
Extracellular Heat Shock Proteins as Therapeutic Targets and Biomarkers in Fibrosing Interstitial Lung Diseases

 ,
,
Abstract
:1. Introduction
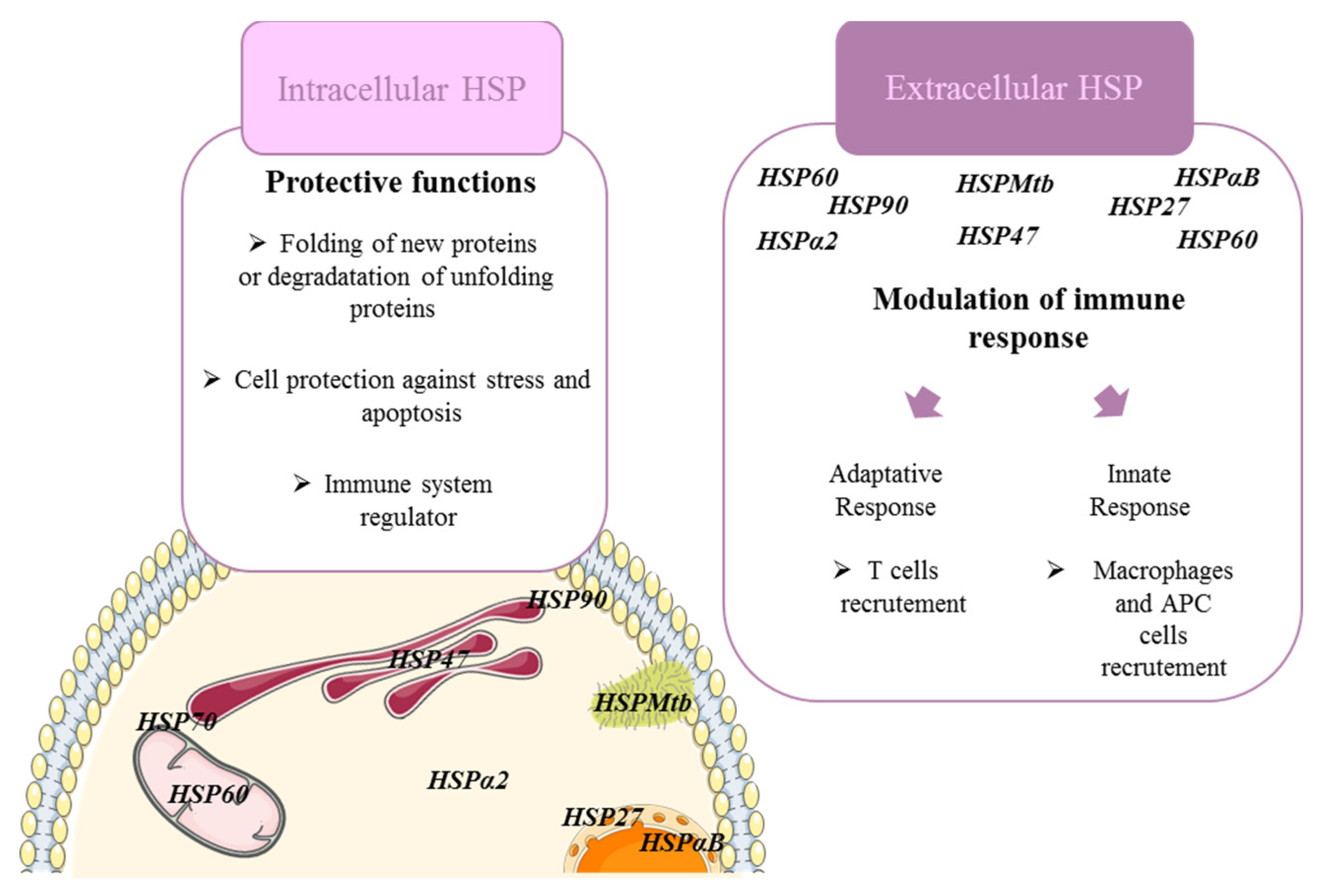
2. Heat Shock Proteins Modulate Immune Cell Functions in Fibrosing ILDs
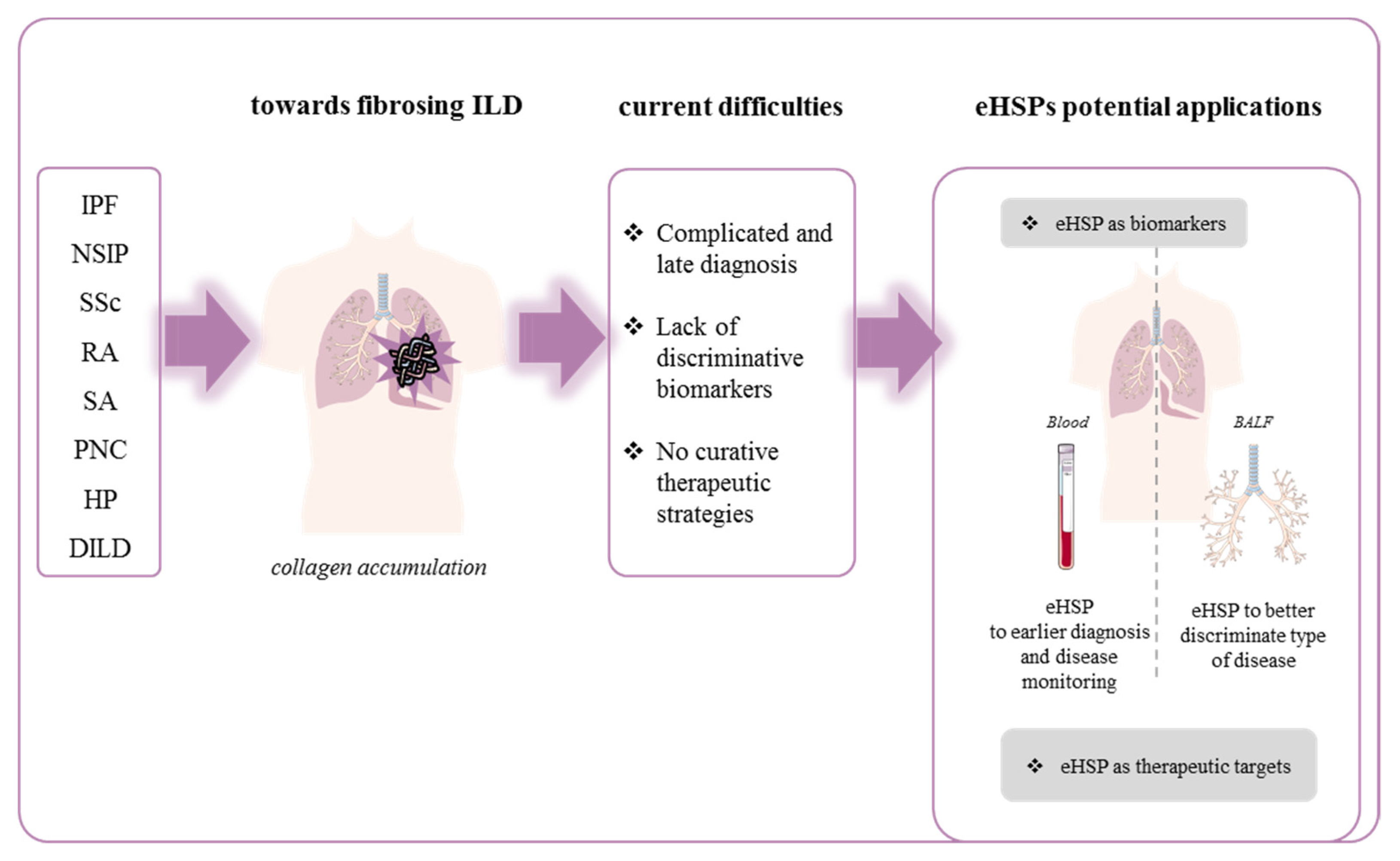
3. Extracellular HSP and Fibrosing Diffuse Interstitial Lung Disease
3.1. Fibrosing Idiopathic Diffuse Interstitial Lung Disease
3.1.1. Idiopathic Pulmonary Fibrosis
3.1.2. Nonspecific Interstitial Pneumonia (NSIP)
3.2. Fibrosing Diffuse Interstitial Lung Disease Associated with Connective Tissue (CTD-FLD)
3.2.1. Systemic Sclerosis-Associated Interstitial Lung Disease (SSc-ILD)
3.2.2. Rheumatoid Arthritis-Associated Interstitial Lung Disease (RA-ILD)
3.3. Sarcoidosis-Associated Interstitial Lung Disease (SA-ILD)
3.4. Fibrosing Environmental Interstitial Lung Disease
3.4.1. Pneumoconiosis (PNC)
3.4.2. Hypersensitivity Pneumonitis (HP)
3.4.3. Drug-Induced ILD
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Olson, A.; Hartmann, N.; Patnaik, P.; Wallace, L.; Schlenker-Herceg, R.; Nasser, M.; Richeldi, L.; Hoffmann-Vold, A.-M.; Cottin, V. Estimation of the Prevalence of Progressive Fibrosing Interstitial Lung Diseases: Systematic Literature Review and Data from a Physician Survey. Adv. Ther. 2021, 38, 854–867. [Google Scholar] [CrossRef]
- Travis, W.D.; Costabel, U.; Hansell, D.M.; King, T.E.; Lynch, D.A.; Nicholson, A.G.; Ryerson, C.J.; Ryu, J.H.; Selman, M.; Wells, A.U.; et al. An Official American Thoracic Society/European Respiratory Society Statement: Update of the International Multidisciplinary Classification of the Idiopathic Interstitial Pneumonias. Am. J. Respir. Crit. Care Med. 2013, 188, 733–748. [Google Scholar] [CrossRef] [PubMed]
- Wijsenbeek, M.; Cottin, V. Spectrum of Fibrotic Lung Diseases. N. Engl. J. Med. 2020, 383, 958–968. [Google Scholar] [CrossRef]
- Wijsenbeek, M.; Kreuter, M.; Olson, A.; Fischer, A.; Bendstrup, E.; Wells, C.D.; Denton, C.P.; Mounir, B.; Zouad-Lejour, L.; Quaresma, M.; et al. Progressive Fibrosing Interstitial Lung Diseases: Current Practice in Diagnosis and Management. Curr. Med. Res. Opin. 2019, 35, 2015–2024. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Llanos, O.; Hamzeh, N. Sarcoidosis. Med. Clin. N. Am. 2019, 103, 527–534. [Google Scholar] [CrossRef] [PubMed]
- Kisseleva, T.; Brenner, D.A. Mechanisms of Fibrogenesis. Exp. Biol. Med. (Maywood) 2008, 233, 109–122. [Google Scholar] [CrossRef] [PubMed]
- Bergeron, A.; Soler, P.; Kambouchner, M.; Loiseau, P.; Milleron, B.; Valeyre, D.; Hance, A.J.; Tazi, A. Cytokine Profiles in Idiopathic Pulmonary Fibrosis Suggest an Important Role for TGF-β and IL-10. Eur. Respir. J. 2003, 22, 69–76. [Google Scholar] [CrossRef] [Green Version]
- Sun, L.; Louie, M.C.; Vannella, K.M.; Wilke, C.A.; LeVine, A.M.; Moore, B.B.; Shanley, T.P. New Concepts of IL-10-Induced Lung Fibrosis: Fibrocyte Recruitment and M 2 Activation in a CCL2/CCR2 Axis. Am. J. Physiol.-Lung Cell. Mol. Physiol. 2011, 300, L341–L353. [Google Scholar] [CrossRef] [Green Version]
