
Hypoxia and Hypoxia-Inducible Factor Signaling in Muscular Dystrophies: Cause and Consequences

 , ,
, , 
Abstract
:1. Introduction
2. Causes of Hypoxia and HIF-1α Pathway Activation in MDs
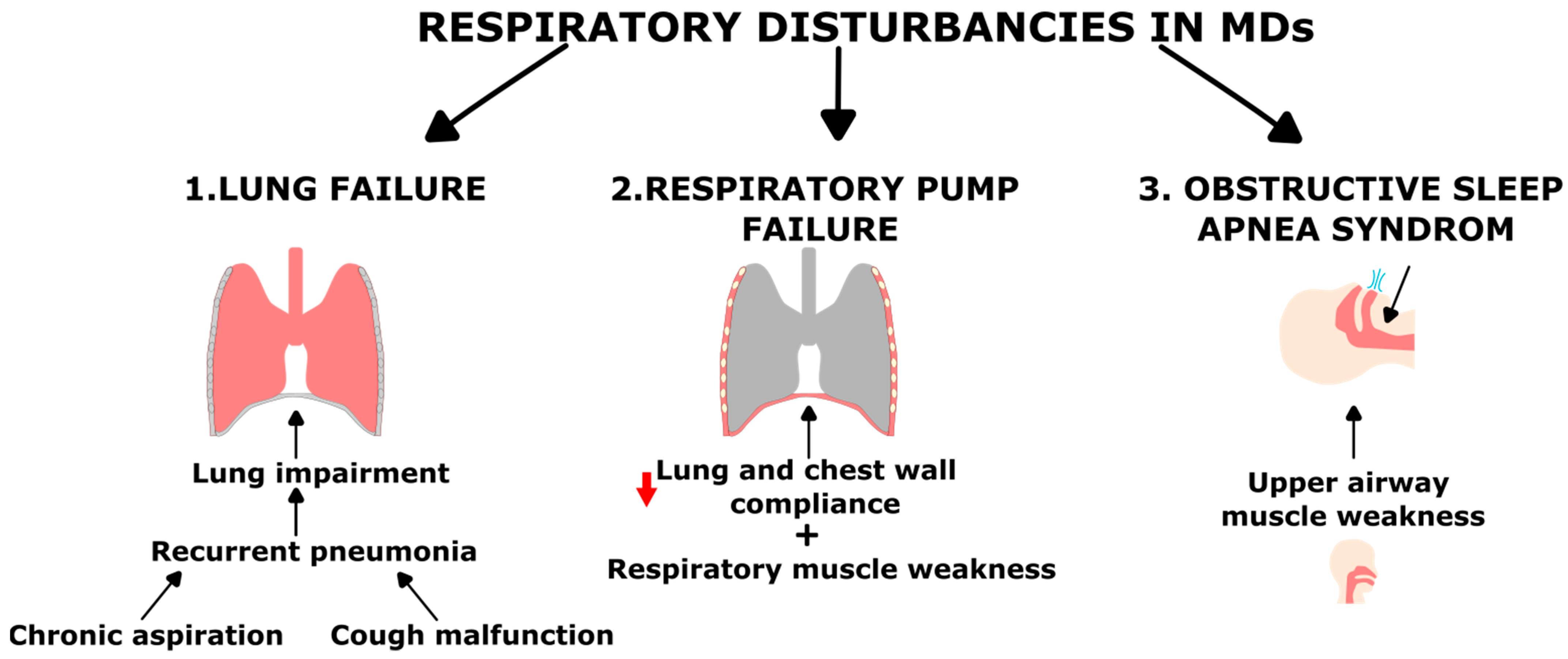
2.1. Respiratory Complications in Muscular Dystrophy
2.2. Muscle Ischemia
2.3. MD Primary Genetic Defect
3. Consequences of Hypoxia and HIF-1α Pathway Activation on Skeletal Muscle
3.1. Impact on Myogenesis and Regeneration
3.2. Ultrastructural Modification
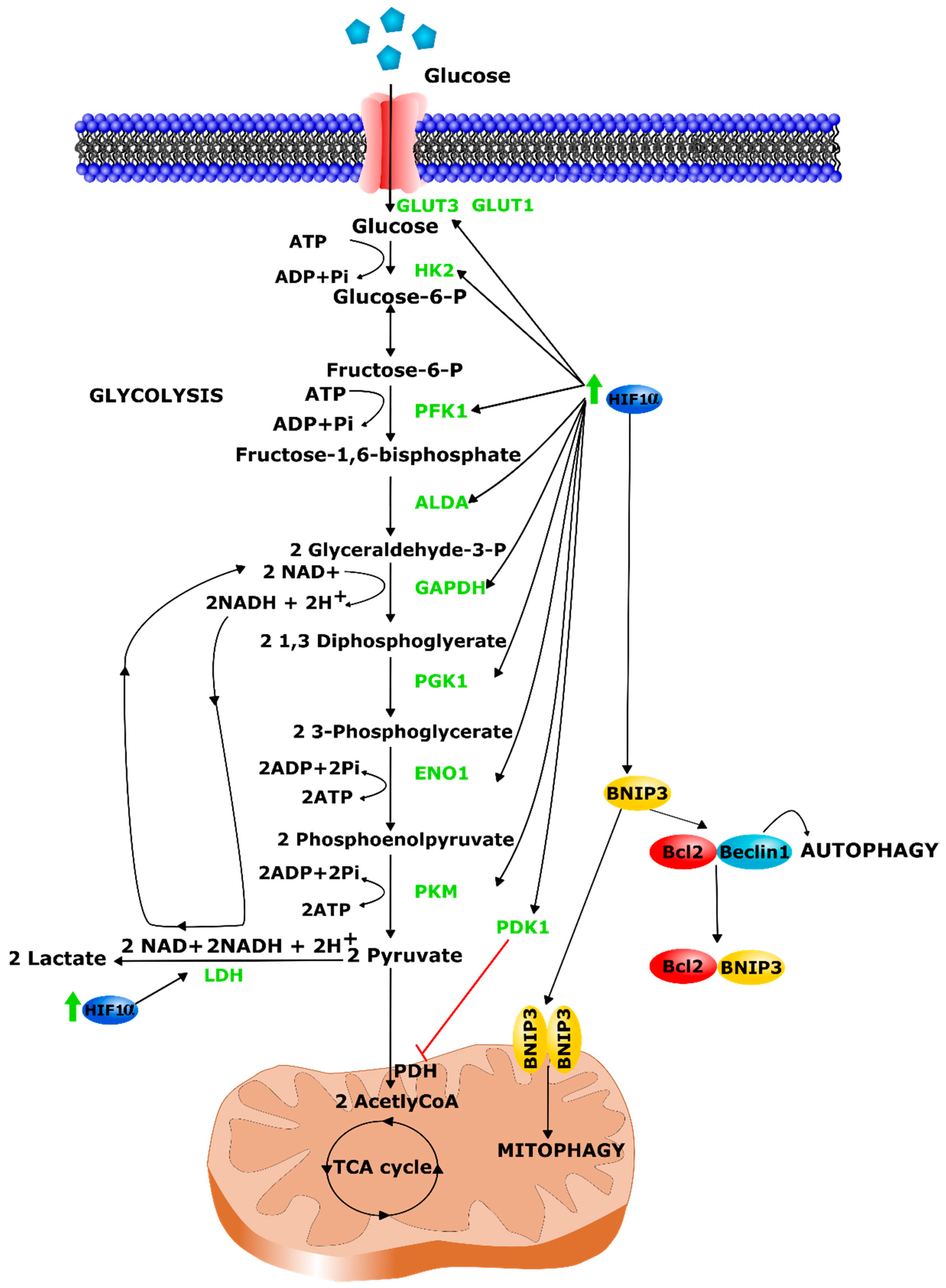
3.3. Metabolic Alterations
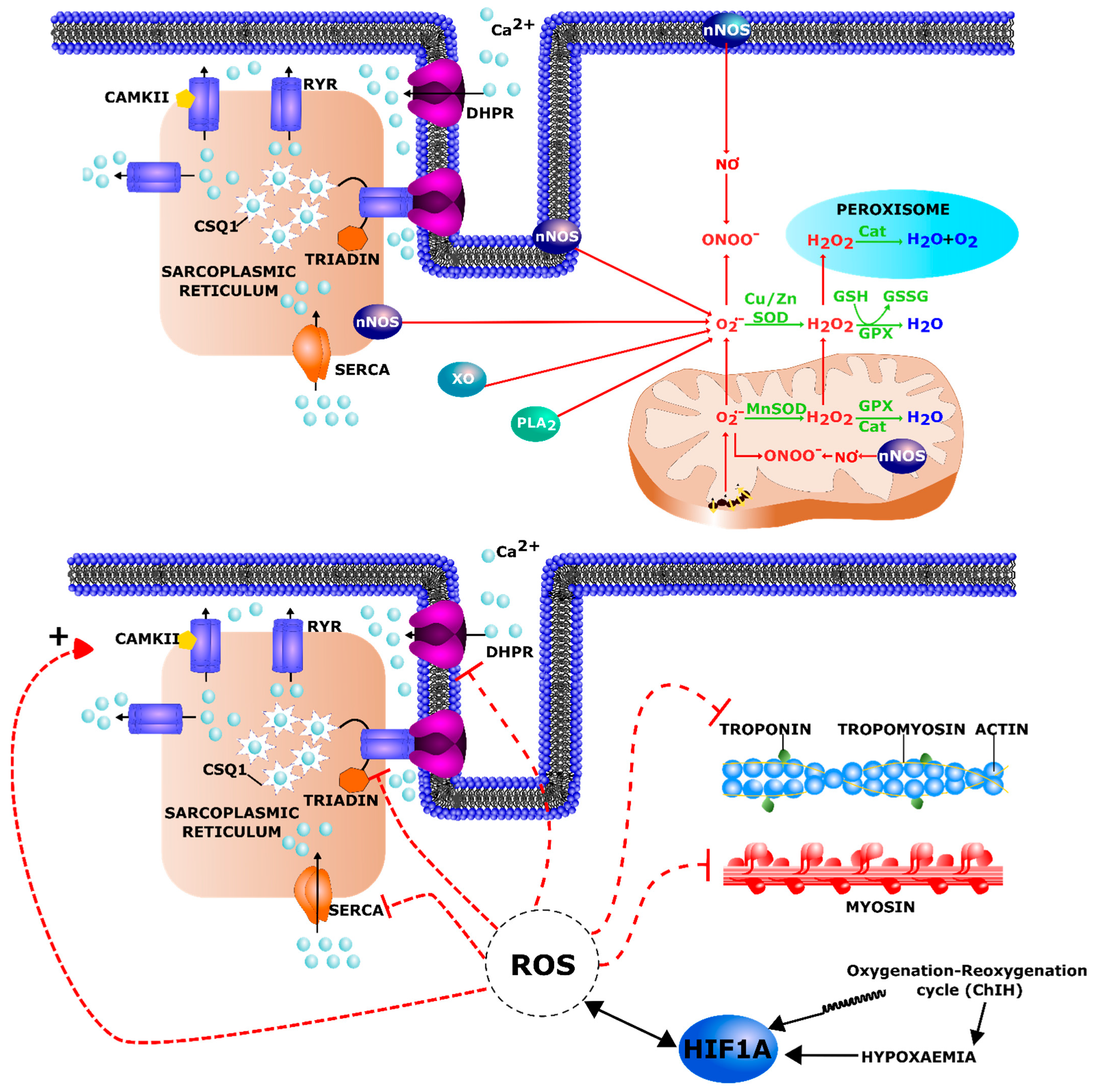
3.4. Oxidative Stress
4. Pharmacological HIF-1α Modulators in MDs
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Shahrizaila, N.; Kinnear, W.J.M.; Wills, A.J. Respiratory involvement in inherited primary muscle conditions. J. Neurol. Neurosurg. Psychiatry 2006, 77, 1108–1115. [Google Scholar] [CrossRef] [PubMed]
- Carter, J.C.; Sheehan, D.W.; Prochoroff, A.; Birnkrant, D.J. Muscular Dystrophies. Clin. Chest Med. 2018, 39, 377–389. [Google Scholar] [CrossRef] [PubMed]
- Mercuri, E.; Bönnemann, C.G.; Muntoni, F. Muscular dystrophies. Lancet 2019, 394, 2025–2038. [Google Scholar] [CrossRef]
- Mensch, A.; Zierz, S. Cellular Stress in the Pathogenesis of Muscular Disorders—From Cause to Consequence. Int. J. Mol. Sci. 2020, 21, 5830. [Google Scholar] [CrossRef]
- The Nobel Prize in Physiology or Medicine. 2019. Available online: https://www.nobelprize.org/prizes/medicine/2019/summary/ (accessed on 27 June 2021).
