
Neurotoxic Effects of Neonicotinoids on Mammals: What Is There beyond the Activation of Nicotinic Acetylcholine Receptors?—A Systematic Review


Abstract
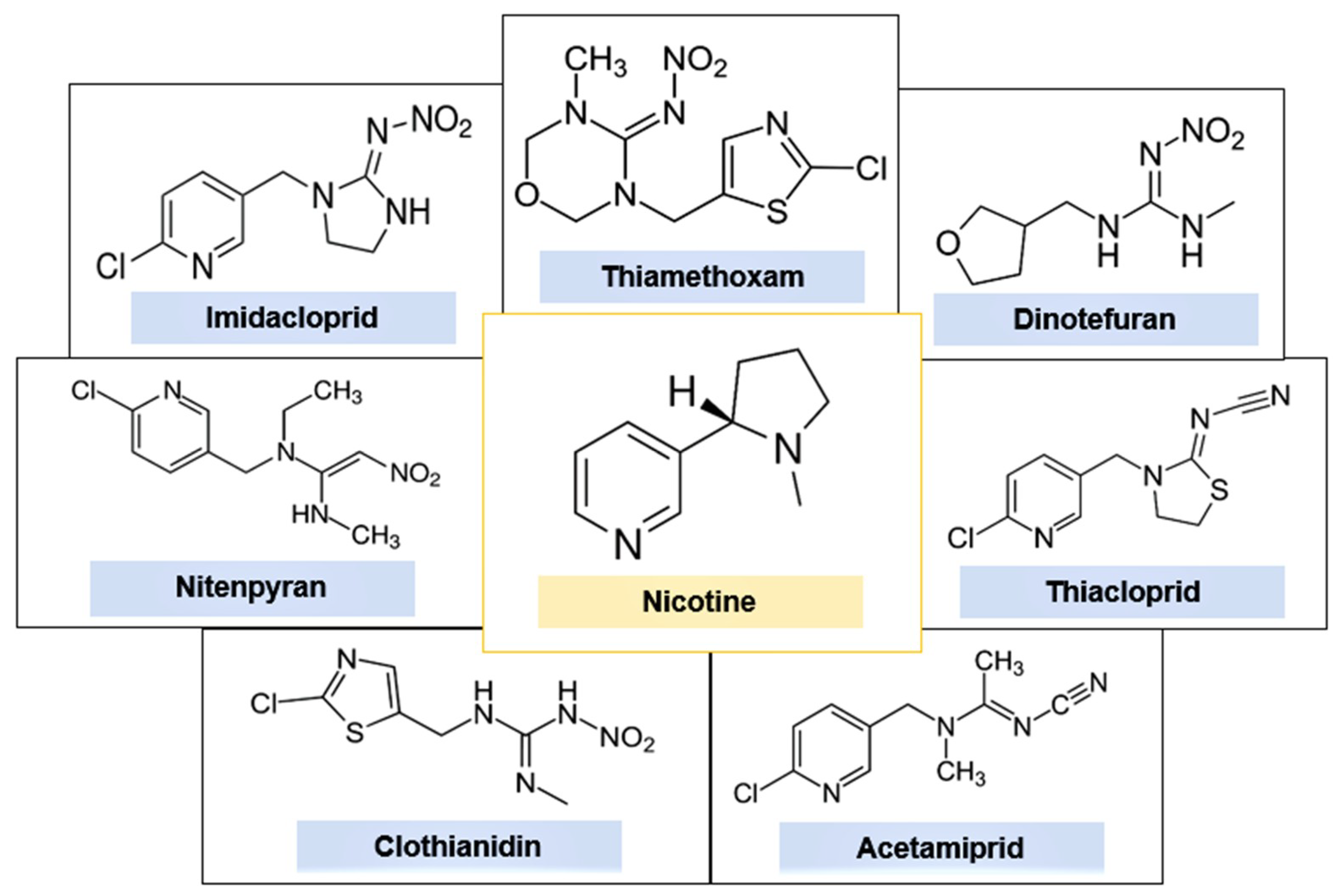
:1. Introduction
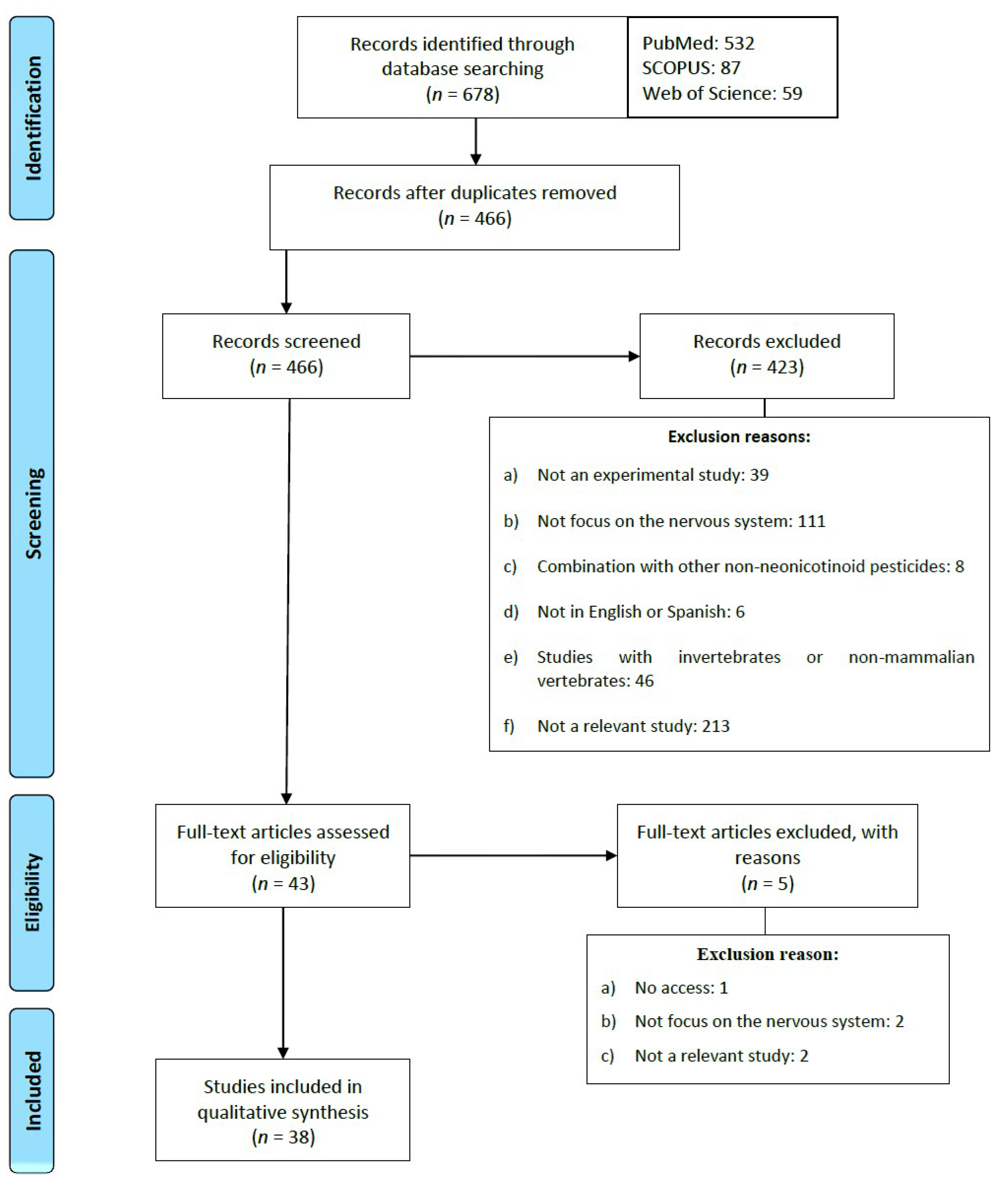
2. Methodology
Exclusion and Inclusion Criteria
3. Results
3.1. Effects of NNs on Rodents
3.1.1. Effects on Nervous System Development
3.1.2. Effects on Behavior and Cognitive Functions
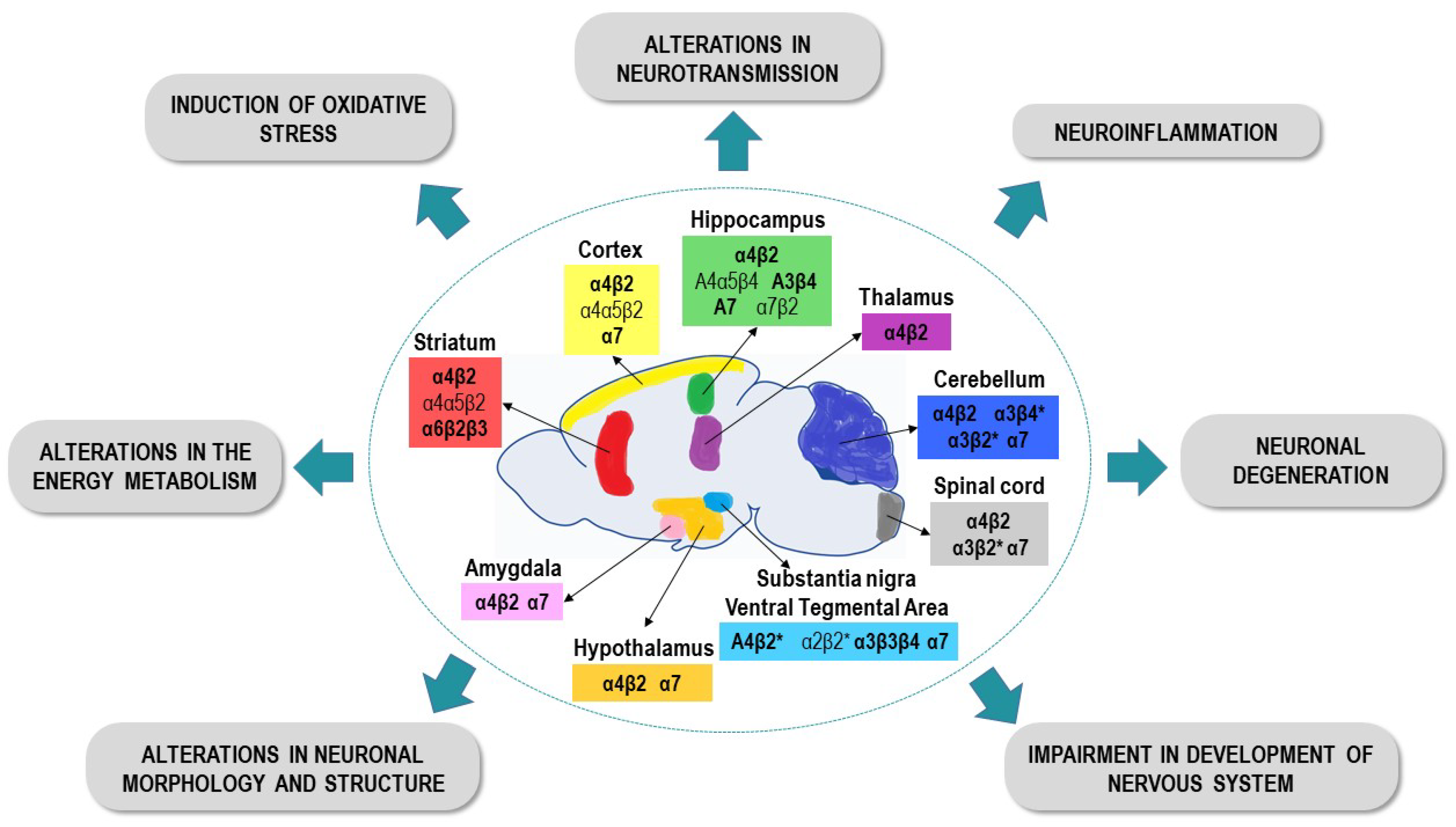
3.1.3. Changes in Neurotransmission
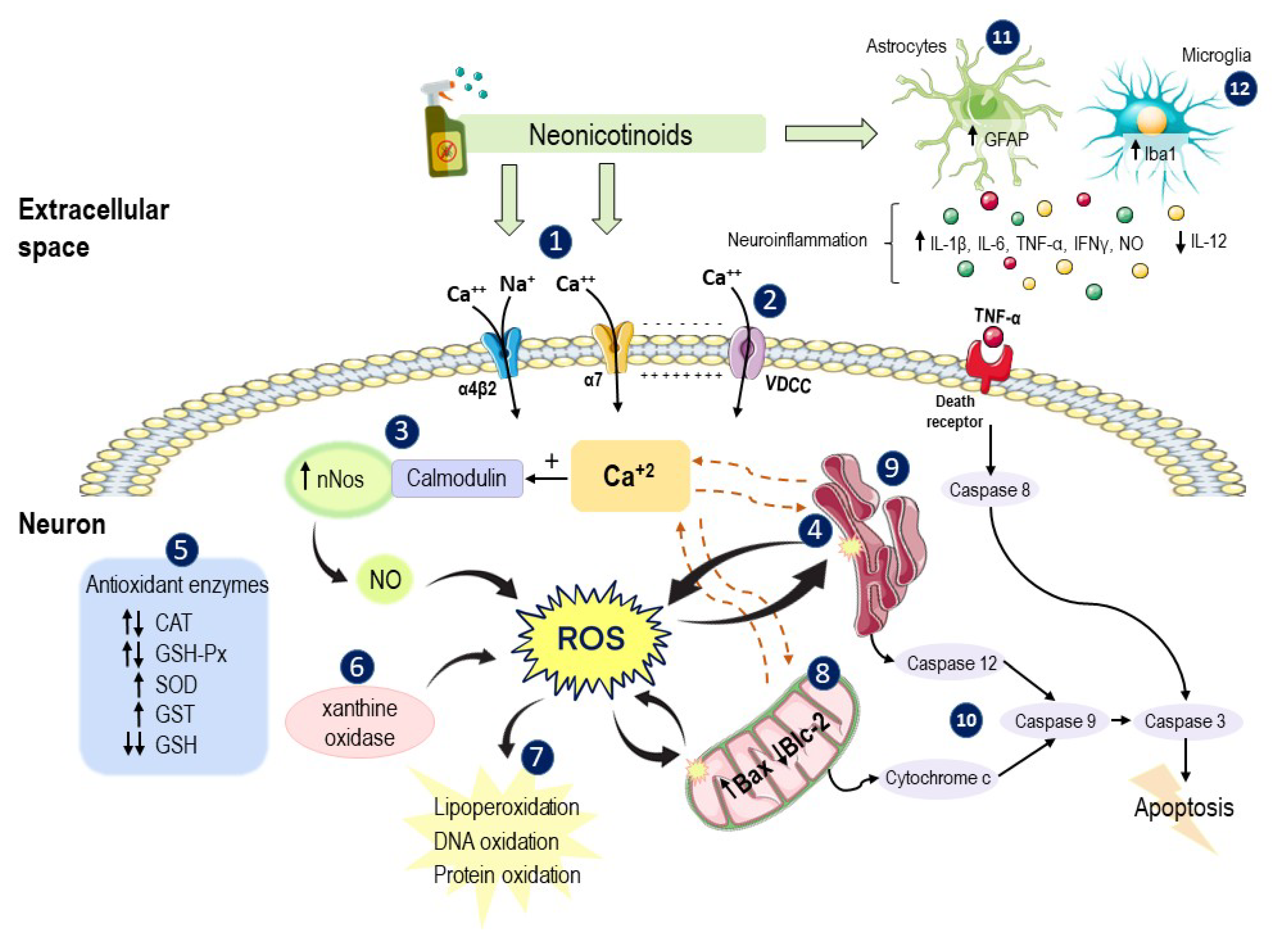
3.1.4. Induction of Oxidative Stress
3.1.5. Induction of Inflammation
3.1.6. Effects on Energy Metabolism
3.1.7. Induction of Apoptosis
3.2. Effect of NNs on Humans
3.2.1. In Vitro Neurotoxic Effects on Human Cell Cultures
3.2.2. Descriptive and Analytical Studies
3.3. Effects of NNs on Other Mammals
3.4. Discussion
Relevance of Daily Human Exposure to Pesticides NNs
4. Limitations
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Availability of Data and Material
Conflicts of Interest
Abbreviations
References
- Jeschke, P.; Nauen, R.; Schindler, M.; Elbert, A. Overview of the status and global strategy for neonicotinoids. J. Agric. Food Chem. 2011, 59, 2897–2908. [Google Scholar] [CrossRef]
- Bass, C.; Denholm, I.; Williamson, M.S.; Nauen, R. The global status of insect resistance to neonicotinoid insecticides. Pestic. Biochem. Physiol. 2015, 121, 78–87. [Google Scholar] [CrossRef] [Green Version]
- Goulson, D. An overview of the environmental risks posed by neonicotinoid insecticides. J. Appl. Ecol. 2013, 50, 977–987. [Google Scholar] [CrossRef]
- Simon-Delso, N.; Amaral-Rogers, V.; Belzunces, L.P.; Bonmatin, J.M.; Chagnon, M.; Downs, C.; Furlan, L.; Gibbons, D.W.; Giorio, C.; Girolami, V.; et al. Systemic insecticides (neonicotinoids and fipronil): Trends, uses, mode of action and metabolites. Environ. Sci. Pollut. Res. Int. 2015, 22, 5–34. [Google Scholar] [CrossRef]
- Tomizawa, M.; Casida, J.E. Selective toxicity of neonicotinoids attributable to specificity of insect and mammalian nicotinic receptors. Annu. Rev. Entomol. 2003, 48, 339–364. [Google Scholar] [CrossRef]
- Jeschke, P.; Nauen, R. Neonicotinoids-from zero to hero in insecticide chemistry. Pest Manag. Sci. 2008, 64, 1084–1098. [Google Scholar] [CrossRef]
- Yamamoto, I.; Tomizawa, M.; Saito, T.; Miyamoto, T.; Walcott, E.C.; Sumikawa, K. Structural factors contributing to insecticidal and selective actions of neonicotinoids. Arch. Insect Biochem. Physiol. 1998, 37, 24–32. [Google Scholar] [CrossRef]
- Breer, H.; Sattelle, D.B. Molecular properties and functions of insect acetylcholine receptors. J. Insect Physiol. 1987, 33, 771–790. [Google Scholar] [CrossRef]
- Dani, J.A. Neuronal nicotinic acetylcholine receptor structure and function and response to nicotine. Int. Rev. Neurobiol. 2015, 124, 3–19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bartlett, A.J.; Hedges, A.M.; Intini, K.D.; Brown, L.R.; Maisonneuve, F.J.; Robinson, S.A.; Gillis, P.L.; de Solla, S.R. Lethal and sublethal toxicity of neonicotinoid and butenolide insecticides to the mayfly, Hexagenia spp. Environ. Pollut. 2018, 238, 63–75. [Google Scholar] [CrossRef] [PubMed]
- Kara, M.; Yumrutas, O.; Demir, C.F.; Ozdemir, H.H.; Bozgeyik, I.; Coskun, S.; Eraslan, E.; Bal, R. Insecticide imidacloprid influences cognitive functions and alters learning performance and related gene expression in a rat model. Int. J. Exp. Pathol. 2015, 96, 332–337. [Google Scholar] [CrossRef]
- Anderson, J.C.; Dubetz, C.; Palace, V.P. Neonicotinoids in the Canadian aquatic environment: A literature review on current use products with a focus on fate, exposure, and biological effects. Sci. Total Environ. 2015, 505, 409–422. [Google Scholar] [CrossRef] [PubMed]
- Mohamed, F.; Gawarammana, I.; Robertson, T.A.; Roberts, M.S.; Palangasinghe, C.; Zawahir, S.; Jayamanne, S.; Kandasamy, J.; Eddleston, M.; Buckley, N.A.; et al. Acute human self-poisoning with imidacloprid compound: A neonicotinoid insecticide. PLoS ONE 2009, 4, e5127. [Google Scholar] [CrossRef]