- Sime, P.J.; Xing, Z.; Graham, F.L.; Csaky, K.G.; Gauldie, J. Adenovector-Mediated Gene Transfer of Active Transforming Growth Factor-Beta1 Induces Prolonged Severe Fibrosis in Rat Lung. J. Clin. Investig. 1997, 100, 768–776. [Google Scholar] [CrossRef] [Green Version]
- Kim, K.K.; Kugler, M.C.; Wolters, P.J.; Robillard, L.; Galvez, M.G.; Brumwell, A.N.; Sheppard, D.; Chapman, H.A. Alveolar Epithelial Cell Mesenchymal Transition Develops in Vivo during Pulmonary Fibrosis and Is Regulated by the Extracellular Matrix. Proc. Natl. Acad. Sci. USA 2006, 103, 13180–13185. [Google Scholar] [CrossRef] [Green Version]
- Coker, R.K.; Laurent, G.J.; Jeffery, P.K.; du Bois, R.M.; Black, C.M.; McAnulty, R.J. Localisation of Transforming Growth Factor Beta1 and Beta3 MRNA Transcripts in Normal and Fibrotic Human Lung. Thorax 2001, 56, 549–556. [Google Scholar] [CrossRef] [Green Version]
- Lanneau, D.; Wettstein, G.; Bonniaud, P.; Garrido, C. Heat Shock Proteins: Cell Protection through Protein Triage. Sci. World J. 2010, 10, 1543–1552. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jego, G.; Hazoumé, A.; Seigneuric, R.; Garrido, C. Targeting Heat Shock Proteins in Cancer. Cancer Lett. 2013, 332, 275–285. [Google Scholar] [CrossRef]
- Schmitt, E.; Maingret, L.; Puig, P.E.; Rerole, A.L.; Ghiringhelli, F.; Hammann, A.; Solary, E.; Kroemer, G.; Garrido, C. Heat Shock Protein 70 Neutralization Exerts Potent Antitumor Effects in Animal Models of Colon Cancer and Melanoma. Cancer Res. 2006, 66, 4191–4197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Almeida, S.; Regimbeau, M.; Jego, G.; Garrido, C.; Girodon, F.; Hermetet, F. Heat Shock Proteins and PD-1/PD-L1 as Potential Therapeutic Targets in Myeloproliferative Neoplasms. Cancers 2020, 12, 2592. [Google Scholar] [CrossRef] [PubMed]
- Bellaye, P.-S.; Burgy, O.; Causse, S.; Garrido, C.; Bonniaud, P. Heat Shock Proteins in Fibrosis and Wound Healing: Good or Evil? Pharmacol. Ther. 2014, 143, 119–132. [Google Scholar] [CrossRef]
- Iwashita, T.; Kadota, J.; Naito, S.; Kaida, H.; Ishimatsu, Y.; Miyazaki, M.; Ozono, Y.; Kohno, S. Involvement of Collagen-Binding Heat Shock Protein 47 and Procollagen Type I Synthesis in Idiopathic Pulmonary Fibrosis: Contribution of Type II Pneumocytes to Fibrosis. Hum. Pathol. 2000, 31, 1498–1505. [Google Scholar] [CrossRef]
- Mishima, Y.; Miyazaki, M.; Abe, K.; Ozono, Y.; Shioshita, K.; Xia, Z.; Harada, T.; Taguchi, T.; Koji, T.; Kohno, S. Enhanced Expression of Heat Shock Protein 47 in Rat Model of Peritoneal Fibrosis. Perit. Dial. Int. J. Int. Soc. Perit. Dial. 2003, 23, 14–22. [Google Scholar] [CrossRef]
- Razzaque, M.S.; Nazneen, A.; Taguchi, T. Immunolocalization of Collagen and Collagen-Binding Heat Shock Protein 47 in Fibrotic Lung Diseases. Mod. Pathol. Off. J. U. S. Can. Acad. Pathol. Inc. 1998, 11, 1183–1188. [Google Scholar]
- Hagiwara, S.; Iwasaka, H.; Matsumoto, S.; Noguchi, T. An Antisense Oligonucleotide to HSP47 Inhibits Paraquat-Induced Pulmonary Fibrosis in Rats. Toxicology 2007, 236, 199–207. [Google Scholar] [CrossRef]
