
C-Type Natriuretic Peptide Ameliorates Vascular Injury and Improves Neurological Outcomes in Neonatal Hypoxic-Ischemic Brain Injury in Mice


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
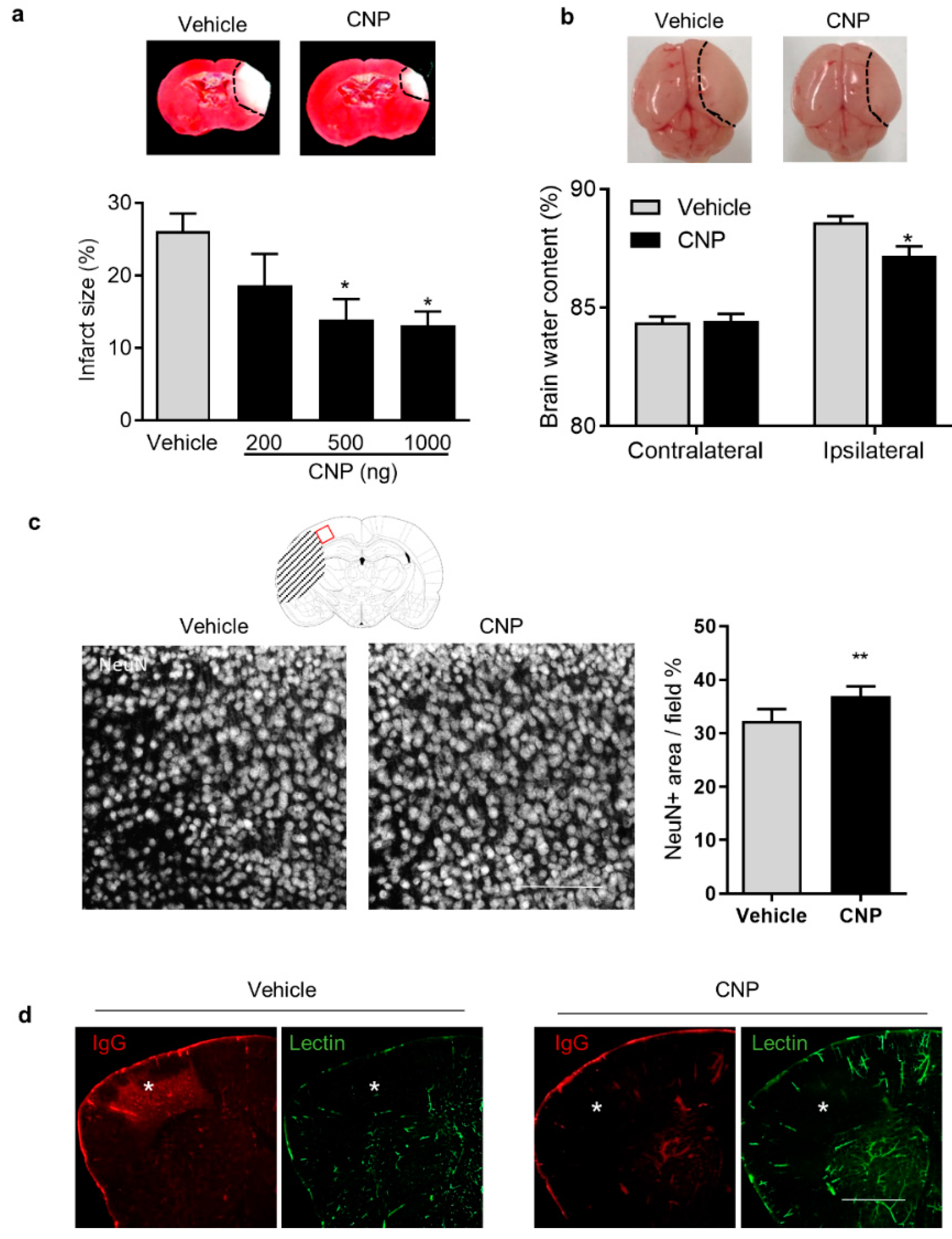
2.1. Recombinant CNP Treatment Decreased Brain Infract, Cerebral Edema and IgG Leakage in Neonatal HI Brain Injury
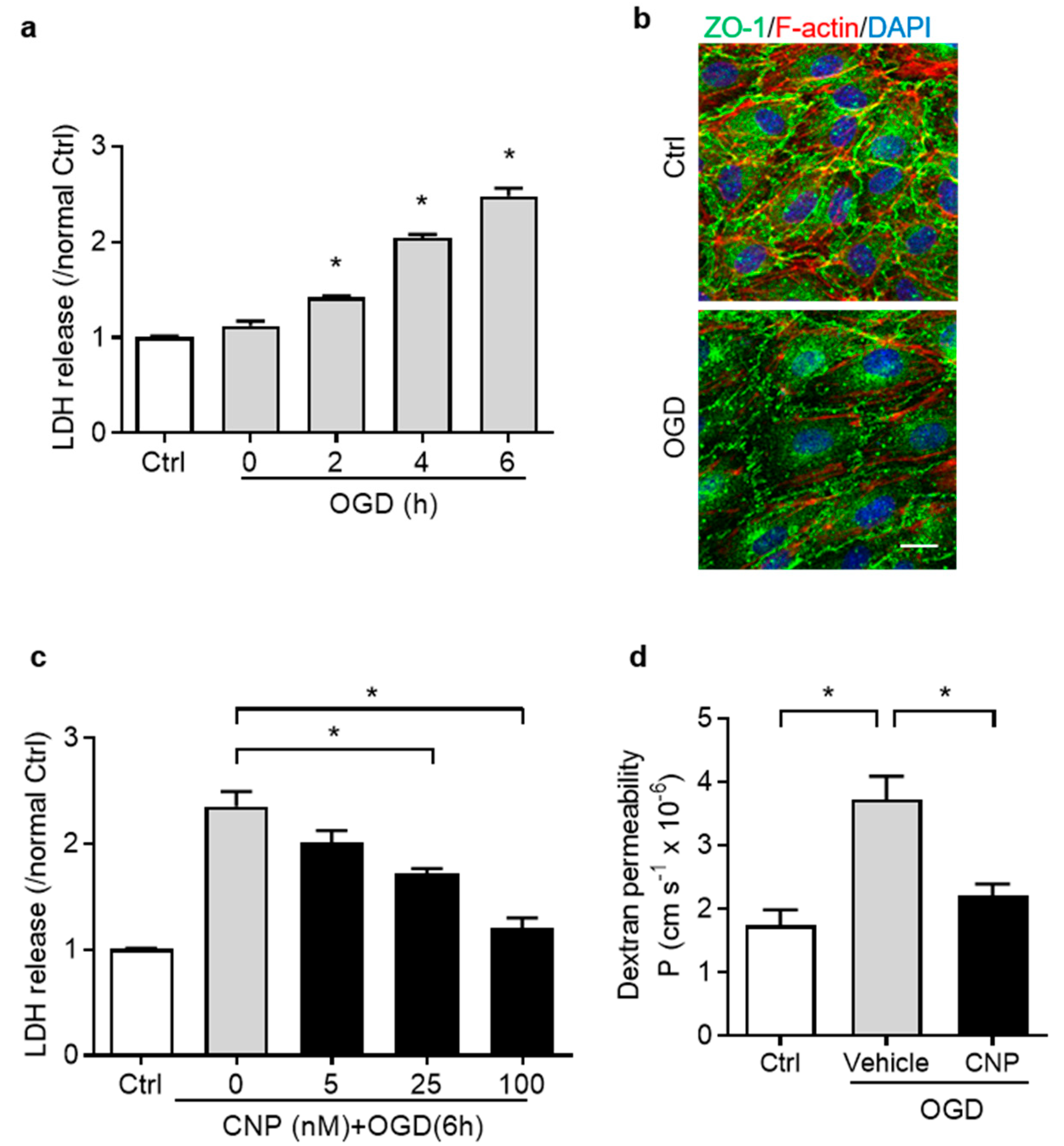
2.2. Recombinant CNP Protected Brain Endothelial Cells from Oxygen–Glucose Deprivation Injury
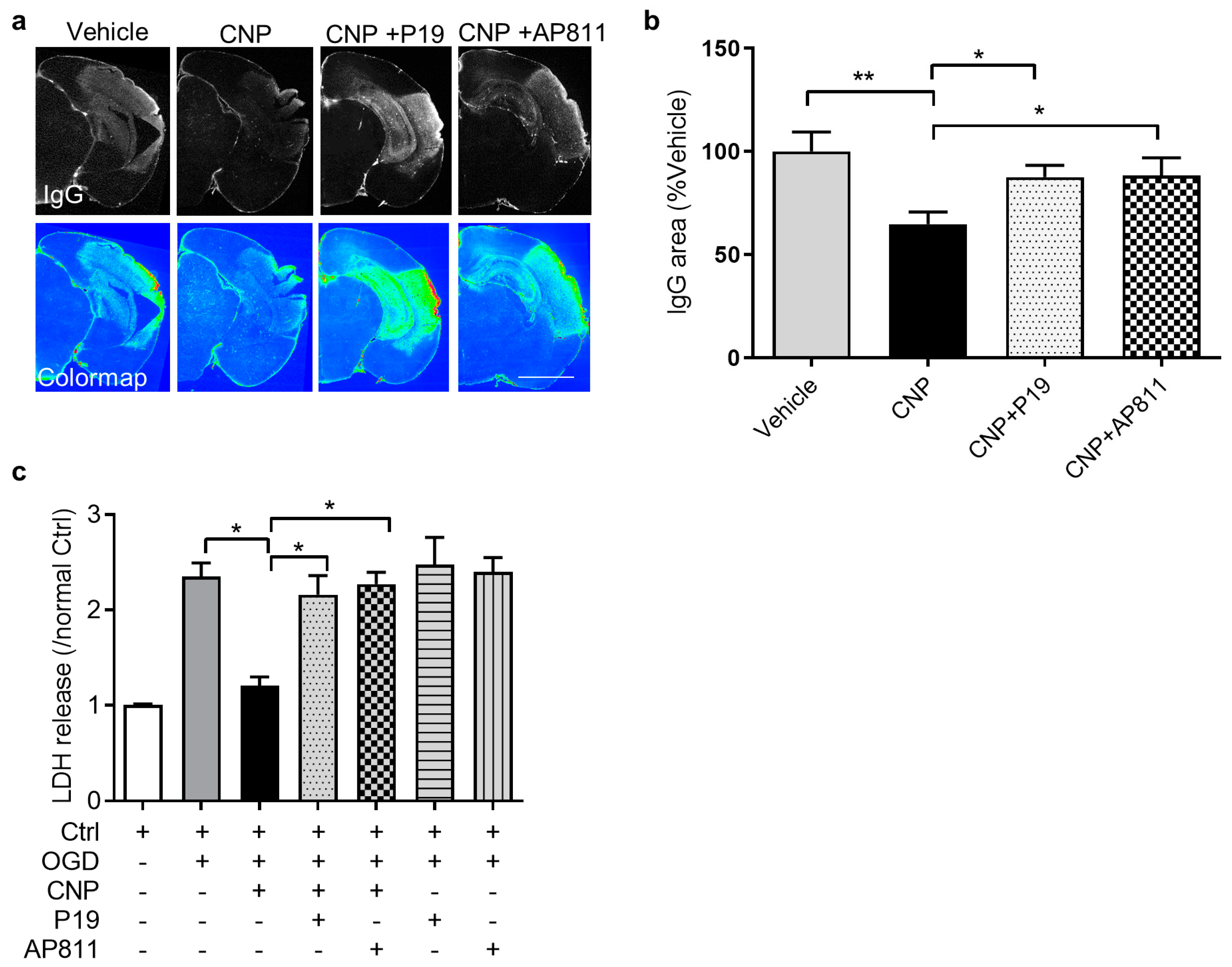
2.3. Both NPR2 and NPR3 Were Involved in the Endothelial Protective Effect of CNP In Vitro
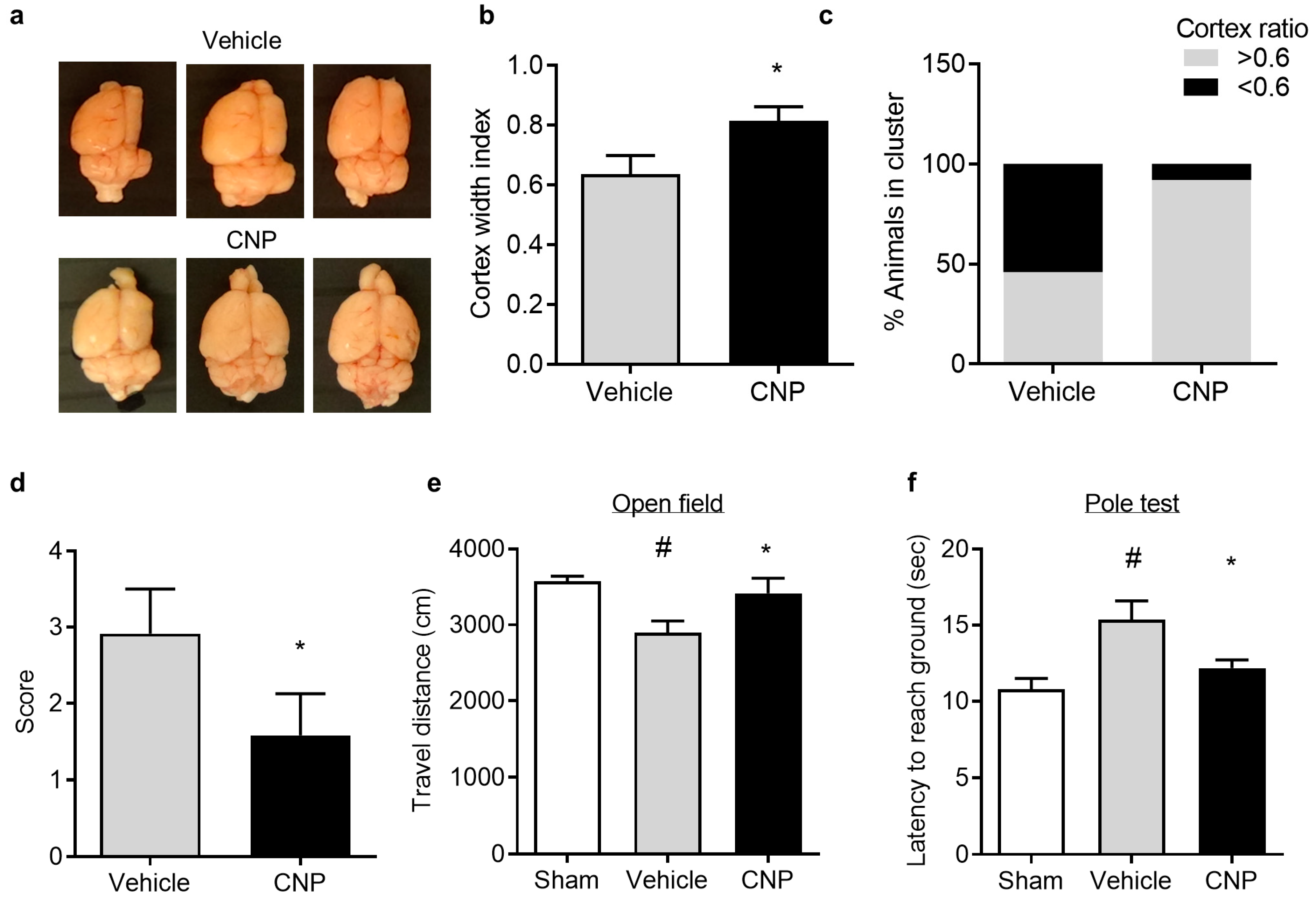
2.4. Recombinant CNP Reduced Cerebral Atrophy and Improved Neurological Outcome in Mice with Hypoxic-Ischemic Encephalopathy
3. Discussion
4. Material and Methods
4.1. Animals and Surgical Procedures
4.2. Intracerebroventricular Injection
4.3. Measurement of Brain Infarct Size
4.4. Brain Water Content Assessment
4.5. Neurological Function Assessment and Brain Atrophy Assay
4.6. Lectin Infusion and Immunofluorescence Staining
4.7. Oxygen–Glucose Deprivation (OGD) and Lactate Dehydrogenase (LDH) Assay
4.8. Dextran Permeability Assessment
4.9. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| BBB | blood–brain barrier |
| CNP | C-type natriuretic peptide |
| HI | hypoxic ischemic |
| HIE | hypoxic-ischemic encephalopathy |
| IgG | immunoglobin G |
| LDH | lactate dehydrogenase |
| NPR2 | natriuretic peptide receptor 2 |
| NPR3 | natriuretic peptide receptor 3 |
| OGD | oxygen glucose deprivation |
| TTC | 2,3,5-triphenyltetrazolium chloride |
| ZO-1 | Zonula occludens-1 |
References
- Allen, K.A.; Brandon, D.H. Hypoxic Ischemic Encephalopathy: Pathophysiology and Experimental Treatments. Newborn Infant Nurs Rev. 2011, 11, 125–133. [Google Scholar] [CrossRef] [Green Version]
- Ferriero, D.M. Neonatal brain injury. N. Engl. J. Med. 2004, 351, 1985–1995. [Google Scholar] [CrossRef]
- Hsu, Y.C.; Chang, Y.C.; Lin, Y.C.; Sze, C.I.; Huang, C.C.; Ho, C.J. Cerebral microvascular damage occurs early after hypoxia-ischemia via nNOS activation in the neonatal brain. J. Cereb. Blood Flow Metab. 2014, 34, 668–676. [Google Scholar] [CrossRef] [Green Version]
- Kumar, A.; Mittal, R.; Khanna, H.D.; Basu, S. Free radical injury and blood-brain barrier permeability in hypoxic-ischemic encephalopathy. Pediatrics 2008, 122, e722–e727. [Google Scholar] [CrossRef] [PubMed]
