9709–9719. [Google Scholar] [CrossRef]
- Li, P.; Zheng, X.; Shou, K.; Niu, Y.; Jian, C.; Zhao, Y.; Yi, W.; Hu, X.; Yu, A. The iron chelator Dp44mT suppresses osteosarcoma’s proliferation, invasion and migration: In vitro and in vivo. Am. J. Transl. Res. 2016, 8, 5370–5385. [Google Scholar]
- Dixon, S.J.; Stockwell, B.R. The role of iron and reactive oxygen species in cell death. Nat. Chem. Biol. 2014, 10, 9–17. [Google Scholar] [CrossRef]
- Toyokuni, S. Iron-induced carcinogenesis: The role of redox regulation. Free Radic. Biol. Med. 1996, 20, 553–566. [Google Scholar] [CrossRef]
- Inoue, S.; Kawanishi, S. Hydroxyl radical production and human DNA damage induced by ferric nitrilotriacetate and hydrogen peroxide. Cancer Res. 1987, 47, 6522–6527. [Google Scholar]
- Zhou, L.; Zhao, B.; Zhang, L.; Wang, S.; Dong, D.; Lv, H.; Shang, P. Alterations in Cellular Iron Metabolism Provide More Therapeutic Opportunities for Cancer. Int. J. Mol. Sci. 2018, 19, 1545. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, B.; Zhang, J.-Y.; Liu, X.-S.; Chen, H.-Z.; Ai, Y.-L.; Cheng, K.; Sun, R.-Y.; Zhou, D.; Han, J.; Wu, Q. Tom20 senses iron-activated ROS signaling to promote melanoma cell pyroptosis. Cell Res. 2018, 28, 1171–1185. [Google Scholar] [CrossRef] [Green Version]
- Mòdol, T.; Brice, N.; de Galarreta, M.R.; Garzón, A.G.; Iraburu, M.J.; Martínez-Irujo, J.; López-Zabalza, M.J. Fibronectin Peptides as Potential Regulators of Hepatic Fibrosis Through Apoptosis of Hepatic Stellate Cells. J. Cell. Physiol. 2014, 230, 546–553. [Google Scholar] [CrossRef]
- Pan, M.-H.; Chiou, Y.-S.; Cheng, A.-C.; Bai, N.; Lo, C.-Y.; Tan, D.; Ho, T.-T. Involvement of MAPK, Bcl-2 family, cytochrome c, and caspases in induction of apoptosis by 1,6-O,O-diacetylbritannilactone in human leukemia cells. J. Mol. Nutr. Food Res. 2007, 51, 229–238. [Google Scholar] [CrossRef] [PubMed]
- El-Najjar, N.; Chatila, M.; Moukadem, H.; Vuorela, H.; Ocker, M.; Gandesiri, M.; Schneider-Stock, R.; Gali-Muhtasib, H. Reactive oxygen species mediate thymoquinone-induced apoptosis and activate ERK and JNK signaling. Apoptosis 2010, 15, 183–195. [Google Scholar] [CrossRef] [PubMed]
- Sakata, S.; Hayashi, S.; Fujishiro, T.; Kawakita, K.; Kanzaki, N.; Hashimoto, S.; Iwasa, K.; Chinzei, N.; Kihara, S.; Haneda, M.; et al. Oxidative stress-induced apoptosis and matrix loss of chondrocytes is inhibited by eicosapentaenoic acid. J. Orthop. Res. 2014, 33, 359–365. [Google Scholar] [CrossRef] [PubMed]
- Ding, L.; Cao, J.; Lin, W.; Chen, H.; Xiong, X.; Ao, H.; Yu, M.; Lin, J.; Cui, Q. The Roles of Cyclin-Dependent Kinases in Cell-Cycle Progression and Therapeutic Strategies in Human Breast Cancer. Int. J. Mol. Sci. 2020, 21, 1960. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beard, R.R.; Iron, J.L.; Bowman, B.A. Present Knowledge in Nutrition, 9th ed.; International Life Sciences Institute: Washington, DC, USA, 2006. [Google Scholar]
- Yang, J.; Zhang, G.; Dong, D.; Shang, P. Effects of Iron Overload and Oxidative Damage on the Musculoskeletal System in the Space Environment: Data from Spaceflights and Ground-Based Simulation Models. Int. J. Mol. Sci. 2018, 19, 2608. [Google Scholar] [CrossRef] [Green Version]
- Tonelli, C.; Chio, I.I.C.; Tuveson, D.A. Transcriptional Regulation by Nrf2. Antioxid. Redox Signal. 2018, 29, 1727–1745. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tury, S.; Assayag, F.; Bonin, F.; Chateau-Joubert, S.; Servely, J.-L.; Vacher, S.; Becette, V.; Caly, M.; Rapinat, A.; Gentien, D.; et al. The iron chelator deferasirox synergises with chemotherapy to treat triple-negative breast cancers. J. Pathol. 2018, 246, 103–114. [Google Scholar] [CrossRef]
- Kim, B.-S.; Yoon, K.-H.; Oh, H.-M.; Choi, E.-Y.; Kim, S.-W.; Han, W.-C.; Kim, E.-A.; Choi, S.-C.; Kim, T.-H.; Yun, K.-J.; et al. Involvement of p38 MAP kinase during iron chelator-mediated apoptotic cell death. Cell. Immunol. 2002, 220, 96–106. [Google Scholar] [CrossRef]
- Stevens, R.G.; Graubard, B.I.; Micozzi, M.S.; Neriishi, K.; Blumberg, B.S. Moderate elevation of body iron level and increased risk of cancer occurrence and death. Int. J. Cancer 1994, 56, 364–369. [Google Scholar] [CrossRef]