- Hagiwara, S.; Iwasaka, H.; Matsumoto, S.; Noguchi, T. Antisense Oligonucleotide Inhibition of Heat Shock Protein (HSP) 47 Improves Bleomycin-Induced Pulmonary Fibrosis in Rats. Respir. Res. 2007, 8, 37. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hagiwara, S.; Iwasaka, H.; Matsumoto, S.; Noguchi, T. Introduction of Antisense Oligonucleotides to Heat Shock Protein 47 Prevents Pulmonary Fibrosis in Lipopolysaccharide-Induced Pneumopathy of the Rat. Eur. J. Pharmacol. 2007, 564, 174–180. [Google Scholar] [CrossRef] [PubMed]
- Sontake, V.; Wang, Y.; Kasam, R.K.; Sinner, D.; Reddy, G.B.; Naren, A.P.; McCormack, F.X.; White, E.S.; Jegga, A.G.; Madala, S.K. Hsp90 Regulation of Fibroblast Activation in Pulmonary Fibrosis. JCI Insight 2017, 2. [Google Scholar] [CrossRef] [Green Version]
- Bonniaud, P.; Burgy, O.; Garrido, C. Heat Shock Protein-90 toward Theranostics: A Breath of Fresh Air in Idiopathic Pulmonary Fibrosis. Eur. Respir. J. 2018, 51, 1702612. [Google Scholar] [CrossRef]
- Sellares, J.; Veraldi, K.L.; Thiel, K.J.; Cárdenes, N.; Alvarez, D.; Schneider, F.; Pilewski, J.M.; Rojas, M.; Feghali-Bostwick, C.A. Intracellular Heat Shock Protein 70 Deficiency in Pulmonary Fibrosis. Am. J. Respir. Cell Mol. Biol. 2019, 60, 629–636. [Google Scholar] [CrossRef]
- Fujibayashi, T.; Hashimoto, N.; Jijiwa, M.; Hasegawa, Y.; Kojima, T.; Ishiguro, N. Protective Effect of Geranylgeranylacetone, an Inducer of Heat Shock Protein 70, against Drug-Induced Lung Injury/Fibrosis in an Animal Model. BMC Pulm Med. 2009, 9, 45. [Google Scholar] [CrossRef] [Green Version]
- Wettstein, G.; Bellaye, P.-S.; Kolb, M.; Hammann, A.; Crestani, B.; Soler, P.; Marchal-Somme, J.; Hazoume, A.; Gauldie, J.; Gunther, A.; et al. Inhibition of HSP27 Blocks Fibrosis Development and EMT Features by Promoting Snail Degradation. FASEB J. 2013, 27, 1549–1560. [Google Scholar] [CrossRef]
- Bellaye, P.-S.; Wettstein, G.; Burgy, O.; Besnard, V.; Joannes, A.; Colas, J.; Causse, S.; Marchal-Somme, J.; Fabre, A.; Crestani, B.; et al. The Small Heat-Shock Protein AB-Crystallin Is Essential for the Nuclear Localization of Smad4: Impact on Pulmonary Fibrosis. J. Pathol. 2014, 232, 458–472. [Google Scholar] [CrossRef]
- Boutanquoi, P.-M.; Burgy, O.; Beltramo, G.; Bellaye, P.-S.; Dondaine, L.; Marcion, G.; Pommerolle, L.; Vadel, A.; Spanjaard, M.; Demidov, O.; et al. TRIM33 Prevents Pulmonary Fibrosis by Impairing TGF-Β1 Signalling. Eur. Respir. J. 2020, 55, 1901346. [Google Scholar] [CrossRef] [PubMed]
- Albakova, Z.; Siam, M.K.S.; Sacitharan, P.K.; Ziganshin, R.H.; Ryazantsev, D.Y.; Sapozhnikov, A.M. Extracellular Heat Shock Proteins and Cancer: New Perspectives. Transl. Oncol. 2021, 14, 100995. [Google Scholar] [CrossRef] [PubMed]
- Hance, M.; Nolan, K.; Isaacs, J. The Double-Edged Sword: Conserved Functions of Extracellular Hsp90 in Wound Healing and Cancer. Cancers 2014, 6, 1065–1097. [Google Scholar] [CrossRef]
- Gunther, S.; Ostheimer, C.; Stangl, S.; Specht, H.M.; Mozes, P.; Jesinghaus, M.; Vordermark, D.; Combs, S.E.; Peltz, F.; Jung, M.P.; et al. Correlation of Hsp70 Serum Levels with Gross Tumor Volume and Composition of Lymphocyte Subpopulations in Patients with Squamous Cell and Adeno Non-Small Cell Lung Cancer. Front. Immunol. 2015, 6, 556. [Google Scholar] [CrossRef] [Green Version]