- Kuiper, C.; Vissers, M.C.M. Ascorbate as a Co-Factor for Fe- and 2-Oxoglutarate Dependent Dioxygenases: Physiological Activity in Tumor Growth and Progression. Front. Oncol. 2014, 4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Safran, M.; Kaelin, W.G. HIF hydroxylation and the mammalian oxygen-sensing pathway. J. Clin. Investig. 2003, 111, 779–783. [Google Scholar] [CrossRef]
- Schofield, C.J.; Ratcliffe, P.J. Oxygen sensing by HIF hydroxylases. Nat. Rev. Mol. Cell Biol. 2004, 5, 343–354. [Google Scholar] [CrossRef]
- Semenza, G.L. Regulation of Mammalian O2Homeostasis by Hypoxia-Inducible Factor. Annu. Rev. Cell Dev. Biol. 1999, 15, 551–578. [Google Scholar] [CrossRef]
- Semenza, G.L. Hypoxia-Inducible Factor 1: Control of Oxygen Homeostasis in Health and Disease. Pediatr. Res. 2001, 49, 614–617. [Google Scholar] [CrossRef] [Green Version]
- Downes, N.L.; Laham-Karam, N.; Kaikkonen, M.U.; Ylä-Herttuala, S. Differential but Complementary HIF1α and HIF2α Transcriptional Regulation. Mol. Ther. 2018, 26, 1735–1745. [Google Scholar] [CrossRef] [Green Version]
- Duan, C. Hypoxia-inducible factor 3 biology: Complexities and emerging themes. Am. J. Physiol. Physiol. 2016, 310, C260–C269. [Google Scholar] [CrossRef] [Green Version]
- Morash, A.J.; Kotwica, A.O.; Murray, A.J. Tissue-specific changes in fatty acid oxidation in hypoxic heart and skeletal muscle. Am. J. Physiol. Integr. Comp. Physiol. 2013, 305, R534–R541. [Google Scholar] [CrossRef]
- Lindholm, M.E.; Rundqvist, H. Skeletal muscle hypoxia-inducible factor-1 and exercise. Exp. Physiol. 2016, 101, 28–32. [Google Scholar] [CrossRef]
- Gan, Z. Hypoxia in skeletal muscles: From physiology to gene expression. Musculoskelet. Regen. 2016, 2. [Google Scholar] [CrossRef] [Green Version]
- Favier, F.B.; Britto, F.A.; Freyssenet, D.G.; Bigard, X.A.; Benoit, H. HIF-1-driven skeletal muscle adaptations to chronic hypoxia: Molecular insights into muscle physiology. Cell. Mol. Life Sci. 2015, 72, 4681–4696. [Google Scholar] [CrossRef] [PubMed]
- Clanton, T.L.; Klawitter, P.F. Invited Review: Adaptive responses of skeletal muscle to intermittent hypoxia: The known and the unknown. J. Appl. Physiol. 2001, 90, 2476–2487. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mauro, M.A.L.; D’Angelo, M.G.; Aliverti, A. Sleep Disordered Breathing in Duchenne Muscular Dystrophy. Curr. Neurol. Neurosci. Rep. 2017, 17, 44. [Google Scholar] [CrossRef]
- Hoque, R. Sleep-Disordered Breathing in Duchenne Muscular Dystrophy: An Assessment of the Literature. J. Clin. Sleep Med. 2016, 12, 905–911. [Google Scholar] [CrossRef] [PubMed]
- Runte, M.; Spiesshoefer, J.; Heidbreder, A.; Dreher, M.; Young, P.; Brix, T.; Boentert, M. Sleep-related breathing disorders in facioscapulohumeral dystrophy. Sleep Breath. 2019, 23, 899–906. [Google Scholar] [CrossRef] [PubMed]
- Allen, J. Pulmonary complications of neuromuscular disease: A Respiratory mechanics perspective. Paediatr. Respir. Rev. 2010, 11, 18–23. [Google Scholar] [CrossRef]
- Lo Mauro, A.; Aliverti, A. Physiology of respiratory disturbances in muscular dystrophies. Breathe 2016, 12, 318–327. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pane, M.; Messina, S.; Vasco, G.; Foley, A.R.; Morandi, L.; Pegoraro, E.; Mongini, T.; D’Amico, A.; Bianco, F.; Lombardo, M.E.; et al. Respiratory and cardiac function in congenital muscular dystrophies with alpha dystroglycan deficiency. Neuromuscul. Disord. 2012, 22, 685–689. [Google Scholar] [CrossRef]
- Johnson, K.; Bertoli, M.; Phillips, L.; Töpf, A.; Van den Bergh, P.; Vissing, J.; Witting, N.; Nafissi, S.; Jamal-Omidi, S.; Łusakowska, A.; et al. Detection of variants in dystroglycanopathy-associated genes through the application of targeted whole-exome sequencing analysis to a large cohort of patients with unexplained limb-girdle muscle weakness. Skelet. Muscle 2018, 8, 23. [Google Scholar] [CrossRef] [Green Version]
- Jimenez-Mallebrera, C.; Brown, S.C.; Sewry, C.A.; Muntoni, F. Congenital muscular dystrophy: Molecular and cellular aspects. Cell. Mol. Life Sci. 2005, 62, 809–823. [Google Scholar] [CrossRef] [PubMed]
- Collins, J.; Van Dyke, R.; Fenchel, M.; McCallum, M.; Völker, S.; Foley, A.R.; Muntoni, F.; Stehling, F.; Schara, U.; Rutkowski, A.; et al. S.P.23 Percent predicted forced vital capacity is a viable outcome measure in Laminin alpha 2—Deficient congenital muscular dystrophy. Neuromuscul. Disord. 2012, 22, 893–894. [Google Scholar] [CrossRef]
- Villar-Quiles, R.N.; Von Der Hagen, M.; Métay, C.; Gonzalez, V.; Donkervoort, S.; Bertini, E.; Castiglioni, C.; Chaigne, D.; Colomer, J.; Cuadrado, M.L.; et al. The clinical, histological, and genotypic spectrum of SEPN1-related myopathy: A case series. Neurology 2020, 95, e1512–e1527. [Google Scholar] [CrossRef]
- Caggiano, S.; Khirani, S.; Dabaj, I.; Cavassa, E.; Amaddeo, A.; Arroyo, J.O.; Desguerre, I.; Richard, P.; Cutrera, R.; Ferreiro, A.; et al. Diaphragmatic dysfunction in SEPN1-related myopathy. Neuromuscul. Disord. 2017, 27, 747–755. [Google Scholar] [CrossRef] [PubMed]
- Yonekawa, T.; Komaki, H.; Okada, M.; Hayashi, Y.K.; Nonaka, I.; Sugai, K.; Sasaki, M.; Nishino, I. Rapidly progressive scoliosis and respiratory deterioration in Ullrich congenital muscular dystrophy. J. Neurol. Neurosurg. Psychiatry 2013, 84, 982–988. [Google Scholar] [CrossRef]
- LoMauro, A.; Romei, M.; Gandossini, S.; Pascuzzo, R.; Vantini, S.; D’Angelo, M.G.; Aliverti, A. Evolution of respiratory function in Duchenne muscular dystrophy from childhood to adulthood. Eur. Respir. J. 2018, 51. [Google Scholar] [CrossRef] [Green Version]
- Taylor, J.; Sewry, C.A.; Dubowitz, V.; Muntoni, F. Early onset, autosomal recessive muscular dystrophy with Emergy-Dreifuss phenotype and normal emerin expression. Neurology 1998, 51, 1116–1120. [Google Scholar] [CrossRef]
- Santos, D.B.; Boussaid, G.; Stojkovic, T.; Orlikowski, D.; Letilly, N.; Béhin, A.; Butel, S.; Lofaso, F.; Prigent, H. Respiratory muscle dysfunction in facioscapulohumeral muscular dystrophy. Neuromuscul. Disord. 2015, 25, 632–639. [Google Scholar] [CrossRef]
- Moreira, S.; Wood, L.; Smith, D.; Marini-Bettolo, C.; Guglieri, M.; McMacken, G.; Bailey, G.; Mayhew, A.; Muni-Lofra, R.; Eglon, G.; et al. Respiratory involvement in ambulant and non-ambulant patients with facioscapulohumeral muscular dystrophy. J. Neurol. 2017, 264, 1271–1280. [Google Scholar] [CrossRef]
- Henke, C.; Spiesshoefer, J.; Kabitz, H.-J.; Herkenrath, S.; Randerath, W.; Brix, T.; Görlich, D.; Young, P.; Boentert, M. Respiratory muscle weakness in facioscapulohumeral muscular dystrophy. Muscle Nerve 2019, 60, 679–686. [Google Scholar] [CrossRef]
- D’Angelo, M.G.; Romei, M.; Lo Mauro, A.; Marchi, E.; Gandossini, S.; Bonato, S.; Comi, G.P.; Magri, F.; Turconi, A.C.; Pedotti, A.; et al. Respiratory pattern in an adult population of dystrophic patients. J. Neurol. Sci. 2011, 306, 54–61. [Google Scholar] [CrossRef]
- Henke, C.; Spiesshoefer, J.; Kabitz, H.-J.; Herkenrath, S.; Randerath, W.; Brix, T.; Görlich, D.; Young, P.; Boentert, M. Characteristics of respiratory muscle involvement in myotonic dystrophy type. Neuromuscul. Disord. 2020, 30, 17–27. [Google Scholar] [CrossRef]
- Hawkins, A.M.; Hawkins, C.L.; Razak, K.A.; Khoo, T.K.; Tran, K.; Jackson, R.V. Respiratory dysfunction in myotonic dystrophy type 1: A systematic review. Neuromuscul. Disord. 2019, 29, 198–212. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perrin, C.; Unterborn, J.N.; Ambrosio, C.D.; Hill, N.S. Pulmonary complications of chronic neuromuscular diseases and their management. Muscle Nerve 2004, 29, 5–27. [Google Scholar] [CrossRef] [PubMed]
- Aliverti, A.; Lo Mauro, A.; D’Angelo, M.G. Assessment and management of respiratory function in patients with Duchenne muscular dystrophy: Current and emerging options. Ther. Clin. Risk Manag. 2015, 11, 1475–1488. [Google Scholar] [CrossRef] [Green Version]
- Kravitz, R.M. Airway Clearance in Duchenne Muscular Dystrophy. Pediatrics 2009, 123, S231–S235. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bach, J.R.; Rajaraman, R.; Ballanger, F.; Tzeng, A.C.; Ishikawa, Y.; Kulessa, R.; Bansal, T. Neuromuscular Ventilatory Insufficiency Effect of Home Mechanical Ventilator Use v Oxygen Therapy on Pneumonia and Hospitalization Rates. Am. J. Phys. Med. Rehabil. 1998, 77, 8–19. [Google Scholar] [CrossRef] [PubMed]
- Eikermann, M.; Vogt, F.M.; Herbstreit, F.; Vahid-Dastgerdi, M.; Zenge, M.O.; Ochterbeck, C.; De Greiff, A.; Peters, J. The Predisposition to Inspiratory Upper Airway Collapse during Partial Neuromuscular Blockade. Am. J. Respir. Crit. Care Med. 2007, 175, 9–15. [Google Scholar] [CrossRef]
- Kurz, L.T.; Mubarak, S.J.; Schultz, P.; Park, S.M.; Leach, J. Correlation of Scoliosis and Pulmonary Function in Duchenne Muscular Dystrophy. J. Pediatr. Orthop. 1983, 3, 347–353. [Google Scholar] [CrossRef]
- Oliveira, J.; Parente Freixo, J.; Santos, M.; Coelho, T. LAMA2 Muscular Dystrophy. In GeneReviews®; Adam, M.P., Ardinger, H.H., Pagon, R.A., Wallace, S.E., Bean, L.J., Mirzaa, G., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 1993. [Google Scholar]
