
MYC Ran Up the Clock: The Complex Interplay between MYC and the Molecular Circadian Clock in Cancer


{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction to MYC and the Molecular Circadian Clock in Normal Cellular Biology
2. Bi-Directional Relationship between MYC and the Molecular Clock in Cancer
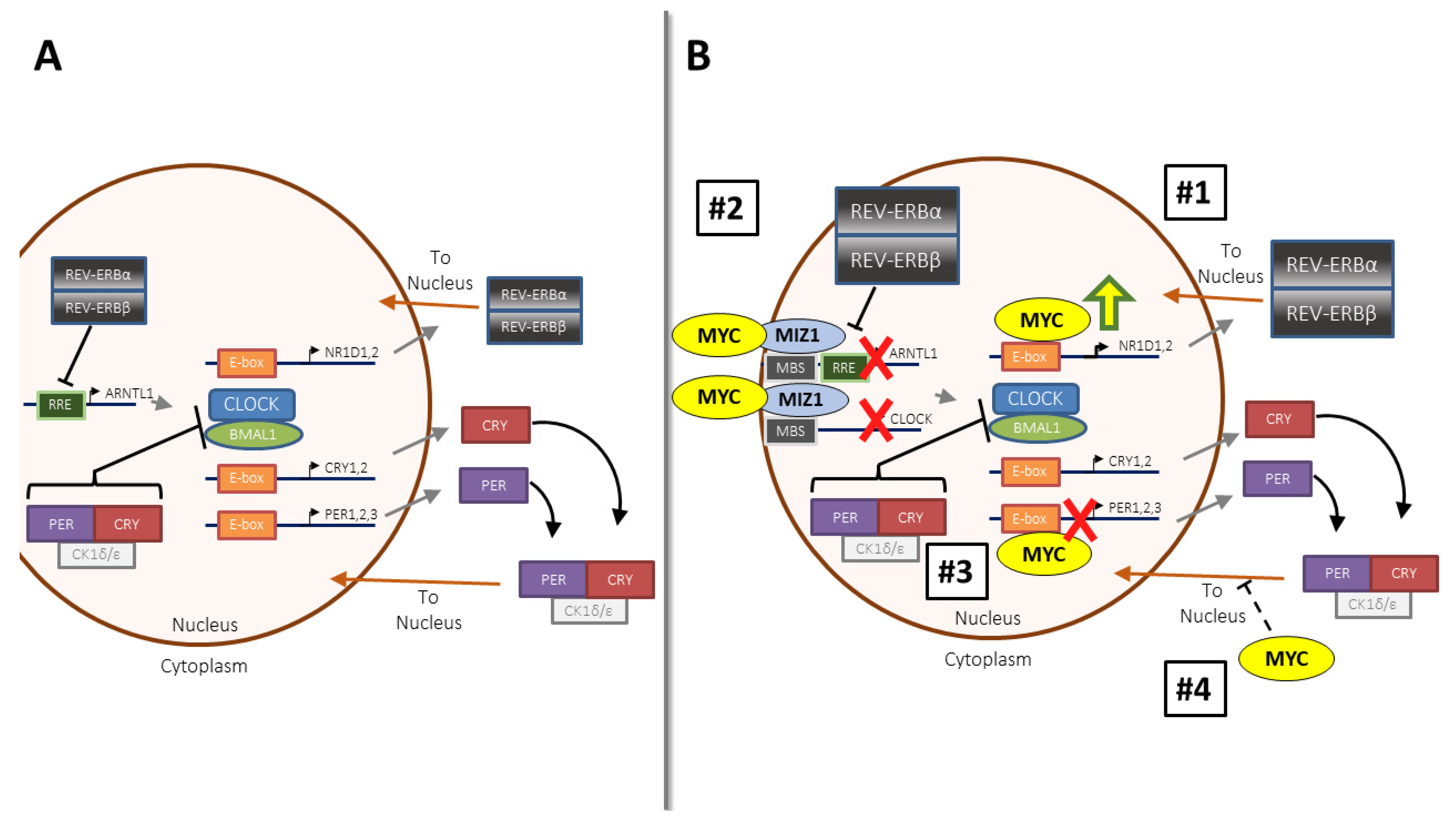
2.1. MYC’s Influence on the Molecular Clock in Somatic Cells and Cancer
2.2. Links between a Deregulated Circadian Clock and Cancer, and Downstream Effects on MYC Expression
2.3. How and When Does MYC Disrupt the Molecular Clock? Context Matters
3. Control of Chromatin State and Pause Release by MYC and the Molecular Clock
3.1. Role of MYC in Chromatin and Global Pause Release
3.2. Molecular Clock Control of Chromatin State and Pause Release
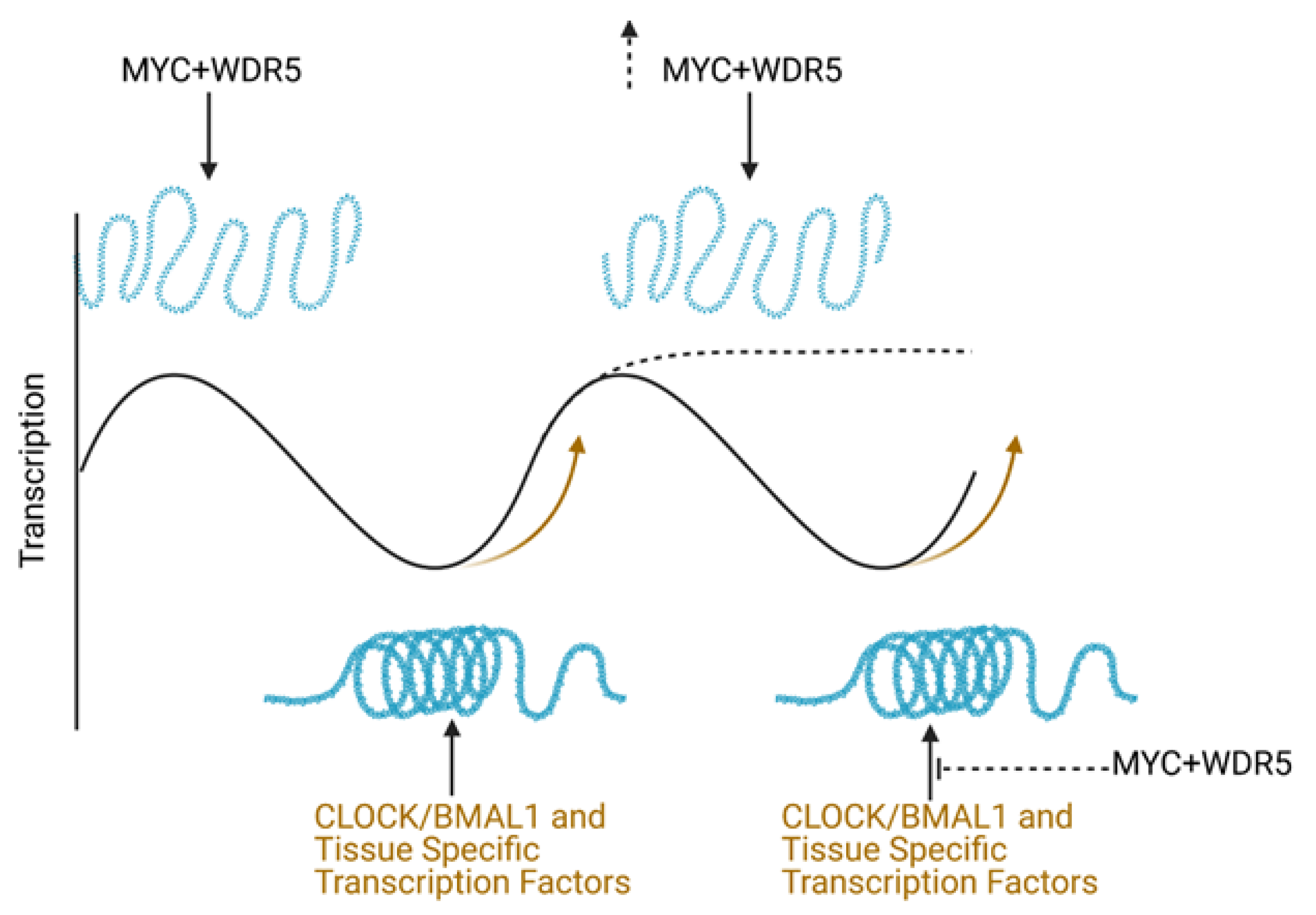
3.3. How Do MYC and the Molecular Clock Interact to Regulate Global Transcriptional Output?
4. MYC and CLOCK Clash for Control of Metabolism
4.1. Brief Overview of MYC-Mediated Metabolic Rewiring
4.2. The Molecular Clock Controls Cell Autonomous Metabolic Oscillation and Metabolic Gene Expression Programs
4.3. How Does MYC-Driven Metabolic Rewiring Interact with Circadian Oscillation of Cell Metabolism?
5. Does Time Stop When MYC Alters the Tumor Immune Microenvironment?
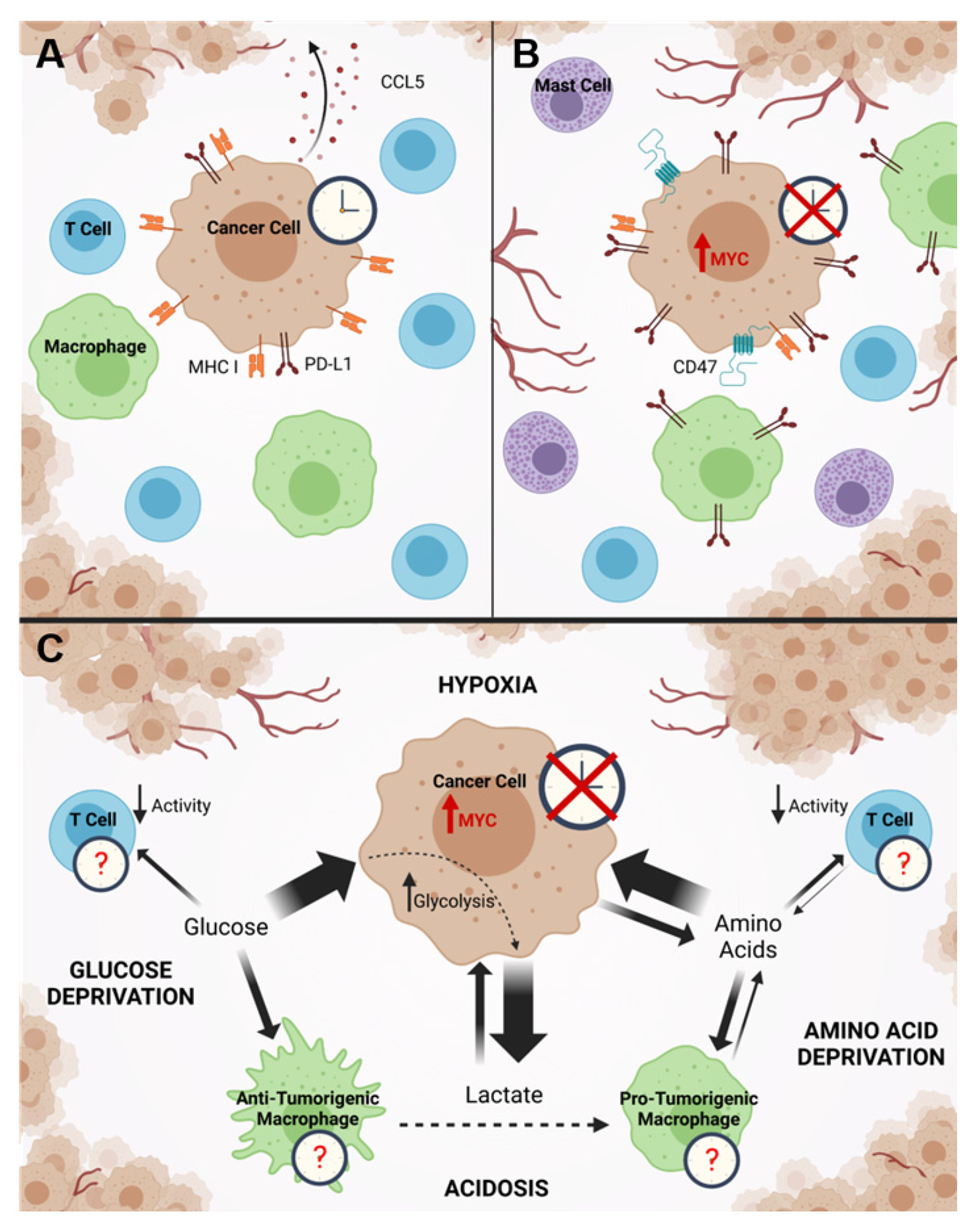
5.1. MYC Modulation of the Tumor Immune Microenvironment
5.2. Connections between MYC and Potential Molecular Clock Disruption in the Tumor Immune Microenvironment
6. Clinical Applications: MYC Inhibition, and Measurement of the Molecular Clock in Patients
6.1. Recent Strategies to Inhibit MYC in Human Cancer
6.2. Restored Rhythms after MYC Inhibition? New Strategies to Detect Molecular Clock Rhythmicity in Tumors and Other Tissues of Cancer Patients
7. Conclusions and Future Outlook
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Carroll, P.A.; Freie, B.W.; Mathsyaraja, H.; Eisenman, R.N. The MYC transcription factor network: Balancing metabolism, proliferation and oncogenesis. Front. Med. 2018, 12, 412–425. [Google Scholar] [CrossRef] [Green Version]
- Wolf, E.; Lin, C.Y.; Eilers, M.; Levens, D.L. Taming of the beast: Shaping Myc-dependent amplification. Trends Cell Biol. 2015, 25, 241–248. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bretones, G.; Delgado, M.D.; León, J. Myc and cell cycle control. Biochim. Biophys. Acta Bioenerg. 2015, 1849, 506–516. [Google Scholar] [CrossRef] [PubMed]
- Schaub, F.X.; Dhankani, V.; Berger, A.C.; Trivedi, M.; Richardson, A.; Shaw, R.; Zhao, W.; Zhang, X.; Ventura, A.; Liu, Y.; et al. Pan-cancer Alterations of the MYC Oncogene and Its Proximal Network across the Cancer Genome Atlas. Cell Syst. 2018, 6, 282–300.e2. [Google Scholar] [CrossRef] [Green Version]
- Stine, Z.E.; Walton, Z.E.; Altman, B.; Hsieh, A.L.; Dang, C.V. MYC, Metabolism, and Cancer. Cancer Discov. 2015, 5, 1024–1039. [Google Scholar] [CrossRef] [Green Version]
- Kelleher, F.C.; Rao, A.; Maguire, A. Circadian molecular clocks and cancer. Cancer Lett. 2014, 342, 9–18. [Google Scholar] [CrossRef]
- Yi, J.S.; Díaz, N.M.; D’Souza, S.; Buhr, E.D. The molecular clockwork of mammalian cells. Semin. Cell Dev. Biol. 2021. [Google Scholar] [CrossRef]
- Brown, S.A.; Azzi, A. Peripheral Circadian Oscillators in Mammals. Organotypic Models Drug Dev. 2013, 217, 45–66. [Google Scholar] [CrossRef]
- Bass, J.; Takahashi, J. Circadian Integration of Metabolism and Energetics. Science 2010, 330, 1349–1354. [Google Scholar] [CrossRef] [Green Version]
- Krishnaiah, S.Y.; Wu, G.; Altman, B.; Growe, J.; Rhoades, S.D.; Coldren, F.; Venkataraman, A.; Olarerin-George, A.O.; Francey, L.J.; Mukherjee, S.; et al. Clock Regulation of Metabolites Reveals Coupling between Transcription and Metabolism. Cell Metab. 2017, 25, 961–974.e4. [Google Scholar] [CrossRef] [Green Version]
- Marcheva, B.; Ramsey, K.M.; Buhr, E.D.; Kobayashi, Y.; Su, H.; Ko, C.H.; Ivanova, G.; Omura, C.; Mo, S.; Vitaterna, M.H.; et al. Disruption of the clock components CLOCK and BMAL1 leads to hypoinsulinaemia and diabetes. Nat. Cell Biol. 2010, 466, 627–631. [Google Scholar] [CrossRef] [Green Version]
- Shi, S.-Q.; Ansari, T.S.; McGuinness, O.P.; Wasserman, D.H.; Johnson, C.H. Circadian Disruption Leads to Insulin Resistance and Obesity. Curr. Biol. 2013, 23, 372–381. [Google Scholar] [CrossRef] [Green Version]
- Depner, C.M.; Stothard, E.R.; Wright, K.P. Metabolic Consequences of Sleep and Circadian Disorders. Curr. Diabetes Rep. 2014, 14, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.; Lahens, N.F.; Ballance, H.I.; Hughes, M.E.; HogenEsch, J.B. A circadian gene expression atlas in mammals: Implications for biology and medicine. Proc. Natl. Acad. Sci. USA 2014, 111, 16219–16224. [Google Scholar]
- Mure, L.S.; Le, H.D.; Benegiamo, G.; Chang, M.W.; Rios, L.; Jillani, N.; Ngotho, M.; Kariuki, T.; Dkhissi-Benyahya, O.; Cooper, H.M.; et al. Diurnal transcriptome atlas of a primate across major neural and peripheral tissues. Science 2018, 359, 0318. [Google Scholar] [CrossRef] [Green Version]
- Aryal, R.P.; Kwak, P.B.; Tamayo, A.G.; Gebert, M.; Chiu, P.-L.; Walz, T.; Weitz, C.J. Macromolecular Assemblies of the Mammalian Circadian Clock. Mol. Cell 2017, 67, 770–782.e6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Levine, D.; Hong, H.; Weidemann, B.J.; Ramsey, K.M.; Affinati, A.H.; Schmidt, M.S.; Cedernaes, J.; Omura, C.; Braun, R.; Lee, C.; et al. NAD+ Controls Circadian Reprogramming through PER2 Nuclear Translocation to Counter Aging. Mol. Cell 2020, 78, 835–849.e7. [Google Scholar] [CrossRef] [PubMed]
- Anafi, R.C.; Francey, L.J.; Hogenesch, J.B.; Kim, J. CYCLOPS reveals human transcriptional rhythms in health and disease. Proc. Natl. Acad. Sci. USA 2017, 114, 5312–5317. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yin, L.; Wu, N.; Curtin, J.C.; Qatanani, M.; Szwergold, N.R.; Reid, R.A.; Waitt, G.M.; Parks, D.J.; Pearce, K.H.; Wisely, G.B.; et al. Rev-erb, a Heme Sensor That Coordinates Metabolic and Circadian Pathways. Science 2007, 318, 1786–1789. [Google Scholar] [CrossRef]