- Banerjee, S.; Lin, C.F.; Skinner, K.A.; Schiffhauer, L.M.; Peacock, J.; Hicks, D.G.; Redmond, E.M.; Morrow, D.; Huston, A.; Shayne, M.; et al. Heat Shock Protein 27 Differentiates Tolerogenic Macrophages That May Support Human Breast Cancer Progression. Cancer Res. 2011, 71, 318–327. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stope, M.B.; Klinkmann, G.; Diesing, K.; Koensgen, D.; Burchardt, M.; Mustea, A. Heat Shock Protein HSP27 Secretion by Ovarian Cancer Cells Is Linked to Intracellular Expression Levels, Occurs Independently of the Endoplasmic Reticulum Pathway and HSP27’s Phosphorylation Status, and Is Mediated by Exosome Liberation. Dis. Markers 2017, 2017, 1575374. [Google Scholar] [CrossRef] [PubMed]
- Thuringer, D.; Berthenet, K.; Cronier, L.; Solary, E.; Garrido, C. Primary Tumor- and Metastasis-Derived Colon Cancer Cells Differently Modulate Connexin Expression and Function in Human Capillary Endothelial Cells. Oncotarget 2015, 6, 28800-15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thuringer, D.; Jego, G.; Wettstein, G.; Terrier, O.; Cronier, L.; Yousfi, N.; Hébrard, S.; Bouchot, A.; Hazoumé, A.; Joly, A.; et al. Extracellular HSP27 Mediates Angiogenesis through Toll-like Receptor 3. FASEB J. 2013, 27, 4169–4183. [Google Scholar] [CrossRef]
- Chanteloup, G.; Cordonnier, M.; Isambert, N.; Bertaut, A.; Hervieu, A.; Hennequin, A.; Luu, M.; Zanetta, S.; Coudert, B.; Bengrine, L.; et al. Monitoring HSP70 Exosomes in Cancer Patients’ Follow up: A Clinical Prospective Pilot Study. J. Extracell Vesicles 2020, 9, 1766192. [Google Scholar] [CrossRef]
- Wallace, W.A.; Roberts, S.N.; Caldwell, H.; Thornton, E.; Greening, A.P.; Lamb, D.; Howie, S.E. Circulating Antibodies to Lung Protein(s) in Patients with Cryptogenic Fibrosing Alveolitis. Thorax 1994, 49, 218–224. [Google Scholar] [CrossRef] [Green Version]
- Kahloon, R.A.; Xue, J.; Bhargava, A.; Csizmadia, E.; Otterbein, L.; Kass, D.J.; Bon, J.; Soejima, M.; Levesque, M.C.; Lindell, K.O.; et al. Patients with Idiopathic Pulmonary Fibrosis with Antibodies to Heat Shock Protein 70 Have Poor Prognoses. Am. J. Respir. Crit. Care Med. 2013, 187, 768–775. [Google Scholar] [CrossRef] [Green Version]
- Mills, R.; Mathur, A.; Nicol, L.M.; Walker, J.J.; Przybylski, A.A.; Mackinnon, A.C.; Howie, S.E.M.; Wallace, W.A.H.; Dransfield, I.; Hirani, N. Intrapulmonary Autoantibodies to HSP72 Are Associated with Improved Outcomes in IPF. J. Immunol. Res. 2019, 2019, 1–11. [Google Scholar] [CrossRef]
- Yokota, S.; Kubota, H.; Matsuoka, Y.; Naitoh, M.; Hirata, D.; Minota, S.; Takahashi, H.; Fujii, N.; Nagata, K. Prevalence of HSP47 Antigen and Autoantibodies to HSP47 in the Sera of Patients with Mixed Connective Tissue Disease. Biochem. Biophys. Res. Commun. 2003, 303, 413–418. [Google Scholar] [CrossRef]
- Kakugawa, T.; Yokota, S.; Mukae, H.; Kubota, H.; Sakamoto, N.; Mizunoe, S.; Matsuoka, Y.; Kadota, J.; Fujii, N.; Nagata, K.; et al. High Serum Concentrations of Autoantibodies to HSP47 in Nonspecific Interstitial Pneumonia Compared with Idiopathic Pulmonary Fibrosis. BMC Pulm Med. 2008, 8, 23. [Google Scholar] [CrossRef] [Green Version]