- Saxena, K.; Jolly, M.K. Acute vs. Chronic vs. Cyclic Hypoxia: Their Differential Dynamics, Molecular Mechanisms, and Effects on Tumor Progression. Biomolecules 2019, 9, 339. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Irfan, M.; Selim, B.; Rabinstein, A.A.; St. Louis, E.K. Neuromuscular Disorders and Sleep in Critically Ill Patients. Crit. Care Clin. 2015, 31, 533–550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Phillips, M.F.; Smith, P.E.; Carroll, N.; Edwards, R.H.; Calverley, P.M. Nocturnal Oxygenation and Prognosis in Duchenne Muscular Dystrophy. Am. J. Respir. Crit. Care Med. 1999, 160, 198–202. [Google Scholar] [CrossRef] [PubMed]
- Wohlgemuth, M.; Van Der Kooi, E.L.; Van Kesteren, R.G.; van der Maarel, S.; Padberg, G.W. Ventilatory support in facioscapulohumeral muscular dystrophy. Neurology 2004, 63, 176–178. [Google Scholar] [CrossRef]
- Della Marca, G.; Pantanali, F.; Frusciante, R.; Scarano, E.; Cianfoni, A.; Calò, L.; Dittoni, S.; Vollono, C.; Losurdo, A.; Testani, E.; et al. Cephalometric findings in facioscapulohumeral muscular dystrophy patients with obstructive sleep apneas. Sleep Breath. 2010, 15, 99–106. [Google Scholar] [CrossRef]
- Della Marca, G.; Frusciante, R.; Dittoni, S.; Vollono, C.; Buccarella, C.; Iannaccone, E.; Rossi, M.; Scarano, E.; Pirronti, T.; Cianfoni, A.; et al. Sleep disordered breathing in facioscapulohumeral muscular dystrophy. J. Neurol. Sci. 2009, 285, 54–58. [Google Scholar] [CrossRef]
- Kiyan, E.; Okumus, G.; Cuhadaroglu, C.; Deymeer, F. Sleep apnea in adult myotonic dystrophy patients who have no excessive daytime sleepiness. Sleep Breath. 2009, 14, 19–24. [Google Scholar] [CrossRef]
- Tennant, D.; Howell, N.J. The role of HIFs in ischemia-reperfusion injury. Hypoxia 2014, 2, 107–115. [Google Scholar] [CrossRef] [Green Version]
- Milkiewicz, M.; Pugh, C.W.; Egginton, S. Inhibition of endogenous HIF inactivation induces angiogenesis in ischaemic skeletal muscles of mice. J. Physiol. 2004, 560, 21–26. [Google Scholar] [CrossRef] [PubMed]
- Yiu, E.M.; Kornberg, A.J. Duchenne muscular dystrophy. J. Paediatr. Child Health 2015, 51, 759–764. [Google Scholar] [CrossRef]
- Thomas, G.D. Functional muscle ischemia in Duchenne and Becker muscular dystrophy. Front. Physiol. 2013, 4, 381. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sicinski, P.; Geng, Y.; Ryder-Cook, A.S.; Barnard, E.A.; Darlison, M.G.; Barnard, P.J. The molecular basis of muscular dystrophy in the mdx mouse: A point mutation. Science 1989, 244, 1578–1580. [Google Scholar] [CrossRef] [PubMed]
- Buckley, A.F.; Bossen, E.H. Skeletal Muscle Microvasculature in the Diagnosis of Neuromuscular Disease. J. Neuropathol. Exp. Neurol. 2013, 72, 906–918. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Loufrani, L.; Dubroca, C.; You, D.; Li, Z.; Levy, B.; Paulin, D.; Henrion, D. Absence of Dystrophin in Mice Reduces NO-Dependent Vascular Function and Vascular Density: Total Recovery After a Treatment with the Aminoglycoside Gentamicin. Arter. Thromb. Vasc. Biol. 2004, 24, 671–676. [Google Scholar] [CrossRef] [Green Version]
- Palladino, M.; Gatto, I.; Neri, V.; Straino, S.; Smith, R.C.; Silver, M.; Gaetani, E.; Marcantoni, M.; Giarretta, I.; Stigliano, E.; et al. Angiogenic Impairment of the Vascular Endothelium. Arter. Thromb. Vasc. Biol. 2013, 33, 2867–2876. [Google Scholar] [CrossRef] [Green Version]
- Rhoads, R.P.; Flann, K.L.; Cardinal, T.R.; Rathbone, C.R.; Liu, X.; Allen, R.E. Satellite cells isolated from aged or dystrophic muscle exhibit a reduced capacity to promote angiogenesis In Vitro. Biochem. Biophys. Res. Commun. 2013, 440, 399–404. [Google Scholar] [CrossRef] [Green Version]
- Shimizu-Motohashi, Y.; Asakura, A. Angiogenesis as a novel therapeutic strategy for Duchenne muscular dystrophy through decreased ischemia and increased satellite cells. Front. Physiol. 2014, 5. [Google Scholar] [CrossRef] [Green Version]
- Podkalicka, P.; Mucha, O.; Dulak, J.; Loboda, A. Targeting angiogenesis in Duchenne muscular dystrophy. Cell. Mol. Life Sci. 2019, 76, 1507–1528. [Google Scholar] [CrossRef] [Green Version]
- Krock, B.L.; Skuli, N.; Simon, M.C. Hypoxia-Induced Angiogenesis: Good and Evil. Genes Cancer 2011, 2, 1117–1133. [Google Scholar] [CrossRef] [Green Version]
- Thurston, G. Complementary actions of VEGF and Angiopoietin-1 on blood vessel growth and leakage. J. Anat. 2002, 200, 575–580. [Google Scholar] [CrossRef]
- Elson, D.A.; Thurston, G.; Huang, L.E.; Ginzinger, D.G.; McDonald, D.M.; Johnson, R.S.; Arbeit, J.M. Induction of hypervascularity without leakage or inflammation in transgenic mice overexpressing hypoxia-inducible factor-1alpha. Genes Dev. 2001, 15, 2520–2532. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Statland, J.M.; Odrzywolski, K.J.; Shah, B.; Henderson, D.J.; Fricke, A.F.; van der Maarel, S.M.; Tapscott, S.J.; Tawil, R. Immunohistochemical Characterization of Facioscapulohumeral Muscular Dystrophy Muscle Biopsies. J. Neuromuscul. Dis. 2015, 2, 291–299. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shields, C.L.; Zahler, J.; Falk, N.; Furuta, M.; Eagle, R.C.; Espinosa, L.E.B.; Fischer, P.R.; Shields, J.A. Neovascular Glaucoma From Advanced Coats Disease as the Initial Manifestation of Facioscapulohumeral Dystrophy in a 2-Year-Old Child. Arch. Ophthalmol. 2007, 125, 840–842. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goselink, R.J.M.; Schreur, V.; Van Kernebeek, C.R.; Padberg, G.W.; Van Der Maarel, S.M.; Van Engelen, B.G.M.; Erasmus, C.E.; Theelen, T. Ophthalmological findings in facioscapulohumeral dystrophy. Brain Commun. 2019, 1, 1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Banerji, C.R.S.; Knopp, P.; Moyle, L.A.; Severini, S.; Orrell, R.W.; Teschendorff, A.E.; Zammit, P.S. β-catenin is central to DUX4 -driven network rewiring in facioscapulohumeral muscular dystrophy. J. R. Soc. Interface 2015, 12, 20140797. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Banerji, C.R.S.; Panamarova, M.; Hebaishi, H.; White, R.B.; Relaix, F.; Severini, S.; Zammit, P.S. PAX7 target genes are globally repressed in facioscapulohumeral muscular dystrophy skeletal muscle. Nat. Commun. 2017, 8, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsumagari, K.; Chang, S.-C.; Lacey, M.; Baribault, C.; Chittur, S.V.; Sowden, J.; Tawil, R.; Crawford, G.E.; Ehrlich, M. Gene expression during normal and FSHD myogenesis. BMC Med. Genom. 2011, 4, 67. [Google Scholar] [CrossRef] [Green Version]
- Lek, A.; Zhang, Y.; Woodman, K.G.; Huang, S.; DeSimone, A.M.; Cohen, J.; Ho, V.; Conner, J.; Mead, L.; Kodani, A.; et al. Applying genome-wide CRISPR-Cas9 screens for therapeutic discovery in facioscapulohumeral muscular dystrophy. Sci. Transl. Med. 2020, 12. [Google Scholar] [CrossRef]
- Geng, L.N.; Yao, Z.; Snider, L.; Fong, A.P.; Cech, J.N.; Young, J.M.; van der Maarel, S.; Ruzzo, W.L.; Gentleman, R.C.; Tawil, R.; et al. DUX4 Activates Germline Genes, Retroelements, and Immune Mediators: Implications for Facioscapulohumeral Dystrophy. Dev. Cell 2012, 22, 38–51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Osborne, R.J.; Welle, S.; Venance, S.L.; Thornton, C.A.; Tawil, R. Expression profile of FSHD supports a link between retinal vasculopathy and muscular dystrophy. Neurology 2006, 68, 569–577. [Google Scholar] [CrossRef]
- Sugino, S.; Miyatake, M.; Ohtani, Y.; Yoshioka, K.; Miike, T.; Uchino, M. Vascular alterations in Fukuyama type congenital muscular dystrophy. Brain Dev. 1991, 13, 77–81. [Google Scholar] [CrossRef]
- Zervos, A.; Hunt, K.E.; Tong, H.-Q.; Avallone, J.; Morales, J.; Friedman, N.; Cohen, B.H.; Clark, B.; Guo, S.; Gazda, H.; et al. Clinical, genetic and histopathologic findings in two siblings with muscle-eye-brain disease. Eur. J. Ophthalmol. 2002, 12, 253–261. [Google Scholar] [CrossRef] [PubMed]
- Hoang, Q.V.; Blair, M.P.; Rahmani, B.; Galasso, J.M.; Shapiro, M.J. Multiple Retinal Holes and Peripheral Nonperfusion in Muscle-Eye-Brain Disease. Arch. Ophthalmol. 2011, 129, 373–375. [Google Scholar] [CrossRef] [Green Version]
- Koh, M.Y.; Spivak-Kroizman, T.R.; Powis, G. HIF-1 regulation: Not so easy come, easy go. Trends Biochem. Sci. 2008, 33, 526–534. [Google Scholar] [CrossRef]
- Muz, B.; de la Puente, P.; Azab, F.; Azab, A.K. The role of hypoxia in cancer progression, angiogenesis, metastasis, and resistance to therapy. Hypoxia 2015, 3, 83–92. [Google Scholar] [CrossRef] [Green Version]
- Macklin, P.S.; Yamamoto, A.; Browning, L.; Hofer, M.; Adam, J.; Pugh, C.W. Recent advances in the biology of tumour hypoxia with relevance to diagnostic practice and tissue-based research. J. Pathol. 2020, 250, 593–611. [Google Scholar] [CrossRef] [Green Version]
- Gabriëls, J.; Beckers, M.-C.; Ding, H.; De Vriese, A.; Plaisance, S.M.; van der Maarel, S.; Padberg, G.W.; Frants, R.R.; Hewitt, J.E.; Collen, D.; et al. Nucleotide sequence of the partially deleted D4Z4 locus in a patient with FSHD identifies a putative gene within each 3.3 kb element. Gene 1999, 236, 25–32. [Google Scholar] [CrossRef]
- Dixit, M.; Ansseau, E.; Tassin, A.; Winokur, S.; Shi, R.; Qian, H.; Sauvage, S.; Matteotti, C.; van Acker, A.M.; Leo, O.; et al. DUX4, a candidate gene of facioscapulohumeral muscular dystrophy, encodes a transcriptional activator of PITX1. Proc. Natl. Acad. Sci. USA 2007, 104, 18157–18162. [Google Scholar] [CrossRef] [Green Version]
- Tassin, A.; Laoudj-Chenivesse, D.; Vanderplanck, C.; Barro, M.; Charron, S.; Ansseau, E.; Chen, Y.-W.; Mercier, J.; Coppée, F.; Belayew, A. DUX4 expression in FSHD muscle cells: How could such a rare protein cause a myopathy? J. Cell. Mol. Med. 2012, 17, 76–89. [Google Scholar] [CrossRef] [PubMed]
