
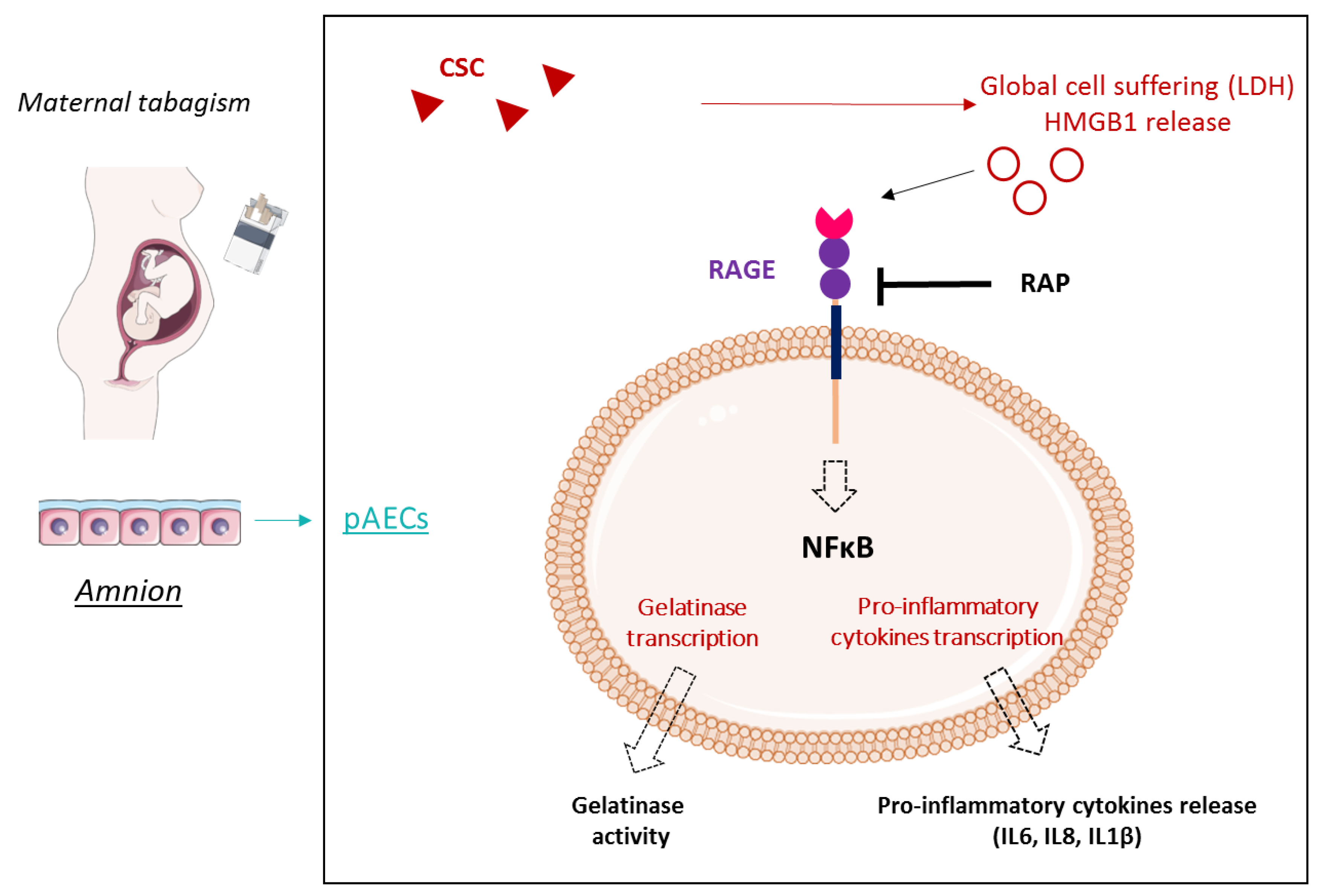
Cigarette Smoke Condensate Exposure Induces Receptor for Advanced Glycation End-Products (RAGE)-Dependent Sterile Inflammation in Amniotic Epithelial Cells

 ,
,
Abstract
:
1. Introduction
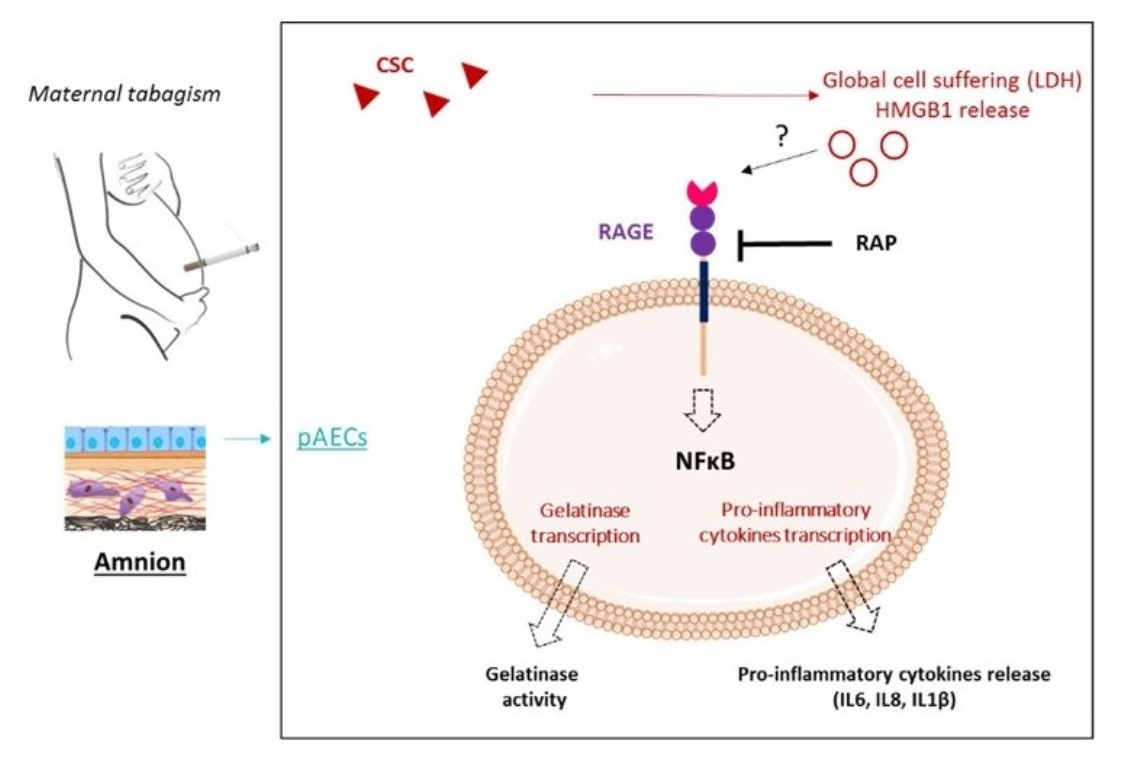
2. Results
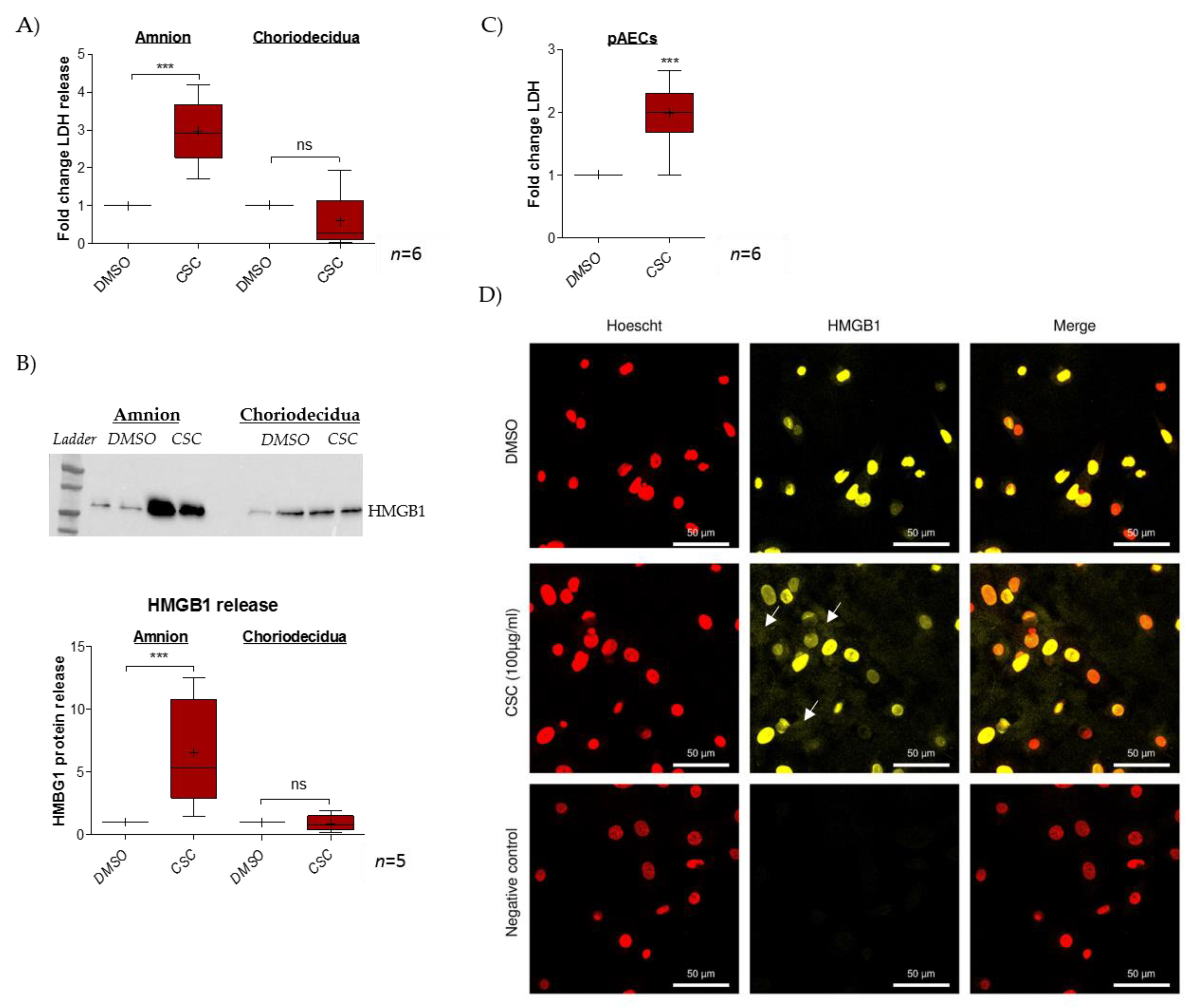
2.1. Cigarette-Smoke Condensate Induces a Danger Response in the Fetal Amnion and Its Cells
2.2. RAGE Axis Actors Are Expressed in Primary Amniotic Epithelial Cells (pAECS)
2.3. Cigarette Smoke Condensate Induces RAGE Signaling Cascade in pAECs
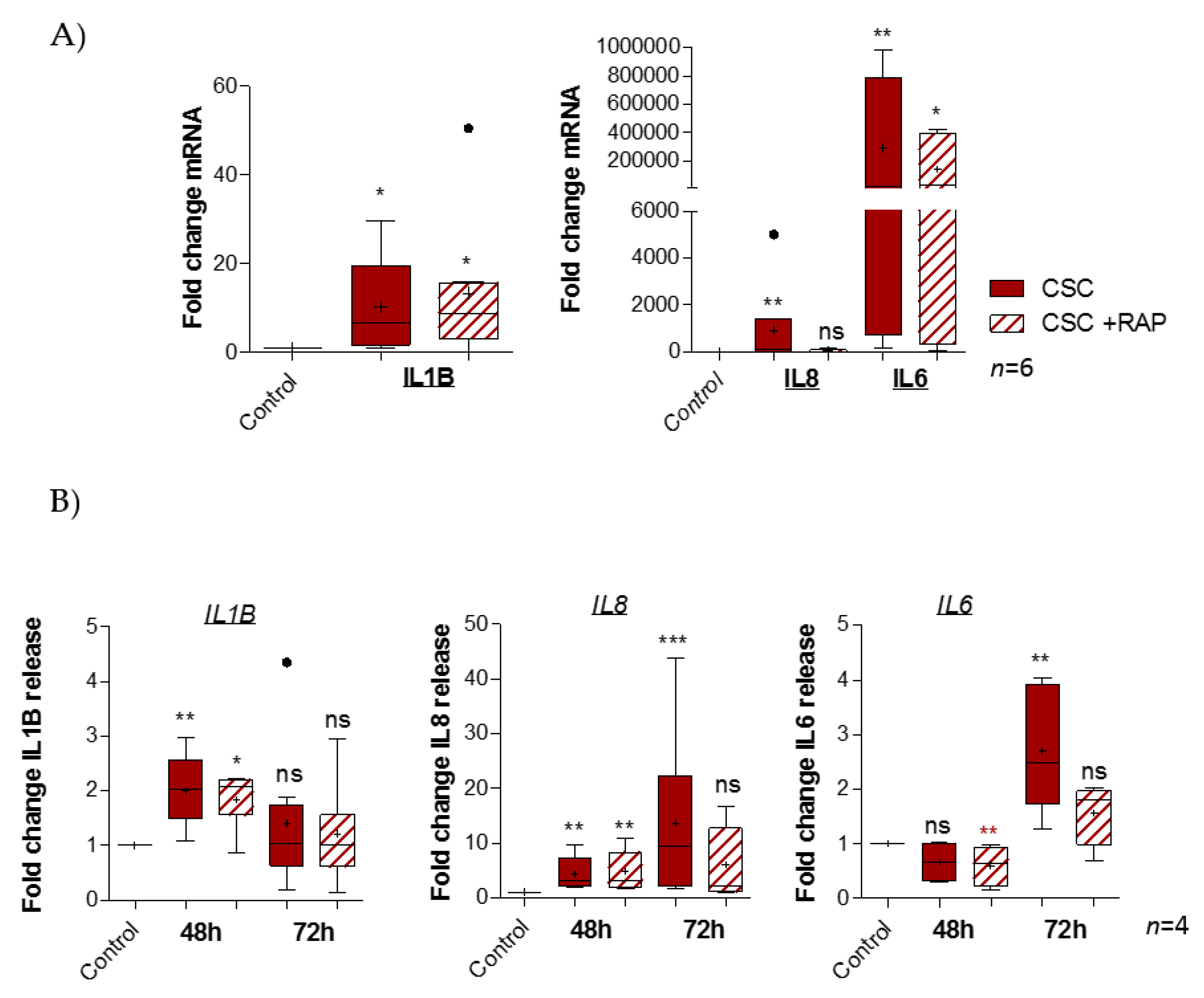
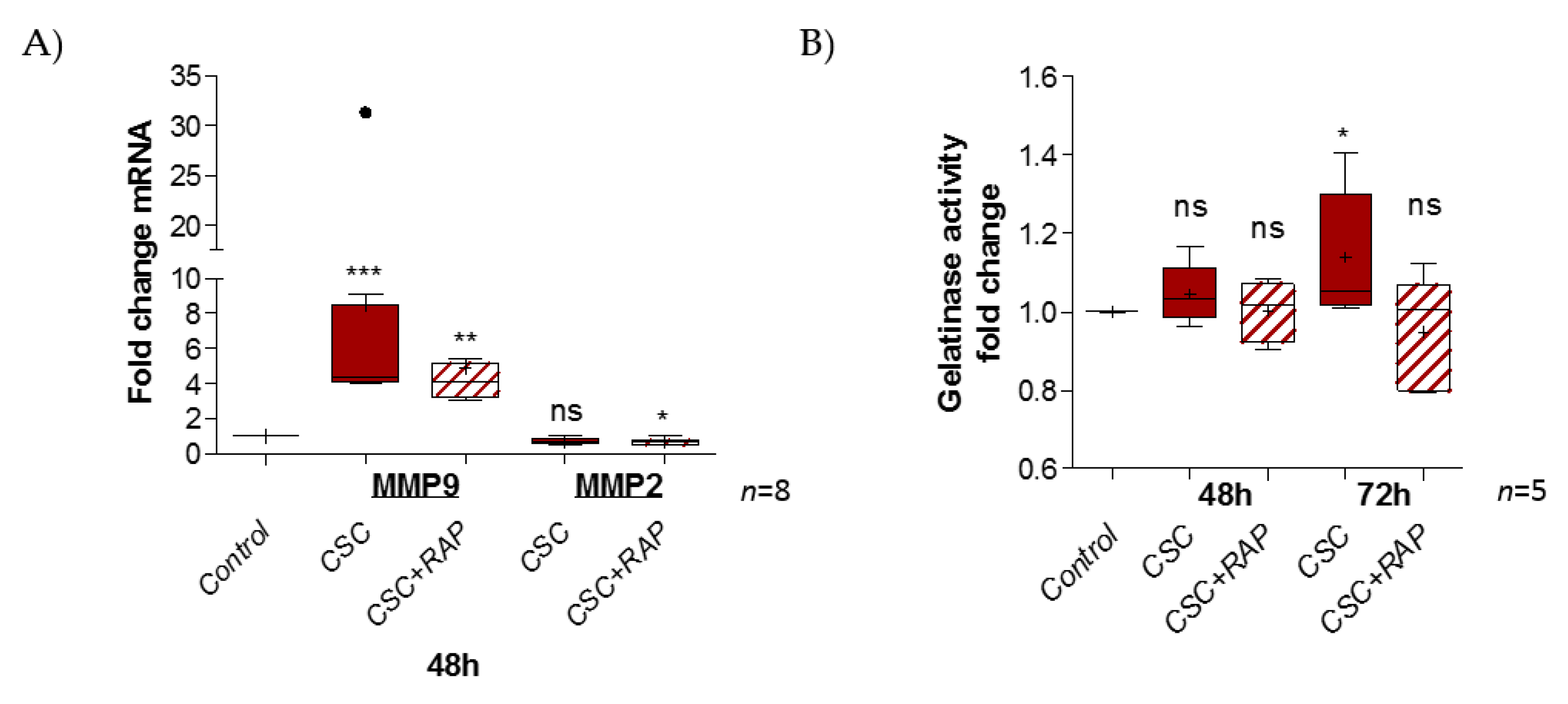
2.4. Cigarette Smoke Condensate Induces Pro-Inflammatory Response in pAECs
3. Discussion
4. Materials and Methods
4.1. Chemicals
4.2. Tissue Collection
4.3. Tissue and Cell Culture
4.4. Tissue and Cell Treatment
4.5. Global Cellular Distress Determination
4.6. Western Blot Analysis of HMGB1 Release
4.7. RT-PCR and Quantitative RT-PCR on Explants and Cells
4.8. Cytokine Release Assay
4.9. Immunofluorescence
4.10. NFκB Gene Reporter Luciferase Assay
4.11. Gelatinase Activity Measurement
4.12. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Nabet, C.; Lelong, N.; Ancel, P.-Y.; Saurel-Cubizolles, M.-J.; Kaminski, M. Smoking during Pregnancy According to Obstetric Complications and Parity: Results of the EUROPOP Study. Eur. J. Epidemiol. 2007, 22, 715–721. [Google Scholar] [CrossRef]
- Lewis, J.; Hirschi, K.; Arroyo, J.; Bikman, B.; Kooyman, D.; Reynolds, P. Plausible Roles for RAGE in Conditions Exacerbated by Direct and Indirect (Secondhand) Smoke Exposure. Int. J. Mol. Sci. 2017, 18, 652. [Google Scholar] [CrossRef] [Green Version]
- Wrześniak, M.; Kepinska, M.; Królik, M.; Milnerowicz, H. The Influence of Tobacco Smoke on Protein and Metal Levels in the Serum of Women during Pregnancy. PLoS ONE 2016, 11, e0161342. [Google Scholar] [CrossRef]
- Witt, S.H.; Frank, J.; Gilles, M.; Lang, M.; Treutlein, J.; Streit, F.; Wolf, I.A.C.; Peus, V.; Scharnholz, B.; Send, T.S.; et al. Impact on Birth Weight of Maternal Smoking throughout Pregnancy Mediated by DNA Methylation. BMC Genom. 2018, 19, 290. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rogers, J.M. Tobacco and Pregnancy. Reprod. Toxicol. 2009, 28, 152–160. [Google Scholar] [CrossRef] [PubMed]