- Tomizawa, M.; Casida, J.E. Neonicotinoid insecticide toxicology: Mechanisms of selective action. Annu. Rev. Pharmacol. Toxicol. 2005, 45, 247–268. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Katić, A.; Kašuba, V.; Kopjar, N.; Lovaković, B.T.; Marjanović Čermak, A.M.; Mendaš, G.; Micek, V.; Milić, M.; Pavičić, I.; Pizent, A.; et al. Effects of low-level imidacloprid oral exposure on cholinesterase activity, oxidative stress responses, and primary DNA damage in the blood and brain of male Wistar rats. Chem. Biol. Interact. 2021, 338, 109287. [Google Scholar] [CrossRef]
- Bonmatin, J.M.; Giorio, C.; Girolami, V.; Goulson, D.; Kreutzweiser, D.P.; Krupke, C.; Liess, M.; Long, E.; Marzaro, M.; Mitchell, E.A.D.; et al. Environmental fate and exposure; neonicotinoids and fipronil. Environ. Sci. Pollut. Res. Int. 2015, 22, 35–67. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.; Tao, L.; McLean, J.; Lu, C. Quantitative analysis of neonicotinoid insecticide residues in foods: Implication for dietary exposures. J. Agric. Food Chem. 2014, 62, 6082–6090. [Google Scholar] [CrossRef] [PubMed]
- Daraghmeh, A.; Shraim, A.; Abulhaj, S.; Sansour, R.; Ng, J.C. Imidacloprid residues in fruits, vegetables and water samples from Palestine. Environ. Geochem. Health 2007, 29, 45–50. [Google Scholar] [CrossRef] [PubMed]
- Frenich, A.G.; Vidal, J.M.; Pastor-Montoro, E.; Romero-González, R. High-throughput determination of pesticide residues in food commodities by use of ultra-performance liquid chromatography–tandem mass spectrometry. Anal. Bioanal. Chem. 2008, 390, 947–959. [Google Scholar] [CrossRef] [PubMed]
- Hladik, M.L.; Kolpin, D.W. First national-scale reconnaissance of neonicotinoid insecticides in streams across the USA. Environ. Chem. 2016, 13, 12–20. [Google Scholar] [CrossRef]
- Iancu, V.; Petre, J.; Galaon, T.; Radu, G.L. Occurrence of neonicotinoid residues in Danube River and tributaries. Rev. Chim. 2019, 70, 313–318. [Google Scholar] [CrossRef]
- Liu, S.; Zheng, Z.; Wei, F.; Ren, Y.; Gui, W.; Wu, H.; Zhu, G. Simultaneous determination of seven neonicotinoid pesticide residues in food by ultraperformance liquid chromatography tandem mass spectrometry. J. Agric. Food Chem. 2010, 58, 3271–3278. [Google Scholar] [CrossRef] [PubMed]
- Niaz, A.; Sial, R.A.; Yaseen, M.; Mand, G.A.; Javed, M.H.; Ahmad, E.; Ahmad, R.; Rahim, M. Determination of imidacloprid residues in rice from various districts of Punjab using high performance liquid chromatography. J. Anim. Plant Sci. 2016, 26, 170–176. [Google Scholar]
- Tanner, G.; Czerwenka, C. LC-MS/MS analysis of neonicotinoid insecticides in honey: Methodology and residue findings in Austrian honeys. J. Agric. Food Chem. 2011, 59, 12271–12277. [Google Scholar] [CrossRef] [PubMed]
- Tišler, T.; Jemec, A.; Mozetič, B.; Trebše, P. Hazard identification of imidacloprid to aquatic environment. Chemosphere 2009, 76, 907–914. [Google Scholar] [CrossRef] [PubMed]
- Xu, M.; Huang, H.; Li, N.; Li, F.; Wang, D.; Luo, Q. Occurrence and ecological risk of pharmaceuticals and personal care products (PPCPs) and pesticides in typical surface watersheds, China. Ecotoxicol. Environ. Saf. 2019, 175, 289–298. [Google Scholar] [CrossRef] [PubMed]
- Tsvetkov, N.; Samson-Robert, O.; Sood, K.; Patel, H.S.; Malena, D.A.; Gajiwala, P.H.; Maciukiewicz, P.; Fournier, V.; Zayed, A. Chronic exposure to neonicotinoids reduces honey bee health near corn crops. Science 2017, 356, 1395–1397. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wood, T.J.; Goulson, D. The environmental risks of neonicotinoid pesticides: A review of the evidence post 2013. Environ. Sci. Pollut. Res. Int. 2017, 24, 17285–17325. [Google Scholar] [CrossRef] [PubMed]
- Woodcock, B.A.; Bullock, J.M.; Shore, R.F.; Heard, M.S.; Pereira, M.G.; Redhead, J.; Ridding, L.; Dean, H.; Sleep, D.; Henrys, P.; et al. Country-specific effects of neonicotinoid pesticides on honey bees and wild bees. Science 2017, 356, 1393–1395. [Google Scholar] [CrossRef] [Green Version]
- Commission Implementing Regulation (EU) 2018/783 of 29 May 2018 Amending Implementing Regulation (EU) No 540/2011 as Regards the Conditions of Approval of the Active Substance Imidacloprid. 2018, Volume 132. Available online: https://eur-lex.europa.eu/legal-content/EN/TXT/?uri=CELEX:32018R0783 (accessed on 2 July 2021).