- Bugge, A.; Feng, D.; Everett, L.J.; Briggs, E.R.; Mullican, S.E.; Wang, F.; Jager, J.; Lazar, M.A. Rev-erb and Rev-erb coordinately protect the circadian clock and normal metabolic function. Genes Dev. 2012, 26, 657–667. [Google Scholar] [CrossRef] [Green Version]
- Cho, H.; Zhao, X.; Hatori, M.; Yu, R.T.; Barish, G.D.; Lam, M.T.; Chong, L.-W.; DiTacchio, L.; Atkins, A.R.; Glass, C.K.; et al. Regulation of circadian behaviour and metabolism by REV-ERB-α and REV-ERB-β. Nature 2012, 485, 123–127. [Google Scholar] [CrossRef] [Green Version]
- Altman, B.J. Cancer Clocks Out for Lunch: Disruption of Circadian Rhythm and Metabolic Oscillation in Cancer. Front. Cell Dev. Biol. 2016, 4, 62. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Avet-Loiseau, H.; Gerson, F.; Magrangeas, F.; Minvielle, S.; Harousseau, J.-L.; Bataille, R. Rearrangements of the c-myc oncogene are present in 15% of primary human multiple myeloma tumors. Blood 2001, 98, 3082–3086. [Google Scholar] [CrossRef] [Green Version]
- Boxer, L.M.; Dang, C.V. Translocations involving c-myc and c-myc function. Oncogene 2001, 20, 5595–5610. [Google Scholar] [CrossRef] [Green Version]
- Ott, G.; Rosenwald, A.; Campo, E. Understanding MYC-driven aggressive B-cell lymphomas: Pathogenesis and classification. Blood 2013, 122, 3884–3891. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Choi, P.; Francis, J.M.; Imielinski, M.; Watanabe, H.; Cherniack, A.D.; Meyerson, M. Identification of focally amplified lineage-specific super-enhancers in human epithelial cancers. Nat. Genet. 2016, 48, 176–182. [Google Scholar] [CrossRef] [PubMed]
- Xie, S.; Lin, H.; Sun, T.; Arlinghaus, R.B. Jak2 is involved in c-Myc induction by Bcr-Abl. Oncogene 2002, 21, 7137–7146. [Google Scholar] [CrossRef] [Green Version]
- Sawyers, C.L.; Callahan, W.; Witte, O.N. Dominant negative MYC blocks transformation by ABL oncogenes. Cell 1992, 70, 901–910. [Google Scholar] [CrossRef]
- Palomero, T.; Lim, W.K.; Odom, D.; Sulis, M.L.; Real, P.; Margolin, A.; Barnes, K.C.; O’Neil, J.; Neuberg, D.; Weng, A.; et al. NOTCH1 directly regulates c-MYC and activates a feed-forward-loop transcriptional network promoting leukemic cell growth. Proc. Natl. Acad. Sci. USA 2006, 103, 18261–18266. [Google Scholar] [CrossRef] [Green Version]
- Weng, A.; Millholland, J.M.; Yashiro-Ohtani, Y.; Arcangeli, M.-L.; Lau, A.; Wai, C.; Del Bianco, C.; Rodriguez, C.G.; Sai, H.; Tobias, J.; et al. c-Myc is an important direct target of Notch1 in T-cell acute lymphoblastic leukemia/lymphoma. Genes Dev. 2006, 20, 2096–2109. [Google Scholar] [CrossRef] [Green Version]
- Ghosh, A.; Grigorieva, I.; Steele, R.; Hoover, R.; Ray, R. PTEN transcriptionally modulates c-myc gene expression in human breast carcinoma cells and is involved in cell growth regulation. Gene 1999, 235, 85–91. [Google Scholar] [CrossRef]
- Zheng, H.; Ying, H.; Yan, H.; Kimmelman, A.; Hiller, D.; Chen, A.-J.; Perry, S.; Tonon, G.; Chu, G.; Ding, Z.; et al. Pten and p53 Converge on c-Myc to Control Differentiation, Self-renewal, and Transformation of Normal and Neoplastic Stem Cells in Glioblastoma. Cold Spring Harb. Symp. Quant. Biol. 2008, 73, 427–437. [Google Scholar] [CrossRef] [Green Version]
- Chou, Y.-T.; Lin, H.-H.; Lien, Y.-C.; Wang, Y.-H.; Hong, C.-F.; Kao, Y.-R.; Lin, S.-C.; Chang, Y.-C.; Lin, S.-Y.; Chen, S.-J.; et al. EGFR Promotes Lung Tumorigenesis by Activating miR-7 through a Ras/ERK/Myc Pathway That Targets the Ets2 Transcriptional Repressor ERF. Cancer Res. 2010, 70, 8822–8831. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Altman, B.J.; Hsieh, A.L.; Gouw, A.; Dang, C.V. Correspondence: Oncogenic MYC persistently upregulates the molecular clock component REV-ERBα. Nat. Commun. 2017, 8, 14862. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Altman, B.; Hsieh, A.L.; Sengupta, A.; Krishnanaiah, S.Y.; Stine, Z.E.; Walton, Z.E.; Gouw, A.; Venkataraman, A.; Li, B.; Goraksha-Hicks, P.; et al. MYC Disrupts the Circadian Clock and Metabolism in Cancer Cells. Cell Metab. 2015, 22, 1009–1019. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shostak, A.; Ruppert, B.; Diernfellner, A.; Brunner, M. Correspondence: Reply to ‘Oncogenic MYC persistently upregulates the molecular clock component REV-ERBα’. Nat. Commun. 2017, 8, 14918. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shostak, A.; Project, I.M.-S.; Ruppert, B.; Ha, N.; Bruns, P.; Toprak, U.H.; Eils, R.; Schlesner, M.; Diernfellner, A.; Brunner, M.; et al. MYC/MIZ1-dependent gene repression inversely coordinates the circadian clock with cell cycle and proliferation. Nat. Commun. 2016, 7, 11807. [Google Scholar] [CrossRef] [Green Version]
- Moreno-Smith, M.; Milazzo, G.; Tao, L.; Fekry, B.; Zhu, B.; Mohammad, M.A.; Di Giacomo, S.; Borkar, R.; Reddy, K.R.K.; Capasso, M.; et al. Restoration of the molecular clock is tumor suppressive in neuroblastoma. Nat. Commun. 2021, 12, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Repouskou, A.; Prombona, A. c-MYC targets the central oscillator gene Per1 and is regulated by the circadian clock at the post-transcriptional level. Biochim. Biophys. Acta (BBA) Bioenerg. 2016, 1859, 541–552. [Google Scholar] [CrossRef]
- Umemura, Y.; Koike, N.; Matsumoto, T.; Yoo, S.-H.; Chen, Z.; Yasuhara, N.; Takahashi, J.S.; Yagita, K. Transcriptional program of Kpna2/Importin-α2 regulates cellular differentiation-coupled circadian clock development in mammalian cells. Proc. Natl. Acad. Sci. USA 2014, 111, E5039–E5048. [Google Scholar] [CrossRef] [Green Version]
- Hsieh, A.L.; Zheng, X.; Yue, Z.; Stine, Z.E.; Mancuso, A.; Rhoades, S.D.; Brooks, R.; Weljie, A.M.; Eisenman, R.N.; Sehgal, A.; et al. Misregulation of Drosophila Myc Disrupts Circadian Behavior and Metabolism. Cell Rep. 2019, 29, 1778–1788.e4. [Google Scholar] [CrossRef] [Green Version]
- Blaževitš, O.; Bolshette, N.; Vecchio, D.; Guijarro, A.; Croci, O.; Campaner, S.; Grimaldi, B. MYC-Associated Factor MAX is a Regulator of the Circadian Clock. Int. J. Mol. Sci. 2020, 21, 2294. [Google Scholar] [CrossRef] [Green Version]
- Schernhammer, E.S.; Feskanich, D.; Liang, G.; Han, J. Rotating Night-Shift Work and Lung Cancer Risk Among Female Nurses in the United States. Am. J. Epidemiol. 2013, 178, 1434–1441. [Google Scholar] [CrossRef] [PubMed]