- von Chamier, M.; Reyes, L.; Hayward, L.F.; Brown, M.B. Nicotine Induces Maternal and Fetal Inflammatory Responses Which Predispose Intrauterine Infection Risk in a Rat Model. Nicotine Tob. Res. 2021, natb080. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.-H.; An, Y.-J.; Jo, S.; Lee, S.-H.; Lee, S.J.; Choi, S.-J.; Lee, K. Comparison of Volatile Organic Compounds between Cigarette Smoke Condensate (CSC) and Extract (CSE) Samples. Environ. Health Toxicol. 2018, 33, e2018012. [Google Scholar] [CrossRef]
- Polettini, J.; Richardson, L.S.; Menon, R. Oxidative Stress Induces Senescence and Sterile Inflammation in Murine Amniotic Cavity. Placenta 2018, 63, 26–31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jin, J.; Richardson, L.; Sheller-Miller, S.; Zhong, N.; Menon, R. Oxidative Stress Induces P38MAPK-Dependent Senescence in the Feto-Maternal Interface Cells. Placenta 2018, 67, 15–23. [Google Scholar] [CrossRef] [PubMed]
- Bredeson, S.; Papaconstantinou, J.; Deford, J.H.; Kechichian, T.; Syed, T.A.; Saade, G.R.; Menon, R. HMGB1 Promotes a P38MAPK Associated Non-Infectious Inflammatory Response Pathway in Human Fetal Membranes. PLoS ONE 2014, 9, e113799. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sheller-Miller, S.; Urrabaz-Garza, R.; Saade, G.; Menon, R. Damage-Associated Molecular Pattern Markers HMGB1 and Cell-Free Fetal Telomere Fragments in Oxidative-Stressed Amnion Epithelial Cell-Derived Exosomes. J. Reprod. Immunol. 2017, 123, 3–11. [Google Scholar] [CrossRef] [PubMed]
- Menon, R.; Peltier, M.R. Novel Insights into the Regulatory Role of Nuclear Factor (Erythroid-Derived 2)-Like 2 in Oxidative Stress and Inflammation of Human Fetal Membranes. Int. J. Mol. Sci. 2020, 21, 6139. [Google Scholar] [CrossRef] [PubMed]
- Menon, R.; Fortunato, S.J.; Yu, J.; Milne, G.L.; Sanchez, S.; Drobek, C.O.; Lappas, M.; Taylor, R.N. Cigarette Smoke Induces Oxidative Stress and Apoptosis in Normal Term Fetal Membranes. Placenta 2011, 32, 317–322. [Google Scholar] [CrossRef]
- Rouzaire, M.; Comptour, A.; Belville, C.; Bouvier, D.; Sapin, V.; Gallot, D.; Blanchon, L. Cigarette Smoke Condensate Affects the Retinoid Pathway in Human Amnion. Placenta 2017, 58, 98–104. [Google Scholar] [CrossRef]