- Badgujar, P.C.; Jain, S.K.; Singh, A.; Punia, J.S.; Gupta, R.P.; Chandratre, G.A. Immunotoxic effects of imidacloprid following 28 days of oral exposure in BALB/c mice. Environ. Toxicol. Pharmacol. 2013, 35, 408–418. [Google Scholar] [CrossRef]
- Bagri, P.; Kumar, V.; Sikka, A.K. Assessment of imidacloprid-induced mutagenic effects in somatic cells of Swiss albino male mice. Drug Chem. Toxicol. 2016, 39, 412–417. [Google Scholar] [CrossRef]
- Bal, R.; Naziroğlu, M.; Türk, G.; Yilmaz, Ö.; Kuloğlu, T.; Etem, E.; Baydas, G. Insecticide imidacloprid induces morphological and DNA damage through oxidative toxicity on the reproductive organs of developing male rats. Cell Biochem. Funct. 2012, 30, 492–499. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhardwaj, S.; Srivastava, M.K.; Kapoor, U.; Srivastava, L.P. A 90 days oral toxicity of imidacloprid in female rats: Morphological, biochemical and histopathological evaluations. Food Chem. Toxicol. 2010, 48, 1185–1190. [Google Scholar] [CrossRef]
- Duzguner, V.; Erdogan, S. Acute oxidant and inflammatory effects of imidacloprid on the mammalian central nervous system and liver in rats. Pestic. Biochem. Physiol. 2010, 97, 13–18. [Google Scholar] [CrossRef]
- Gibbons, D.; Morrissey, C.; Mineau, P. A review of the direct and indirect effects of neonicotinoids and fipronil on vertebrate wildlife. Environ. Sci. Pollut. Res. Int. 2015, 22, 103–118. [Google Scholar] [CrossRef] [Green Version]
- Kapoor, U.; Srivastava, M.K.; Srivastava, L.P. Toxicological impact of technical imidacloprid on ovarian morphology, hormones and antioxidant enzymes in female rats. Food Chem. Toxicol. 2011, 49, 3086–3089. [Google Scholar] [CrossRef] [PubMed]
- Mikolić, A.; Karačonji, I.B. Imidacloprid as reproductive toxicant and endocrine disruptor: Investigations in laboratory animals. Arh. Hig. Rada. Toksikol. 2018, 69, 103–108. [Google Scholar] [CrossRef] [Green Version]
- Rodrigues, K.J.A.; Santana, M.B.; Do Nascimento, J.L.M.; Picanco-Diniz, D.L.W.; Maues, L.A.L.; Santos, S.N.; Ferreira, V.M.M.; Alfonso, M.; Durán, R.; Faro, L.R.F. Behavioral and biochemical effects of neonicotinoid thiamethoxam on the cholinergic system in rats. Ecotoxicol. Environ. Saf. 2010, 73, 101–107. [Google Scholar] [CrossRef]
- Abou-Donia, M.B.; Goldstein, L.B.; Bullman, S.; Tu, T.; Khan, W.A.; Dechkovskaia, A.M.; Abdel-Rahman, A.A. Imidacloprid induces neurobehavioral deficits and increases expression of glial fibrillary acidic protein in the motor cortex and hippocampus in offspring rats following in utero exposure. J. Toxicol. Environ. Health A 2008, 71, 119–130. [Google Scholar] [CrossRef] [PubMed]
- Osaka, A.; Ueyama, J.; Kondo, T.; Nomura, H.; Sugiura, Y.; Saito, I.; Nakane, K.; Takaishi, A.; Ogi, H.; Wakusawa, S.; et al. Exposure characterization of three major insecticide lines in urine of young children in Japan-neonicotinoids, organophosphates, and pyrethroids. Environ. Res. 2016, 147, 89–96. [Google Scholar] [CrossRef]
- Ospina, M.; Wong, L.Y.; Baker, S.E.; Serafim, A.B.; Morales-Agudelo, P.; Calafat, A.M. Exposure to neonicotinoid insecticides in the US general population: Data from the 2015-2016 national health and nutrition examination survey. Environ. Res. 2019, 176, 108555. [Google Scholar] [CrossRef]
- Tao, Y.; Dong, F.; Xu, J.; Phung, D.; Liu, Q.; Li, R.; Liu, X.; Wu, X.; He, M.; Zheng, Y. Characteristics of neonicotinoid imidacloprid in urine following exposure of humans to orchards in China. Environ. Int. 2019, 132, 105079. [Google Scholar] [CrossRef]
- Ueyama, J.; Harada, K.H.; Koizumi, A.; Sugiura, Y.; Kondo, T.; Saito, I.; Kamijima, M. Temporal levels of urinary neonicotinoid and dialkylphosphate concentrations in Japanese women between 1994 and 2011. Environ. Sci. Technol. 2015, 49, 14522–14528. [Google Scholar] [CrossRef]
- Tomizawa, M.; Casida, J.E. Minor structural changes in nicotinoid insecticides confer differential subtype selectivity for mammalian nicotinic acetylcholine receptors. Br. J. Pharmacol. 1999, 127, 115–122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cimino, A.M.; Boyles, A.L.; Thayer, K.A.; Perry, M.J. Effects of neonicotinoid pesticide exposure on human health: A systematic review. Environ. Health Perspect. 2017, 125, 155–162. [Google Scholar] [CrossRef] [Green Version]
- Forrester, M.B. Neonicotinoid insecticide exposures reported to six poison centers in Texas. Hum. Exp. Toxicol. 2014, 33, 568–573. [Google Scholar] [CrossRef] [PubMed]
- Imamura, T.; Yanagawa, Y.; Nishikawa, K.; Matsumoto, N.; Sakamoto, T. Two cases of acute poisoning with acetamiprid in humans. Clin. Toxicol. 2010, 48, 851–853. [Google Scholar] [CrossRef] [PubMed]
- Phua, D.H.; Lin, C.C.; Wu, M.L.; Deng, J.F.; Yang, C.C. Neonicotinoid insecticides: An emerging cause of acute pesticide poisoning. Clin. Toxicol. 2009, 47, 336–341. [Google Scholar] [CrossRef]
- Taira, K.; Aoyama, Y.; Kawakami, T.; Kamata, M.; Aoi, T. Detection of chloropyridinyl neonicotinoid insecticide metabolite 6-chloronicotinic acid in the urine: Six cases with subacute nicotinic symptoms. Chudoku Kenkyu 2011, 24, 222–230. [Google Scholar]
- Todani, M.; Kaneko, T.; Hayashida, H.; Kaneda, K.; Tsuruta, R.; Kasaoka, S.; Maekawa, T. Acute poisoning with neonicotinoid insecticide acetamiprid. Chudoku Kenkyu 2008, 21, 387–390. [Google Scholar]
- Iyyadurai, R.; George, I.A.; Peter, J.V. Imidacloprid poisoning-newer insecticide and fatal toxicity. J. Med. Toxicol. 2010, 6, 77–78. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Proença, P.; Teixeira, H.; Castanheira, F.; Pinheiro, J.; Monsanto, P.V.; Marques, E.P.; Vieira, D.N. Two fatal intoxication cases with imidacloprid: LC/MS analysis. Forensic Sci. Int. 2005, 153, 75–80. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shadnia, S.; Moghaddam, H.H. Fatal intoxication with imidacloprid insecticide. Am. J. Emerg. Med. 2008, 26, 634.e1–634.e4. [Google Scholar] [CrossRef] [PubMed]
- Moher, D.; Liberati, A.; Tetzlaff, J.; Altman, D.G. The PRISMA Group. Preferred reporting items for systematic reviews and meta-analyses: The PRISMA statement. PLoS Med. 2009, 6, e1000097. [Google Scholar] [CrossRef] [Green Version]
- Von Wedel-Parlow, M.; Wölte, P.; Galla, H.J. Regulation of major efflux transporters under inflammatory conditions at the blood-brain barrier in vitro. J. Neurochem. 2009, 111, 111–118. [Google Scholar] [CrossRef]
- Burke, A.P.; Niibori, Y.; Terayama, H.; Ito, M.; Pidgeon, C.; Arsenault, J.; Camarero, P.R.; Cummins, C.L.; Mateo, R.; Sakabe, K.; et al. Mammalian susceptibility to a neonicotinoid insecticide after fetal and early postnatal exposure. Sci. Rep. 2018, 8, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Hirano, T.; Miyata, Y.; Kubo, S.; Ohno, S.; Onaru, K.; Maeda, M.; Kitauchi, S.; Nishi, M.; Tabuchi, Y.; Ikenaka, Y.; et al. Aging-related changes in the sensitivity of behavioral effects of the neonicotinoid pesticide clothianidin in male mice. Toxicol. Lett. 2021, 342, 95–103. [Google Scholar] [CrossRef]
- Kapoor, U.; Srivastava, M.K.; Trivedi, P.; Garg, V.; Srivastava, L.P. Disposition and acute toxicity of imidacloprid in female rats after single exposure. Food Chem. Toxicol. 2014, 68, 190–195. [Google Scholar] [CrossRef]
- Shamsi, M.; Soodi, M.; Shahbazi, S.; Omidi, A. Effect of Acetamiprid on spatial memory and hippocampal glutamatergic system. Environ. Sci. Pollut. Res. Int. 2021, 28, 27933–27941. [Google Scholar] [CrossRef]
- Terayama, H.; Endo, H.; Tsukamoto, H.; Matsumoto, K.; Umezu, M.; Kanazawa, T.; Ito, M.; Sato, T.; Naito, M.; Kawakami, S.; et al. Acetamiprid accumulates in different amounts in murine brain regions. Int. J. Environ. Res. Public Health 2016, 13, 937. [Google Scholar] [CrossRef] [Green Version]
- Jin, X.; Wang, F.; Liu, X.; Liang, B.; Chen, Z.; He, J.; Zhang, H.; Zhang, J. Negative correlation of CD34+ cells with blood–brain barrier permeability following traumatic brain injury in a rat model. Microcirculation 2014, 21, 696–702. [Google Scholar] [CrossRef]
- Christen, V.; Rusconi, M.; Crettaz, P.; Fent, K. Developmental neurotoxicity of different pesticides in PC-12 cells In Vitro. Toxicol. Appl. Pharmacol. 2017, 325, 25–36. [Google Scholar] [CrossRef]
- Kimura-Kuroda, J.; Nishito, Y.; Yanagisawa, H.; Kuroda, Y.; Komuta, Y.; Kawano, H.; Hayashi, M. Neonicotinoid insecticides alter the gene expression profile of neuron-enriched cultures from neonatal rat cerebellum. Int. J. Environ. Res. Public Health 2016, 13, 987. [Google Scholar] [CrossRef] [Green Version]
- Kagawa, N.; Nagao, T. Neurodevelopmental toxicity in the mouse neocortex following prenatal exposure to acetamiprid. J. Appl. Toxicol. 2018, 38, 1521–1528. [Google Scholar] [CrossRef]
- Nakayama, A.; Yoshida, M.; Kagawa, N.; Nagao, T. The neonicotinoids acetamiprid and imidacloprid impair neurogenesis and alter the microglial profile in the hippocampal dentate gyrus of mouse neonates. J. Appl. Toxicol. 2019, 39, 877–887. [Google Scholar] [CrossRef] [PubMed]
- Shideler, K.K.; Yan, J. M1 muscarinic receptor for the development of auditory cortical function. Mol. Brain 2010, 3, 29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tanaka, T. Reproductive and neurobehavioral effects of clothianidin administered to mice in the diet. Birth Defects Res. B Dev. Reprod. Toxicol. 2012, 95, 151–159. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, T. Effects of maternal clothianidin exposure on behavioral development in F1 generation mice. Toxicol. Ind. Health 2012, 28, 697–707. [Google Scholar] [CrossRef] [PubMed]
- Abd-Elhakim, Y.M.; Mohammed, H.H.; Mohamed, W.A. Imidacloprid impacts on neurobehavioral performance, oxidative stress, and apoptotic events in the brain of adolescent and adult rats. J. Agric. Food Chem. 2018, 66, 13513–13524. [Google Scholar] [CrossRef]