- Schernhammer, E.S.; Laden, F.; Speizer, F.E.; Willett, W.C.; Hunter, D.J.; Kawachi, I.; Colditz, G.A. Rotating night shifts and risk of breast cancer in women participating in the nurses’ health study. J. Natl. Cancer Inst. 2001, 93, 1563–1568. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schernhammer, E.S.; Laden, F.; Speizer, F.E.; Willett, W.C.; Hunter, D.J.; Kawachi, I.; Fuchs, C.S.; Colditz, G.A. Night-shift work and risk of colorectal cancer in the nurses’ health study. J. Natl. Cancer Inst. 2003, 95, 825–828. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ward, E.M.; Germolec, D.; Kogevinas, M.; McCormick, D.; Vermeulen, R.; Anisimov, V.N.; Aronson, K.J.; Bhatti, P.; Cocco, P.; Costa, G.; et al. Carcinogenicity of night shift work. Lancet Oncol. 2019, 20, 1058–1059. [Google Scholar] [CrossRef]
- Sephton, S.E.; Lush, E.; Dedert, E.A.; Floyd, A.R.; Rebholz, W.; Dhabhar, F.S.; Spiegel, D.; Salmon, P. Diurnal cortisol rhythm as a predictor of lung cancer survival. Brain, Behav. Immun. 2013, 30, S163–S170. [Google Scholar] [CrossRef]
- Sephton, S.E.; Sapolsky, R.M.; Kraemer, H.C.; Spiegel, D. Diurnal Cortisol Rhythm as a Predictor of Breast Cancer Survival. J. Natl. Cancer Inst. 2000, 92, 994–1000. [Google Scholar] [CrossRef] [PubMed]
- Innominato, P.F.; Komarzynski, S.; Palesh, O.G.; Dallmann, R.; Bjarnason, G.A.; Giacchetti, S.; Ulusakarya, A.; Bouchahda, M.; Haydar, M.; Ballesta, A.; et al. Circadian rest-activity rhythm as an objective biomarker of patient-reported outcomes in patients with advanced cancer. Cancer Med. 2018, 7, 4396–4405. [Google Scholar] [CrossRef] [PubMed]
- Innominato, P.F.; Focan, C.; Gorlia, T.; Moreau, T.; Garufi, C.; Waterhouse, J.; Giacchetti, S.; Coudert, B.; Iacobelli, S.; Genet, D.; et al. Circadian Rhythm in Rest and Activity: A Biological Correlate of Quality of Life and a Predictor of Survival in Patients with Metastatic Colorectal Cancer. Cancer Res. 2009, 69, 4700–4707. [Google Scholar] [CrossRef] [Green Version]
- Mormont, M.C.; Waterhouse, J.; Bleuzen, P.; Giacchetti, S.; Jami, A.; Bogdan, A.; Lellouch, J.; Misset, J.L.; Touitou, Y.; Lévi, F. Marked 24-h rest/activity rhythms are associated with better quality of life, better response, and longer survival in patients with metastatic colorectal cancer and good performance status. Clin. Cancer Res. 2000, 6, 3038. [Google Scholar] [PubMed]
- Levi, F.; Dugué, P.-A.; Innominato, P.; Karaboué, A.; Dispersyn, G.; Parganiha, A.; Giacchetti, S.; Moreau, T.; Focan, C.; Waterhouse, J.; et al. Wrist actimetry circadian rhythm as a robust predictor of colorectal cancer patients survival. Chrono- Int. 2014, 31, 891–900. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Filipski, E.; Delaunay, F.; King, V.M.; Wu, M.-W.; Claustrat, B.; Gréchez-Cassiau, A.; Guettier, C.; Hastings, M.H.; Francis, L. Effects of Chronic Jet Lag on Tumor Progression in Mice. Cancer Res. 2004, 64, 7879–7885. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Filipski, E.; Subramanian, P.; Carrière, J.; Guettier, C.; Barbason, H.; Lévi, F. Circadian disruption accelerates liver carcinogenesis in mice. Mutat. Res. Toxicol. Environ. Mutagen. 2009, 680, 95–105. [Google Scholar] [CrossRef]
- Logan, R.W.; Zhang, C.; Murugan, S.; O’Connell, S.; Levitt, D.; Rosenwasser, A.M.; Sarkar, D.K. Chronic Shift-Lag Alters the Circadian Clock of NK Cells and Promotes Lung Cancer Growth in Rats. J. Immunol. 2012, 188, 2583–2591. [Google Scholar] [CrossRef] [Green Version]
- Kettner, N.; Voicu, H.; Finegold, M.J.; Coarfa, C.; Sreekumar, A.; Putluri, N.; Katchy, C.A.; Lee, C.; Moore, D.D.; Fu, L. Circadian Homeostasis of Liver Metabolism Suppresses Hepatocarcinogenesis. Cancer Cell 2016, 30, 909–924. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Papagiannakopoulos, T.; Bauer, M.R.; Davidson, S.M.; Heimann, M.; Subbaraj, L.; Bhutkar, A.; Bartlebaugh, J.; Heiden, M.G.V.; Jacks, T. Circadian Rhythm Disruption Promotes Lung Tumorigenesis. Cell Metab. 2016, 24, 324–331. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, Y.; Lahens, N.F.; Zhang, S.; Bedont, J.; Field, J.M.; Sehgal, A. G1/S cell cycle regulators mediate effects of circadian dysregulation on tumor growth and provide targets for timed anticancer treatment. PLoS Biol. 2019, 17, e3000228. [Google Scholar] [CrossRef] [Green Version]
- Aiello, I.; Fedele, M.L.M.; Román, F.; Marpegan, L.; Caldart, C.; Chiesa, J.J.; Golombek, D.A.; Finkielstein, C.V.; Paladino, N. Circadian disruption promotes tumor-immune microenvironment remodeling favoring tumor cell proliferation. Sci. Adv. 2020, 6, eaaz4530. [Google Scholar] [CrossRef]
- Fu, L.; Pelicano, H.; Liu, J.; Huang, P.; Lee, C.C. The circadian gene period2 plays an important role in tumor suppression and DNA damage response in vivo. Cell 2002, 111, 41–50. [Google Scholar] [CrossRef] [Green Version]
- Huber, A.-L.; Papp, S.J.; Chan, A.B.; Henriksson, E.; Jordan, S.D.; Kriebs, A.; Nguyen, M.; Wallace, M.; Li, Z.; Metallo, C.M.; et al. CRY2 and FBXL3 Cooperatively Degrade c-MYC. Mol. Cell 2016, 64, 774–789. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kiessling, S.; Beaulieu-Laroche, L.; Blum, I.D.; Landgraf, D.; Welsh, D.K.; Storch, K.-F.; Labrecque, N.; Cermakian, N. Enhancing circadian clock function in cancer cells inhibits tumor growth. BMC Biol. 2017, 15, 1–18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, Y.; Fong, S.Y.; Shon, J.; Zhang, S.L.; Brooks, R.; Lahens, N.F.; Chen, D.; Van Dang, C.; Field, J.M.; Sehgal, A. Time-of-day specificity of anticancer drugs may be mediated by circadian regulation of the cell cycle. Sci. Adv. 2021, 7, 2645. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Selby, C.P.; Yang, Y.; Lindsey-Boltz, L.A.; Cao, X.; Eynullazada, K.; Sancar, A. Circadian regulation of c-MYC in mice. Proc. Natl. Acad. Sci. USA 2020, 117, 21609–21617. [Google Scholar] [CrossRef] [PubMed]
- Repouskou, A.; Sourlingas, T.G.; Sekeri-Pataryas, K.E.; Prombona, A. The circadian expression of c-myc is modulated by the histone deacetylase inhibitor trichostatin a in synchronized murine neuroblastoma cells. Chronobiol. Int. 2010, 27, 722–741. [Google Scholar] [CrossRef]