- Cerami, C.; Founds, H.; Nicholl, I.; Mitsuhashi, T.; Giordano, D.; Vanpatten, S.; Lee, A.; Al-Abed, Y.; Vlassara, H.; Bucala, R.; et al. Tobacco Smoke Is a Source of Toxic Reactive Glycation Products. Proc. Natl. Acad. Sci. USA 1997, 94, 13915–13920. [Google Scholar] [CrossRef] [Green Version]
- Nicholl, I.D.; Bucala, R. Advanced Glycation Endproducts and Cigarette Smoking. Cell Mol. Biol. 1998, 44, 1025–1033. [Google Scholar]
- Tsai, C.-Y.; Chou, H.-C.; Chen, C.-M. Perinatal Nicotine Exposure Alters Lung Development and Induces HMGB1-RAGE Expression in Neonatal Mice. Birth Defects Res. 2020, 113, 570–578. [Google Scholar] [CrossRef]
- Railwah, C.; Lora, A.; Zahid, K.; Goldenberg, H.; Campos, M.; Wyman, A.; Jundi, B.; Ploszaj, M.; Rivas, M.; Dabo, A.; et al. Cigarette Smoke Induction of S100A9 Contributes to Chronic Obstructive Pulmonary Disease. Am. J. Physiol. Lung Cell. Mol. Physiol. 2020, 319, L1021–L1035. [Google Scholar] [CrossRef]
- Zhang, S.-P.; Wu, Y.-W.; Wu, Z.-Z.; Liu, H.-Y.; Nie, J.-H.; Tong, J. Up-Regulation of RAGE and S100A6 in Rats Exposed to Cigarette Smoke. Environ. Toxicol. Pharmacol. 2009, 28, 259–264. [Google Scholar] [CrossRef]
- Wood, T.T.; Winden, D.R.; Marlor, D.R.; Wright, A.J.; Jones, C.M.; Chavarria, M.; Rogers, G.D.; Reynolds, P.R. Acute Secondhand Smoke-Induced Pulmonary Inflammation Is Diminished in RAGE Knockout Mice. Am. J. Physiol. Lung Cell. Mol. Physiol. 2014, 307, L758–L764. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, H.; Lee, J.; Hong, S.-H.; Rahman, I.; Yang, S.-R. Inhibition of RAGE Attenuates Cigarette Smoke-Induced Lung Epithelial Cell Damage via RAGE-Mediated Nrf2/DAMP Signaling. Front. Pharmacol. 2018, 9, 684. [Google Scholar] [CrossRef]
- Wolf, L.; Herr, C.; Niederstraßer, J.; Beisswenger, C.; Bals, R. Receptor for Advanced Glycation Endproducts (RAGE) Maintains Pulmonary Structure and Regulates the Response to Cigarette Smoke. PLoS ONE 2017, 12, e0180092. [Google Scholar] [CrossRef] [Green Version]