- Khalil, S.R.; Awad, A.; Mohammed, H.H.; Nassan, M.A. Imidacloprid insecticide exposure induces stress and disrupts glucose homeostasis in male rats. Environ. Toxicol. Pharmacol. 2017, 55, 165–174. [Google Scholar] [CrossRef]
- Drago, J.; McColl, C.D.; Horne, M.K.; Finkelstein, D.I.; Ross, S.A. Neuronal nicotinic receptors: Insights gained from gene knockout an knocking mutant mice. Cell. Mol. Life Sci. 2003, 60, 1267–1280. [Google Scholar] [CrossRef]
- McGranahan, T.M.; Patzlaff, N.E.; Grady, S.R.; Heinemann, S.F.; Booker, T.K. α4β2 nicotinic acetylcholine receptors on dopaminergic neurons mediate nicotine reward and anxiety relief. J. Neurosci. 2011, 31, 10891–10902. [Google Scholar] [CrossRef] [Green Version]
- Yoneda, N.; Takada, T.; Hirano, T.; Yanai, S.; Yamamoto, A.; Mantani, Y.; Yokoyama, T.; Kitagawa, H.; Tabuchi, Y.; Hoshi, N. Peripubertal exposure to the neonicotinoid pesticide dinotefuran affects dopaminergic neurons and causes hyperactivity in male mice. J. Vet. Med. Sci. 2018, 80, 634–637. [Google Scholar] [CrossRef] [Green Version]
- Byrne, F.J.; Daugherty, M.P.; Grafton-Cardwell, E.E.; Bethke, J.A.; Morse, J.G. Evaluation of systemic neonicotinoid insecticides for the management of the Asian citrus psyllid Diaphorina citri on containerized citrus. Pest Manag. Sci. 2017, 73, 506–514. [Google Scholar] [CrossRef]
- Kobashi, K.; Harada, T.; Adachi, Y.; Mori, M.; Ihara, M.; Hayasaka, D. Comparative ecotoxicity of imidacloprid and dinotefuran to aquatic insects in rice mesocosms. Ecotoxicol. Environ. Saf. 2017, 138, 122–129. [Google Scholar] [CrossRef]
- Sánchez, C. Stress-induced vocalisation in adult animals. A valid model of anxiety? Eur. J. Pharmacol. 2003, 463, 133–143. [Google Scholar] [CrossRef]
- Hirano, T.; Yanai, S.; Takada, T.; Yoneda, N.; Omotehara, T.; Kubota, N.; Minami, K.; Yamamoto, A.; Mantani, Y.; Yokoyama, T.; et al. NOAEL-dose of a neonicotinoid pesticide, clothianidin, acutely induce anxiety-related behavior with human-audible vocalizations in male mice in a novel environment. Toxicol. Lett. 2018, 282, 57–63. [Google Scholar] [CrossRef] [PubMed]
- Brioni, J.D.; O’Neill, A.B.; Kim, D.J.; Decker, M.W. Nicotinic receptor agonists exhibit anxiolytic-like effects on the elevated plus-maze test. Eur. J. Pharmacol. 1993, 238, 1–8. [Google Scholar] [CrossRef]
- Costall, B.; Kelly, M.E.; Naylor, R.J.; Onaivi, E.S. The actions of nicotine and cocaine in a mouse model of anxiety. Pharmacol. Biochem. Behav. 1989, 33, 197–203. [Google Scholar] [CrossRef]
- Gilbert, D.G.; Robinson, J.H.; Chamberlin, C.L.; Spielberger, C.D. Effects of smoking/nicotine on anxiety, heart rate, and lateralization of EEG during a stressful movie. Psychophysiology 1989, 26, 311–320. [Google Scholar] [CrossRef] [PubMed]
- Pomerleau, O.F.; Turk, D.C.; Fertig, J.B. The effects of cigarette smoking on pain and anxiety. Addict. Behav. 1984, 9, 265–271. [Google Scholar] [CrossRef]
- Herbert, M.; Foulds, J.; Fife-Schaw, C. No effect of cigarette smoking on attention or mood in non-deprived smokers. Addiction 2001, 96, 1349–1356. [Google Scholar] [CrossRef]
- Perkins, K.A.; Sexton, J.E.; Reynolds, W.A.; Grobe, J.E.; Fonte, C.; Stiller, R.L. Comparison of acute subjective and heart rate effects of nicotine intake via tobacco smoking versus nasal spray. Pharmacol. Biochem. Behav. 1994, 47, 295–299. [Google Scholar] [CrossRef]
- Casarrubea, M.; Davies, C.; Faulisi, F.; Pierucci, M.; Colangeli, R.; Partridge, L.; Chambers, S.; Cassar, D.; Valentino, M.; Muscat, R.; et al. Acute nicotine induces anxiety and disrupts temporal pattern organization of rat exploratory behavior in hole-board: A potential role for the lateral habenula. Front. Cell. Neurosci. 2015, 9, 197. [Google Scholar] [CrossRef] [Green Version]
- Foulds, J.; Stapleton, J.A.; Bell, N.; Swettenham, J.; Jarvis, M.J.; Russell, M.A. Mood and physiological effects of subcutaneous nicotine in smokers and never-smokers. Drug Alcohol Depend. 1997, 44, 105–115. [Google Scholar] [CrossRef]
- Takada, T.; Yoneda, N.; Hirano, T.; Yanai, S.; Yamamoto, A.; Mantani, Y.; Yokoyama, T.; Kitagawa, H.; Tabuchi, Y.; Hoshi, N. Verification of the causal relationship between subchronic exposures to dinotefuran and depression-related phenotype in juvenile mice. J. Vet. Med. Sci. 2018, 80, 720–724. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McClernon, F.J.; Hiott, F.B.; Westman, E.C.; Rose, J.E.; Levin, E.D. Transdermal nicotine attenuates depression symptoms in nonsmokers: A double-blind, placebo-controlled trial. Psychopharmacology 2006, 189, 125–133. [Google Scholar] [CrossRef] [PubMed]
- Salín-Pascual, R.J.; Rosas, M.; Jimenez-Genchi, A.; Rivera-Meza, B.L. Antidepressant effect of transdermal nicotine patches in nonsmoking patients with major depression. J. Clin. Psychiatry 1996, 57, 387–389. [Google Scholar]
- Semba, J.I.; Mataki, C.; Yamada, S.; Nankai, M.; Toru, M. Antidepressantlike effects of chronic nicotine on learned helplessness paradigm in rats. Biol. Psychiatry 1998, 43, 389–391. [Google Scholar] [CrossRef]
- Janowsky, D.; Davis, J.; El-Yousef, M.K.; Sekerke, H.J. A cholinergic-adrenergic hypothesis of mania and depression. Lancet 1972, 300, 632–635. [Google Scholar] [CrossRef]
- Fenster, C.P.; Rains, M.F.; Noerager, B.; Quick, M.W.; Lester, R.A. Influence of subunit composition on desensitization of neuronal acetylcholine receptors at low concentrations of nicotine. J. Neurosci. 1997, 17, 5747–5759. [Google Scholar] [CrossRef]
- Fenster, C.P.; Whitworth, T.L.; Sheffield, E.B.; Quick, M.W.; Lester, R.A. Upregulation of surface α4β2 nicotinic receptors is initiated by receptor desensitization after chronic exposure to nicotine. J. Neurosci. 1999, 19, 4804–4814. [Google Scholar] [CrossRef] [PubMed]
- Mineur, Y.S.; Picciotto, M.R. Biological basis for the co-morbidity between smoking and mood disorders. J. Dual Diagn. 2009, 5, 122–130. [Google Scholar] [CrossRef] [PubMed]
- Paradiso, K.G.; Steinbach, J.H. Nicotine is highly effective at producing desensitization of rat α4β2 neuronal nicotinic receptors. J. Physiol. 2003, 553, 857–871. [Google Scholar] [CrossRef] [PubMed]
- Quick, M.W.; Lester, R.A. Desensitization of neuronal nicotinic receptors. J. Neurobiol. 2002, 53, 457–478. [Google Scholar] [CrossRef]
- Han, Z.Y.; Le Novère, N.; Zoli, M.; Hill Jr, J.A.; Champtiaux, N.; Changeux, J.P. Localization of nAChR subunit mRNAs in the brain of Macaca mulatta. Eur. J. Neurosci. 2000, 12, 3664–3674. [Google Scholar] [CrossRef] [PubMed]
- Philip, N.S.; Carpenter, L.L.; Tyrka, A.R.; Price, L.H. Nicotinic acetylcholine receptors and depression: A review of the preclinical and clinical literature. Psychopharmacology 2010, 212, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berntson, G.G.; Beattie, M.S.; Walker, J.M. Effects of nicotinic and muscarinic compounds on biting attack in the cat. Pharmacol. Biochem. Behav. 1976, 5, 235–239. [Google Scholar] [CrossRef]
- Driscoll, P.; Baettig, K. Selective inhibition by nicotine of shock-induced fighting in the rat. Pharmacol. Biochem. Behav. 1981, 14, 175–179. [Google Scholar] [CrossRef]
- Rodgers, R.J. Effect of nicotine, mecamylamine, and hexamethonium on shock-induced fighting, pain reactivity, and locomotor behaviour in rats. Psychopharmacology 1979, 66, 93–98. [Google Scholar] [CrossRef]
- Silverman, A.P. Behaviour of rats given a ‘smoking dose’ of nicotine. Anim. Behav. 1971, 19, 67–74. [Google Scholar] [CrossRef]
- Waldbillig, R.J. Suppressive effects of intraperitoneal and intraventricular injections of nicotine on muricide and shock-induced attack on conspecifics. Pharmacol. Biochem. Behav. 1980, 12, 619–623. [Google Scholar] [CrossRef]
- Coura, R.S.; Cressant, A.; Xia, J.; de Chaumont, F.; Olivo-Marin, J.C.; Pelloux, Y.; Dalley, J.W.; Granon, S. Nonaggressive and adapted social cognition is controlled by the interplay between noradrenergic and nicotinic receptor mechanisms in the prefrontal cortex. FASEB J. 2013, 27, 4343–4354. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abrous, D.N.; Adriani, W.; Montaron, M.F.; Aurousseau, C.; Rougon, G.; Le Moal, M.; Piazza, P.V. Nicotine self-administration impairs hippocampal plasticity. J. Neurosci. 2002, 22, 3656–3662. [Google Scholar] [CrossRef] [Green Version]
- Park, S.; Knopick, C.; McGurk, S.; Meltzer, H.Y. Nicotine impairs spatial working memory while leaving spatial attention intact. Neuropsychopharmacology 2000, 22, 200–209. [Google Scholar] [CrossRef]
- Haam, J.; Yakel, J.L. Cholinergic modulation of the hippocampal region and memory function. J. Neurochem. 2017, 142, 111–121. [Google Scholar] [CrossRef] [Green Version]
- Dineley, K.T.; Pandya, A.A.; Yakel, J.L. Nicotinic ACh receptors as therapeutic targets in CNS disorders. Trends Pharmacol. Sci. 2015, 36, 96–108. [Google Scholar] [CrossRef] [Green Version]
- Bannerman, D.M. Fractionating spatial memory with glutamate receptor subunit-knockout mice. Biochem. Soc. Trans. 2009, 37, 1323–1327. [Google Scholar] [CrossRef] [Green Version]
- Cheng, Q.; Yakel, J.L. The effect of α7 nicotinic receptor activation on glutamatergic transmission in the hippocampus. Biochem. Pharmacol. 2015, 97, 439–444. [Google Scholar] [CrossRef] [Green Version]
- Hsiao, C.J.; Lin, C.L.; Lin, T.Y.; Wang, S.E.; Wu, C.H. Imidacloprid toxicity impairs spatial memory of echolocation bats through neural apoptosis in hippocampal CA1 and medial entorhinal cortex areas. Neuroreport 2016, 27, 462–468. [Google Scholar] [CrossRef] [PubMed]
- Zheng, M.; Qin, Q.; Zhou, W.; Liu, Q.; Zeng, S.; Xiao, H.; Bai, Q.; Gao, J. Metabolic disturbance in hippocampus and liver of mice: A primary response to imidacloprid exposure. Sci. Rep. 2020, 10, 5713. [Google Scholar] [CrossRef]