- Janich, P.; Pascual, G.; Merlos-Suárez, A.; Batlle, E.; Ripperger, J.; Albrecht, U.; Cheng, H.-Y.M.; Obrietan, K.; Di Croce, L.; Benitah, S.A. The circadian molecular clock creates epidermal stem cell heterogeneity. Nat. Cell Biol. 2011, 480, 209–214. [Google Scholar] [CrossRef] [Green Version]
- Puram, R.V.; Kowalczyk, M.S.; de Boer, C.; Schneider, R.K.; Miller, P.; McConkey, M.; Tothova, Z.; Tejero, H.; Heckl, D.; Järås, M.; et al. Core Circadian Clock Genes Regulate Leukemia Stem Cells in AML. Cell 2016, 165, 303–316. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dong, Z.; Zhang, G.; Qu, M.; Gimple, R.C.; Wu, Q.; Qiu, Z.; Prager, B.C.; Wang, X.; Kim, L.J.; Morton, A.; et al. Targeting Glioblastoma Stem Cells through Disruption of the Circadian Clock. Cancer Discov. 2019, 9, 1556–1573. [Google Scholar] [CrossRef] [Green Version]
- Takahashi, K.; Yamanaka, S. A decade of transcription factor-mediated reprogramming to pluripotency. Nat. Rev. Mol. Cell Biol. 2016, 17, 183–193. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Wang, H.; Li, Z.; Wu, Q.; Lathia, J.D.; McLendon, R.E.; Hjelmeland, A.B.; Rich, J.N. c-Myc Is Required for Maintenance of Glioma Cancer Stem Cells. PLoS ONE 2008, 3, e3769. [Google Scholar] [CrossRef]
- Kieffer-Kwon, K.-R.; Nimura, K.; Rao, S.S.; Xu, J.; Jung, S.; Pekowska, A.; Dose, M.; Stevens, E.; Mathe, E.; Dong, P.; et al. Myc Regulates Chromatin Decompaction and Nuclear Architecture during B Cell Activation. Mol. Cell 2017, 67, 566–578.e10. [Google Scholar] [CrossRef] [PubMed]
- Tesi, A.; de Pretis, S.; Furlan, M.; Filipuzzi, M.; Morelli, M.; Andronache, A.; Doni, M.; Verrecchia, A.; Pelizzola, M.; Amati, B.; et al. An early Myc-dependent transcriptional program orchestrates cell growth during B-cell activation. EMBO Rep. 2019, 20, e47987. [Google Scholar] [CrossRef] [PubMed]
- Guccione, E.; Martinato, F.; Finocchiaro, G.; Luzi, L.; Tizzoni, L.; Dall’ Olio, V.; Zardo, G.; Nervi, C.; Bernard, L.; Amati, B. Myc-binding-site recognition in the human genome is determined by chromatin context. Nat. Cell Biol. 2006, 8, 764–770. [Google Scholar] [CrossRef]
- Lorenzin, F.; Benary, U.; Baluapuri, A.; Walz, S.; Jung, L.A.; von Eyss, B.; Kisker, C.; Wolf, J.; Eilers, M.; Wolf, E. Different promoter affinities account for specificity in MYC-dependent gene regulation. eLife 2016, 5. [Google Scholar] [CrossRef]
- Lourenco, C.; Resetca, D.; Redel, C.; Lin, P.; MacDonald, A.S.; Ciaccio, R.; Kenney, T.M.G.; Wei, Y.; Andrews, D.W.; Sunnerhagen, M.; et al. MYC protein interactors in gene transcription and cancer. Nat. Rev. Cancer 2021, 1–13. [Google Scholar] [CrossRef]
- Guarnaccia, A.D.; Tansey, W.P. Moonlighting with WDR5: A Cellular Multitasker. J. Clin. Med. 2018, 7, 21. [Google Scholar] [CrossRef] [Green Version]
- Thomas, L.R.; Adams, C.M.; Wang, J.; Weissmiller, A.M.; Creighton, J.; Lorey, S.L.; Liu, Q.; Fesik, S.W.; Eischen, C.M.; Tansey, W.P. Interaction of the oncoprotein transcription factor MYC with its chromatin cofactor WDR5 is essential for tumor maintenance. Proc. Natl. Acad. Sci. USA 2019, 116, 25260–25268. [Google Scholar] [CrossRef] [Green Version]
- Brown, S.A.; Ripperger, J.; Kadener, S.; Fleury-Olela, F.; Vilbois, F.; Rosbash, M.; Schibler, U. PERIOD1-Associated Proteins Modulate the Negative Limb of the Mammalian Circadian Oscillator. Science 2005, 308, 693–696. [Google Scholar] [CrossRef] [Green Version]
- Saunders, A.; Core, L.J.; Lis, J.T. Breaking barriers to transcription elongation. Nat. Rev. Mol. Cell Biol. 2006, 7, 557–567. [Google Scholar] [CrossRef]
- Rahl, P.B.; Lin, C.Y.; Seila, A.C.; Flynn, R.; McCuine, S.; Burge, C.B.; Sharp, P.A.; Young, R.A. c-Myc Regulates Transcriptional Pause Release. Cell 2010, 141, 432–445. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, C.Y.; Lovén, J.; Rahl, P.B.; Paranal, R.M.; Burge, C.B.; Bradner, J.E.; Lee, T.I.; Young, R.A. Transcriptional Amplification in Tumor Cells with Elevated c-Myc. Cell 2012, 151, 56–67. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nie, Z.; Hu, G.; Wei, G.; Cui, K.; Yamane, A.; Resch, W.; Wang, R.; Green, D.R.; Tessarollo, L.; Casellas, R.; et al. c-Myc Is a Universal Amplifier of Expressed Genes in Lymphocytes and Embryonic Stem Cells. Cell 2012, 151, 68–79. [Google Scholar] [CrossRef] [Green Version]
- Guo, J.; Li, T.; Schipper, J.; A Nilson, K.; Fordjour, F.K.; Cooper, J.J.; Gordân, R.; Price, D.H. Sequence specificity incompletely defines the genome-wide occupancy of Myc. Genome Biol. 2014, 15, 482. [Google Scholar] [CrossRef]
- Durbin, A.D.; Zimmerman, M.W.; Dharia, N.V.; Abraham, B.J.; Iniguez, A.B.; Weichert-Leahey, N.; He, S.; Krill-Burger, J.M.; Root, D.E.; Vazquez, F.; et al. Selective gene dependencies in MYCN-amplified neuroblastoma include the core transcriptional regulatory circuitry. Nat. Genet. 2018, 50, 1240–1246. [Google Scholar] [CrossRef]
- Liang, K.; Smith, E.R.; Aoi, Y.; Stoltz, K.L.; Katagi, H.; Woodfin, A.R.; Rendleman, E.J.; Marshall, S.A.; Murray, D.C.; Wang, L.; et al. Targeting Processive Transcription Elongation via SEC Disruption for MYC-Induced Cancer Therapy. Cell 2018, 175, 766–779.e17. [Google Scholar] [CrossRef] [Green Version]
- Walz, S.; Lorenzin, F.; Morton, J.P.; Wiese, K.E.; Von Eyss, B.; Herold, S.; Rycak, L.; Dumay-Odelot, H.; Karim, S.; Bartkuhn, M.; et al. Activation and repression by oncogenic MYC shape tumour-specific gene expression profiles. Nat. Cell Biol. 2014, 511, 483–487. [Google Scholar] [CrossRef]
- Sabò, A.; Kress, T.R.; Pelizzola, M.; De Pretis, S.; Gorski, M.M.; Tesi, A.; Morelli, M.J.; Bora, P.; Doni, M.; Verrecchia, A.; et al. Selective transcriptional regulation by Myc in cellular growth control and lymphomagenesis. Nat. Cell Biol. 2014, 511, 488–492. [Google Scholar] [CrossRef]
- Herold, S.; Kalb, J.; Büchel, G.; Ade, C.P.; Baluapuri, A.; Xu, J.; Koster, J.; Solvie, D.; Carstensen, A.; Klotz, C.; et al. Recruitment of BRCA1 limits MYCN-driven accumulation of stalled RNA polymerase. Nat. Cell Biol. 2019, 567, 545–549. [Google Scholar] [CrossRef] [PubMed]