- Lim, K.R.Q.; Nguyen, Q.; Yokota, T. DUX4 Signalling in the Pathogenesis of Facioscapulohumeral Muscular Dystrophy. Int. J. Mol. Sci. 2020, 21, 729. [Google Scholar] [CrossRef] [Green Version]
- Wagner, K.R. Facioscapulohumeral Muscular Dystrophies. Contin. Lifelong Learn. Neurol. 2019, 25, 1662–1681. [Google Scholar] [CrossRef]
- Banerji, C.R.S. PAX7 target gene repression associates with FSHD progression and pathology over 1 year. Hum. Mol. Genet. 2020, 29, 2124–2133. [Google Scholar] [CrossRef]
- Banerji, C.R.S.; Zammit, P.S. Pathomechanisms and biomarkers in facioscapulohumeral muscular dystrophy: Roles of DUX4 and PAX7. EMBO Mol. Med. 2021, e13695. [Google Scholar] [CrossRef]
- Valle-Tenney, R.; Rebolledo, D.; Acuña, M.J.; Brandan, E. HIF-hypoxia signaling in skeletal muscle physiology and fibrosis. J. Cell Commun. Signal. 2020, 14, 147–158. [Google Scholar] [CrossRef]
- Majmundar, A.J.; Wong, W.J.; Simon, M.C. Hypoxia-Inducible Factors and the Response to Hypoxic Stress. Mol. Cell 2010, 40, 294–309. [Google Scholar] [CrossRef] [Green Version]
- Chaillou, T.; Lanner, J.T. Regulation of myogenesis and skeletal muscle regeneration: Effects of oxygen levels on satellite cell activity. FASEB J. 2016, 30, 3929–3941. [Google Scholar] [CrossRef] [Green Version]
- Dumont, N.A.; Bentzinger, C.F.; Sincennes, M.-C.; Rudnicki, M.A. Satellite Cells and Skeletal Muscle Regeneration. In Comprehensive Physiology; John Wiley & Sons: Hoboken, NJ, USA, 2015; pp. 1027–1059. ISBN 978-0-470-65071-4. [Google Scholar]
- Chaillou, T. Skeletal Muscle Fiber Type in Hypoxia: Adaptation to High-Altitude Exposure and Under Conditions of Pathological Hypoxia. Front. Physiol. 2018, 9. [Google Scholar] [CrossRef]
- Bentzinger, C.F.; Wang, Y.X.; Dumont, N.A.; Rudnicki, M.A. Cellular dynamics in the muscle satellite cell niche. EMBO Rep. 2013, 14, 1062–1072. [Google Scholar] [CrossRef] [Green Version]
- Chargé, S.B.P.; Rudnicki, M.A. Cellular and Molecular Regulation of Muscle Regeneration. Physiol. Rev. 2004, 84, 209–238. [Google Scholar] [CrossRef]
- Meadows, E.; Cho, J.-H.; Flynn, J.M.; Klein, W.H. Myogenin regulates a distinct genetic program in adult muscle stem cells. Dev. Biol. 2008, 322, 406–414. [Google Scholar] [CrossRef] [Green Version]
- Hasty, P.; Bradley, A.; Morris, J.H.; Edmondson, D.G.; Venuti, J.M.; Olson, E.N.; Klein, W.H. Muscle deficiency and neonatal death in mice with a targeted mutation in the myogenin gene. Nat. Cell Biol. 1993, 364, 501–506. [Google Scholar] [CrossRef]
- Lepper, C.; Conway, S.J.; Fan, C.-M. Adult satellite cells and embryonic muscle progenitors have distinct genetic requirements. Nat. Cell Biol. 2009, 460, 627–631. [Google Scholar] [CrossRef] [Green Version]
- Dort, J.; Fabre, P.; Molina, T.; Dumont, N.A. Macrophages Are Key Regulators of Stem Cells during Skeletal Muscle Regeneration and Diseases. Stem Cells Int. 2019, 2019, 4761427. [Google Scholar] [CrossRef]
- Yang, X.; Yang, S.; Wang, C.; Kuang, S. The hypoxia-inducible factors HIF1α and HIF2α are dispensable for embryonic muscle development but essential for postnatal muscle regeneration. J. Biol. Chem. 2017, 292, 5981–5991. [Google Scholar] [CrossRef] [Green Version]
- Settelmeier, S.; Schreiber, T.; Mäki, J.; Byts, N.; Koivunen, P.; Myllyharju, J.; Fandrey, J.; Winning, S. Prolyl hydroxylase domain 2 reduction enhances skeletal muscle tissue regeneration after soft tissue trauma in mice. PLoS ONE 2020, 15. [Google Scholar] [CrossRef]
- Cirillo, F.; Resmini, G.; Ghiroldi, A.; Piccoli, M.; Bergante, S.; Tettamanti, G.; Anastasia, L. Activation of the hypoxia-inducible factor 1a promotes myogenesis through the noncanonical Wnt pathway, leading to hypertrophic myotubes. FASEB J. 2017, 31, 2146–2156. [Google Scholar] [CrossRef] [Green Version]
- Park, A.M.; Sanders, T.A.; Maltepe, E. Hypoxia-inducible factor (HIF) and HIF-stabilizing agents in neonatal care. Semin. Fetal Neonatal Med. 2010, 15, 196–202. [Google Scholar] [CrossRef] [Green Version]
- Dias, I.B.; Bouma, H.R.; Henning, R.H. Unraveling the Big Sleep: Molecular Aspects of Stem Cell Dormancy and Hibernation. Front. Physiol. 2021, 12, 624950. [Google Scholar] [CrossRef]
- Pisani, D.F.; Dechesne, C.A. Skeletal Muscle HIF-1α Expression Is Dependent on Muscle Fiber Type. J. Gen. Physiol. 2005, 126, 173–178. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scheerer, N.; Dehne, N.; Stockmann, C.; Swoboda, S.; Baba, H.A.; Neugebauer, A.; Johnson, R.S.; Fandrey, J. Myeloid hypoxia-inducible factor-1α is essential for skeletal muscle regeneration in mice. J. Immunol. 2013, 191, 407–414. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yun, Z.; Lin, Q.; Giaccia, A.J. Adaptive Myogenesis under Hypoxia. Mol. Cell. Biol. 2005, 25, 3040–3055. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Majmundar, A.J.; Skuli, N.; Mesquita, R.C.; Kim, M.N.; Yodh, A.G.; Nguyen-McCarty, M.; Simon, M.C. O2 Regulates Skeletal Muscle Progenitor Differentiation through Phosphatidylinositol 3-Kinase/AKT Signaling. Mol. Cell. Biol. 2012, 32, 36–49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gustafsson, M.V.; Zheng, X.; Pereira, T.; Gradin, K.; Jin, S.; Lundkvist, J.L.; Ruas, J.; Poellinger, L.; Lendahl, U.; Bondesson, M. Hypoxia Requires Notch Signaling to Maintain the Undifferentiated Cell State. Dev. Cell 2005, 9, 617–628. [Google Scholar] [CrossRef] [Green Version]
- Wang, C.; Liu, W.; Liu, Z.; Chen, L.; Liu, X.; Kuang, S. Hypoxia Inhibits Myogenic Differentiation through p53 Protein-dependent Induction of Bhlhe40 Protein. J. Biol. Chem. 2015, 290, 29707–29716. [Google Scholar] [CrossRef] [Green Version]
- Launay, T.; Hagstrãm, L.; Lottin-Divoux, S.; Marchant, D.; Quidu, P.; Favret, F.; Duvallet, A.; Darribãre, T.; Richalet, J.P.; Beaudry, M. Blunting effect of hypoxia on the proliferation and differentiation of human primary and rat L6 myoblasts is not counteracted by Epo. Cell Prolif. 2010, 43, 1–8. [Google Scholar] [CrossRef]
- Di Carlo, A.; De Mori, R.; Martelli, F.; Pompilio, G.; Capogrossi, M.C.; Germani, A. Hypoxia Inhibits Myogenic Differentiation through Accelerated MyoD Degradation. J. Biol. Chem. 2004, 279, 16332–16338. [Google Scholar] [CrossRef] [Green Version]
- Wagatsuma, A.; Arakawa, M.; Matsumoto, H.; Matsuda, R.; Hoshino, T.; Mabuchi, K. Cobalt chloride, a chemical hypoxia-mimicking agent, suppresses myoblast differentiation by downregulating myogenin expression. Mol. Cell. Biochem. 2020, 470, 199–214. [Google Scholar] [CrossRef]
- Sakushima, K.; Yoshikawa, M.; Osaki, T.; Miyamoto, N.; Hashimoto, T. Moderate hypoxia promotes skeletal muscle cell growth and hypertrophy in C2C12 cells. Biochem. Biophys. Res. Commun. 2020, 525, 921–927. [Google Scholar] [CrossRef]
- Muñoz-Sánchez, J.; Chánez-Cárdenas, M.E. The use of cobalt chloride as a chemical hypoxia model. J. Appl. Toxicol. 2019, 39, 556–570. [Google Scholar] [CrossRef]
- Kook, S.-H.; Son, Y.-O.; Lee, K.-Y.; Lee, H.-J.; Chung, W.-T.; Choi, K.-C.; Lee, J.-C. Hypoxia affects positively the proliferation of bovine satellite cells and their myogenic differentiation through up-regulation of MyoD. Cell Biol. Int. 2008, 32, 871–878. [Google Scholar] [CrossRef]
- Khanna, S.; Roy, S.; Maurer, M.; Ratan, R.R.; Sen, C.K. Oxygen-sensitive reset of hypoxia-inducible factor transactivation response: Prolyl hydroxylases tune the biological normoxic set point. Free Radic. Biol. Med. 2006, 40, 2147–2154. [Google Scholar] [CrossRef] [Green Version]
- Carreau, A.; El Hafny-Rahbi, B.; Matejuk, A.; Grillon, C.; Kieda, C. Why is the partial oxygen pressure of human tissues a crucial parameter? Small molecules and hypoxia. J. Cell. Mol. Med. 2011, 15, 1239–1253. [Google Scholar] [CrossRef] [Green Version]
- Boekstegers, P.; Riessen, R.; Seyde, W. Oxygen Partial Pressure Distribution Within Skeletal Muscle: Indicator of Whole Body Oxygen Delivery in Patients? Chem. Biol. Pteridines Folates 1990, 277, 507–514. [Google Scholar] [CrossRef]
- Ikossi, D.G.; Knudson, M.M.; Morabito, D.J.; Cohen, M.J.; Wan, J.J.; Khaw, L.; Stewart, C.J.; Hemphill, C.; Manley, G.T. Continuous Muscle Tissue Oxygenation in Critically Injured Patients: A Prospective Observational Study. J. Trauma Inj. Infect. Crit. Care 2006, 61, 780–790. [Google Scholar] [CrossRef]
- Bylund-Fellenius, A.C.; Walker, P.M.; Elander, A.; Holm, S.; Holm, J.; Scherstén, T. Energy metabolism in relation to oxygen partial pressure in human skeletal muscle during exercise. Biochem. J. 1981, 200, 247–255. [Google Scholar] [CrossRef] [Green Version]
- Dubinin, M.V.; Talanov, E.Y.; Tenkov, K.S.; Starinets, V.S.; Mikheeva, I.B.; Sharapov, M.G.; Belosludtsev, K.N. Duchenne muscular dystrophy is associated with the inhibition of calcium uniport in mitochondria and an increased sensitivity of the organelles to the calcium-induced permeability transition. Biochim. Biophys. Acta Mol. Basis Dis. 2020, 1866, 165674. [Google Scholar] [CrossRef]
- Jash, S.; Adhya, S. Effects of Transient Hypoxia versus Prolonged Hypoxia on Satellite Cell Proliferation and Differentiation In Vivo. Stem Cells Int. 2015, 2015. [Google Scholar] [CrossRef]