- Kakugawa, T.; Yokota, S.; Ishimatsu, Y.; Hayashi, T.; Nakashima, S.; Hara, S.; Sakamoto, N.; Kubota, H.; Mine, M.; Matsuoka, Y.; et al. Serum Heat Shock Protein 47 Levels Are Elevated in Acute Exacerbation of Idiopathic Pulmonary Fibrosis. Cell Stress Chaperones 2013, 18, 581–590. [Google Scholar] [CrossRef] [Green Version]
- Luzina, I.G.; Kopach, P.; Lockatell, V.; Kang, P.H.; Nagarsekar, A.; Burke, A.P.; Hasday, J.D.; Todd, N.W.; Atamas, S.P. Interleukin-33 Potentiates Bleomycin-Induced Lung Injury. Am. J. Respir. Cell Mol. Biol. 2013, 49, 999–1008. [Google Scholar] [CrossRef] [PubMed]
- Ogawa, F.; Shimizu, K.; Hara, T.; Muroi, E.; Hasegawa, M.; Takehara, K.; Sato, S. Serum Levels of Heat Shock Protein 70, a Biomarker of Cellular Stress, Are Elevated in Patients with Systemic Sclerosis: Association with Fibrosis and Vascular Damage. Clin. Exp. Rheumatol. 2008, 26, 659–662. [Google Scholar] [PubMed]
- Štorkánová, H.; Oreská, S.; Špiritović, M.; Heřmánková, B.; Bubová, K.; Komarc, M.; Pavelka, K.; Vencovský, J.; Distler, J.H.W.; Šenolt, L.; et al. Plasma Hsp90 Levels in Patients with Systemic Sclerosis and Relation to Lung and Skin Involvement: A Cross-Sectional and Longitudinal Study. Sci. Rep. 2021, 11, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Matsuda, K.M.; Yoshizaki, A.; Kotani, H.; Norimatsu, Y.; Kuzumi, A.; Fukayama, M.; Fukasawa, T.; Ebata, S.; Yoshizaki-Ogawa, A.; Asano, Y.; et al. Serum Heat Shock Protein 27 Levels in Patients with Systemic Sclerosis: A Possible Biomarker of Skin Sclerosis. J. Eur. Acad. Dermatol. Venereol. 2021, 35, e157–e159. [Google Scholar] [CrossRef] [PubMed]
- Shirahama, R.; Miyazaki, Y.; Okamoto, T.; Inase, N.; Yoshizawa, Y. Proteome Analysis of Bronchoalveolar Lavage Fluid in Lung Fibrosis Associated with Systemic Sclerosis. Allergol. Int. 2010, 59, 409–415. [Google Scholar] [CrossRef] [Green Version]
- Harlow, L.; Rosas, I.O.; Gochuico, B.R.; Mikuls, T.R.; Dellaripa, P.F.; Oddis, C.V.; Ascherman, D.P. Identification of Citrullinated Hsp90 Isoforms as Novel Autoantigens in Rheumatoid Arthritis-Associated Interstitial Lung Disease: Citrullinated Hsp90 in RA-ILD. Arthritis Rheum. 2013, 65, 869–879. [Google Scholar] [CrossRef] [PubMed]
- Mantej, J.; Polasik, K.; Piotrowska, E.; Tukaj, S. Autoantibodies to Heat Shock Proteins 60, 70, and 90 in Patients with Rheumatoid Arthritis. Cell Stress Chaperones 2019, 24, 283–287. [Google Scholar] [CrossRef] [PubMed]
- Hrycaj, P.; Wurm, K.; Mennet, P.; Müller, W. Antibodies to Heat Shock Proteins in Patients with Pulmonary Sarcoidosis. Sarcoidosis 1995, 12, 124–130. [Google Scholar] [PubMed]
- Dubaniewicz, A.; Holownia, A.; Kalinowski, L.; Wybieralska, M.; Dobrucki, I.T.; Singh, M. Is Mycobacterial Heat Shock Protein 16kDa, a Marker of the Dormant Stage of Mycobacterium Tuberculosis, a Sarcoid Antigen? Hum. Immunol. 2013, 74, 45–51. [Google Scholar] [CrossRef] [PubMed]
- Staton, J.M.; Dench, J.E.; Currie, B.; Fitzpatrick, D.R.; Himbeck, R.P.; Allen, R.; Bruce, J.; Robinson, B.W.S.; Bielefeldt-Ohmann, H. Expression and Immune Recognition of Stress Proteins in Sarcoidosis and Other Chronic Interstitial Lung Diseases. Immunol. Cell Biol. 1995, 73, 23–32. [Google Scholar] [CrossRef]