- Prasad, K.; Dhar, I.; Caspar-Bell, G. Role of Advanced Glycation End Products and Its Receptors in the Pathogenesis of Cigarette Smoke-Induced Cardiovascular Disease. Int. J. Angiol. 2015, 24, 75–80. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lewis, J.B.; Mejia, C.; Jordan, C.; Monson, T.D.; Bodine, J.S.; Dunaway, T.M.; Egbert, K.M.; Lewis, A.L.; Wright, T.J.; Ogden, K.C.; et al. Inhibition of the Receptor for Advanced Glycation End-Products (RAGE) Protects from Secondhand Smoke (SHS)-Induced Intrauterine Growth Restriction IUGR in Mice. Cell Tissue Res. 2017, 370, 513–521. [Google Scholar] [CrossRef] [PubMed]
- Xie, J.; Méndez, J.D.; Méndez-Valenzuela, V.; Aguilar-Hernández, M.M. Cellular Signalling of the Receptor for Advanced Glycation End Products (RAGE). Cell. Signal. 2013, 25, 2185–2197. [Google Scholar] [CrossRef] [PubMed]
- Barbezier, N.; Tessier, F.J.; Chango, A. Receptor of advanced glycation endproducts RAGE/AGER: An integrative view for clinical applications. Ann. Biol. Clin. 2014, 72, 669–680. [Google Scholar] [CrossRef] [PubMed]
- Sakaguchi, M.; Kinoshita, R.; Putranto, E.W.; Ruma, I.M.W.; Sumardika, I.W.; Youyi, C.; Tomonobu, N.; Yamamoto, K.-I.; Murata, H. Signal Diversity of Receptor for Advanced Glycation End Products. Acta Med. Okayama 2017, 71, 459–465. [Google Scholar] [CrossRef]
- Sakaguchi, M.; Murata, H.; Yamamoto, K.; Ono, T.; Sakaguchi, Y.; Motoyama, A.; Hibino, T.; Kataoka, K.; Huh, N. TIRAP, an Adaptor Protein for TLR2/4, Transduces a Signal from RAGE Phosphorylated upon Ligand Binding. PLoS ONE 2011, 6, e23132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hudson, B.I.; Kalea, A.Z.; del Mar Arriero, M.; Harja, E.; Boulanger, E.; D’Agati, V.; Schmidt, A.M. Interaction of the RAGE Cytoplasmic Domain with Diaphanous-1 Is Required for Ligand-Stimulated Cellular Migration through Activation of Rac1 and Cdc42. J. Biol. Chem. 2008, 283, 34457–34468. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, V.; Fleming, T.; Terjung, S.; Gorzelanny, C.; Gebhardt, C.; Agrawal, R.; Mall, M.A.; Ranzinger, J.; Zeier, M.; Madhusudhan, T.; et al. Homeostatic Nuclear RAGE-ATM Interaction Is Essential for Efficient DNA Repair. Nucleic Acids Res. 2017, 45, 10595–10613. [Google Scholar] [CrossRef]
- Williams, M.A.; Mittendorf, R.; Stubblefield, P.G.; Lieberman, E.; Schoenbaum, S.C.; Monson, R.R. Cigarettes, Coffee, and Preterm Premature Rupture of the Membranes. Am. J. Epidemiol. 1992, 135, 895–903. [Google Scholar] [CrossRef]