- Ek, C.J.; D’Angelo, B.; Baburamani, A.A.; Lehner, C.; Leverin, A.L.; Smith, P.L.; Nilsson, H.; Svedin, P.; Hagberg, H.; Mallard, C. Brain barrier properties and cerebral blood flow in neonatal mice exposed to cerebral hypoxia-ischemia. J. Cereb. Blood Flow Metab. 2015, 35, 818–827. [Google Scholar] [CrossRef] [Green Version]
- Lee, W.L.A.; Michael-Titus, A.T.; Shah, D.K. Hypoxic-Ischaemic Encephalopathy and the Blood-Brain Barrier in Neonates. Dev. Neurosci. 2017, 39, 49–58. [Google Scholar] [CrossRef] [PubMed]
- Wang, N.; Yi, H.; Fang, L.; Jin, J.; Ma, Q.; Shen, Y.; Li, J.; Liang, S.; Xiong, J.; Li, Z.; et al. CD226 Attenuates Treg Proliferation via Akt and Erk Signaling in an EAE Model. Front. Immunol. 2020, 11, 1883. [Google Scholar] [CrossRef]
- Bashir, R.A.; Vayalthrikkovil, S.; Espinoza, L.; Irvine, L.; Scott, J.; Mohammad, K. Prevalence and Characteristics of Intracranial Hemorrhages in Neonates with Hypoxic Ischemic Encephalopathy. Am. J. Perinatol. 2018, 35, 676–681. [Google Scholar] [CrossRef]
- Williams, C.E.; Gunn, A.; Gluckman, P.D. Time course of intracellular edema and epileptiform activity following prenatal cerebral ischemia in sheep. Stroke 1991, 22, 516–521. [Google Scholar] [CrossRef] [Green Version]
- Lupton, B.A.; Hill, A.; Roland, E.H.; Whitfield, M.F.; Flodmark, O. Brain swelling in the asphyxiated term newborn: Pathogenesis and outcome. Pediatrics 1988, 82, 139–146. [Google Scholar]
- Goasdoue, K.; Chand, K.K.; Miller, S.M.; Lee, K.M.; Colditz, P.B.; Wixey, J.A.; Bjorkman, S.T. Seizures Are Associated with Blood-Brain Barrier Disruption in a Piglet Model of Neonatal Hypoxic-Ischaemic Encephalopathy. Dev. Neurosci. 2019, 40, 1–16. [Google Scholar] [CrossRef]
- Ferrari, D.C.; Nesic, O.; Perez-Polo, J.R. Perspectives on neonatal hypoxia/ischemia-induced edema formation. Neurochem. Res. 2010, 35, 1957–1965. [Google Scholar] [CrossRef] [PubMed]
- Kaneko, T.; Shirakami, G.; Nakao, K.; Nagata, I.; Nakagawa, O.; Hama, N.; Suga, S.; Miyamoto, S.; Kubo, H.; Hirai, O.; et al. C-type natriuretic peptide (CNP) is the major natriuretic peptide in human cerebrospinal fluid. Brain Res. 1993, 612, 104–109. [Google Scholar] [CrossRef]
- Scotland, R.S.; Ahluwalia, A.; Hobbs, A.J. C-type natriuretic peptide in vascular physiology and disease. Pharmacol. Ther. 2005, 105, 85–93. [Google Scholar] [CrossRef]