- Jonas, P.; Lisman, J. Structure, function, and plasticity of hippocampal dentate gyrus microcircuits. Front. Neural Circuits 2014, 8, 107. [Google Scholar] [CrossRef] [Green Version]
- Hainmueller, T.; Bartos, M. Dentate gyrus circuits for encoding, retrieval and discrimination of episodic memories. Nat. Rev. Neurosci. 2020, 21, 153–168. [Google Scholar] [CrossRef] [PubMed]
- Hampel, H.; Mesulam, M.M.; Cuello, A.C.; Farlow, M.R.; Giacobini, E.; Grossberg, G.T.; Khachaturian, A.S.; Vergallo, A.; Cavedo, E.; Snyder, P.J.; et al. The cholinergic system in the pathophysiology and treatment of Alzheimer’s disease. Brain 2018, 141, 1917–1933. [Google Scholar] [CrossRef]
- Maurer, S.V.; Williams, C.L. The cholinergic system modulates memory and hippocampal plasticity via its interactions with non-neuronal cells. Front. Immunol. 2017, 8, 1489. [Google Scholar] [CrossRef] [Green Version]
- Vohra, P.; Khera, K.S.; Sangha, G.K. Physiological, biochemical and histological alterations induced by administration of imidacloprid in female albino rats. Pestic. Biochem. Physiol. 2014, 110, 50–56. [Google Scholar] [CrossRef] [PubMed]
- Houchat, J.N.; Cartereau, A.; Le Mauff, A.; Taillebois, E.; Thany, S.H. An overview on the effect of neonicotinoid insecticides on mammalian cholinergic functions through the activation of neuronal nicotinic acetylcholine receptors. Int. J. Environ. Res. Public Health 2020, 17, 3222. [Google Scholar] [CrossRef] [PubMed]
- Park, J.; Taly, A.; Bourreau, J.; De Nardi, F.; Legendre, C.; Henrion, D.; Guérineau, N.C.; Legros, C.; Mattei, C.; Tricoire-Leignel, H. Partial Agonist Activity of Neonicotinoids on Rat Nicotinic Receptors: Consequences over Epinephrine Secretion and In Vivo Blood Pressure. Int. J. Mol. Sci. 2021, 22, 5106. [Google Scholar] [CrossRef]
- Kimura-Kuroda, J.; Komuta, Y.; Kuroda, Y.; Hayashi, M.; Kawano, H. Nicotine-like effects of the neonicotinoid insecticides acetamiprid and imidacloprid on cerebellar neurons from neonatal rats. PLoS ONE 2012, 7, e32432. [Google Scholar] [CrossRef] [PubMed]
- Brini, M.; Calì, T.; Ottolini, D.; Carafoli, E. Neuronal calcium signaling: Function and dysfunction. Cell. Mol. Life Sci. 2014, 71, 2787–2814. [Google Scholar] [CrossRef] [PubMed]
- Grienberger, C.; Konnerth, A. Imaging calcium in neurons. Neuron 2012, 73, 862–885. [Google Scholar] [CrossRef] [Green Version]
- Simons, T.J. Calcium and neuronal function. Neurosurg. Rev. 1988, 11, 119–129. [Google Scholar] [CrossRef] [PubMed]
- Aoyagi, M.; Arvai, A.S.; Tainer, J.A.; Getzoff, E.D. Structural basis for endothelial nitric oxide synthase binding to calmodulin. EMBO J. 2003, 22, 766–775. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Griffith, O.W.; Stuehr, D.J. Nitric oxide synthases: Properties and catalytic mechanism. Annu. Rev. Physiol. 1995, 57, 707–734. [Google Scholar] [CrossRef] [PubMed]
- Marletta, M.A. Nitric oxide synthase: Aspects concerning structure and catalysis. Cell 1994, 78, 927–930. [Google Scholar] [CrossRef] [Green Version]
- McMurry, J.L.; Chrestensen, C.A.; Scott, I.M.; Lee, E.W.; Rahn, A.M.; Johansen, A.M.; Forsberg, B.J.; Harris, K.D.; Salerno, J.C. Rate, affinity and calcium dependence of nitric oxide synthase isoform binding to the primary physiological regulator calmodulin. FEBS J. 2011, 278, 4943–4954. [Google Scholar] [CrossRef] [Green Version]
- Duzguner, V.; Erdogan, S. Chronic exposure to imidacloprid induces inflammation and oxidative stress in the liver & central nervous system of rats. Pestic. Biochem. Physiol. 2012, 104, 58–64. [Google Scholar] [CrossRef]
- Gaimarri, A.; Moretti, M.; Riganti, L.; Zanardi, A.; Clementi, F.; Gotti, C. Regulation of neuronal nicotinic receptor traffic and expression. Brain Res. Rev. 2007, 55, 134–143. [Google Scholar] [CrossRef]
- Perry, D.C.; Dávila-García, M.I.; Stockmeier, C.A.; Kellar, K.J. Increased nicotinic receptors in brains from smokers: Membrane binding and autoradiography studies. J. Pharmacol. Exp. Ther. 1999, 289, 1545–1552. [Google Scholar]
- Alcaro, A.; Huber, R.; Panksepp, J. Behavioral functions of the mesolimbic dopaminergic system: An affective neuroethological perspective. Brain Res. Rev. 2007, 56, 283–321. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bissonette, G.B.; Roesch, M.R. Development and function of the midbrain dopamine system: What we know and what we need to. Genes Brain Behav. 2016, 15, 62–73. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Juárez Olguín, H.; Calderon Guzman, D.; Hernandez Garcia, E.; Barragan Mejia, G. The role of dopamine and its dysfunction as a consequence of oxidative stress. Oxid. Med. Cell. Longev. 2016, 2016, 9730467. [Google Scholar] [CrossRef] [Green Version]
- Faro, L.R.F.; Oliveira, I.M.; Durán, R.; Alfonso, M. In vivo neurochemical characterization of clothianidin induced striatal dopamine release. Toxicology 2012, 302, 197–202. [Google Scholar] [CrossRef] [PubMed]
- Faro, L.R.F.; Tak-Kim, H.; Alfonso, M.; Durán, R. Clothianidin, a neonicotinoid insecticide, activates α4β2, α7 and muscarinic receptors to induce in vivo dopamine release from rat striatum. Toxicology 2019, 426, 152285. [Google Scholar] [CrossRef]
- Kawahata, I.; Yamakuni, T. Imidacloprid, a neonicotinoid insecticide, facilitates tyrosine hydroxylase transcription and phenylethanolamine N-methyltransferase mRNA expression to enhance catecholamine synthesis and its nicotine-evoked elevation in PC12D cells. Toxicology 2018, 394, 84–92. [Google Scholar] [CrossRef] [PubMed]
- Daubner, S.C.; Le, T.; Wang, S. Tyrosine hydroxylase and regulation of dopamine synthesis. Arch. Biochem. Biophys. 2011, 508, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Danbolt, N.C. Glutamate uptake. Prog. Neurobiol. 2001, 65, 1–105. [Google Scholar] [CrossRef]
- Li, C.T.; Yang, K.C.; Lin, W.C. Glutamatergic dysfunction and glutamatergic compounds for major psychiatric disorders: Evidence from clinical neuroimaging studies. Front. Psychiatry 2019, 9, 767. [Google Scholar] [CrossRef] [Green Version]
- Lewerenz, J.; Maher, P. Chronic glutamate toxicity in neurodegenerative diseases—What is the evidence? Front. Neurosci. 2015, 9, 469. [Google Scholar] [CrossRef]
- Ohgi, Y.; Futamura, T.; Hashimoto, K. Glutamate signaling in synaptogenesis and NMDA receptors as potential therapeutic targets for psychiatric disorders. Curr. Mol. Med. 2015, 15, 206–221. [Google Scholar] [CrossRef]
- Reiner, A.; Levitz, J. Glutamatergic signaling in the central nervous system: Ionotropic and metabotropic receptors in concert. Neuron 2018, 98, 1080–1098. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mann, J.J. Role of the serotonergic system in the pathogenesis of major depression and suicidal behavior. Neuropsychopharmacology 1999, 21, 99–105. [Google Scholar] [CrossRef] [Green Version]
- Štrac, D.Š.; Pivac, N.; Mück-Šeler, D. The serotonergic system and cognitive function. Transl. Neurosci. 2016, 7, 35–49. [Google Scholar] [CrossRef] [Green Version]
- Calvo-Flores Guzmán, B.; Vinnakota, C.; Govindpani, K.; Waldvogel, H.J.; Faull, R.L.; Kwakowsky, A. The GABAergic system as a therapeutic target for Alzheimer′s disease. J. Neurochem. 2018, 146, 649–669. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kilb, W. Development of the GABAergic system from birth to adolescence. Neuroscientist 2012, 18, 613–630. [Google Scholar] [CrossRef]
- Meltzer, H. Serotonergic dysfunction in depression. Br. J. Psychiatry Suppl. 1989, 155, 25–31. [Google Scholar] [CrossRef]
- Wassef, A.; Baker, J.; Kochan, L.D. GABA and schizophrenia: A review of basic science and clinical studies. J. Clin. Psychopharmacol. 2003, 23, 601–640. [Google Scholar] [CrossRef]
- Abdollahi, M.; Ranjbar, A.; Shadnia, S.; Nikfar, S.; Rezaiee, A. Pesticides and oxidative stress: A review. Med. Sci. Monit. 2004, 10, RA141–RA147. [Google Scholar]
- Agrawal, A.; Sharma, B. Pesticides induced oxidative stress in mammalian systems. Int. J. Biol. Med. Res. 2010, 1, 90–104. [Google Scholar]
- Hayashi, M. Oxidative stress in developmental brain disorders. Neuropathology 2009, 29, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Gilgun-Sherki, Y.; Melamed, E.; Offen, D. Oxidative stress induced-neurodegenerative diseases: The need for antioxidants that penetrate the blood brain barrier. Neuropharmacology 2001, 40, 959–975. [Google Scholar] [CrossRef]
- Gandhi, S.; Abramov, A.Y. Mechanism of oxidative stress in neurodegeneration. Oxid. Med. Cell. Longev. 2012, 2012, 428010. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salim, S. Oxidative stress and the central nervous system. J. Pharmacol. Exp. Ther. 2017, 360, 201–205. [Google Scholar] [CrossRef] [PubMed]
- Annabi, E.; Ben Salem, I.; Abid-Essefi, S. Acetamiprid, a neonicotinoid insecticide, induced cytotoxicity and genotoxicity in PC12 cells. Toxicol. Mech. Methods 2019, 29, 580–586. [Google Scholar] [CrossRef]
- Gasmi, S.; Kebieche, M.; Rouabhi, R.; Touahria, C.; Lahouel, A.; Lakroun, Z.; Henine, S.; Soulimani, R. Alteration of membrane integrity and respiratory function of brain mitochondria in the rats chronically exposed to a low dose of acetamiprid. Environ. Sci. Pollut. Res. Int. 2017, 24, 22258–22264. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.H.; Cha, M.; Lee, B.H. Neuroprotective Effect of Antioxidants in the Brain. Int. J. Mol. Sci. 2020, 21, 7152. [Google Scholar] [CrossRef]
- Pisoschi, A.M.; Pop, A. The role of antioxidants in the chemistry of oxidative stress: A review. Eur. J. Med. Chem. 2015, 97, 55–74. [Google Scholar] [CrossRef]
- Halliwell, B. Free radicals and antioxidants–quo vadis? Trends Pharmacol. Sci. 2011, 32, 125–130. [Google Scholar] [CrossRef] [PubMed]
- Poljsak, B.; Šuput, D.; Milisav, I. Achieving the balance between ROS and antioxidants: When to use the synthetic antioxidants. Oxid. Med. Cell. Longev. 2013, 2013, 956792. [Google Scholar] [CrossRef]