- England, M.; Benjamin, A.; Abenhaim, H. Increased Risk of Preterm Premature Rupture of Membranes at Early Gestational Ages among Maternal Cigarette Smokers. Am. J. Perinatol. 2013, 30, 821–826. [Google Scholar] [CrossRef]
- Shea, A.K.; Steiner, M. Cigarette Smoking during Pregnancy. Nicotine Tob. Res. 2008, 10, 267–278. [Google Scholar] [CrossRef]
- Pouwels, S.D.; Zijlstra, G.J.; van der Toorn, M.; Hesse, L.; Gras, R.; Ten Hacken, N.H.T.; Krysko, D.V.; Vandenabeele, P.; de Vries, M.; van Oosterhout, A.J.M.; et al. Cigarette Smoke-Induced Necroptosis and DAMP Release Trigger Neutrophilic Airway Inflammation in Mice. Am. J. Physiol. Lung Cell. Mol. Physiol. 2016, 310, L377–L386. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pouwels, S.D.; Hesse, L.; Faiz, A.; Lubbers, J.; Bodha, P.K.; ten Hacken, N.H.T.; van Oosterhout, A.J.M.; Nawijn, M.C.; Heijink, I.H. Susceptibility for Cigarette Smoke-Induced DAMP Release and DAMP-Induced Inflammation in COPD. Am. J. Physiol. Lung Cell. Mol. Physiol. 2016, 311, L881–L892. [Google Scholar] [CrossRef] [Green Version]
- Sharma, A.; Kaur, S.; Sarkar, M.; Sarin, B.C.; Changotra, H. The AGE-RAGE Axis and RAGE Genetics in Chronic Obstructive Pulmonary Disease. Clin. Rev. Allergy Immunol. 2020, 60, 244–258. [Google Scholar] [CrossRef] [PubMed]
- Choltus, H.; Lavergne, M.; Belville, C.; Gallot, D.; Minet-Quinard, R.; Durif, J.; Blanchon, L.; Sapin, V. Occurrence of a RAGE-Mediated Inflammatory Response in Human Fetal Membranes. Front. Physiol. 2020, 11, 581. [Google Scholar] [CrossRef] [PubMed]
- Filipovich, Y.; Lu, S.-J.; Akira, S.; Hirsch, E. The Adaptor Protein MyD88 Is Essential for E Coli-Induced Preterm Delivery in Mice. Am. J. Obstet. Gynecol. 2009, 200, 93.e1–93.e8. [Google Scholar] [CrossRef]
- Lee, Y.; Allport, V.; Sykes, A.; Lindstrom, T.; Slater, D.; Bennett, P. The Effects of Labour and of Interleukin 1 Beta upon the Expression of Nuclear Factor Kappa B Related Proteins in Human Amnion. Mol. Hum. Reprod. 2003, 9, 213–218. [Google Scholar] [CrossRef] [Green Version]