- Langub, M.C., Jr.; Watson, R.E., Jr.; Herman, J.P. Distribution of natriuretic peptide precursor mRNAs in the rat brain. J. Comp. Neurol. 1995, 356, 183–199. [Google Scholar] [CrossRef] [PubMed]
- Potter, L.R.; Abbey-Hosch, S.; Dickey, D.M. Natriuretic peptides, their receptors, and cyclic guanosine monophosphate-dependent signaling functions. Endocr. Rev. 2006, 27, 47–72. [Google Scholar] [CrossRef]
- Vigne, P.; Frelin, C. C-type natriuretic peptide is a potent activator of guanylate cyclase in endothelial cells from brain microvessels. Biochem. Biophys. Res. Commun. 1992, 183, 640–644. [Google Scholar] [CrossRef]
- Yeung, V.T.; Ho, S.K.; Cockram, C.S.; Lee, C.M.; Nicholls, M.G. Activation of protein kinase C attenuates the cyclic GMP responses to C-type natriuretic peptide in cultured mouse astrocytes. FEBS Lett. 1992, 308, 301–304. [Google Scholar] [CrossRef] [Green Version]
- Levin, E.R.; Frank, H.J. Natriuretic peptides inhibit rat astroglial proliferation: Mediation by C receptor. Am. J. Physiol. 1991, 261 Pt 2, R453–R457. [Google Scholar] [CrossRef]
- Miyajima, M.; Arai, H.; Okuda, O.; Hishii, M.; Nakanishi, H.; Ishii, H.; Sato, K. Effect of C-type natriuretic peptide (CNP) on water channel aquaporin-4 (AQP4) expression in cultured astrocytes. Brain Res. Mol. Brain Res. 2004, 122, 109–115. [Google Scholar] [CrossRef]
- Zhao, Z.; Ma, L. Regulation of axonal development by natriuretic peptide hormones. Proc. Natl. Acad. Sci. USA 2009, 106, 18016–18021. [Google Scholar] [CrossRef] [Green Version]
- Espiner, E.A.; Dalrymple-Alford, J.C.; Prickett, T.C.; Alamri, Y.; Anderson, T.J. C-type natriuretic peptide in Parkinson’s disease: Reduced secretion and response to deprenyl. J. Neural. Transm. 2014, 121, 371–378. [Google Scholar] [CrossRef]
- Ma, Q.; Zhang, L. C-type natriuretic peptide functions as an innate neuroprotectant in neonatal hypoxic-ischemic brain injury in mouse via natriuretic peptide receptor 2. Exp. Neurol. 2018, 304, 58–66. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Z.; Sagare, A.P.; Ma, Q.; Halliday, M.R.; Kong, P.; Kisler, K.; Winkler, E.A.; Ramanathan, A.; Kanekiyo, T.; Bu, G.; et al. Central role for PICALM in amyloid-beta blood-brain barrier transcytosis and clearance. Nat. Neurosci. 2015, 18, 978–987. [Google Scholar] [CrossRef] [Green Version]
- Levin, E.R.; Gardner, D.G.; Samson, W.K. Natriuretic peptides. N. Engl. J. Med. 1998, 339, 321–328. [Google Scholar]