- Chaillou, T.; Koulmann, N.; Meunier, A.; Pugnière, P.; McCarthy, J.J.; Beaudry, M.; Bigard, X. Ambient hypoxia enhances the loss of muscle mass after extensive injury. Pflügers Arch. Eur. J. Physiol. 2013, 466, 587–598. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chaillou, T.; Koulmann, N.; Meunier, A.; Chapot, R.; Serrurier, B.; Beaudry, M.; Bigard, X. Effect of hypoxia exposure on the recovery of skeletal muscle phenotype during regeneration. Mol. Cell. Biochem. 2014, 390, 31–40. [Google Scholar] [CrossRef]
- Yanay, N.; Rabie, M.; Nevo, Y. Impaired Regeneration in Dystrophic Muscle-New Target for Therapy. Front. Neurosci. 2020, 13, 69. [Google Scholar]
- Banerji, C.R.S.; Henderson, D.; Tawil, R.N.; Zammit, P.S. Skeletal muscle regeneration in facioscapulohumeral muscular dystrophy is correlated with pathological severity. Hum. Mol. Genet. 2020, 29, 2746–2760. [Google Scholar] [CrossRef]
- Carregari, V.C.; Monforte, M.; Di Maio, G.; Pieroni, L.; Urbani, A.; Ricci, E.; Tasca, G. Proteomics of Muscle Microdialysates Identifies Potential Circulating Biomarkers in Facioscapulohumeral Muscular Dystrophy. Int. J. Mol. Sci. 2020, 22, 290. [Google Scholar] [CrossRef]
- Guiraud, S.; Davies, K.E. Regenerative biomarkers for Duchenne muscular dystrophy. Neural Regen. Res. 2019, 14, 1317–1320. [Google Scholar] [CrossRef]
- Dadgar, S.; Wang, Z.; Johnston, H.; Kesari, A.; Nagaraju, K.; Chen, Y.-W.; Hill, D.A.; Partridge, T.A.; Giri, M.; Freishtat, R.; et al. Asynchronous remodeling is a driver of failed regeneration in Duchenne muscular dystrophy. J. Cell Biol. 2014, 207, 139–158. [Google Scholar] [CrossRef]
- Bhattacharyya, S.; Kelley, K.; Melichian, D.S.; Tamaki, Z.; Fang, F.; Su, Y.; Feng, G.; Pope, R.M.; Budinger, G.S.; Mutlu, G.M.; et al. Toll-Like Receptor 4 Signaling Augments Transforming Growth Factor-β Responses: A Novel Mechanism for Maintaining and Amplifying Fibrosis in Scleroderma. Am. J. Pathol. 2013, 182, 192–205. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rhoads, R.; Johnson, R.M.; Rathbone, C.R.; Liu, X.; Temm-Grove, C.; Sheehan, S.M.; Hoying, J.B.; Allen, R.E. Satellite cell-mediated angiogenesis In Vitro coincides with a functional hypoxia-inducible factor pathway. Am. J. Physiol. Physiol. 2009, 296, C1321–C1328. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morgan, J.E.; Zammit, P.S. Direct effects of the pathogenic mutation on satellite cell function in muscular dystrophy. Exp. Cell Res. 2010, 316, 3100–3108. [Google Scholar] [CrossRef]
- Schiaffino, S.; Reggiani, C. Fiber Types in Mammalian Skeletal Muscles. Physiol. Rev. 2011, 91, 1447–1531. [Google Scholar] [CrossRef] [Green Version]
- Mason, S.D.; Howlett, R.A.; Kim, M.J.; Olfert, I.M.; Hogan, M.C.; McNulty, W.; Hickey, R.P.; Wagner, P.D.; Kahn, C.R.; Giordano, F.J.; et al. Loss of Skeletal Muscle HIF-1α Results in Altered Exercise Endurance. PLoS Biol. 2004, 2, e288. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mason, S.D.; Rundqvist, H.; Papandreou, I.; Duh, R.; McNulty, W.J.; Howlett, R.A.; Olfert, I.M.; Sundberg, C.J.; Denko, N.C.; Poellinger, L.; et al. HIF-1α in endurance training: Suppression of oxidative metabolism. Am. J. Physiol. Integr. Comp. Physiol. 2007, 293, R2059–R2069. [Google Scholar] [CrossRef]
- Shin, J.; Nunomiya, A.; Kitajima, Y.; Dan, T.; Miyata, T.; Nagatomi, R. Prolyl hydroxylase domain 2 deficiency promotes skeletal muscle fiber-type transition via a calcineurin/NFATc1-dependent pathway. Skelet. Muscle 2015, 6, 5. [Google Scholar] [CrossRef] [Green Version]
- Nunomiya, A.; Shin, J.; Kitajima, Y.; Dan, T.; Miyata, T.; Nagatomi, R. Activation of the hypoxia-inducible factor pathway induced by prolyl hydroxylase domain 2 deficiency enhances the effect of running training in mice. Acta Physiol. 2017, 220, 99–112. [Google Scholar] [CrossRef] [PubMed]
- Rasbach, K.A.; Gupta, R.K.; Ruas, J.; Wu, J.; Naseri, E.; Estall, J.; Spiegelman, B.M. PGC-1 regulates a HIF2 -dependent switch in skeletal muscle fiber types. Proc. Natl. Acad. Sci. USA 2010, 107, 21866–21871. [Google Scholar] [CrossRef] [Green Version]
- Glaser, J.; Suzuki, M. Skeletal Muscle Fiber Types in Neuromuscular Diseases. In Muscle Cell and Tissue—Current Status of Research Field; InTech Open: London, UK, 2018. [Google Scholar]
- Talbot, J.; Maves, L. Skeletal muscle fiber type: Using insights from muscle developmental biology to dissect targets for susceptibility and resistance to muscle disease. Wiley Interdiscip. Rev. Dev. Biol. 2016, 5, 518–534. [Google Scholar] [CrossRef] [Green Version]
- Lassche, S.; Stienen, G.J.M.; Irving, T.C.; Van Der Maarel, S.M.; Voermans, N.C.; Padberg, G.W.; Granzier, H.; Van Engelen, B.G.; Ottenheijm, C.A. Sarcomeric dysfunction contributes to muscle weakness in facioscapulohumeral muscular dystrophy. Neurology 2013, 80, 733–737. [Google Scholar] [CrossRef] [Green Version]
- Vihola, A.; Bassez, G.; Meola, G.; Zhang, S.; Haapasalo, H.; Paetau, A.; Mancinelli, E.; Rouche, A.; Hogrel, J.; Laforet, P.; et al. Histopathological differences of myotonic dystrophy type 1 (DM1) and PROMM/DM2. Neurology 2003, 60, 1854–1857. [Google Scholar] [CrossRef] [Green Version]
- Webster, C.; Silberstein, L.; Hays, A.P.; Blau, H.M. Fast muscle fibers are preferentially affected in Duchenne muscular dystrophy. Cell 1988, 52, 503–513. [Google Scholar] [CrossRef]
- Lin, J.; Wu, H.; Tarr, P.T.; Zhang, C.-Y.; Wu, Z.; Boss, O.; Michael, L.F.; Puigserver, P.; Isotani, E.; Olson, E.N.; et al. Transcriptional co-activator PGC-1α drives the formation of slow-twitch muscle fibres. Nat. Cell Biol. 2002, 418, 797–801. [Google Scholar] [CrossRef]
- Handschin, C.; Kobayashi, Y.M.; Chin, S.; Seale, P.; Campbell, K.P.; Spiegelman, B.M. PGC-1 regulates the neuromuscular junction program and ameliorates Duchenne muscular dystrophy. Genes Dev. 2007, 21, 770–783. [Google Scholar] [CrossRef] [Green Version]
- Knobloch, M.; Pilz, G.-A.; Ghesquière, B.; Kovacs, W.; Wegleiter, T.; Moore, D.; Hruzova, M.; Zamboni, N.; Carmeliet, P.; Jessberger, S. A Fatty Acid Oxidation-Dependent Metabolic Shift Regulates Adult Neural Stem Cell Activity. Cell Rep. 2017, 20, 2144–2155. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ryall, J.G.; Dell’Orso, S.; Derfoul, A.; Juan, A.; Zare, H.; Feng, X.; Clermont, D.; Koulnis, M.; Gutierrez-Cruz, G.; Fulco, M.; et al. The NAD+-Dependent SIRT1 Deacetylase Translates a Metabolic Switch into Regulatory Epigenetics in Skeletal Muscle Stem Cells. Cell Stem Cell 2015, 16, 171–183. [Google Scholar] [CrossRef] [Green Version]
- Bose, S.; Zhang, C.; Le, A. Glucose Metabolism in Cancer: The Warburg Effect and Beyond. In The Heterogeneity of Cancer Metabolism; Le, A., Ed.; Springer: Chem, Switzerland, 2021; Volume 1311, pp. 3–15. [Google Scholar]
- Mason, S.; Johnson, R.S. The role of HIF-1 in Hypoxic Response in the Skeletal Muscle. In Hypoxia and the Circulation; Roach, R.C., Wagner, P.D., Hackett, P.H., Eds.; Springer Science and Business Media LLC: Boston, MA, USA, 2007; Volume 618, pp. 229–244. [Google Scholar]
- Egan, B.; Zierath, J.R. Exercise Metabolism and the Molecular Regulation of Skeletal Muscle Adaptation. Cell Metab. 2013, 17, 162–184. [Google Scholar] [CrossRef] [Green Version]
- Semenza, G.L. O2, and the 3 PHDs: How Animal Cells Signal Hypoxia to the Nucleus. Cell 2001, 107, 1–3. [Google Scholar] [CrossRef] [Green Version]
- Horscroft, J.A.; Murray, A.J. Skeletal muscle energy metabolism in environmental hypoxia: Climbing towards consensus. Extrem. Physiol. Med. 2014, 3, 1–17. [Google Scholar] [CrossRef] [Green Version]
- De Palma, S.; Ripamonti, M.; Viganò, A.; Moriggi, M.; Capitanio, D.; Samaja, M.; Milano, G.; Cerretelli, P.; Wait, R.; Gelfi, C. Metabolic Modulation Induced by Chronic Hypoxia in Rats Using a Comparative Proteomic Analysis of Skeletal Muscle Tissue. J. Proteome Res. 2007, 6, 1974–1984. [Google Scholar] [CrossRef] [PubMed]
- Lindholm, M.E.; Fischer, H.; Poellinger, L.; Johnson, R.S.; Gustafsson, T.; Sundberg, C.J.; Rundqvist, H. Negative regulation of HIF in skeletal muscle of elite endurance athletes: A tentative mechanism promoting oxidative metabolism. Am. J. Physiol. Integr. Comp. Physiol. 2014, 307, R248–R255. [Google Scholar] [CrossRef] [Green Version]
- Green, H.J.; Sutton, J.R.; Wolfel, E.E.; Reeves, J.T.; Butterfield, G.E.; Brooks, G.A. Altitude acclimatization and energy metabolic adaptations in skeletal muscle during exercise. J. Appl. Physiol. 1992, 73, 2701–2708. [Google Scholar] [CrossRef] [PubMed]
- Brooks, G.A.; Wolfel, E.E.; Butterfield, G.E.; Cymerman, A.; Roberts, A.C.; Mazzeo, R.S.; Reeves, J.T. Poor relationship between arterial [lactate] and leg net release during exercise at 4300 m altitude. Am. J. Physiol. Integr. Comp. Physiol. 1998, 275, R1192–R1201. [Google Scholar] [CrossRef]
- Liu, Y.; Ma, Z.; Zhao, C.; Wang, Y.; Wu, G.; Xiao, J.; McClain, C.J.; Li, X.; Feng, W. HIF-1α and HIF-2α are critically involved in hypoxia-induced lipid accumulation in hepatocytes through reducing PGC-1α-mediated fatty acid β-oxidation. Toxicol. Lett. 2014, 226, 117–123. [Google Scholar] [CrossRef] [PubMed]
- Chicco, A.J.; Le, C.H.; Gnaiger, E.; Dreyer, H.C.; Muyskens, J.B.; D’Alessandro, A.; Nemkov, T.; Hocker, A.D.; Prenni, J.E.; Wolfe, L.M.; et al. Adaptive remodeling of skeletal muscle energy metabolism in high-altitude hypoxia: Lessons from AltitudeOmics. J. Biol. Chem. 2018, 293, 6659–6671. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, H.; Bosch-Marce, M.; Shimoda, L.A.; Tan, Y.S.; Baek, J.H.; Wesley, J.B.; Gonzalez, F.J.; Semenza, G.L. Mitochondrial Autophagy Is an HIF-1-dependent Adaptive Metabolic Response to Hypoxia. J. Biol. Chem. 2008, 283, 10892–10903. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bloemberg, D.; Quadrilatero, J. Autophagy, apoptosis, and mitochondria: Molecular integration and physiological relevance in skeletal muscle. Am. J. Physiol. Physiol. 2019, 317, C111–C130. [Google Scholar] [CrossRef]