- Nie, Z.; Guo, C.; Das, S.K.; Chow, C.C.; Batchelor, E.; Simons, S.S.; Levens, D. Dissecting transcriptional amplification by MYC. eLife 2020, 9. [Google Scholar] [CrossRef] [PubMed]
- Koike, N.; Yoo, S.-H.; Huang, H.-C.; Kumar, V.; Lee, C.; Kim, T.-K.; Takahashi, J.S. Transcriptional Architecture and Chromatin Landscape of the Core Circadian Clock in Mammals. Science 2012, 338, 349–354. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Menet, J.S.; Pescatore, S.; Rosbash, M. CLOCK:BMAL1 is a pioneer-like transcription factor. Genes Dev. 2014, 28, 8–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, Y.H.; Marhon, S.A.; Zhang, Y.; Steger, D.J.; Won, K.-J.; Lazar, M.A. Rev-erbα dynamically modulates chromatin looping to control circadian gene transcription. Science 2018, 359, 1274–1277. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mermet, J.; Yeung, J.; Hurni, C.; Mauvoisin, D.; Gustafson, K.; Jouffe, C.; Nicolas, D.; Emmenegger, Y.; Gobet, C.; Franken, P.; et al. Clock-dependent chromatin topology modulates circadian transcription and behavior. Genes Dev. 2018, 32, 347–358. [Google Scholar] [CrossRef] [Green Version]
- Trott, A.J.; Menet, J.S. Regulation of circadian clock transcriptional output by CLOCK:BMAL1. PLoS Genet. 2018, 14, e1007156. [Google Scholar] [CrossRef] [PubMed]
- Yeung, J.; Mermet, J.; Jouffe, C.; Marquis, J.; Charpagne, A.; Gachon, F.; Naef, F. Transcription factor activity rhythms and tissue-specific chromatin interactions explain circadian gene expression across organs. Genome Res. 2018, 28, 182–191. [Google Scholar] [CrossRef] [Green Version]
- Le Martelot, G.; Canella, D.; Symul, L.; Migliavacca, E.; Gilardi, F.; Liechti, R.; Martin, O.; Harshman, K.; Delorenzi, M.; Desvergne, B.; et al. Genome-Wide RNA Polymerase II Profiles and RNA Accumulation Reveal Kinetics of Transcription and Associated Epigenetic Changes During Diurnal Cycles. PLoS Biol. 2012, 10, e1001442. [Google Scholar] [CrossRef] [Green Version]
- Zhu, J.; Li, C.; Gong, C.; Li, X. Regulation of Pol II Pausing Is Involved in Daily Gene Transcription in the Mouse Liver. J. Biol. Rhythm. 2018, 33, 350–362. [Google Scholar] [CrossRef]
- Petkau, N.; Budak, H.; Zhou, X.; Oster, H.; Eichele, G. Acetylation of BMAL1 by TIP60 controls BRD4-P-TEFb recruitment to circadian promoters. eLife 2019, 8. [Google Scholar] [CrossRef]
- Chalishazar, M.D.; Wait, S.J.; Huang, F.; Ireland, A.S.; Mukhopadhyay, A.; Lee, Y.; Schuman, S.S.; Guthrie, M.R.; Berrett, K.C.; Vahrenkamp, J.M.; et al. MYC-Driven Small-Cell Lung Cancer is Metabolically Distinct and Vulnerable to Arginine Depletion. Clin. Cancer Res. 2019, 25, 5107–5121. [Google Scholar] [CrossRef] [Green Version]
- Carroll, P.; Diolaiti, D.; McFerrin, L.; Gu, H.; Djukovic, D.; Du, J.; Cheng, P.F.; Anderson, S.; Ulrich, M.; Hurley, J.B.; et al. Deregulated Myc Requires MondoA/Mlx for Metabolic Reprogramming and Tumorigenesis. Cancer Cell 2015, 27, 271–285. [Google Scholar] [CrossRef] [Green Version]
- Gouw, A.; Margulis, K.; Liu, N.S.; Raman, S.J.; Mancuso, A.; Toal, G.G.; Tong, L.; Mosley, A.; Hsieh, A.L.; Sullivan, D.; et al. The MYC Oncogene Cooperates with Sterol-Regulated Element-Binding Protein to Regulate Lipogenesis Essential for Neoplastic Growth. Cell Metab. 2019, 30, 556–572.e5. [Google Scholar] [CrossRef] [PubMed]
- Ch, R.; Rey, G.; Ray, S.; Jha, P.K.; Driscoll, P.C.; Dos Santos, M.S.; Malik, D.M.; Lach, R.; Weljie, A.M.; MacRae, J.I.; et al. Rhythmic glucose metabolism regulates the redox circadian clockwork in human red blood cells. Nat. Commun. 2021, 12, 1–14. [Google Scholar] [CrossRef]
- O’Neill, J.; Reddy, A.B. Circadian clocks in human red blood cells. Nat. Cell Biol. 2011, 469, 498–503. [Google Scholar] [CrossRef] [Green Version]
- Schmitt, K.; Grimm, A.; Dallmann, R.; Oettinghaus, B.; Restelli, L.M.; Witzig, M.; Ishihara, N.; Mihara, K.; Ripperger, J.A.; Albrecht, U.; et al. Circadian Control of DRP1 Activity Regulates Mitochondrial Dynamics and Bioenergetics. Cell Metab. 2018, 27, 657–666.e5. [Google Scholar] [CrossRef] [Green Version]
- Aviram, R.; Manella, G.; Kopelman, N.; Neufeld-Cohen, A.; Zwighaft, Z.; Elimelech, M.; Adamovich, Y.; Golik, M.; Wang, C.; Han, X.; et al. Lipidomics Analyses Reveal Temporal and Spatial Lipid Organization and Uncover Daily Oscillations in Intracellular Organelles. Mol. Cell 2016, 62, 636–648. [Google Scholar] [CrossRef]
- Early, J.O.; Menon, D.; Wyse, C.A.; Cervantes-Silva, M.P.; Zaslona, Z.; Carroll, R.G.; Palsson-McDermott, E.M.; Angiari, S.; Ryan, D.G.; Corcoran, S.E.; et al. Circadian clock protein BMAL1 regulates IL-1β in macrophages via NRF2. Proc. Natl. Acad. Sci. USA 2018, 115, E8460–E8468. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peek, C.B.; Levine, D.; Cedernaes, J.; Taguchi, A.; Kobayashi, Y.; Tsai, S.J.; Bonar, N.A.; McNulty, M.R.; Ramsey, K.M.; Bass, J. Circadian Clock Interaction with HIF1α Mediates Oxygenic Metabolism and Anaerobic Glycolysis in Skeletal Muscle. Cell Metab. 2017, 25, 86–92. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, Y.; Tang, D.; Liu, N.; Xiong, W.; Huang, H.; Li, Y.; Ma, Z.; Zhao, H.; Chen, P.; Qi, X.; et al. Reciprocal Regulation between the Circadian Clock and Hypoxia Signaling at the Genome Level in Mammals. Cell Metab. 2017, 25, 73–85. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adamovich, Y.; Ladeuix, B.; Golik, M.; Koeners, M.; Asher, G. Rhythmic Oxygen Levels Reset Circadian Clocks through HIF1α. Cell Metab. 2017, 25, 93–101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Collins, E.J.; Cervantes-Silva, M.P.; Timmons, G.A.; O’Siorain, J.R.; Curtis, A.M.; Hurley, J.M. Post-transcriptional circadian regulation in macrophages organizes temporally distinct immunometabolic states. Genome Res. 2021, 31, 171–185. [Google Scholar] [CrossRef] [PubMed]
- Wise, D.R.; DeBerardinis, R.J.; Mancuso, A.; Sayed, N.; Zhang, X.-Y.; Pfeiffer, H.K.; Nissim, I.; Daikhin, E.; Yudkoff, M.; McMahon, S.B.; et al. Myc regulates a transcriptional program that stimulates mitochondrial glutaminolysis and leads to glutamine addiction. Proc. Natl. Acad. Sci. USA 2008, 105, 18782–18787. [Google Scholar] [CrossRef] [Green Version]