- Khambata, R.S.; Panayiotou, C.M.; Hobbs, A.J. Natriuretic peptide receptor-3 underpins the disparate regulation of endothelial and vascular smooth muscle cell proliferation by C-type natriuretic peptide. Br. J. Pharm. 2011, 164, 584–597. [Google Scholar] [CrossRef] [Green Version]
- Ma, J.; Yu, W.; Wang, Y.; Cao, G.; Cai, S.; Chen, X.; Yan, N.; Yuan, Y.; Zeng, H.; Fleenor, D.L.; et al. Neuroprotective effects of C-type natriuretic peptide on rat retinal ganglion cells. Invest. Ophthalmol. Vis. Sci. 2010, 51, 3544–3553. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moyes, A.J.; Khambata, R.S.; Villar, I.; Bubb, K.J.; Baliga, R.S.; Lumsden, N.G.; Xiao, F.; Gane, P.J.; Rebstock, A.S.; Worthington, R.J.; et al. Endothelial C-type natriuretic peptide maintains vascular homeostasis. J. Clin. Investig. 2014, 124, 4039–4051. [Google Scholar] [CrossRef] [PubMed]
- Sangaralingham, S.J.; Burnett, J.C., Jr. Relaxing With C-Type Natriuretic Peptide, the Guanylyl Cyclase B Receptor, and Pericytes. Circulation 2018, 138, 509–512. [Google Scholar] [CrossRef]
- Sangaralingham, S.J.; Chen, Y.; Burnett, J.C. C-type natriuretic peptide: The heart’s guardian angel. Eur. Heart J. 2020, 41, 1021–1023. [Google Scholar] [CrossRef] [PubMed]
- Bubb, K.J.; Aubdool, A.A.; Moyes, A.J.; Lewis, S.; Drayton, J.P.; Tang, O.; Mehta, V.; Zachary, I.C.; Abraham, D.J.; Tsui, J.; et al. Endothelial C-Type Natriuretic Peptide Is a Critical Regulator of Angiogenesis and Vascular Remodeling. Circulation 2019, 139, 1612–1628. [Google Scholar] [CrossRef]
- Klinger, J.R.; Warburton, R.; Carino, G.P.; Murray, J.; Murphy, C.; Napier, M.; Harrington, E.O. Natriuretic peptides differentially attenuate thrombin-induced barrier dysfunction in pulmonary microvascular endothelial cells. Exp. Cell Res. 2006, 312, 401–410. [Google Scholar] [CrossRef]
- Chen, G.; Zhao, J.; Yin, Y.; Wang, B.; Liu, Q.; Li, P.; Zhao, L.; Zhou, H. C-type natriuretic peptide attenuates LPS-induced endothelial activation: Involvement of p38, Akt, and NF-kappaB pathways. Amino Acids 2014, 46, 2653–2663. [Google Scholar] [CrossRef]
- Vannucci, R.C. Hypoxic-ischemic encephalopathy. Am. J. Perinatol. 2000, 17, 113–120. [Google Scholar] [CrossRef]