- Band, M.; Joel, A.; Hernandez, A.; Avivi, A. Hypoxia-induced BNIP3 expression and mitophagy: In Vivo comparison of the rat and the hypoxia-tolerant mole rat, Spalax ehrenbergi. FASEB J. 2009, 23, 2327–2335. [Google Scholar] [CrossRef]
- Gamboa, J.L.; García-Cazarín, M.L.; Andrade, F.H. Chronic hypoxia increases insulin-stimulated glucose uptake in mouse soleus muscle. Am. J. Physiol. Integr. Comp. Physiol. 2011, 300, R85–R91. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Theije, C.C.; Langen, R.C.J.; Lamers, W.H.; Schols, A.M.W.J.; Köhler, S.E. Distinct responses of protein turnover regulatory pathways in hypoxia- and semistarvation-induced muscle atrophy. Am. J. Physiol. Cell. Mol. Physiol. 2013, 305, L82–L91. [Google Scholar] [CrossRef] [Green Version]
- Chen, R.; Jiang, T.; She, Y.; Xu, J.; Li, C.; Zhou, S.; Shen, H.; Shi, H.; Liu, S. Effects of Cobalt Chloride, a Hypoxia-Mimetic Agent, on Autophagy and Atrophy in Skeletal C2C12 Myotubes. BioMed. Res. Int. 2017, 2017. [Google Scholar] [CrossRef] [Green Version]
- Dreyfus, J.-C.; Schapira, G.; Schapira, F.; Demos, J. Activités enzymatiques du muscle humain: Recherches sur la biochimie comparée de l’homme normal et myopathique, et du rat. Clin. Chim. Acta 1956, 1, 434–449. [Google Scholar] [CrossRef]
- Pant, M.; Sopariwala, D.H.; Bal, N.C.; Lowe, J.; Delfín, D.A.; Rafael-Fortney, J.; Periasamy, M. Metabolic Dysfunction and Altered Mitochondrial Dynamics in the Utrophin-Dystrophin Deficient Mouse Model of Duchenne Muscular Dystrophy. PLoS ONE 2015, 10, e0123875. [Google Scholar] [CrossRef] [Green Version]
- Nghiem, P.P.; Bello, L.; Stoughton, W.B.; López, S.M.; Vidal, A.; Hernandez, B.V.; Hulbert, K.N.; Gourley, T.R.; Bettis, A.K.; Balog-Alvarez, C.J.; et al. Changes in Muscle Metabolism are Associated with Phenotypic Variability in Golden Retriever Muscular Dystrophy. Yale J. Biol. Med. 2017, 90, 351–360. [Google Scholar]
- Schneider, S.M.; Sridhar, V.; Bettis, A.K.; Heath-Barnett, H.; Balog-Alvarez, C.J.; Guo, L.-J.; Johnson, R.; Jaques, S.; Vitha, S.; Glowcwski, A.C.; et al. Glucose Metabolism as a Pre-clinical Biomarker for the Golden Retriever Model of Duchenne Muscular Dystrophy. Mol. Imaging Biol. 2018, 20, 780–788. [Google Scholar] [CrossRef] [Green Version]
- Rodríiguez-Cruz, M.; Sanchez, R.; Escobar, R.E.; Del Cruz-Guzmáan, O.R.; Lóopez-Alarcóon, M.; Bernabe-Garcíia, M.; Coral-Váazquez, R.M.; Matute, G.; Wong, A.C.V. Evidence of Insulin Resistance and Other Metabolic Alterations in Boys with Duchenne or Becker Muscular Dystrophy. Int. J. Endocrinol. 2015, 2015. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matsumura, T.; Iwahashi, H.; Funahashi, T.; Takahashi, M.P.; Saito, T.; Yasui, K.; Saito, T.; Iyama, A.; Toyooka, K.; Fujimura, H.; et al. A cross-sectional study for glucose intolerance of myotonic dystrophy. J. Neurol. Sci. 2009, 276, 60–65. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, N.K.; Yadav, R.; Mukherjee, S.; Sinha, N. Perturbation of muscle metabolism in patients with muscular dystrophy in early or acute phase of disease: In Vitro, high resolution NMR spectroscopy based analysis. Clin. Chim. Acta 2018, 478, 171–181. [Google Scholar] [CrossRef] [PubMed]
- Turki, A.; Hayot, M.; Carnac, G.; Pillard, F.; Passerieux, E.; Bommart, S.; de Mauverger, E.R.; Hugon, G.; Pincemail, J.; Pietri, S.; et al. Functional muscle impairment in facioscapulohumeral muscular dystrophy is correlated with oxidative stress and mitochondrial dysfunction. Free Radic. Biol. Med. 2012, 53, 1068–1079. [Google Scholar] [CrossRef] [PubMed]
- Banerji, C.R.S.; Panamarova, M.; Pruller, J.; Figeac, N.; Hebaishi, H.; Fidanis, E.; Saxena, A.; Contet, J.; Sacconi, S.; Severini, S.; et al. Dynamic transcriptomic analysis reveals suppression of PGC1α/ERRα drives perturbed myogenesis in facioscapulohumeral muscular dystrophy. Hum. Mol. Genet. 2019, 28, 1244–1259. [Google Scholar] [CrossRef]
- Orsucci, D.; Ienco, E.C.; Rossi, A.; Siciliano, G.; Mancuso, M. Mitochondrial Syndromes Revisited. J. Clin. Med. 2021, 10, 1249. [Google Scholar] [CrossRef]
- Barbieri, E.; Sestili, P. Reactive Oxygen Species in Skeletal Muscle Signaling. J. Signal. Transduct. 2012, 2012. [Google Scholar] [CrossRef] [Green Version]
- Mason, S.; Wadley, G.D. Skeletal muscle reactive oxygen species: A target of good cop/bad cop for exercise and disease. Redox Rep. 2014, 19, 97–106. [Google Scholar] [CrossRef] [Green Version]
- He, F.; Li, J.; Liu, Z.; Chuang, C.-C.; Yang, W.; Zuo, L. Redox Mechanism of Reactive Oxygen Species in Exercise. Front. Physiol. 2016, 7. [Google Scholar] [CrossRef] [Green Version]
- Pan, J.-S.; Hong, M.-Z.; Ren, J.-L. Reactive oxygen species: A double-edged sword in oncogenesis. World J. Gastroenterol. 2009, 15, 1702–1707. [Google Scholar] [CrossRef]
- Singh, S.N.; Vats, P.; Kumria, M.M.L.; Ranganathan, S.; Shyam, R.; Arora, M.P.; Jain, C.L.; Sridharan, K. Effect of high altitude (7620 m) exposure on glutathione and related metabolism in rats. Graefe’s Arch. Clin. Exp. Ophthalmol. 2001, 84, 233–237. [Google Scholar] [CrossRef]
- Magalhães, J.; Ascensão, A.; Soares, J.M.C.; Ferreira, R.; Neuparth, M.J.; Marques, F.; Duarte, J.A. Acute and severe hypobaric hypoxia increases oxidative stress and impairs mitochondrial function in mouse skeletal muscle. J. Appl. Physiol. 2005, 99, 1247–1253. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neuparth, M.J.; Ferreira, R.; Amado, F.; Magalhães, J.; Ascensão, A.; Soares, J.M.C.; Oliveira, J.; Duarte, J.A. Acute and severe hypobaric hypoxia-induced muscle oxidative stress in mice: The role of glutathione against oxidative damage. Graefe’s Arch. Clin. Exp. Ophthalmol. 2003, 91, 185–191. [Google Scholar] [CrossRef]
- Faiss, R.; Pialoux, V.; Sartori, C.; Faes, C.; Dériaz, O.; Millet, G. Ventilation, Oxidative Stress, and Nitric Oxide in Hypobaric versus Normobaric Hypoxia. Med. Sci. Sports Exerc. 2013, 45, 253–260. [Google Scholar] [CrossRef]
- Pialoux, V.; Mounier, R.; Rock, E.; Mazur, A.; Schmitt, L.; Richalet, J.-P.; Robach, P.; Coudert, J.; Fellmann, N. Effects of Acute Hypoxic Exposure on Prooxidant/Antioxidant Balance in Elite Endurance Athletes. Int. J. Sports Med. 2009, 30, 87–93. [Google Scholar] [CrossRef]
- Magalhães, J.; Ascensão, A.; Viscor, G.; Soares, J.; Oliveira, J.; Marques, F.; Duarte, J. Oxidative stress in humans during and after 4 hours of hypoxia at a simulated altitude of 5500 m. Aviat. Space Environ. Med. 2004, 75, 16–22. [Google Scholar]
- Dosek, A.; Ohno, H.; Acs, Z.; Taylor, A.W.; Radak, Z. High altitude and oxidative stress. Respir. Physiol. Neurobiol. 2007, 158, 128–131. [Google Scholar] [CrossRef]
- Debevec, T.; Pialoux, V.; Saugy, J.J.; Schmitt, L.; Cejuela, R.; Mury, P.; Ehrström, S.; Faiss, R.; Millet, G.P. Prooxidant/Antioxidant Balance in Hypoxia: A Cross-Over Study on Normobaric vs. Hypobaric “Live High-Train Low”. PLoS ONE 2015, 10, e0137957. [Google Scholar] [CrossRef] [Green Version]
- Debevec, T.; Pialoux, V.; Mekjavic, I.; Eiken, O.; Mury, P.; Millet, G. Moderate Exercise Blunts Oxidative Stress Induced by Normobaric Hypoxic Confinement. Med. Sci. Sports Exerc. 2014, 46, 33–41. [Google Scholar] [CrossRef]
- Wright, V.P.; Klawitter, P.F.; Iscru, D.F.; Merola, A.J.; Clanton, T.L. Superoxide scavengers augment contractile but not energetic responses to hypoxia in rat diaphragm. J. Appl. Physiol. 2005, 98, 1753–1760. [Google Scholar] [CrossRef] [Green Version]
- Mohanraj, P.; Merola, A.J.; Wright, V.P.; Clanton, T. Antioxidants protect rat diaphragmatic muscle function under hypoxic conditions. J. Appl. Physiol. 1998, 84, 1960–1966. [Google Scholar] [CrossRef]
- Quindry, J.; Dumke, C.; Slivka, D.; Ruby, B. Impact of extreme exercise at high altitude on oxidative stress in humans. J. Physiol. 2016, 594, 5093–5104. [Google Scholar] [CrossRef] [Green Version]
- Cofta, S.; Winiarska, H.M.; Płóciniczak, A.; Bielawska, L.; Brożek, A.; Piorunek, T.; Kostrzewska, T.M.; Wysocka, E. Oxidative Stress Markers and Severity of Obstructive Sleep Apnea. Adv. Exp. Med. Biol. 2019, 1222, 27–35. [Google Scholar] [CrossRef]
- Conotte, S.; Tassin, A.; Conotte, R.; Colet, J.-M.; Boudjeltia, K.Z.; Legrand, A. Metabonomic profiling of chronic intermittent hypoxia in a mouse model. Respir. Physiol. Neurobiol. 2018, 256, 157–173. [Google Scholar] [CrossRef]
- Dunleavy, M.; Bradford, A.; O’Halloran, K.D. Oxidative Stress Impairs Upper Airway Muscle Endurance in an Animal Model of Sleep-Disordered Breathing. In Integration in Respiratory Control: From Genes to Systems; Poulin, M.J., Wilson, R.J.A., Eds.; Springer: New York, NY, USA, 2008; pp. 458–462. ISBN 978-0-387-73693-8. [Google Scholar]
- Dutta, A.; Ray, K.; Singh, V.K.; Vats, P.; Singh, S.N.; Singh, S.B. l-carnitine supplementation attenuates intermittent hypoxia-induced oxidative stress and delays muscle fatigue in rats. Exp. Physiol. 2008, 93, 1139–1146. [Google Scholar] [CrossRef] [PubMed]