- Khan, J.Y.; Black, S.M. Developmental changes in murine brain antioxidant enzymes. Pediatr. Res. 2003, 54, 77–82. [Google Scholar] [CrossRef] [Green Version]
- Dwivedi, D.; Megha, K.; Mishra, R.; Mandal, P.K. Glutathione in brain: Overview of Its conformations, functions, biochemical characteristics, quantitation and potential therapeutic role in brain disorders. Neurochem. Res. 2020, 45, 1461–1480. [Google Scholar] [CrossRef]
- Nihei, H.; Kanemitsu, H.; Tamura, A.; Oka, H.; Sano, K. Cerebral uric acid, xanthine, and hypoxanthine after ischemia: The effect of allopurinol. Neurosurgery 1989, 25, 613–617. [Google Scholar] [CrossRef]
- Kaminsky, Y.; Kosenko, E. Brain purine metabolism and xanthine dehydrogenase/oxidase conversion in hyperammonemia are under control of NMDA receptors and nitric oxide. Brain Res. 2009, 1294, 193–201. [Google Scholar] [CrossRef] [PubMed]
- Kökoglu, E.; Belce, A.; Özyurt, E.; Tepeler, Z. Xanthine oxidase levels in human brain tumors. Cancer Lett. 1990, 50, 179–181. [Google Scholar] [CrossRef]
- Veljković, A.; Hadži-Dokić, J.; Sokolović, D.; Bašić, D.; Veličković-Janković, L.; Stojanović, M.; Popović, D.; Kocić, G. Xanthine Oxidase/Dehydrogenase Activity as a Source of Oxidative Stress in Prostate Cancer Tissue. Diagnostics 2020, 10, 668. [Google Scholar] [CrossRef]
- Ott, M.; Gogvadze, V.; Orrenius, S.; Zhivotovsky, B. Mitochondria, oxidative stress and cell death. Apoptosis 2007, 12, 913–922. [Google Scholar] [CrossRef] [PubMed]
- Guo, C.; Sun, L.; Chen, X.; Zhang, D. Oxidative stress, mitochondrial damage and neurodegenerative diseases. Neural Regen. Res. 2013, 8, 2003–2014. [Google Scholar] [CrossRef] [PubMed]
- Nicolson, G.L. Mitochondrial dysfunction and chronic disease: Treatment with natural supplements. Integr. Med. (Encinitas) 2014, 13, 35–43. [Google Scholar] [PubMed]
- Lakroun, Z.; Kebieche, M.; Lahouel, A.; Zama, D.; Desor, F.; Soulimani, R. Oxidative stress and brain mitochondria swelling induced by endosulfan and protective role of quercetin in rat. Environ. Sci. Pollut. Res. Int. 2015, 22, 7776–7781. [Google Scholar] [CrossRef]
- Aquilano, K.; Baldelli, S.; Rotilio, G.; Ciriolo, M.R. Role of nitric oxide synthases in Parkinson’s disease: A review on the antioxidant and anti-inflammatory activity of polyphenols. Neurochem. Res. 2008, 33, 2416–2426. [Google Scholar] [CrossRef]
- DiSabato, D.J.; Quan, N.; Godbout, J.P. Neuroinflammation: The devil is in the details. J. Neurochem. 2016, 139, 136–153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Norden, D.M.; Trojanowski, P.J.; Villanueva, E.; Navarro, E.; Godbout, J.P. Sequential activation of microglia and astrocyte cytokine expression precedes increased iba-1 or GFAP immunoreactivity following systemic immune challenge. Glia 2016, 64, 300–316. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ayoub, A.E.; Salm, A.K. Increased morphological diversity of microglia in the activated hypothalamic supraoptic nucleus. J. Neurosci. 2003, 23, 7759–7766. [Google Scholar] [CrossRef]
- Stratoulias, V.; Venero, J.L.; Tremblay, M.È.; Joseph, B. Microglial subtypes: Diversity within the microglial community. EMBO J. 2019, 38, e101997. [Google Scholar] [CrossRef] [PubMed]
- Zhou, B.; Zuo, Y.X.; Jiang, R.T. Astrocyte morphology: Diversity, plasticity, and role in neurological diseases. CNS Neurosci. Ther. 2019, 25, 665–673. [Google Scholar] [CrossRef] [PubMed]
- Blandini, F. Neural and immune mechanisms in the pathogenesis of Parkinson’s disease. J. Neuroimmune Pharmacol. 2013, 8, 189–201. [Google Scholar] [CrossRef]
- Kigerl, K.A.; Gensel, J.C.; Ankeny, D.P.; Alexander, J.K.; Donnelly, D.J.; Popovich, P.G. Identification of two distinct macrophage subsets with divergent effects causing either neurotoxicity or regeneration in the injured mouse spinal cord. J. Neurosci. 2009, 29, 13435–13444. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Akhmetzyanova, E.; Kletenkov, K.; Mukhamedshina, Y.; Rizvanov, A. Different approaches to modulation of microglia phenotypes after spinal cord injury. Front. Syst. Neurosci. 2019, 13, 37. [Google Scholar] [CrossRef] [Green Version]
- Franco, R.; Fernandez-Suarez, D. Alternatively activated microglia and macrophages in the central nervous system. Prog. Neurobiol. 2015, 131, 65–86. [Google Scholar] [CrossRef]
- Sonar, S.A.; Lal, G. The iNOS activity during an immune response controls the CNS pathology in experimental autoimmune encephalomyelitis. Front. Immunol. 2019, 10, 710. [Google Scholar] [CrossRef] [Green Version]
- Ghasemi, M.; Fatemi, A. Pathologic role of glial nitric oxide in adult and pediatric neuroinflammatory diseases. Neurosci. Biobehav. Rev. 2014, 45, 168–182. [Google Scholar] [CrossRef]
- Jaramillo, M.; Gowda, D.C.; Radzioch, D.; Olivier, M. Hemozoin increases IFN-γ-inducible macrophage nitric oxide generation through extracellular signal-regulated kinase-and NF-κB-dependent pathways. J. Immunol. 2003, 171, 4243–4253. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sheng, W.; Zong, Y.; Mohammad, A.; Ajit, D.; Cui, J.; Han, D.; Hamilton, J.L.; Simonyi, A.; Sun, A.Y.; Gu, Z.; et al. Pro-inflammatory cytokines and lipopolysaccharide induce changes in cell morphology, and upregulation of ERK1/2, iNOS and sPLA 2-IIA expression in astrocytes and microglia. J. Neuroinflamm. 2011, 8, 121. [Google Scholar] [CrossRef] [Green Version]
- Choi, J.Y.; Nam, S.A.; Jin, D.C.; Kim, J.; Cha, J.H. Expression and cellular localization of inducible nitric oxide synthase in lipopolysaccharide-treated rat kidneys. J. Histochem. Cytochem. 2012, 60, 301–315. [Google Scholar] [CrossRef] [PubMed]
- Jha, M.K.; Morrison, B.M. Glia-neuron energy metabolism in health and diseases: New insights into the role of nervous system metabolic transporters. Exp. Neurol. 2018, 309, 23–31. [Google Scholar] [CrossRef] [PubMed]
- Camandola, S.; Mattson, M.P. Brain metabolism in health, aging, and neurodegeneration. EMBO J. 2017, 36, 1474–1492. [Google Scholar] [CrossRef] [PubMed]
- Watts, M.E.; Pocock, R.; Claudianos, C. Brain energy and oxygen metabolism: Emerging role in normal function and disease. Front. Mol. Neurosci. 2018, 11, 216. [Google Scholar] [CrossRef] [PubMed]
- Fotopoulou, E.; Lykogianni, M.; Papadimitriou, E.; Mavrikou, S.; Machera, K.; Kintzios, S.; Thomaidou, D.; Aliferis, K.A. Mining the effect of the neonicotinoids imidacloprid and clothianidin on the chemical homeostasis and energy equilibrium of primary mouse neural stem/progenitor cells using metabolomics. Pestic. Biochem. Physiol. 2020, 168, 104617. [Google Scholar] [CrossRef] [PubMed]
- Bélanger, M.; Allaman, I.; Magistretti, P.J. Brain energy metabolism: Focus on astrocyte-neuron metabolic cooperation. Cell Metab. 2011, 14, 724–738. [Google Scholar] [CrossRef] [Green Version]
- Kempermann, G.; Song, H.; Gage, F.H. Neurogenesis in the adult hippocampus. Cold Spring Harb. Perspect. Biol. 2015, 7, a018812. [Google Scholar] [CrossRef] [Green Version]
- Toda, T.; Parylak, S.L.; Linker, S.B.; Gage, F.H. The role of adult hippocampal neurogenesis in brain health and disease. Mol. Psychiatry 2019, 24, 67–87. [Google Scholar] [CrossRef] [PubMed]
- Baptista, P.; Andrade, J.P. Adult hippocampal neurogenesis: Regulation and possible functional and clinical correlates. Front. Neuroanat. 2018, 12, 44. [Google Scholar] [CrossRef] [PubMed]
- Mu, Y.; Gage, F.H. Adult hippocampal neurogenesis and its role in Alzheimer’s disease. Mol. Neurodegener. 2011, 6, 85. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Becerra, L.V.; Pimienta, H.J. Apoptosis neuronal: La diversidad de señales y de tipos celulares. Colomb. Med. 2009, 40, 124–133. [Google Scholar]
- Fricker, M.; Tolkovsky, A.M.; Borutaite, V.; Coleman, M.; Brown, G.C. Neuronal cell death. Physiol. Rev. 2018, 98, 813–880. [Google Scholar] [CrossRef] [PubMed]
- Méndez-Armenta, M.; Nava-Ruíz, C.; Juárez-Rebollar, D.; Rodríguez-Martínez, E.; Yescas Gómez, P. Oxidative stress associated with neuronal apoptosis in experimental models of epilepsy. Oxid. Med. Cell. Longev. 2014, 2014, 293689. [Google Scholar] [CrossRef]
- Hollville, E.; Romero, S.E.; Deshmukh, M. Apoptotic cell death regulation in neurons. FEBS J. 2019, 286, 3276–3298. [Google Scholar] [CrossRef] [Green Version]
- Blanquie, O.; Yang, J.W.; Kilb, W.; Sharopov, S.; Sinning, A.; Luhmann, H.J. Electrical activity controls area-specific expression of neuronal apoptosis in the mouse developing cerebral cortex. eLife 2017, 6, e27696. [Google Scholar] [CrossRef]
- Hirano, T.; Minagawa, S.; Furusawa, Y.; Yunoki, T.; Ikenaka, Y.; Yokoyama, T.; Hoshi, N.; Tabuchi, Y. Growth and neurite stimulating effects of the neonicotinoid pesticide clothianidin on human neuroblastoma SH-SY5Y cells. Toxicol. Appl. Pharmacol. 2019, 383, 114777. [Google Scholar] [CrossRef]
- Chuderland, D.; Seger, R. Calcium regulates ERK signaling by modulating its protein-protein interactions. Commun. Integr. Biol. 2008, 1, 4–5. [Google Scholar] [CrossRef] [Green Version]
- Cheng, P.; Alberts, I.; Li, X. The role of ERK1/2 in the regulation of proliferation and differentiation of astrocytes in developing brain. Int. J. Dev. Neurosci. 2013, 31, 783–789. [Google Scholar] [CrossRef] [PubMed]
- Tomizawa, M.; Casida, J.E. Desnitro-imidacloprid activates the extracellular signal-regulated kinase cascade via the nicotinic receptor and intracellular calcium mobilization in N1E-115 cells. Toxicol. Appl. Pharmacol. 2002, 184, 180–186. [Google Scholar] [CrossRef]
- Cheng, L.; Lu, Y.; Zhao, Z.; Hoogenboom, R.L.; Zhang, Q.; Liu, X.; Song, W.; Guan, S.; Song, W.; Rao, Q. Assessing the combined toxicity effects of three neonicotinoid pesticide mixtures on human neuroblastoma SK-N-SH and lepidopteran Sf-9 cells. Food Chem. Toxicol. 2020, 145, 111632. [Google Scholar] [CrossRef] [PubMed]