- Sheller-Miller, S.; Radnaa, E.; Yoo, J.-K.; Kim, E.; Choi, K.; Kim, Y.; Kim, Y.N.; Richardson, L.; Choi, C.; Menon, R. Exosomal Delivery of NF-ΚB Inhibitor Delays LPS-Induced Preterm Birth and Modulates Fetal Immune Cell Profile in Mouse Models. Sci. Adv. 2021, 7, eabd3865. [Google Scholar] [CrossRef]
- Takbiri Osgoei, L.; Parivar, K.; Ebrahimi, M.; Mortaz, E. Nicotine Modulates the Release of Inflammatory Cytokines and Expression of TLR2, TLR4 of Cord Blood Mononuclear Cells. Iran. J. Allergy Asthma Immunol. 2018, 17, 372–378. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, Y.; Wang, D.; Wang, B.; Li, H.; Xiong, J.; Xu, S.; Chen, Q.; Tao, K.; Yang, X.; Zhu, Y.; et al. HMGB1 Translocation and Release Mediate Cigarette Smoke–Induced Pulmonary Inflammation in Mice through a TLR4/MyD88-Dependent Signaling Pathway. MBoC 2017, 28, 201–209. [Google Scholar] [CrossRef]
- Allam, V.S.R.R.; Faiz, A.; Lam, M.; Rathnayake, S.N.H.; Ditz, B.; Pouwels, S.D.; Brandsma, C.-A.; Timens, W.; Hiemstra, P.S.; Tew, G.W.; et al. RAGE and TLR4 Differentially Regulate Airway Hyperresponsiveness: Implications for COPD. Allergy 2020, 76, 1123–1135. [Google Scholar] [CrossRef]
- Winden, D.R.; Barton, D.B.; Betteridge, B.C.; Bodine, J.S.; Jones, C.M.; Rogers, G.D.; Chavarria, M.; Wright, A.J.; Jergensen, Z.R.; Jimenez, F.R. Antenatal Exposure of Maternal Secondhand Smoke (SHS) Increases Fetal Lung Expression of RAGE and Induces RAGE-Mediated Pulmonary Inflammation. Respir. Res. 2014, 15, 129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, H.J.; Park, K.H.; Kim, Y.M.; Joo, E.; Ahn, K.; Shin, S. A Protein Microarray Analysis of Amniotic Fluid Proteins for the Prediction of Spontaneous Preterm Delivery in Women with Preterm Premature Rupture of Membranes at 23 to 30 Weeks of Gestation. PLoS ONE 2020, 15, e0244720. [Google Scholar] [CrossRef] [PubMed]
- Romero, R.; Chaiworapongsa, T.; Espinoza, J.; Gomez, R.; Yoon, B.H.; Edwin, S.; Mazor, M.; Maymon, E.; Berry, S. Fetal Plasma MMP-9 Concentrations Are Elevated in Preterm Premature Rupture of the Membranes. Am. J. Obstet. Gynecol. 2002, 187, 1125–1130. [Google Scholar] [CrossRef]
- Ferrand, P.E.; Parry, S.; Sammel, M.; Macones, G.A.; Kuivaniemi, H.; Romero, R.; Strauss, J.F. A Polymorphism in the Matrix Metalloproteinase-9 Promoter Is Associated with Increased Risk of Preterm Premature Rupture of Membranes in African Americans. Mol. Hum. Reprod. 2002, 8, 494–501. [Google Scholar] [CrossRef] [Green Version]