- Lee, A.C.; Kozuki, N.; Blencowe, H.; Vos, T.; Bahalim, A.; Darmstadt, G.L.; Niermeyer, S.; Ellis, M.; Robertson, N.J.; Cousens, S.; et al. Intrapartum-related neonatal encephalopathy incidence and impairment at regional and global levels for 2010 with trends from 1990. Pediatr. Res. 2013, 74 (Suppl. S1), 50–72. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Chen, K.; Sloan, S.A.; Bennett, M.L.; Scholze, A.R.; O’Keeffe, S.; Phatnani, H.P.; Guarnieri, P.; Caneda, C.; Ruderisch, N.; et al. An RNA-sequencing transcriptome and splicing database of glia, neurons, and vascular cells of the cerebral cortex. J. Neurosci. 2014, 34, 11929–11947. [Google Scholar] [CrossRef] [PubMed]
- Ahluwalia, A.; MacAllister, R.J.; Hobbs, A.J. Vascular actions of natriuretic peptides. Cyclic GMP-dependent and -independent mechanisms. Basic Res. Cardiol. 2004, 99, 83–89. [Google Scholar] [CrossRef] [PubMed]
- Balkaya, M.; Krober, J.M.; Rex, A.; Endres, M. Assessing post-stroke behavior in mouse models of focal ischemia. J. Cereb. Blood Flow Metab. 2013, 33, 330–338. [Google Scholar] [CrossRef] [Green Version]
- Huang, L.; Ma, Q.; Li, Y.; Li, B.; Zhang, L. Inhibition of microRNA-210 suppresses pro-inflammatory response and reduces acute brain injury of ischemic stroke in mice. Exp. Neurol. 2018, 300, 41–50. [Google Scholar] [CrossRef]
- Shen, Y.H.; Wang, X.L.; Wilcken, D.E. Nitric oxide induces and inhibits apoptosis through different pathways. FEBS Lett. 1998, 433, 125–131. [Google Scholar] [CrossRef] [Green Version]
- Kolluru, G.K.; Tamilarasan, K.P.; Rajkumar, A.S.; Geetha Priya, S.; Rajaram, M.; Saleem, N.K.; Majumder, S.; Jaffar Ali, B.M.; Illavazagan, G.; Chatterjee, S. Nitric oxide/cGMP protects endothelial cells from hypoxia-mediated leakiness. Eur. J. Cell Biol. 2008, 87, 147–161. [Google Scholar] [CrossRef] [PubMed]
- Kotamraju, S.; Matalon, S.; Matsunaga, T.; Shang, T.; Hickman-Davis, J.M.; Kalyanaraman, B. Upregulation of immunoproteasomes by nitric oxide: Potential antioxidative mechanism in endothelial cells. Free Radic. Biol. Med. 2006, 40, 1034–1044. [Google Scholar] [CrossRef]
- Zhou, H.; Murthy, K.S. Identification of the G protein-activating sequence of the single-transmembrane natriuretic peptide receptor C (NPR-C). Am. J. Physiol. Cell Physiol. 2003, 284, C1255–C1261. [Google Scholar] [CrossRef] [Green Version]