- Şenyildiz, M.; Kilinc, A.; Ozden, S. Investigation of the genotoxic and cytotoxic effects of widely used neonicotinoid insecticides in HepG2 and SH-SY5Y cells. Toxicol. Ind. Health 2018, 34, 375–383. [Google Scholar] [CrossRef]
- Mucchietto, V.; Fasoli, F.; Pucci, S.; Moretti, M.; Benfante, R.; Maroli, A.; Di Lascio, S.; Bolchi, C.; Pallavicini, M.; Dowell, C.; et al. α9-and α7-containing receptors mediate the pro-proliferative effects of nicotine in the A549 adenocarcinoma cell line. Br. J. Pharmacol. 2018, 175, 1957–1972. [Google Scholar] [CrossRef] [PubMed]
- Wong, H.P.S.; Yu, L.; Lam, E.K.Y.; Tai, E.K.K.; Wu, W.K.K.; Cho, C.H. Nicotine promotes cell proliferation via α7-nicotinic acetylcholine receptor and catecholamine-synthesizing enzymes-mediated pathway in human colon adenocarcinoma HT-29 cells. Toxicol. Appl. Pharmacol. 2007, 221, 261–267. [Google Scholar] [CrossRef]
- Vinod, K.V.; Srikant, S.; Thiruvikramaprakash, G.; Dutta, T.K. A fatal case of thiacloprid poisoning. Am. J. Emerg. Med. 2015, 33, 310–e5. [Google Scholar] [CrossRef]
- Marfo, J.T.; Fujioka, K.; Ikenaka, Y.; Nakayama, S.M.; Mizukawa, H.; Aoyama, Y.; Ishizuka, M.; Taira, K. Relationship between urinary N-desmethyl-acetamiprid and typical symptoms including neurological findings: A prevalence case-control study. PLoS ONE 2015, 10, e0142172. [Google Scholar] [CrossRef] [Green Version]
- Agarwal, R.; Srinivas, R. Severe neuropsychiatric manifestations and rhabdomyolysis in a patient with imidacloprid poisoning. Am. J. Emerg. Med. 2007, 25, 844–845. [Google Scholar] [CrossRef]
- Huang, N.C.; Lin, S.L.; Chou, C.H.; Hung, Y.M.; Chung, H.M.; Huang, S.T. Fatal ventricular fibrillation in a patient with acute imidacloprid poisoning. Am. J. Emerg. Med. 2006, 24, 883–885. [Google Scholar] [CrossRef]
- Wu, I.W.; Lin, J.L.; Cheng, E.T. Acute poisoning with the neonicotinoid insecticide imidacloprid in N-methyl pyrrolidone. J. Toxicol. Clin. Toxicol. 2001, 39, 617–621. [Google Scholar] [CrossRef]
- Gunier, R.B.; Bradman, A.; Harley, K.G.; Kogut, K.; Eskenazi, B. Prenatal residential proximity to agricultural pesticide use and IQ in 7-year-old children. Environ. Health Perspect. 2017, 125, 057002. [Google Scholar] [CrossRef] [Green Version]
- Keil, A.P.; Daniels, J.L.; Hertz-Picciotto, I. Autism spectrum disorder, flea and tick medication, and adjustments for exposure misclassification: The CHARGE (CHildhood Autism Risks from Genetics and Environment) case–control study. Environ. Health 2014, 13, 3. [Google Scholar] [CrossRef] [Green Version]
- National Research Council (US) Committee on Developmental Toxicology. Scientific Frontiers in Developmental Toxicology and Risk Assessment; National Academies Press: Washington, DC, USA, 2000. [Google Scholar]
- Shelton, J.F.; Geraghty, E.M.; Tancredi, D.J.; Delwiche, L.D.; Schmidt, R.J.; Ritz, B.; Hansen, R.L.; Hertz-Picciotto, I. Neurodevelopmental disorders and prenatal residential proximity to agricultural pesticides: The CHARGE study. Environ. Health Perspect. 2014, 122, 1103–1109. [Google Scholar] [CrossRef] [Green Version]
- Landrigan, P.J.; Goldman, L.R. Protecting children from pesticides and other toxic chemicals. J. Expo. Sci. Environ. Epidemiol. 2011, 21, 119–120. [Google Scholar] [CrossRef]
- Pascale, A.; Laborde, A. Impact of pesticide exposure in childhood. Rev. Environ. Health 2020, 35, 221–227. [Google Scholar] [CrossRef] [PubMed]
- Berheim, E.H.; Jenks, J.A.; Lundgren, J.G.; Michel, E.S.; Grove, D.; Jensen, W.F. Effects of neonicotinoid insecticides on physiology and reproductive characteristics of captive female and fawn white-tailed deer. Sci. Rep. 2019, 9, 4534. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, C.H.; Lin, C.L.; Wang, S.E.; Lu, C.W. Effects of imidacloprid, a neonicotinoid insecticide, on the echolocation system of insectivorous bats. Pestic. Biochem. Physiol. 2020, 163, 94–101. [Google Scholar] [CrossRef] [PubMed]
- Fenton, M.B. Echolocation: Implications for ecology and evolution of bats. Q. Rev. Biol. 1984, 59, 33–53. [Google Scholar] [CrossRef]
- Jakobsen, L.; Brinkløv, S.; Surlykke, A. Intensity and directionality of bat echolocation signals. Front. Physiol. 2013, 4, 89. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wenstrup, J.J.; Portfors, C.V. Neural processing of target distance by echolocating bats: Functional roles of the auditory midbrain. Neurosci. Biobehav. Rev. 2011, 35, 2073–2083. [Google Scholar] [CrossRef] [Green Version]
- Wang, C.; Youle, R.J. The role of mitochondria in apoptosis. Annu. Rev. Genet. 2009, 43, 95–118. [Google Scholar] [CrossRef] [Green Version]
- Korsmeyer, S.J.; Wei, M.C.; Saito, M.T.; Weiler, S.; Oh, K.J.; Schlesinger, P.H. Pro-apoptotic cascade activates BID, which oligomerizes BAK or BAX into pores that result in the release of cytochrome c. Cell Death Differ. 2000, 7, 1166–1173. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Liu, J.; Chen, S.; Liu, J.; Liu, L.; Liu, G.; Wang, F.; Jiang, W.; Zhang, C.; Wang, S.; et al. Caspase-12 is involved in stretch-induced apoptosis mediated endoplasmic reticulum stress. Apoptosis 2016, 21, 432–442. [Google Scholar] [CrossRef]
- Szegezdi, E.V.A.; Fitzgerald, U.N.A.; Samali, A. Caspase-12 and ER-stress-mediated apoptosis: The story so far. Ann. N. Y. Acad. Sci. 2003, 1010, 186–194. [Google Scholar] [CrossRef] [PubMed]
- Ndebele, K.; Gona, P.; Jin, T.G.; Benhaga, N.; Chalah, A.; Degli-Esposti, M.; Khosravi-Far, R. Tumor necrosis factor (TNF)-related apoptosis-inducing ligand (TRAIL) induced mitochondrial pathway to apoptosis and caspase activation is potentiated by phospholipid scramblase-3. Apoptosis 2008, 13, 845–856. [Google Scholar] [CrossRef] [Green Version]
- Hams, N.; Padmanarayana, M.; Qiu, W.; Johnson, C.P. Otoferlin is a multivalent calcium-sensitive scaffold linking SNAREs and calcium channels. Proc. Natl. Acad. Sci. USA 2017, 114, 8023–8028. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schug, N.; Braig, C.; Zimmermann, U.; Engel, J.; Winter, H.; Ruth, P.; Blin, N.; Pfister, M.; Kalbacher, H.; Knipper, M. Differential expression of otoferlin in brain, vestibular system, immature and mature cochlea of the rat. Eur. J. Neurosci. 2006, 24, 3372–3380. [Google Scholar] [CrossRef]
- Shen, Y.Y.; Liang, L.; Li, G.S.; Murphy, R.W.; Zhang, Y.P. Parallel evolution of auditory genes for echolocation in bats and toothed whales. PLoS Genet. 2012, 8, e1002788. [Google Scholar] [CrossRef] [Green Version]
- Dallos, P. Cochlear amplification, outer hair cells and prestin. Curr. Opin. Neurobiol. 2008, 18, 370–376. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morell, M.; Vogl, A.W.; IJsseldijk, L.L.; Piscitelli-Doshkov, M.; Tong, L.; Ostertag, S.; Ferreira, M.; Fraija-Fernandez, N.; Colegrove, K.M.; Puel, J.L.; et al. Echolocating whales and bats express the motor protein prestin in the inner ear: A potential marker for hearing loss. Front. Vet. Sci. 2020, 7, 429. [Google Scholar] [CrossRef] [PubMed]
- Zheng, J.; Shen, W.; He, D.Z.; Long, K.B.; Madison, L.D.; Dallos, P. Prestin is the motor protein of cochlear outer hair cells. Nature 2000, 405, 149–155. [Google Scholar] [CrossRef]
- Staes, N.; Sherwood, C.C.; Wright, K.; De Manuel, M.; Guevara, E.E.; Marques-Bonet, T.; Krützen, M.; Massiah, M.; Hopkins, W.D.; Ely, J.J.; et al. FOXP2 variation in great ape populations offers insight into the evolution of communication skills. Sci. Rep. 2017, 7, 16866. [Google Scholar] [CrossRef] [Green Version]
- Li, G.; Wang, J.; Rossiter, S.J.; Jones, G.; Zhang, S. Accelerated FoxP2 evolution in echolocating bats. PLoS ONE 2007, 2, e900. [Google Scholar] [CrossRef]
- Yin, J.X.; Ruan, Y.N.; Liu, J.L.; Zhang, S.Y.; Racey, P. FoxP2 expression in an echolocating bat (Rhinolophus ferrumequinum): Functional implications. Mamm. Biol. 2017, 85, 24–29. [Google Scholar] [CrossRef]
- Zoli, M.; Pistillo, F.; Gotti, C. Diversity of native nicotinic receptor subtypes in mammalian brain. Neuropharmacology 2015, 96, 302–311. [Google Scholar] [CrossRef]
- Zoli, M.; Pucci, S.; Vilella, A.; Gotti, C. Neuronal and extraneuronal nicotinic acetylcholine receptors. Curr. Neuropharmacol. 2018, 16, 338–349. [Google Scholar] [CrossRef]
- Nair, L.R.; Liu, X. Targeting the α4β2-and α7-Subtypes of Nicotinic Acetylcholine Receptors for Smoking Cessation Medication Development. J. Addict. Res. Ther. 2019, 10, 381. [Google Scholar]
- Wooltorton, J.R.; Pidoplichko, V.I.; Broide, R.S.; Dani, J.A. Differential desensitization and distribution of nicotinic acetylcholine receptor subtypes in midbrain dopamine areas. J. Neurosci. 2003, 23, 3176–3185. [Google Scholar] [CrossRef] [PubMed]
- Zeid, D.; Kutlu, M.G.; Gould, T.J. Differential effects of nicotine exposure on the hippocampus across lifespan. Curr. Neuropharmacol. 2018, 16, 388–402. [Google Scholar] [CrossRef] [PubMed]
- Wojda, U.; Salinska, E.; Kuznicki, J. Calcium ions in neuronal degeneration. IUBMB Life 2008, 60, 575–590. [Google Scholar] [CrossRef]
- Bano, D.; Nicotera, P. Ca2+ signals and neuronal death in brain ischemia. Stroke 2007, 38, 674–676. [Google Scholar] [CrossRef] [Green Version]
- Giorgi, C.; Baldassari, F.; Bononi, A.; Bonora, M.; De Marchi, E.; Marchi, S.; Missiroli, S.; Patergnani, S.; Rimessi, A.; Suski, J.M.; et al. Mitochondrial Ca2+ and apoptosis. Cell Calcium 2012, 52, 36–43. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morrissey, C.A.; Mineau, P.; Devries, J.H.; Sanchez-Bayo, F.; Liess, M.; Cavallaro, M.C.; Liber, K. Neonicotinoid contamination of global surface waters and associated risk to aquatic invertebrates: A review. Environ. Int. 2015, 74, 291–303. [Google Scholar] [CrossRef] [PubMed]
- Sánchez-Bayo, F.; Goka, K.; Hayasaka, D. Contamination of the aquatic environment with neonicotinoids and its implication for ecosystems. Front. Environ. Sci. 2016, 4, 71. [Google Scholar] [CrossRef]