- Dubaniewicz, A.; Trzonkowski, P.; Dubaniewicz-Wybieralska, M.; Dubaniewicz, A.; Singh, M.; Myśliwski, A. Mycobacterial Heat Shock Protein-Induced Blood T Lymphocytes Subsets and Cytokine Pattern: Comparison of Sarcoidosis with Tuberculosis and Healthy Controls. Respirology 2007, 12, 346–354. [Google Scholar] [CrossRef]
- Amenomori, M.; Mukae, H.; Sakamoto, N.; Kakugawa, T.; Hayashi, T.; Hara, A.; Hara, S.; Fujita, H.; Ishimoto, H.; Ishimatsu, Y.; et al. HSP47 in Lung Fibroblasts Is a Predictor of Survival in Fibrotic Nonspecific Interstitial Pneumonia. Respir. Med. 2010, 104, 895–901. [Google Scholar] [CrossRef] [Green Version]
- Kakugawa, T.; Mukae, H.; Hayashi, T.; Ishii, H.; Nakayama, S.; Sakamoto, N.; Yoshioka, S.; Sugiyama, K.; Mine, M.; Mizuta, Y.; et al. Expression of HSP47 in Usual Interstitial Pneumonia and Nonspecific Interstitial Pneumonia. Respir. Res. 2005, 6, 57. [Google Scholar] [CrossRef] [Green Version]
- Yang, X.; Zheng, J.; Bai, Y.; Tian, F.; Yuan, J.; Sun, J.; Liang, H.; Guo, L.; Tan, H.; Chen, W.; et al. Using Lymphocyte and Plasma Hsp70 as Biomarkers for Assessing Coke Oven Exposure among Steel Workers. Environ. Health Perspect. 2007, 115, 1573–1577. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Yuan, J.; Sun, J.; Wang, H.; Liang, H.; Bai, Y.; Guo, L.; Tan, H.; Yang, M.; Wang, J.; et al. Association between Heat-Shock Protein 70 Gene Polymorphisms and DNA Damage in Peripheral Blood Lymphocytes among Coke-Oven Workers. Mutat. Res. 2008, 649, 221–229. [Google Scholar] [CrossRef]
- Zhang, J.H.; Zhang, J.; Sun, J.Y.; Tian, L.; Niu, Q. Relationship between the Expression of Heat Shock Protein and Genetic Damage in Peripheral Blood of Workers Exposed to Coke Oven Emissions. Zhonghua Lao Dong Wei Sheng Zhi Ye Bing Za Zhi 2008, 26, 16–19. [Google Scholar]
- Racine, C.; Israël-Assayag, E.; Cormier, Y. Expression of Heat Shock Protein 72 by Alveolar Macrophages in Hypersensitivity Pneumonitis. Am. J. Physiol. Lung Cell. Mol. Physiol. 1999, 276, L501–L505. [Google Scholar] [CrossRef] [Green Version]
- Okamoto, T. Proteome Analysis of Bronchoalveolar Lavage Fluid in Chronic Hypersensitivity Pneumonitis. Allergol. Int. 2012, 61, 10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kakugawa, T.; Yokota, S.; Ishimatsu, Y.; Hayashi, T.; Nakashima, S.; Hara, S.; Sakamoto, N.; Matsuoka, Y.; Kubota, H.; Mine, M.; et al. Serum Heat Shock Protein 47 Levels in Patients with Drug-Induced Lung Disease. Respir. Res. 2013, 14, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bagnato, G.; Harari, S. Cellular Interactions in the Pathogenesis of Interstitial Lung Diseases. Eur. Respir. Rev. 2015, 24, 102–114. [Google Scholar] [CrossRef]
- Desai, O.; Winkler, J.; Minasyan, M.; Herzog, E.L. The Role of Immune and Inflammatory Cells in Idiopathic Pulmonary Fibrosis. Front. Med. 2018, 5, 43. [Google Scholar] [CrossRef] [PubMed]