- Skelly, J.R.; Edge, D.; Shortt, C.M.; Jones, J.F.X.; Bradford, A.; O’Halloran, K.D. Tempol Ameliorates Pharyngeal Dilator Muscle Dysfunction in a Rodent Model of Chronic Intermittent Hypoxia. Am. J. Respir. Cell Mol. Biol. 2012, 46, 139–148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bradford, A.; McGuire, M.; O’Halloran, K.D. Does episodic hypoxia affect upper airway dilator muscle function? Implications for the pathophysiology of obstructive sleep apnoea. Respir. Physiol. Neurobiol. 2005, 147, 223–234. [Google Scholar] [CrossRef] [PubMed]
- Shortt, C.M.; Fredsted, A.; Chow, H.B.; Williams, R.; Skelly, J.R.; Edge, D.; Bradford, A.; O’Halloran, K.D. Reactive oxygen species mediated diaphragm fatigue in a rat model of chronic intermittent hypoxia. Exp. Physiol. 2014, 99, 688–700. [Google Scholar] [CrossRef] [Green Version]
- McGuire, M.; MacDermott, M.; Bradford, A. Effects of Chronic Intermittent Asphyxia on Rat Diaphragm and Limb Muscle Contractility. Chest 2003, 123, 875–881. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.; Jin, L.; Shen, J.; Shang, P.; Jiang, X.; Wang, X. Electrical stimulation influences chronic intermittent hypoxia-hypercapnia induction of muscle fibre transformation by regulating the microRNA/Sox6 pathway. Sci. Rep. 2016, 6, 26415. [Google Scholar] [CrossRef] [Green Version]
- Domínguez-Álvarez, M.; Gea, J.; Barreiro, E. Inflammatory Events and Oxidant Production in the Diaphragm, Gastrocnemius, and Blood of Rats Exposed to Chronic Intermittent Hypoxia: Therapeutic Strategies. J. Cell. Physiol. 2017, 232, 1165–1175. [Google Scholar] [CrossRef] [Green Version]
- Petrillo, S.; Pelosi, L.; Piemonte, F.; Travaglini, L.; Forcina, L.; Catteruccia, M.; Petrini, S.; Verardo, M.; D’Amico, A.; Musarò, A.; et al. Oxidative stress in Duchenne muscular dystrophy: Focus on the NRF2 redox pathway. Hum. Mol. Genet. 2017, 26, 2781–2790. [Google Scholar] [CrossRef]
- Passerieux, E.; Hayot, M.; Jaussent, A.; Carnac, G.; Gouzi, F.; Pillard, F.; Picot, M.-C.; Böcker, K.; Hugon, G.; Pincemail, J.; et al. Effects of vitamin C, vitamin E, zinc gluconate, and selenomethionine supplementation on muscle function and oxidative stress biomarkers in patients with facioscapulohumeral dystrophy: A double-blind randomized controlled clinical trial. Free Radic. Biol. Med. 2015, 81, 158–169. [Google Scholar] [CrossRef] [PubMed]
- Laoudj-Chenivesse, D.; Carnac, G.; Bisbal, C.; Hugon, G.; Bouillot, S.; Desnuelle, C.; Vassetzky, Y.; Fernandez, A. Increased levels of adenine nucleotide translocator 1 protein and response to oxidative stress are early events in facioscapulohumeral muscular dystrophy muscle. J. Mol. Med. 2004, 83, 216–224. [Google Scholar] [CrossRef] [PubMed]
- Denny, A.P.; Heather, A.K. Are Antioxidants a Potential Therapy for FSHD? A Review of the Literature. Oxidative Med. Cell. Longev. 2017, 2017, 7020295. [Google Scholar] [CrossRef] [Green Version]
- Arbogast, S.; Beuvin, M.; Fraysse, B.; Zhou, H.; Muntoni, F.; Ferreiro, A. Oxidative stress inSEPN1-related myopathy: From pathophysiology to treatment. Ann. Neurol. 2009, 65, 677–686. [Google Scholar] [CrossRef]
- Terrill, J.R.; Radley-Crabb, H.G.; Iwasaki, T.; Lemckert, F.A.; Arthur, P.G.; Grounds, M.D. Oxidative stress and pathology in muscular dystrophies: Focus on protein thiol oxidation and dysferlinopathies. FEBS J. 2013, 280, 4149–4164. [Google Scholar] [CrossRef]
- Sieprath, T.; Darwiche, R.; De Vos, W.H. Lamins as mediators of oxidative stress. Biochem. Biophys. Res. Commun. 2012, 421, 635–639. [Google Scholar] [CrossRef]
- Dudley, R.W.R.; Khairallah, M.; Mohammed, S.; Lands, L.; Des Rosiers, C.; Petrof, B.J. Dynamic responses of the glutathione system to acute oxidative stress in dystrophic mouse (mdx) muscles. Am. J. Physiol. Integr. Comp. Physiol. 2006, 291, R704–R710. [Google Scholar] [CrossRef] [Green Version]
- Renjini, R.; Gayathri, N.; Nalini, A.; Bharath, M.M.S. Oxidative Damage in Muscular Dystrophy Correlates with the Severity of the Pathology: Role of Glutathione Metabolism. Neurochem. Res. 2012, 37, 885–898. [Google Scholar] [CrossRef]
- Kaczor, J.; Hall, J.E.; Payne, E.; Tarnopolsky, M.A. Low intensity training decreases markers of oxidative stress in skeletal muscle of mdx mice. Free Radic. Biol. Med. 2007, 43, 145–154. [Google Scholar] [CrossRef]
- Selsby, J.T. Increased catalase expression improves muscle function in mdx mice. Exp. Physiol. 2010, 96, 194–202. [Google Scholar] [CrossRef] [Green Version]
- Hori, Y.S.; Kuno, A.; Hosoda, R.; Tanno, M.; Miura, T.; Shimamoto, K.; Horio, Y. Resveratrol Ameliorates Muscular Pathology in the Dystrophic mdx Mouse, a Model for Duchenne Muscular Dystrophy. J. Pharmacol. Exp. Ther. 2011, 338, 784–794. [Google Scholar] [CrossRef] [Green Version]
- Call, J.A.; Voelker, K.A.; Wolff, A.V.; McMillan, R.P.; Evans, N.P.; Hulver, M.W.; Talmadge, R.J.; Grange, R.W. Endurance capacity in maturing mdx mice is markedly enhanced by combined voluntary wheel running and green tea extract. J. Appl. Physiol. 2008, 105, 923–932. [Google Scholar] [CrossRef] [Green Version]
- Buetler, T.M.; Renard, M.; Offord, E.A.; Schneider, H.; Ruegg, U.T. Green tea extract decreases muscle necrosis in mdx mice and protects against reactive oxygen species. Am. J. Clin. Nutr. 2002, 75, 749–753. [Google Scholar] [CrossRef] [Green Version]
- Buyse, G.M.; Voit, T.; Schara, U.; Straathof, C.S.M.; D’Angelo, M.G.; Bernert, G.; Cuisset, J.-M.; Finkel, R.S.; Goemans, N.; McDonald, C.M.; et al. Efficacy of idebenone on respiratory function in patients with Duchenne muscular dystrophy not using glucocorticoids (DELOS): A double-blind randomised placebo-controlled phase 3 trial. Lancet 2015, 385, 1748–1757. [Google Scholar] [CrossRef] [Green Version]
- Buyse, G.M.; Goemans, N.; van den Hauwe, M.; Meier, T. Effects of glucocorticoids and idebenone on respiratory function in patients with duchenne muscular dystrophy. Pediatr. Pulmonol. 2013, 48, 912–920. [Google Scholar] [CrossRef]
- Buyse, G.M.; Goemans, N.; van den Hauwe, M.; Thijs, D.; de Groot, I.J.; Schara, U.; Ceulemans, B.; Meier, T.; Mertens, L. Idebenone as a novel, therapeutic approach for Duchenne muscular dystrophy: Results from a 12 month, double-blind, randomized placebo-controlled trial. Neuromuscul. Disord. 2011, 21, 396–405. [Google Scholar] [CrossRef]
- Winokur, S.T.; Barrett, K.; Martin, J.H.; Forrester, J.R.; Simon, M.; Tawil, R.; Chung, S.-A.; Masny, P.S.; Figlewicz, D.A. Facioscapulohumeral muscular dystrophy (FSHD) myoblasts demonstrate increased susceptibility to oxidative stress. Neuromuscul. Disord. 2003, 13, 322–333. [Google Scholar] [CrossRef]
- Wilson, V.D.; Thomas, C.; Passerieux, E.; Hugon, G.; Pillard, F.; Andrade, A.G.P.; Bommart, S.; Picot, M.-C.; Pincemail, J.; Mercier, J.; et al. Impaired oxygen demand during exercise is related to oxidative stress and muscle function in Facioscapulohumeral Muscular Dystrophy. JCSM Rapid Commun. 2018, 1, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Sasaki-Honda, M.; Jonouchi, T.; Arai, M.; Hotta, A.; Mitsuhashi, S.; Nishino, I.; Matsuda, R.; Sakurai, H. A patient-derived iPSC model revealed oxidative stress increases facioscapulohumeral muscular dystrophy-causative DUX4. Hum. Mol. Genet. 2018, 27, 4024–4035. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karpukhina, A.; Galkin, I.; Ma, Y.; Dib, C.; Zinovkin, R.; Pletjushkina, O.; Chernyak, B.; Popova, E.; Vassetzky, Y. Analysis of genes regulated by DUX4 via oxidative stress reveals potential therapeutic targets for treatment of facioscapulohumeral dystrophy. Redox Biol. 2021, 43, 102008. [Google Scholar] [CrossRef] [PubMed]
- Rudolf, R.; Magalhães, P.J.; Pozzan, T. Direct in vivo monitoring of sarcoplasmic reticulum Ca2+ and cytosolic cAMP dynamics in mouse skeletal muscle. J. Cell Biol. 2006, 173, 187–193. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stoyanovsky, D.; Murphy, T.; Anno, P.R.; Kim, Y.-M.; Salama, G. Nitric oxide activates skeletal and cardiac ryanodine receptors. Cell Calcium 1997, 21, 19–29. [Google Scholar] [CrossRef]
- Coirault, C.; Guellich, A.; Barbry, T.; Samuel, J.-L.; Riou, B.; LeCarpentier, Y. Oxidative stress of myosin contributes to skeletal muscle dysfunction in rats with chronic heart failure. Am. J. Physiol. Circ. Physiol. 2007, 292, H1009–H1017. [Google Scholar] [CrossRef] [PubMed]
- Yamada, T.; Mishima, T.; Sakamoto, M.; Sugiyama, M.; Matsunaga, S.; Wada, M. Oxidation of myosin heavy chain and reduction in force production in hyperthyroid rat soleus. J. Appl. Physiol. 2006, 100, 1520–1526. [Google Scholar] [CrossRef] [Green Version]
- De Brotto, M.P.; Van Leyen, S.; Brotto, L.S.; Jin, J.-P.; Nosek, C.M.; Nosek, T.M. Hypoxia/fatigue-induced degradation of troponin I and troponin C: New insights into physiologic muscle fatigue. Pflügers Arch. Eur. J. Physiol. 2001, 442, 738–744. [Google Scholar] [CrossRef]
- Kaneko, M.; Suzuki, H.; Masuda, H.; Yuan, G.; Hayashi, H.; Kobayashi, A.; Yamazaki, N. Effects of oxygen free radicals on Ca2+ binding to cardiac troponin. Jpn. Circ. J. 1992, 56, 1288–1290. [Google Scholar] [CrossRef] [Green Version]
- Kanatous, S.B.; Mammen, P.P.A.; Rosenberg, P.B.; Martin, C.M.; White, M.D.; DiMaio, J.M.; Huang, G.; Muallem, S.; Garry, D.J. Hypoxia reprograms calcium signaling and regulates myoglobin expression. Am. J. Physiol. Physiol. 2009, 296, C393–C402. [Google Scholar] [CrossRef] [Green Version]
- Aley, P.K.; Porter, K.E.; Boyle, J.P.; Kemp, P.J.; Peers, C. Hypoxic Modulation of Ca2+ Signaling in Human Venous Endothelial Cells. J. Biol. Chem. 2005, 280, 13349–13354. [Google Scholar] [CrossRef] [Green Version]