- Macarie, R.D.; Vadana, M.; Ciortan, L.; Tucureanu, M.M.; Ciobanu, A.; Vinereanu, D.; Manduteanu, I.; Simionescu, M.; Butoi, E. The Expression of MMP-1 and MMP-9 Is up-Regulated by Smooth Muscle Cells after Their Cross-Talk with Macrophages in High Glucose Conditions. J. Cell. Mol. Med. 2018, 22, 4366–4376. [Google Scholar] [CrossRef] [Green Version]
- Yan, W.; Fan, W.; Chen, C.; Wu, Y.; Fan, Z.; Chen, J.; Chen, Z.; Chen, H. IL-15 up-Regulates the MMP-9 Expression Levels and Induces Inflammatory Infiltration of Macrophages in Polymyositis through Regulating the NF-KB Pathway. Gene 2016, 591, 137–147. [Google Scholar] [CrossRef]
- Sindhu, S.; Al-Roub, A.; Koshy, M.; Thomas, R.; Ahmad, R. Palmitate-Induced MMP-9 Expression in the Human Monocytic Cells Is Mediated through the TLR4-MyD88 Dependent Mechanism. Cell. Physiol. Biochem. 2016, 39, 889–900. [Google Scholar] [CrossRef]
- Dwir, D.; Giangreco, B.; Xin, L.; Tenenbaum, L.; Cabungcal, J.-H.; Steullet, P.; Goupil, A.; Cleusix, M.; Jenni, R.; Chtarto, A.; et al. MMP9/RAGE Pathway Overactivation Mediates Redox Dysregulation and Neuroinflammation, Leading to Inhibitory/Excitatory Imbalance: A Reverse Translation Study in Schizophrenia Patients. Mol. Psychiatry 2020, 25, 2889–2904. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bouvier, D.; Rouzaire, M.; Marceau, G.; Prat, C.; Pereira, B.; Lemarié, R.; Deruelle, P.; Fajardy, I.; Gallot, D.; Blanchon, L.; et al. Aquaporins and Fetal Membranes From Diabetic Parturient Women: Expression Abnormalities and Regulation by Insulin. J. Clin. Endocrinol. Metab. 2015, 100, E1270–E1279. [Google Scholar] [CrossRef] [Green Version]
- Gilda, J.E.; Gomes, A.V. Stain-Free Total Protein Staining Is a Superior Loading Control to β-Actin for Western Blots. Anal. Biochem. 2013, 440, 186–188. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bustin, S.A.; Benes, V.; Garson, J.A.; Hellemans, J.; Huggett, J.; Kubista, M.; Mueller, R.; Nolan, T.; Pfaffl, M.W.; Shipley, G.L.; et al. The MIQE Guidelines: Minimum Information for Publication of Quantitative Real-Time PCR Experiments. Clin. Chem. 2009, 55, 611–622. [Google Scholar] [CrossRef] [PubMed] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Sequence 5′-3′ (F: Forward, r: Reverse) | Product Size (bp) | Annealing Temperature (°C) |