- Murthy, K.S.; Teng, B.Q.; Zhou, H.; Jin, J.G.; Grider, J.R.; Makhlouf, G.M. G(i-1)/G(i-2)-dependent signaling by single-transmembrane natriuretic peptide clearance receptor. Am. J. Physiol. Gastrointest Liver Physiol. 2000, 278, G974–G980. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- El Andalousi, J.; Li, Y.; Anand-Srivastava, M.B. Natriuretic peptide receptor-C agonist attenuates the expression of cell cycle proteins and proliferation of vascular smooth muscle cells from spontaneously hypertensive rats: Role of Gi proteins and MAPkinase/PI3kinase signaling. PLoS ONE 2013, 8, e76183. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bolanos, J.P.; Almeida, A. Roles of nitric oxide in brain hypoxia-ischemia. Biochim. Biophys. Acta 1999, 1411, 415–436. [Google Scholar] [CrossRef] [Green Version]
- Fulton, D.; Gratton, J.P.; McCabe, T.J.; Fontana, J.; Fujio, Y.; Walsh, K.; Franke, T.F.; Papapetropoulos, A.; Sessa, W.C. Regulation of endothelium-derived nitric oxide production by the protein kinase Akt. Nature 1999, 399, 597–601. [Google Scholar] [CrossRef] [PubMed]
- Shen, B.; Gao, L.; Hsu, Y.T.; Bledsoe, G.; Hagiwara, M.; Chao, L.; Chao, J. Kallistatin attenuates endothelial apoptosis through inhibition of oxidative stress and activation of Akt-eNOS signaling. Am. J. Physiol. Heart Circ. Physiol. 2010, 299, H1419–H1427. [Google Scholar] [CrossRef] [Green Version]
- Bellis, A.; Castaldo, D.; Trimarco, V.; Monti, M.G.; Chivasso, P.; Sadoshima, J.; Trimarco, B.; Morisco, C. Cross-talk between PKA and Akt protects endothelial cells from apoptosis in the late ischemic preconditioning. Arter. Thromb. Vasc. Biol. 2009, 29, 1207–1212. [Google Scholar] [CrossRef]
- Lin, H.Y.; Wu, C.L.; Huang, C.C. The Akt-endothelial nitric oxide synthase pathway in lipopolysaccharide preconditioning-induced hypoxic-ischemic tolerance in the neonatal rat brain. Stroke 2010, 41, 1543–1551. [Google Scholar] [CrossRef]
- van Schie, P.E.; Schijns, J.; Becher, J.G.; Barkhof, F.; van Weissenbruch, M.M.; Vermeulen, R.J. Long-term motor and behavioral outcome after perinatal hypoxic-ischemic encephalopathy. Eur. J. Paediatr. Neurol. 2015, 19, 354–359. [Google Scholar] [CrossRef]
- Fragopoulou, A.F.; Qian, Y.; Heijtz, R.D.; Forssberg, H. Can Neonatal Systemic Inflammation and Hypoxia Yield a Cerebral Palsy-Like Phenotype in Periadolescent Mice? Mol. Neurobiol. 2019, 56, 6883–6900. [Google Scholar] [CrossRef] [Green Version]