- Jiang, D.; Liang, J.; Fan, J.; Yu, S.; Chen, S.; Luo, Y.; Prestwich, G.D.; Mascarenhas, M.M.; Garg, H.G.; Quinn, D.A.; et al. Regulation of Lung Injury and Repair by Toll-like Receptors and Hyaluronan. Nat. Med. 2005, 11, 1173–1179. [Google Scholar] [CrossRef]
- Kral, J.B.; Kuttke, M.; Schrottmaier, W.C.; Birnecker, B.; Warszawska, J.; Wernig, C.; Paar, H.; Salzmann, M.; Sahin, E.; Brunner, J.S.; et al. Sustained PI3K Activation Exacerbates BLM-Induced Lung Fibrosis via Activation of pro-Inflammatory and pro-Fibrotic Pathways. Sci. Rep. 2016, 6, 23034. [Google Scholar] [CrossRef]
- Siwik, D.A.; Chang, D.L.-F.; Colucci, W.S. Interleukin-1β and Tumor Necrosis Factor-α Decrease Collagen Synthesis and Increase Matrix Metalloproteinase Activity in Cardiac Fibroblasts In Vitro. Circ. Res. 2000, 86, 1259–1265. [Google Scholar] [CrossRef] [Green Version]
- Raghu, G.; Brown, K.K.; Costabel, U.; Cottin, V.; du Bois, R.M.; Lasky, J.A.; Thomeer, M.; Utz, J.P.; Khandker, R.K.; McDermott, L.; et al. Treatment of Idiopathic Pulmonary Fibrosis with Etanercept: An Exploratory, Placebo-Controlled Trial. Am. J. Respir. Crit. Care Med. 2008, 178, 948–955. [Google Scholar] [CrossRef]
- Song, L.; Weng, D.; Dai, W.; Tang, W.; Chen, S.; Li, C.; Chen, Y.; Liu, F.; Chen, J. Th17 Can Regulate Silica-Induced Lung Inflammation through an IL-1β-Dependent Mechanism. J. Cell Mol. Med. 2014, 18, 1773–1784. [Google Scholar] [CrossRef] [PubMed]
- Homer, R.J.; Herzog, E.L. Recent Advances in Pulmonary Fibrosis: Implications for Scleroderma. Curr. Opin. Rheumatol. 2010, 22, 683–689. [Google Scholar] [CrossRef]
- Chizzolini, C. T Cells, B Cells, and Polarized Immune Response in the Pathogenesis of Fibrosis and Systemic Sclerosis. Curr. Opin. Rheumatol. 2008, 20, 707–712. [Google Scholar] [CrossRef]
- O’Reilly, S.; Hugle, T.; van Laar, J.M. T Cells in Systemic Sclerosis: A Reappraisal. Rheumatology 2012, 51, 1540–1549. [Google Scholar] [CrossRef] [Green Version]
- Belperio, J.A.; Dy, M.; Murray, L.; Burdick, M.D.; Xue, Y.Y.; Strieter, R.M.; Keane, M.P. The Role of the Th2 CC Chemokine Ligand CCL17 in Pulmonary Fibrosis. J. Immunol. 2004, 173, 4692–4698. [Google Scholar] [CrossRef] [Green Version]
- Zininga, T.; Ramatsui, L.; Shonhai, A. Heat Shock Proteins as Immunomodulants. Molecules 2018, 23, 2846. [Google Scholar] [CrossRef] [Green Version]
- Calderwood, S.K.; Gong, J.; Murshid, A. Extracellular HSPs: The Complicated Roles of Extracellular HSPs in Immunity. Front. Immunol. 2016, 7. [Google Scholar] [CrossRef] [Green Version]
- Borges, T.J.; Wieten, L.; van Herwijnen, M.J.C.; Broere, F.; van der Zee, R.; Bonorino, C.; van Eden, W. The Anti-Inflammatory Mechanisms of Hsp70. Front. Immunol. 2012, 3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kottke, T.; Sanchez-Perez, L.; Diaz, R.M.; Thompson, J.; Chong, H.; Harrington, K.; Calderwood, S.K.; Pulido, J.; Georgopoulos, N.; Selby, P.; et al. Induction of Hsp70-Mediated Th17 Autoimmunity Can Be Exploited as Immunotherapy for Metastatic Prostate Cancer. Cancer Res. 2007, 67, 11970–11979. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gobbo, J.; Marcion, G.; Cordonnier, M.; Dias, A.M.M.; Pernet, N.; Hammann, A.; Richaud, S.; Mjahed, H.; Isambert, N.; Clausse, V.; et al. Restoring Anticancer Immune Response by Targeting Tumor-Derived Exosomes With a HSP70 Peptide Aptamer. JNCI J. 2016, 108, djv330. [Google Scholar] [CrossRef] [PubMed]