- Gao, P.; Tchernyshyov, I.; Chang, T.-C.; Lee, Y.-S.; Kita, K.; Ochi, T.; Zeller, K.I.; De Marzo, A.M.; Van Eyk, J.E.; Mendell, J.T.; et al. c-Myc suppression of miR-23a/b enhances mitochondrial glutaminase expression and glutamine metabolism. Nat. Cell Biol. 2009, 458, 762–765. [Google Scholar] [CrossRef] [Green Version]
- Qing, G.; Li, B.; Vu, A.; Skuli, N.; Walton, Z.E.; Liu, X.; Mayes, P.A.; Wise, D.; Thompson, C.B.; Maris, J.M.; et al. ATF4 Regulates MYC-Mediated Neuroblastoma Cell Death upon Glutamine Deprivation. Cancer Cell 2012, 22, 631–644. [Google Scholar] [CrossRef] [Green Version]
- Shroff, E.H.; Eberlin, L.S.; Dang, V.M.; Gouw, A.; Gabay, M.; Adam, S.J.; Bellovin, D.I.; Tran, P.T.; Philbrick, W.M.; Garcia-Ocana, A.; et al. MYC oncogene overexpression drives renal cell carcinoma in a mouse model through glutamine metabolism. Proc. Natl. Acad. Sci. USA 2015, 112, 6539–6544. [Google Scholar] [CrossRef] [Green Version]
- Yuneva, M.O.; Fan, T.W.; Allen, T.; Higashi, R.M.; Ferraris, D.; Tsukamoto, T.; Matés, J.M.; Alonso, F.J.; Wang, C.; Seo, Y.; et al. The Metabolic Profile of Tumors Depends on Both the Responsible Genetic Lesion and Tissue Type. Cell Metab. 2012, 15, 157–170. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spranger, S.; Gajewski, T.F. Mechanisms of Tumor Cell–Intrinsic Immune Evasion. Annu. Rev. Cancer Biol. 2018, 2, 213–228. [Google Scholar] [CrossRef]
- Gerber, S.A.; Lim, J.Y.H.; Connolly, K.A.; Sedlacek, A.; Barlow, M.L.; Murphy, S.P.; Egilmez, N.K.; Lord, E.M. Radio-responsive tumors exhibit greater intratumoral immune activity than nonresponsive tumors. Int. J. Cancer 2014, 134, 2383–2392. [Google Scholar] [CrossRef] [Green Version]
- Lim, J.Y.H.; Gerber, S.A.; Murphy, S.P.; Lord, E.M. Type I interferons induced by radiation therapy mediate recruitment and effector function of CD8+ T cells. Cancer Immunol. Immunother. 2014, 63, 259–271. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zitvogel, L.; Apetoh, L.; Ghiringhelli, F.; Kroemer, G. Immunological aspects of cancer chemotherapy. Nat. Rev. Immunol. 2008, 8, 59–73. [Google Scholar] [CrossRef]
- Garrido, F.; Aptsiauri, N.; Doorduijn, E.M.; Lora, A.M.G.; Van Hall, T. The urgent need to recover MHC class I in cancers for effective immunotherapy. Curr. Opin. Immunol. 2016, 39, 44–51. [Google Scholar] [CrossRef] [Green Version]
- Schrier, P.I.; Peltenburg, L.T. Relationship Between myc Oncogene Activation and MHC Class I Expression. In Advances in Cancer Research; Elsevier: Amsterdam, The Netherlands, 1992; Volume 60, pp. 181–246. [Google Scholar]
- Bernards, R.; Dessain, S.K.; Weinberg, R.A. N-myc amplification causes down-modulation of MHC class I antigen expression in neuroblastoma. Cell 1986, 47, 667–674. [Google Scholar] [CrossRef] [Green Version]
- Kortlever, R.M.; Sodir, N.M.; Wilson, C.; Burkhart, D.L.; Pellegrinet, L.; Swigart, L.B.; Littlewood, T.D.; Evan, G.I. Myc Cooperates with Ras by Programming Inflammation and Immune Suppression. Cell 2017, 171, 1301–1315.e14. [Google Scholar] [CrossRef]
- Casey, S.C.; Tong, L.; Li, Y.; Do, R.; Walz, S.; Fitzgerald, K.N.; Gouw, A.; Baylot, V.; Gütgemann, I.; Eilers, M.; et al. MYC regulates the antitumor immune response through CD47 and PD-L1. Science 2016, 352, 227–231. [Google Scholar] [CrossRef] [Green Version]
- Topper, M.J.; Vaz, M.; Chiappinelli, K.B.; Shields, C.E.D.; Niknafs, N.; Yen, R.-W.C.; Wenzel, A.; Hicks, J.; Ballew, M.; Stone, M.; et al. Epigenetic Therapy Ties MYC Depletion to Reversing Immune Evasion and Treating Lung Cancer. Cell 2017, 171, 1284–1300.e21. [Google Scholar] [CrossRef] [Green Version]
- Rakhra, K.; Bachireddy, P.; Zabuawala, T.; Zeiser, R.; Xu, L.; Kopelman, A.; Fan, A.C.; Yang, Q.; Braunstein, L.; Crosby, E.; et al. CD4+ T Cells Contribute to the Remodeling of the Microenvironment Required for Sustained Tumor Regression upon Oncogene Inactivation. Cancer Cell 2010, 18, 485–498. [Google Scholar] [CrossRef] [Green Version]
- Melaiu, O.; Mina, M.; Chierici, M.; Boldrini, R.; Jurman, G.; Romania, P.; D’Alicandro, V.; Benedetti, M.C.; Castellano, A.; Liu, T.; et al. PD-L1 Is a Therapeutic Target of the Bromodomain Inhibitor JQ1 and, Combined with HLA Class I, a Promising Prognostic Biomarker in Neuroblastoma. Clin. Cancer Res. 2017, 23, 4462–4472. [Google Scholar] [CrossRef] [Green Version]
- Sodir, N.M.; Swigart, L.B.; Karnezis, A.N.; Hanahan, D.; Evan, G.I.; Soucek, L. Endogenous Myc maintains the tumor microenvironment. Genes Dev. 2011, 25, 907–916. [Google Scholar] [CrossRef] [Green Version]
- Soucek, L.; Lawlor, E.R.; Soto, D.; Shchors, K.; Swigart, L.B.; I Evan, G. Mast cells are required for angiogenesis and macroscopic expansion of Myc-induced pancreatic islet tumors. Nat. Med. 2007, 13, 1211–1218. [Google Scholar] [CrossRef] [PubMed]
- Scheiermann, C.; Gibbs, J.; Ince, L.; Loudon, A. Clocking in to immunity. Nat. Rev. Immunol. 2018, 18, 423–437. [Google Scholar] [CrossRef] [PubMed]
- Man, K.; Loudon, A.; Chawla, A. Immunity around the clock. Science 2016, 354, 999–1003. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Curtis, A.M.; Fagundes, C.T.; Yang, G.; Palsson-McDermott, E.M.; Wochal, P.; McGettrick, A.F.; Foley, N.H.; Early, J.O.; Chen, L.; Zhang, H.; et al. Circadian control of innate immunity in macrophages by miR-155 targeting Bmal1. Proc. Natl. Acad. Sci. USA 2015, 112, 7231–7236. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kitchen, G.B.; Cunningham, P.; Poolman, T.; Iqbal, M.; Maidstone, R.; Baxter, M.; Bagnall, J.; Begley, N.; Saer, B.; Hussell, T.; et al. The clock gene Bmal1 inhibits macrophage motility, phagocytosis, and impairs defense against pneumonia. Proc. Natl. Acad. Sci. USA 2020, 117, 1543–1551. [Google Scholar] [CrossRef] [Green Version]
- Silver, A.C.; Arjona, A.; Walker, W.E.; Fikrig, E. The Circadian Clock Controls Toll-like Receptor 9-Mediated Innate and Adaptive Immunity. Immunity 2012, 36, 251–261. [Google Scholar] [CrossRef] [Green Version]