- Ten, V.S.; Wu, E.X.; Tang, H.; Bradley-Moore, M.; Fedarau, M.V.; Ratner, V.I.; Stark, R.I.; Gingrich, J.A.; Pinsky, D.J. Late measures of brain injury after neonatal hypoxia-ischemia in mice. Stroke 2004, 35, 2183–2188. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Corcelli, M.; Hawkins, K.; Vlahova, F.; Hunjan, A.; Dowding, K.; De Coppi, P.; David, A.L.; Peebles, D.; Gressens, P.; Hagberg, H.; et al. Neuroprotection of the hypoxic-ischemic mouse brain by human CD117(+)CD90(+)CD105(+) amniotic fluid stem cells. Sci. Rep. 2018, 8, 2425. [Google Scholar] [CrossRef] [Green Version]
- Reinboth, B.S.; Koster, C.; Abberger, H.; Prager, S.; Bendix, I.; Felderhoff-Muser, U.; Herz, J. Endogenous hypothermic response to hypoxia reduces brain injury: Implications for modeling hypoxic-ischemic encephalopathy and therapeutic hypothermia in neonatal mice. Exp. Neurol. 2016, 283 Pt A, 264–275. [Google Scholar] [CrossRef] [Green Version]
- Tress, E.E.; Clark, R.S.; Foley, L.M.; Alexander, H.; Hickey, R.W.; Drabek, T.; Kochanek, P.M.; Manole, M.D. Blood brain barrier is impermeable to solutes and permeable to water after experimental pediatric cardiac arrest. Neurosci. Lett. 2014, 578, 17–21. [Google Scholar] [CrossRef] [Green Version]
- Feng, Y.; Rhodes, P.G.; Bhatt, A.J. Neuroprotective effects of vascular endothelial growth factor following hypoxic ischemic brain injury in neonatal rats. Pediatr Res. 2008, 64, 370–374. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garcia, J.H.; Wagner, S.; Liu, K.F.; Hu, X.J. Neurological deficit and extent of neuronal necrosis attributable to middle cerebral artery occlusion in rats. Statistical validation. Stroke 1995, 26, 627–634. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Ma, Q.; Dasgupta, C.; Halavi, S.; Hartman, R.E.; Xiao, D.; Zhang, L. Inhibition of DNA Methylation in the Developing Rat Brain Disrupts Sexually Dimorphic Neurobehavioral Phenotypes in Adulthood. Mol. Neurobiol. 2017, 54, 3988–3999. [Google Scholar] [CrossRef]
- Shen, G.; Hu, S.; Zhao, Z.; Zhang, L.; Ma, Q. Antenatal Hypoxia Accelerates the Onset of Alzheimer’s Disease Pathology in 5xFAD Mouse Model. Front. Aging Neurosci. 2020, 12, 251. [Google Scholar] [CrossRef]
- Zhao, B.Q.; Wang, S.; Kim, H.Y.; Storrie, H.; Rosen, B.R.; Mooney, D.J.; Wang, X.; Lo, E.H. Role of matrix metalloproteinases in delayed cortical responses after stroke. Nat. Med. 2006, 12, 441–445. [Google Scholar] [CrossRef] [PubMed]