- Zhang, P.; Ren, C.; Sun, H.; Min, L. Sorption, desorption and degradation of neonicotinoids in four agricultural soils and their effects on soil microorganisms. Sci. Total Environ. 2018, 615, 59–69. [Google Scholar] [CrossRef]
- Han, W.; Tian, Y.; Shen, X. Human exposure to neonicotinoid insecticides and the evaluation of their potential toxicity: An overview. Chemosphere 2018, 192, 59–65. [Google Scholar] [CrossRef] [PubMed]
- Ichikawa, G.; Kuribayashi, R.; Ikenaka, Y.; Ichise, T.; Nakayama, S.M.; Ishizuka, M.; Taira, K.; Fujioka, K.; Sairenchi, T.; Kobashi, G.; et al. LC-ESI/MS/MS analysis of neonicotinoids in urine of very low birth weight infants at birth. PLoS ONE 2019, 14, e0219208. [Google Scholar] [CrossRef]
- Kavvalakis, M.P.; Tzatzarakis, M.N.; Theodoropoulou, E.P.; Barbounis, E.G.; Tsakalof, A.K.; Tsatsakis, A.M. Development and application of LC–APCI–MS method for biomonitoring of animal and human exposure to imidacloprid. Chemosphere 2013, 93, 2612–2620. [Google Scholar] [CrossRef]
- Ohno, S.; Ikenaka, Y.; Onaru, K.; Kubo, S.; Sakata, N.; Hirano, T.; Mantani, Y.; Yokoyama, T.; Takahashi, K.; Kato, K.; et al. Quantitative elucidation of maternal-to-fetal transfer of neonicotinoid pesticide clothianidin and its metabolites in mice. Toxicol. Lett. 2020, 322, 32–38. [Google Scholar] [CrossRef]
- Ikenaka, Y.; Miyabara, Y.; Ichise, T.; Nakayama, S.; Nimako, C.; Ishizuka, M.; Tohyama, C. Exposures of children to neonicotinoids in pine wilt disease control areas. Environ. Toxicol. Chem. 2019, 38, 71–79. [Google Scholar] [CrossRef] [PubMed]
- Casida, J.E. Neonicotinoid metabolism: Compounds, substituents, pathways, enzymes, organisms, and relevance. J. Agric. Food Chem. 2011, 59, 2923–2931. [Google Scholar] [CrossRef] [PubMed]
- Dwyer, J.B.; McQuown, S.C.; Leslie, F.M. The dynamic effects of nicotine on the developing brain. Pharmacol. Ther. 2009, 122, 125–139. [Google Scholar] [CrossRef] [PubMed] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Species | Dose and Time Exposition | Objectives | Results | Reference |
|---|---|---|---|---|
| Wistar rats | IMI: 0.5, 2 or 8 mg/kg/day orally for 3 months | Evaluate the effects of different doses of IMI on learning and memory in infant and adult rats |
| [11] |
| Wistar rats | IMI: 0.06, 0.8 or 2.25 mg/kg/day orally for 28 days | Investigate the effects of IMI on cholinesterase activities, oxidative stress biomarkers and primary DNA damage in blood and brain tissue |
| [15] |
| Wistar rats | IMI: 20 mg/kg orally Single dose | Evaluate pharmacokinetic and pharmacodynamic responses after single oral exposure |
| [59] |
| Wistar rats | IMI: 10 or 20 mg/kg/day orally for 60 days | Assess the alterations induced by IMI in the biochemical, histopathological and protein profile in plasma and brain |
| [117] |
| Wistar rats | IMI: 1 mg/kg/day orally for 30 days | Assess the effects of chronic exposure to IMI on the induction of oxidative stress and inflammation |
| [128] |
| Sprague-Dawley rats | IMI: 1 mg/kg/day orally for 60 days | Evaluate the effects of IMI on neurobehavioral performance, oxidative stress and the induction of apoptosis in the brain of adult or adolescent rats |
| [70] |
| Sprague-Dawley rats | IMI: 0.5 or 1 mg/kg/day orally for 60 days | Study the effects of IMI on stress by assessing cortisone and catecholamine levels, with a focus on behavioral alterations |
| [71] |
| CD-1 mice | IMI: 0.5 mg/kg/day infusion through an osmotic pump From GD4 until PND21 | Assess the effects of IMI after an intrauterine and early postnatal exposure |
| [57] |
| KM mice | IMI: 5 or 20 mg/kg/day orally for 28 days | Examine the histopathological, biochemical and metabolic alterations induced by IMI in the hippocampus and liver |
| [112] |
| Wistar rats | ACE: 10, 20 or 40 mg/kg/day orally for 28 days | Investigate the effect of ACE on spatial memory and the vulnerability of the hippocampal glutamatergic system |
| [60] |
| Wistar rats | ACE: 3.14 mg/kg/day orally for 6 months | Assess the effects of ACE on membrane integrity and mitochondrial potential |
| [156] |
| A/J mice | ACE: 71 or 710 μg/g/day orally for 3 and 7 days | Investigate the accumulation of ACE and expression of nAChRs in different areas of the brain |
| [61] |
| ICR mice | ACE: 5 mg/kg/day orally. From GD6 until GD18 | Evaluate the effects of repeated maternal exposure to ACE on the neurodevelopment of the offspring |
| [65] |
| ICR mice | ACE, IMI: 5 mg/kg/day orally From PND12 until PND26 | Evaluate the effects of ACE and IMI exposure on neurogenesis and microglial profiles in the dentate gyrus of the developing hippocampus |
| [66] |
| Sprague-Dawley rats | CLO: 3.5 mM by local administration through a microdialysis probe | Determine the neurochemical effects and mechanisms of action of CLO on striatal dopamine release |
| [134] |
| Sprague-Dawley rats | CLO: 150 or 300 μmol by local administration through a microdialysis probe | Evaluate the role of some subtypes of nAChRs and mAChRs in CLO-induced striatal dopamine release |
| [135] |
| CD-1 mice | CLO: 0.003%, 0.006% or 0.012% orally. From 5 weeks of age of the F0 generation to 11 weeks of age of the F1 generation | Assess the effects of CLO exposure on reproduction and behavior over different generations |
| [68] |
| CD-1 mice | CLO: 0.002%, 0.006% or 0.018% orally. Gestation and lactation periods | Assess the neurobehavioral effects of maternal exposure to CLO |
| [69] |
| C57BL/6J mice | CLO: 5 mg/kg orally Single dose | Investigate the role of aging in CLO-induced behavioral effects |
| [58] |
| C57BL/6N mice | CLO: 5 or 50 mg/kg orally Single dose | Evaluate the neurobehavioral effects of CLO and explore the brain regions targeted by neonicotinoids in mammals |
| [78] |
| C57BL/6N mice | DIN: 100, 500 or 2500 mg/kg/day orally for 6 weeks | Analyze the biochemical and behavioral effects of DIN exposure during the peripubertal period on the nigrostriatal pathway |
| [74] |
| C57BL/6NCrSlc mice | DIN: 100, 500 or 2500 mg/kg/day orally for 5 weeks | Investigate the relationship between subchronic DIN exposure and a depression-related phenotype |
| [87] |
| Cellular Line | Dose and Time of Exposure | Objective | Results | Reference |
|---|---|---|---|---|
| Neuron-enriched cultures from neonatal rat cerebellum | ACE, IMI: 1 μM for 14 days | Examine the neurotoxic effects of long-term and low-dose exposure on cultures enriched with cerebellar neurons |
| [64] |
| Primary cultures of cerebellar neurons from neonatal rats | ACE, IMI: 1, 10 or 100 µM | Determine the effects of the two NNs on neuronal nAChRs and compare their effects with nicotine |
| [120] |
| PC12 cells | IMI, ACE, CLO, TMX: 1–100 μM for 5 days | Evaluate the neurotoxic effects on the development of several commonly used pesticides |
| [63] |
| PC12 cells | IMI: 1, 3, 30 or 100 μM for 24 and 48 h | Assess the effects of IMI on the catecholaminergic function of chromaffin cells |
| [136] |
| PC12 cells | ACE: 100–700 μM for 24 h | Investigate the toxic effects of ACE on PC12 cells |
| [155] |
| NSPC y N2a | IMI, CLO: 0–4000 μΜ for 48 h | Investigate the toxic effects and metabolic changes induced by IMI, CLO and their mixture in cell cultures |
| [189] |
| Type of Study | Toxic Agent | Exposure Mode | Results | Reference |
|---|---|---|---|---|
| Case study | THI | Oral (attempted suicide) |
| [208] |
| Case-control study | ACE, IMI, CLO, NIT, THI, and TMX | Consumption of food with pesticides |
| [209] |
| Cohort study | IMI | Residential exposure |
| [213] |
| Case-control study | IMI | Use of IMI on pets |
| [214] |
| Cellular Line | Dose and Time of Exposure | Objective | Results | Reference |
|---|---|---|---|---|
| SH-SY5Y | CLO: 1–100 μM for 24 h | Assess whether CLO could affect the structure or function of the human nervous system |
| [200] |
| SK-N-SH | IMI, ACE, TMX: 10–2000 mg/L for 24 h | Study the toxicity of NNs alone or in combination |
| [204] |
| SH-SY5Y | IMI, ACE, CLO, THI, TMX: 0.05–4 mM for 24 and 48 h | Investigate the possible effects of common NN insecticides on cytotoxicity and DNA damage |
| [205] |
| Species | Dose and Time Exposition | Objectives | Results | Reference |
|---|---|---|---|---|
| Deer (Odocoileus virginianus) | IMI: 1.500, 3.000 or 15.000 ng/L orally From May to October | Assess the toxic effects of IMI in adult female white-tailed deer |
| [219] |
| Bat (Hipposiderosarmiger terasensis) | IMI: 20 mg/kg orally for 5 days | Compare spatial memory of bats before and after chronic treatment with a low dose of IMI |
| [111] |
| Bat (Hipposideros armiger terasensis) | IMI: 0.5 mg orally for 5 days | Examine whether IMI toxicity can interfere with the echolocation system of bats |
| [220] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Costas-Ferreira, C.; Faro, L.R.F. Neurotoxic Effects of Neonicotinoids on Mammals: What Is There beyond the Activation of Nicotinic Acetylcholine Receptors?—A Systematic Review. Int. J. Mol. Sci. 2021, 22, 8413. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22168413
Costas-Ferreira C, Faro LRF. Neurotoxic Effects of Neonicotinoids on Mammals: What Is There beyond the Activation of Nicotinic Acetylcholine Receptors?—A Systematic Review. International Journal of Molecular Sciences. 2021; 22(16):8413. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22168413
Chicago/Turabian StyleCostas-Ferreira, Carmen, and Lilian R. F. Faro. 2021. "Neurotoxic Effects of Neonicotinoids on Mammals: What Is There beyond the Activation of Nicotinic Acetylcholine Receptors?—A Systematic Review" International Journal of Molecular Sciences 22, no. 16: 8413. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22168413