- Long, J.E.; Drayson, M.; Taylor, A.E.; Toellner, K.-M.; Lord, J.; Phillips, A.C. Morning vaccination enhances antibody response over afternoon vaccination: A cluster-randomised trial. Vaccine 2016, 34, 2679–2685. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adrover, J.M.; Aroca-Crevillén, A.; Crainiciuc, G.; Ostos, F.; Rojas-Vega, Y.; Rubio-Ponce, A.; Cilloniz, C.; Bonzón-Kulichenko, E.; Calvo, E.; Rico, D.; et al. Programmed ‘disarming’ of the neutrophil proteome reduces the magnitude of inflammation. Nat. Immunol. 2020, 21, 135–144. [Google Scholar] [CrossRef] [PubMed]
- Alexander, R.K.; Liou, Y.-H.; Knudsen, N.H.; A Starost, K.; Xu, C.; Hyde, A.L.; Liu, S.; Jacobi, D.; Liao, N.-S.; Lee, C.-H. Bmal1 integrates mitochondrial metabolism and macrophage activation. eLife 2020, 9. [Google Scholar] [CrossRef]
- Hadadi, E.; Taylor, W.; Li, X.-M.; Aslan, Y.; Villote, M.; Rivière, J.; Duvallet, G.; Auriau, C.; Dulong, S.; Raymond-Letron, I.; et al. Chronic circadian disruption modulates breast cancer stemness and immune microenvironment to drive metastasis in mice. Nat. Commun. 2020, 11, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Lim, A.R.; Rathmell, W.K.; Rathmell, J.C. The tumor microenvironment as a metabolic barrier to effector T cells and immunotherapy. eLife 2020, 9. [Google Scholar] [CrossRef] [PubMed]
- Dejure, F.R.; Eilers, M. MYC and tumor metabolism: Chicken and egg. EMBO J. 2017, 36, 3409–3420. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.-H.; Qiu, J.; O’Sullivan, D.; Buck, M.D.; Noguchi, T.; Curtis, J.D.; Chen, Q.; Gindin, M.; Gubin, M.M.; Van Der Windt, G.J.W.; et al. Metabolic Competition in the Tumor Microenvironment Is a Driver of Cancer Progression. Cell 2015, 162, 1229–1241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walton, Z.E.; Patel, C.H.; Brooks, R.C.; Yu, Y.; Ibrahim-Hashim, A.; Riddle, M.; Porcu, A.; Jiang, T.; Ecker, B.L.; Tameire, F.; et al. Acid Suspends the Circadian Clock in Hypoxia through Inhibition of mTOR. Cell 2018, 174, 72–87.e32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ho, P.-C.; Bihuniak, J.D.; Macintyre, A.; Staron, M.; Liu, X.; Amezquita, R.; Tsui, Y.-C.; Cui, G.; Micevic, G.; Perales, J.C.; et al. Phosphoenolpyruvate Is a Metabolic Checkpoint of Anti-tumor T Cell Responses. Cell 2015, 162, 1217–1228. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reinfeld, B.I.; Madden, M.Z.; Wolf, M.M.; Chytil, A.; Bader, J.E.; Patterson, A.R.; Sugiura, A.; Cohen, A.S.; Ali, A.; Do, B.T.; et al. Cell-programmed nutrient partitioning in the tumour microenvironment. Nat. Cell Biol. 2021, 593, 282–288. [Google Scholar] [CrossRef]
- Colegio, O.R.; Chu, N.-Q.; Szabo, A.L.; Chu, T.; Rhebergen, A.M.; Jairam, V.; Cyrus, N.; Brokowski, C.E.; Eisenbarth, S.C.; Phillips, G.M.; et al. Functional polarization of tumour-associated macrophages by tumour-derived lactic acid. Nature 2014, 513, 559–563. [Google Scholar] [CrossRef] [PubMed]
- Bian, Y.; Li, W.; Kremer, D.M.; Sajjakulnukit, P.; Li, S.; Crespo, J.; Nwosu, Z.C.; Zhang, L.; Czerwonka, A.; Pawłowska, A.; et al. Cancer SLC43A2 alters T cell methionine metabolism and histone methylation. Nat. Cell Biol. 2020, 585, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Lamia, K.A.; Sachdeva, U.M.; DiTacchio, L.; Williams, E.C.; Alvarez, J.G.; Egan, D.F.; Vasquez, D.S.; Juguilon, H.; Panda, S.; Shaw, R.J.; et al. AMPK regulates the circadian clock by cryptochrome phosphorylation and degradation. Science 2009, 326, 437–440. [Google Scholar] [CrossRef] [Green Version]
- Dimitrov, S.; Benedict, C.; Heutling, D.; Westermann, J.; Born, J.; Lange, T. Cortisol and epinephrine control opposing circadian rhythms in T cell subsets. Blood 2009, 113, 5134–5143. [Google Scholar] [CrossRef] [Green Version]
- Nobis, C.C.; Laramée, G.D.; Kervezee, L.; De Sousa, D.M.; Labrecque, N.; Cermakian, N. The circadian clock of CD8 T cells modulates their early response to vaccination and the rhythmicity of related signaling pathways. Proc. Natl. Acad. Sci. USA 2019, 116, 20077–20086. [Google Scholar] [CrossRef] [Green Version]
- Madden, S.K.; de Araujo, A.D.; Gerhardt, M.; Fairlie, D.P.; Mason, J.M. Taking the Myc out of cancer: Toward therapeutic strategies to directly inhibit c-Myc. Mol. Cancer 2021, 20, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Whitfield, J.; Beaulieu, M.-E.; Soucek, L. Strategies to Inhibit Myc and Their Clinical Applicability. Front. Cell Dev. Biol. 2017, 5, 10. [Google Scholar] [CrossRef] [Green Version]
- Truica, M.I.; Burns, M.C.; Han, H.; Abdulkadir, S.A. Turning Up the Heat on MYC: Progress in Small-Molecule Inhibitors. Cancer Res. 2021, 81, 248–253. [Google Scholar] [CrossRef] [PubMed]
- Dang, C.V.; Reddy, E.P.; Shokat, K.M.; Soucek, L. Drugging the ’undruggable’ cancer targets. Nat. Rev. Cancer 2017, 17, 502–508. [Google Scholar] [CrossRef]
- Jung, L.A.; Gebhardt, A.; Koelmel, W.; Ade, C.P.; Walz, S.; Kuper, J.; von Eyss, B.; Letschert, S.; Redel, C.; D’Artista, L.; et al. OmoMYC blunts promoter invasion by oncogenic MYC to inhibit gene expression characteristic of MYC-dependent tumors. Oncogene 2016, 36, 1911–1924. [Google Scholar] [CrossRef] [Green Version]
- Soucek, L.; Helmer-Citterich, M.; Sacco, A.; Jucker, R.; Cesareni, G.; Nasi, S. Design and properties of a Myc derivative that efficiently homodimerizes. Oncogene 1998, 17, 2463–2472. [Google Scholar] [CrossRef] [Green Version]
- Soucek, L.; Jucker, R.; Panacchia, L.; Ricordy, R.; Tatò, F.; Nasi, S. Omomyc, a potential Myc dominant negative, enhances Myc-induced apoptosis. Cancer Res. 2002, 62, 3507–3510. [Google Scholar] [PubMed]
- Soucek, L.; Whitfield, J.; Martins, C.P.; Finch, A.J.; Murphy, D.; Sodir, N.M.; Karnezis, A.N.; Swigart, L.B.; Nasi, S.; Evan, G.I. Modelling Myc inhibition as a cancer therapy. Nat. Cell Biol. 2008, 455, 679–683. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beaulieu, M.-E.; Jauset, T.; Massó-Vallés, D.; Martínez-Martín, S.; Rahl, P.; Maltais, L.; Zacarias-Fluck, M.F.; Casacuberta-Serra, S.; Del Pozo, E.S.; Fiore, C.; et al. Intrinsic cell-penetrating activity propels Omomyc from proof of concept to viable anti-MYC therapy. Sci. Transl. Med. 2019, 11, 5012. [Google Scholar] [CrossRef] [PubMed]