- Ma, Q.; Dasgupta, C.; Li, Y.; Bajwa, N.M.; Xiong, F.; Harding, B.; Hartman, R.; Zhang, L. Inhibition of microRNA-210 provides neuroprotection in hypoxic-ischemic brain injury in neonatal rats. Neurobiol. Dis 2016, 89, 202–212. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thagard, A.S.; Slack, J.L.; Estrada, S.M.; Kazanjian, A.A.; Chan, S.; Burd, I.; Napolitano, P.G.; Ieronimakis, N. Long-term impact of intrauterine neuroinflammation and treatment with magnesium sulphate and betamethasone: Sex-specific differences in a preterm labor murine model. Sci. Rep. 2017, 7, 17883. [Google Scholar] [CrossRef] [Green Version]
- Yawno, T.; Sutherland, A.E.; Pham, Y.; Castillo-Melendez, M.; Jenkin, G.; Miller, S.L. Fetal Growth Restriction Alters Cerebellar Development in Fetal and Neonatal Sheep. Front. Physiol. 2019, 10, 560. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Zhang, L.; Pu, H.; Mao, L.; Hu, X.; Jiang, X.; Xu, N.; Stetler, R.A.; Zhang, F.; Liu, X.; et al. Rapid endothelial cytoskeletal reorganization enables early blood-brain barrier disruption and long-term ischaemic reperfusion brain injury. Nat. Commun. 2016, 7, 10523. [Google Scholar] [CrossRef] [PubMed]
- Hind, W.H.; Tufarelli, C.; Neophytou, M.; Anderson, S.I.; England, T.J.; O’Sullivan, S.E. Endocannabinoids modulate human blood-brain barrier permeability in vitro. Br. J. Pharm. 2015, 172, 3015–3027. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, Q.; Dasgupta, C.; Li, Y.; Huang, L.; Zhang, L. MicroRNA-210 Downregulates ISCU and Induces Mitochondrial Dysfunction and Neuronal Death in Neonatal Hypoxic-Ischemic Brain Injury. Mol. Neurobiol. 2019, 56, 5608–5625. [Google Scholar] [CrossRef]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shen, G.; Hu, S.; Zhao, Z.; Zhang, L.; Ma, Q. C-Type Natriuretic Peptide Ameliorates Vascular Injury and Improves Neurological Outcomes in Neonatal Hypoxic-Ischemic Brain Injury in Mice. Int. J. Mol. Sci. 2021, 22, 8966. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22168966
Shen G, Hu S, Zhao Z, Zhang L, Ma Q. C-Type Natriuretic Peptide Ameliorates Vascular Injury and Improves Neurological Outcomes in Neonatal Hypoxic-Ischemic Brain Injury in Mice. International Journal of Molecular Sciences. 2021; 22(16):8966. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22168966
Chicago/Turabian StyleShen, Guofang, Shirley Hu, Zhen Zhao, Lubo Zhang, and Qingyi Ma. 2021. "C-Type Natriuretic Peptide Ameliorates Vascular Injury and Improves Neurological Outcomes in Neonatal Hypoxic-Ischemic Brain Injury in Mice" International Journal of Molecular Sciences 22, no. 16: 8966. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22168966