- Chen, L.; Lu, X.-Y.; Li, J.; Fu, J.-D.; Zhou, Z.-N.; Yang, H.-T. Intermittent hypoxia protects cardiomyocytes against ischemia-reperfusion injury-induced alterations in Ca2+ homeostasis and contraction via the sarcoplasmic reticulum and Na+/Ca2+ exchange mechanisms. Am. J. Physiol. Cell Physiol. 2006, 290, C1221–C1229. [Google Scholar] [CrossRef]
- Briguet, A.; Erb, M.; Courdier-Fruh, I.; Barzaghi, P.; Santos, G.; Herzner, H.; Lescop, C.; Siendt, H.; Henneboehle, M.; Weyermann, P.; et al. Effect of calpain and proteasome inhibition on Ca2+-dependent proteolysis and muscle histopathology in the mdx mouse. FASEB J. 2008, 22, 4190–4200. [Google Scholar] [CrossRef]
- Tidball, J.G.; Spencer, M.J. Calpains and muscular dystrophies. Int. J. Biochem. Cell Biol. 2000, 32, 1–5. [Google Scholar] [CrossRef]
- Lohan, J.; Ohlendieck, K. Drastic reduction in the luminal Ca2+-binding proteins calsequestrin and sarcalumenin in dystrophin-deficient cardiac muscle. Biochim. Biophys. Acta Mol. Basis Dis. 2004, 1689, 252–258. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Culligan, K.; Banville, N.; Dowling, P.; Ohlendieck, K. Drastic reduction of calsequestrin-like proteins and impaired calcium binding in dystrophic mdx muscle. J. Appl. Physiol. 2002, 92, 435–445. [Google Scholar] [CrossRef]
- Gyawali, B.; Shimokata, T.; Honda, K.; Kondoh, C.; Hayashi, N.; Yoshino, Y.; Sassa, N.; Nakano, Y.; Gotoh, M.; Ando, Y. Muscle wasting associated with the long-term use of mTOR inhibitors. Mol. Clin. Oncol. 2016, 5, 641–646. [Google Scholar] [CrossRef] [Green Version]
- Cohen, J.; DeSimone, A.; Lek, M.; Lek, A. Therapeutic Approaches in Facioscapulohumeral Muscular Dystrophy. Trends Mol. Med. 2021, 27, 123–137. [Google Scholar] [CrossRef]
- Bosnakovski, D.; Xu, Z.; Gang, E.J.; Galindo, C.L.; Liu, M.; Simsek, T.; Garner, H.R.; Agha-Mohammadi, S.; Tassin, A.; Coppée, F.; et al. An isogenetic myoblast expression screen identifies DUX4-mediated FSHD-associated molecular pathologies. EMBO J. 2008, 27, 2766–2779. [Google Scholar] [CrossRef] [Green Version]
- Bosnakovski, D.; Choi, S.H.; Strasser, J.M.; Toso, E.A.; Walters, M.A.; Kyba, M. High-throughput screening identifies inhibitors of DUX4-induced myoblast toxicity. Skelet. Muscle 2014, 4, 4. [Google Scholar] [CrossRef] [PubMed] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Regulated Pathways | HIF-1α Target Genes |
|---|---|
| Angiogenesis and erythropoiesis | VEGF and VEGF receptor FLT1, heme oxygenase 1, NOS 2 and 3, PDGF |
| Metabolism | ALDA, ALDOC, ENO1, GAPDH, HK1, HK2, LDH A, PFKL, PGK1, PKM, and TPI GLUT-1, 3, 4, PDK1 |
| Proliferation and survival | Cyclin G2, TGFα and β3, IGF-2 |
| Apoptosis | BNIP3/3L, P53 |
| Myogenesis | WNT signaling |
| Muscular Dystrophy | Pathogenetic Factors | Clinical Characteristics | Respiratory Impairment | ||||
|---|---|---|---|---|---|---|---|
| Inheritance | Affected Gene(s) | Muscle Distribution | Extra-Muscle Manifestations | Frequency | Type | ||
| Early onset | |||||||
| Dystroglycanopathies (Walker–Warburg, Fukuyama muscular dystrophy, muscle–eye–brain disease) | AR | Dystroglycan and glycosy transferase enzymes genes | Primarily axial and limb muscles | Structural brain anomalies | Uncommon (12% in a study on 115 patients) [23] | Nocturnal hypoventilation and acute respiratory failure ↘ FVC (27 patients) [24] | |
| Laminin-deficient muscular dystrophy | AR | LAMA2 | Primarily upper limbs | Diffuse white matter hyperintensities on brain MRI and seizures | Frequent (30% of patients with complete laminin-a2 deficiency) [25] | Skeletal muscle weakness (including intercostal and accessory muscles), scoliosis and decreased chest wall compliance. Alveolar hypoventilation, mucus plugs with bronchial obstruction and atelectasis ↘ FVC (59 patients) [26] | |
| SEPN1 myopathy (muscular dystrophy with rigid spine syndrome) | AR | SEPN1 | Early rigidity of the spine and joint contractures of the ankle and elbow | Rigid spine, scoliosis | Frequent; early 81.7% requiring ventilation (132 patients) [27] | Diaphragmatic weakness ↘ FVC by 24 ± 7% (7 patients) [28] | |
| Ullrich muscular dystrophy | AR | COL6A1, COL6A2, COL6A3 | Primarily axial and limb muscles | Rigid spine, laxity of distal joints | Frequent; early | Diaphragmatic weakness ↘ %VC (40 patients) [29] | |
| Childhood and Adult | |||||||
| Duchenne muscular dystrophy | X-linked R | Dystrophin | Proximal lower limb and truncal weakness, followed by of upper limb and distal muscle weakness | Educational and psychosocial issue, scoliosis, cardiomyopathy and arrhythmias | Frequent | Vital capacity (% predicted) decreases linearly, due to inspiratory and expiratory muscle weakness. Obstructive sleep apnea and hypoventilation. Nocturnal desaturation correlated to the severity of scoliosis. ↘ FVC, FEV1 and PEF (115 subjects) [30] | |
| Becker muscular dystrophy | X-linked R | Dystrophin | Same as DMD but with a milder phenotype | Less common than in DMD | Rare | Lung restriction sometimes occurs but less severe than in DMD | |
| Emery–Dreifuss muscular dystrophy | Variable depending on type | EMD, FHL1, LMNA, SYNE1, SYNE2 | Slowly and progressive humeroperoneal pattern | Cardiac conduction block, insulin resistance, rigid spine | Frequent; typically in adulthood | Restrictive pattern of respiratory impairment ↘ FVC to 60 and 45%p.v. (measured in 2 patients) [31] | |
| Facioscapulohumeral dystrophy | FSHD1 | AD | DUX4, | Facial, shoulder, scapular, arm progressive and asymmetric weakness | Retinal vasculopathy and symptomatic sensorineural hearing loss | First described as uncommon, 1–3% require NIV [32]. ↘ FVC in 38.3% and severely restrictive in 14.9% [33]. | Expiratory and diaphragmatic muscle weakness and obstructive sleep apnea ↘ mean FVC to 69%p.v. in non-mild disease (40.2%p.v. in early onset), minimum 33%p.v. (adult) and 11%p.v. (early onset) [33] ↘ MIP (69%c.v.), MEP (53%c.v.) and PCF (60%c.v.) [34] |
| FSHD2 | Digenic: DUX4 + either SCHMD1, DNMT3B or LRIF1 | ||||||
| Limb girdle muscular dystrophies | AR more frequent than AD | Sarcoglycan, Dystroglycan, Telethonin, Titin, etc. | Variable but mostly proximal weakness | Cardiomoypathy (common in sarcoglycan deficiency and dystroglycano pathy) | Common in sarcoglycan | Respiratory insufficiency due to diaphragmatic weakness Restrictive pulmonary syndrome indicated by TLC < 80%p.v. (13/38 patients) [35] FVC below 40%p.v. (20/38 patients) ↘ PEF (38 patient study) | |
| Myotonic dystrophy | AD | DMPK, CNBP | Distal slowly progressive weakness | Cardiac dysrhythmia, particularly heart block | Common | Sleep apnea syndrome and excessive daytime sleepiness ↘ MEP (21 patient study) [36] ↘ FVC, VC, TLC, RV, FEV1 [37] | |
| Experiments In Vitro | ||||
|---|---|---|---|---|
| Species | Cell Type | Way of HIF-1α Stabilization | Effect on Myogenesis | Ref. |
| Mouse | C2C12 | Hypoxia at 5% O2 | No effect | [106] |
| Hypoxia at 2% O2 | ↘ differentiation with ↘ Myod and Myog expression | |||
| Hypoxia at 0.5% O2 | ||||
| Hypoxia at 0.01% O2 | ||||
| Mouse | C2C12 | Hypoxia at 0.5% O2 | ↘ differentiation with ↘ Myod, Myog and Mhc expression | [107] |
| Mouse | C2C12 | Hypoxia at 1% O2 | ↘ differentiation with ↘ Mhc expression dependent on notch signaling | [108] |
| Mouse | Primary myoblast | Hypoxia at 1% O2 | ↘ differentiation through p53-dependent induction of Bhlhe40 | [109] |
| Mouse | C2C12 | Cobalt chloride | ↘ differentiation with ↘ myoblast proliferation, ↘ Myog expression | [112] |
| Mouse | C2C12 | Hypoxia at 5% O2 | ↘ differentiation with ↘ Myod, Myog and Mhc expression | [113] |
| Hypoxia at 10% O2 | ↗ differentiation with hypertrophy and ↗ Myog and Mhc expression | |||
| Hypoxia at 15% O2 | ||||
| Mouse | C2C12 | Hypoxia at 1% O2 | ↘ differentiation with ↘ Myod, Myf5, Myog and Mhc expression | [111] |
| Rat | L6 | Hypoxia at 1% O2 | ↘ differentiation with ↘ myoblast proliferation and ↘ myogenic index | [110] |
| Rat | L6E9 | Hypoxia at 1% O2 | ↘ differentiation with ↘ Myod, Myf5, Myog and Mhc expression | [111] |
| Human | Primary myoblasts | Hypoxia at 1% O2 | ↘ differentiation with ↘ myoblast proliferation and ↘ myogenic index | [110] |
| Bovine | SCs | Hypoxia at 1% O2 | ↗ differentiation with ↗ SC proliferation and ↗ Myod, Myog and Mhc expression | [115] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nguyen, T.-H.; Conotte, S.; Belayew, A.; Declèves, A.-E.; Legrand, A.; Tassin, A. Hypoxia and Hypoxia-Inducible Factor Signaling in Muscular Dystrophies: Cause and Consequences. Int. J. Mol. Sci. 2021, 22, 7220. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22137220
Nguyen T-H, Conotte S, Belayew A, Declèves A-E, Legrand A, Tassin A. Hypoxia and Hypoxia-Inducible Factor Signaling in Muscular Dystrophies: Cause and Consequences. International Journal of Molecular Sciences. 2021; 22(13):7220. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22137220
Chicago/Turabian StyleNguyen, Thuy-Hang, Stephanie Conotte, Alexandra Belayew, Anne-Emilie Declèves, Alexandre Legrand, and Alexandra Tassin. 2021. "Hypoxia and Hypoxia-Inducible Factor Signaling in Muscular Dystrophies: Cause and Consequences" International Journal of Molecular Sciences 22, no. 13: 7220. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22137220