- Demma, M.J.; Hohn, M.J.; Sun, A.; Mapelli, C.; Hall, B.; Walji, A.; O’Neil, J. Inhibition of Myc transcriptional activity by a mini-protein based upon Mxd1. FEBS Lett. 2020, 594, 1467–1476. [Google Scholar] [CrossRef] [PubMed]
- Cochran, A.G.; Conery, A.R.; Sims, R.J. Bromodomains: A new target class for drug development. Nat. Rev. Drug Discov. 2019, 18, 609–628. [Google Scholar] [CrossRef] [PubMed]
- Delmore, J.E.; Issa, G.C.; Lemieux, M.; Rahl, P.B.; Shi, J.; Jacobs, H.M.; Kastritis, E.; Gilpatrick, T.; Paranal, R.M.; Qi, J.; et al. BET Bromodomain Inhibition as a Therapeutic Strategy to Target c-Myc. Cell 2011, 146, 904–917. [Google Scholar] [CrossRef] [Green Version]
- Henssen, A.G.; Althoff, K.; Odersky, A.; Beckers, A.; Koche, R.; Speleman, F.; Schäfers, S.; Bell, E.; Nortmeyer, M.; Westermann, F.; et al. Targeting MYCN-driven transcription by BET-bromodomain inhibition. Clin. Cancer Res. 2015, 22, 2470–2481. [Google Scholar] [CrossRef] [Green Version]
- Devaiah, B.N.; Mu, J.; Akman, B.; Uppal, S.; Weissman, J.D.; Cheng, D.; Baranello, L.; Nie, Z.; Levens, D.; Singer, D.S. MYC protein stability is negatively regulated by BRD4. Proc. Natl. Acad. Sci. USA 2020, 117, 13457–13467. [Google Scholar] [CrossRef]
- Boike, L.; Cioffi, A.G.; Majewski, F.C.; Co, J.; Henning, N.J.; Jones, M.D.; Liu, G.; McKenna, J.M.; Tallarico, J.A.; Schirle, M.; et al. Discovery of a Functional Covalent Ligand Targeting an Intrinsically Disordered Cysteine within MYC. Cell Chem. Biol. 2021, 28, 4–13.e17. [Google Scholar] [CrossRef]
- Ballesta, A.; Innominato, P.F.; Dallmann, R.; Rand, D.A.; Lévi, F.A. Systems Chronotherapeutics. Pharmacol. Rev. 2017, 69, 161–199. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Selfridge, J.M.; Gotoh, T.; Schiffhauer, S.; Liu, J.; Stauffer, P.E.; Li, A.; Capelluto, D.G.S.; Finkielstein, C.V. Chronotherapy: Intuitive, Sound, Founded…But Not Broadly Applied. Drugs 2016, 76, 1507–1521. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giacchetti, S.; Dugue, P.-A.; Innominato, P.F.; Bjarnason, G.A.; Focan, C.; Garufi, C.; Tumolo, S.; Coudert, B.; Iacobelli, S.; Smaaland, R.; et al. Sex moderates circadian chemotherapy effects on survival of patients with metastatic colorectal cancer: A meta-analysis. Ann. Oncol. 2012, 23, 3110–3116. [Google Scholar] [CrossRef]
- Cederroth, C.R.; Albrecht, U.; Bass, J.; Brown, S.A.; Dyhrfjeld-Johnsen, J.; Gachon, F.; Green, C.B.; Hastings, M.H.; Helfrich-Förster, C.; Hogenesch, J.B.; et al. Medicine in the Fourth Dimension. Cell Metab. 2019, 30, 238–250. [Google Scholar] [CrossRef]
- Hill, R.J.W.; Innominato, P.F.; Lévi, F.; Ballesta, A. Optimizing circadian drug infusion schedules towards personalized cancer chronotherapy. PLoS Comput. Biol. 2020, 16, e1007218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boivin, D.B.; James, F.O.; Wu, A.; Cho-Park, P.F.; Xiong, H.; Sun, Z. Circadian clock genes oscillate in human peripheral blood mononuclear cells. Blood 2003, 102, 4143–4145. [Google Scholar] [CrossRef] [Green Version]
- Koritala, B.S.C.; Porter, K.I.; Arshad, O.A.; Gajula, R.P.; Mitchell, H.D.; Arman, T.; Manjanatha, M.G.; Teeguarden, J.; Van Dongen, H.P.A.; McDermott, J.E.; et al. Night shift schedule causes circadian dysregulation of DNA repair genes and elevated DNA damage in humans. J. Pineal Res. 2021, 70, e12726. [Google Scholar] [CrossRef]
- Shilts, J.; Chen, G.; Hughey, J.J. Evidence for widespread dysregulation of circadian clock progression in human cancer. PeerJ 2018, 6, e4327. [Google Scholar] [CrossRef]
- Masri, S.; Papagiannakopoulos, T.; Kinouchi, K.; Liu, Y.; Cervantes, M.; Baldi, P.; Jacks, T.; Sassone-Corsi, P. Lung Adenocarcinoma Distally Rewires Hepatic Circadian Homeostasis. Cell 2016, 165, 896–909. [Google Scholar] [CrossRef] [Green Version]
- Verlande, A.; Chun, S.K.; Goodson, M.O.; Fortin, B.M.; Bae, H.; Jang, C.; Masri, S. Glucagon regulates the stability of REV-ERBα to modulate hepatic glucose production in a model of lung cancer–associated cachexia. Sci. Adv. 2021, 7, 3885. [Google Scholar] [CrossRef] [PubMed]
- Ueda, H.R.; Chen, W.; Minami, Y.; Honma, S.; Honma, K.; Iino, M.; Hashimoto, S. Molecular-timetable methods for detection of body time and rhythm disorders from single-time-point genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2004, 101, 11227–11232. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- E Laing, E.; Möller-Levet, C.S.; Poh, N.; Santhi, N.; Archer, S.N.; Dijk, D.-J. Blood transcriptome based biomarkers for human circadian phase. eLife 2017, 6, e20214. [Google Scholar] [CrossRef] [Green Version]
- Hughey, J.J. Machine learning identifies a compact gene set for monitoring the circadian clock in human blood. Genome Med. 2017, 9, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Braun, R.; Kath, W.L.; Iwanaszko, M.; Kula-Eversole, E.; Abbott, S.M.; Reid, K.J.; Zee, P.C.; Allada, R. Universal method for robust detection of circadian state from gene expression. Proc. Natl. Acad. Sci. USA 2018, 115, E9247–E9256. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wittenbrink, N.; Ananthasubramaniam, B.; Münch, M.; Koller, B.; Maier, B.; Weschke, C.; Bes, F.; De Zeeuw, J.; Nowozin, C.; Wahnschaffe, A.; et al. High-accuracy determination of internal circadian time from a single blood sample. J. Clin. Investig. 2018, 128, 3826–3839. [Google Scholar] [CrossRef]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Burchett, J.B.; Knudsen-Clark, A.M.; Altman, B.J. MYC Ran Up the Clock: The Complex Interplay between MYC and the Molecular Circadian Clock in Cancer. Int. J. Mol. Sci. 2021, 22, 7761. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22147761
Burchett JB, Knudsen-Clark AM, Altman BJ. MYC Ran Up the Clock: The Complex Interplay between MYC and the Molecular Circadian Clock in Cancer. International Journal of Molecular Sciences. 2021; 22(14):7761. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22147761
Chicago/Turabian StyleBurchett, Jamison B., Amelia M. Knudsen-Clark, and Brian J. Altman. 2021. "MYC Ran Up the Clock: The Complex Interplay between MYC and the Molecular Circadian Clock in Cancer" International Journal of Molecular Sciences 22, no. 14: 7761. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22147761