- Richardson, D. Iron chelators as therapeutic agents for the treatment of cancer. Crit. Rev. Oncol. Hematol. 2002, 42, 267–281. [Google Scholar] [CrossRef]
- Moon, J.-H.; Jeong, J.-K.; Park, S.-Y. Deferoxamine inhibits TRAIL-mediated apoptosis via regulation of autophagy in human colon cancer cells. Oncol. Rep. 2014, 33, 1171–1176. [Google Scholar] [CrossRef]
- Hann, H.-W.L.; Stahlhut, M.W.; Rubin, R.; Maddrey, W.C. Antitumor effect of deferoxamine on human hepatocellular carcinoma growing in athymic nude mice. Cancer 1992, 70, 2051–2056. [Google Scholar] [CrossRef]
- Estrov, Z.; Tawa, A.; Wang, X.H.; Dubé, I.D.; Sulh, H.; Cohen, A.; Gelfand, E.W.; Freedman, M.H. In vitro and in vivo effects of deferoxamine in neonatal acute leukemia. Blood 1987, 69, 757–761. [Google Scholar] [CrossRef] [Green Version]
- Cappellini, M.D.; Cohen, A.; Piga, A.; Bejaoui, M.; Perrotta, S.; Agaoglu, L.; Aydinok, Y.; Kattamis, A.; Kilinc, Y.; Porter, J.; et al. A phase 3 study of deferasirox (ICL670), a once-daily oral iron chelator, in patients with beta-thalassemia. Blood 2006, 107, 3455–3462. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bedford, M.R.; Ford, S.J.; Horniblow, R.D.; Iqbal, T.H.; Tselepis, C. Iron Chelation in the Treatment of Cancer: A New Role for Deferasirox? J. Clin. Pharmacol. 2013, 53, 885–891. [Google Scholar] [CrossRef]
- Bajbouj, K.; Shafarin, J.; Hamad, M. High-Dose Deferoxamine Treatment Disrupts Intracellular Iron Homeostasis, Reduces Growth, and Induces Apoptosis in Metastatic and Nonmetastatic Breast Cancer Cell Lines. Technol. Cancer Res. Treat. 2018, 17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Corcé, V.; Gouin, S.; Renaud, S.; Gaboriau, F.; Deniaud, D. Recent advances in cancer treatment by iron chelators. Bioorganic Med. Chem. Lett. 2016, 26, 251–256. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, C.; Wang, S.; Liu, P. Deferoxamine Enhanced Mitochondrial Iron Accumulation and Promoted Cell Migration in Triple-Negative MDA-MB-231 Breast Cancer Cells Via a ROS-Dependent Mechanism. Int. J. Mol. Sci. 2019, 20, 4952. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, S.-F.; Cheng, S.-D.; Chuang, W.-Y.; Chen, I.-H.; Liao, C.-T.; Wang, H.-M.; Hsieh, L.-L. Cyclin D1 overexpression and poor clinical outcomes in Taiwanese oral cavity squamous cell carcinoma. World J. Surg. Oncol. 2012, 10. [Google Scholar] [CrossRef] [Green Version]
- Fukuchi, K.; Tomoyasu, S.; Watanabe, H.; Kaetsu, S.; Tsuruoka, N.; Gomi, K. Iron deprivation results in an increase in p53 ex-pression. Biol. Chem. Hoppe Seyler 1995, 376, 627–630. [Google Scholar]
- Brodie, C.; Siriwardana, G.; Lucas, J.; Schleicher, R.; Terada, N.; Szepesi, A.; Gelfand, E.; Seligman, P. Neuroblastoma sensitivity to growth inhibition by deferrioxamine: Evidence for a block in G1 phase of the cell cycle. Cancer Res. 1993, 53, 3968–3975. [Google Scholar]
- Renton, F.J.; Jeitner, T.M. Cell cycle-dependent inhibition of the proliferation of human neural tumor cell lines by iron chelators. Biochem. Pharmacol. 1996, 51, 1553–1561. [Google Scholar] [CrossRef]









Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xue, Y.; Zhang, G.; Zhou, S.; Wang, S.; Lv, H.; Zhou, L.; Shang, P. Iron Chelator Induces Apoptosis in Osteosarcoma Cells by Disrupting Intracellular Iron Homeostasis and Activating the MAPK Pathway. Int. J. Mol. Sci. 2021, 22, 7168. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22137168
Xue Y, Zhang G, Zhou S, Wang S, Lv H, Zhou L, Shang P. Iron Chelator Induces Apoptosis in Osteosarcoma Cells by Disrupting Intracellular Iron Homeostasis and Activating the MAPK Pathway. International Journal of Molecular Sciences. 2021; 22(13):7168. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22137168
Chicago/Turabian StyleXue, Yanru, Gejing Zhang, Shoujie Zhou, Shenghang Wang, Huanhuan Lv, Liangfu Zhou, and Peng Shang. 2021. "Iron Chelator Induces Apoptosis in Osteosarcoma Cells by Disrupting Intracellular Iron Homeostasis and Activating the MAPK Pathway" International Journal of Molecular Sciences 22, no. 13: 7168. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22137168