|---|---|---|---|
| hsRAGE | F: TGTGCTGATCCTCCCTGAGA | 139 | 61 |
| R: TGCAGTTGGCCCCTCCTCG | |||
| hs36B4 | F: AGGCTTTAGGTATCACCACT | 219 | 61 |
| R: GCAGAGTTTCCTCTGTGATA | |||
| hsRSP17 | F: TGCGAGGAGATCGCCATTATC | 169 | 61 |
| R: AAGGCTGAGACCTCAGGAAC | |||
| hsMyD88 | F: GCAGGAGGAGGCTGAGAAGC | 177 | 66 |
| R: CGGATCATCTCCTGCACAAACT | |||
| hsTIRAP | F: AAGTACCAGATGCTGCAGGCC | 200 | 66 |
| R: AGTGTCAACTGAGTGTCTGCAG | |||
| hsDia-1 | F: AGAGCCACACTTCCTTTCCATC | 167 | 66 |
| R: TCAATCTCAATCTGGAGGTGCC | |||
| hsIL6 | F: AATGAGGAGACTTGCCTGGTG | 143 | 61 |
| R: AGGAACTGGATCAGGACTTTTG | |||
| hsIL8 | F: TGATTTCTGCAGCTCTGTGTG | 154 | 61 |
| R: TCTGTGTTGGCGCAGTGTGG | |||
| hsIL1β | F: AATCTCCGACCACCACTACAG | 174 | 62 |
| R: TCCCATGTGTCGAAGAAGATAG | |||
| hsMMP9 | F: ATTGACGACGCCTTTGCCCG | 201 | 61 |
| R: ATGGGCGTCTCCCTGAATGC | |||
| hsMMP2 | F: AGCTCATCGCAGATGCCTGG | 199 | 61 |
| R: AAGGGCCTGTGGGAGCCAG |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Choltus, H.; Minet-Quinard, R.; Belville, C.; Durif, J.; Gallot, D.; Blanchon, L.; Sapin, V. Cigarette Smoke Condensate Exposure Induces Receptor for Advanced Glycation End-Products (RAGE)-Dependent Sterile Inflammation in Amniotic Epithelial Cells. Int. J. Mol. Sci. 2021, 22, 8345. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22158345
Choltus H, Minet-Quinard R, Belville C, Durif J, Gallot D, Blanchon L, Sapin V. Cigarette Smoke Condensate Exposure Induces Receptor for Advanced Glycation End-Products (RAGE)-Dependent Sterile Inflammation in Amniotic Epithelial Cells. International Journal of Molecular Sciences. 2021; 22(15):8345. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22158345
Chicago/Turabian StyleCholtus, Helena, Régine Minet-Quinard, Corinne Belville, Julie Durif, Denis Gallot, Loic Blanchon, and Vincent Sapin. 2021. "Cigarette Smoke Condensate Exposure Induces Receptor for Advanced Glycation End-Products (RAGE)-Dependent Sterile Inflammation in Amniotic Epithelial Cells" International Journal of Molecular Sciences 22, no. 15: 8345. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22158345