- Multhoff, G.; Pockley, A.G.; Streffer, C.; Gaipl, U.S. Dual Role of Heat Shock Proteins (HSPs) in Anti-Tumor Immunity. CMM 2012, 12, 1174–1182. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Sahu, D.; Tsen, F. Secreted Heat Shock Protein-90 (Hsp90) in Wound Healing and Cancer. Biochim. Biophys. Acta (BBA)-Mol. Cell Res. 2012, 1823, 730–741. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tukaj, S.; Węgrzyn, G. Anti-Hsp90 Therapy in Autoimmune and Inflammatory Diseases: A Review of Preclinical Studies. Cell Stress Chaperones 2016, 21, 213–218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zügeli, U.; Kaufmann, S.H.E. Immune Response against Heat Shock Proteins in Infectious Diseases. Immunobiology 1999, 201, 22–35. [Google Scholar] [CrossRef]
- Maher, T.M.; Bendstrup, E.; Dron, L.; Langley, J.; Smith, G.; Khalid, J.M.; Patel, H.; Kreuter, M. Global Incidence and Prevalence of Idiopathic Pulmonary Fibrosis. Respir. Res. 2021, 22, 197. [Google Scholar] [CrossRef] [PubMed]
- Raghu, G.; Remy-Jardin, M.; Myers, J.L.; Richeldi, L.; Ryerson, C.J.; Lederer, D.J.; Behr, J.; Cottin, V.; Danoff, S.K.; Morell, F.; et al. Diagnosis of Idiopathic Pulmonary Fibrosis. An Official ATS/ERS/JRS/ALAT Clinical Practice Guideline. Am. J. Respir. Crit. Care Med. 2018, 198, e44–e68. [Google Scholar] [CrossRef]
- Harari, S.; Raghu, G.; Caminati, A.; Cruciani, M.; Franchini, M.; Mannucci, P. Fibrotic Interstitial Lung Diseases and Air Pollution: A Systematic Literature Review. Eur. Respir. Rev. 2020, 29, 200093. [Google Scholar] [CrossRef]
- Lamb, Y.N. Nintedanib: A Review in Fibrotic Interstitial Lung Diseases. Drugs 2021, 81, 575. [Google Scholar] [CrossRef]
- Dong, H.; Luo, L.; Zou, M.; Huang, C.; Wan, X.; Hu, Y.; Le, Y.; Zhao, H.; Li, W.; Zou, F.; et al. Blockade of Extracellular Heat Shock Protein 90α by 1G6-D7 Attenuates Pulmonary Fibrosis through Inhibiting ERK Signaling. Am. J. Physiol. Lung Cell Mol. Physiol. 2017, 313, L1006–L1015. [Google Scholar] [CrossRef] [PubMed]
- Bellaye, P.-S.; Shimbori, C.; Yanagihara, T.; Carlson, D.A.; Hughes, P.; Upagupta, C.; Sato, S.; Wheildon, N.; Haystead, T.; Ask, K.; et al. Synergistic Role of HSP90α and HSP90β to Promote Myofibroblast Persistence in Lung Fibrosis. Eur. Respir. J. 2018, 51, 1700386. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Juarez, M.M.; Chan, A.L.; Norris, A.G.; Morrissey, B.M.; Albertson, T.E. Acute Exacerbation of Idiopathic Pulmonary Fibrosis-a Review of Current and Novel Pharmacotherapies. J. Thorac. Dis. 2015, 7, 499–519. [Google Scholar] [CrossRef]
- Belloli, E.A.; Beckford, R.; Hadley, R.; Flaherty, K.R. Idiopathic Non-specific Interstitial Pneumonia. Respirology 2016, 21, 259–268. [Google Scholar] [CrossRef]
- Churg, A.; Bilawich, A. Confluent Fibrosis and Fibroblast Foci in Fibrotic Non-Specific Interstitial Pneumonia. Histopathology 2016, 69, 128–135. [Google Scholar] [CrossRef]
- Poletti, V.; Romagnoli, M.; Piciucchi, S.; Chilosi, M. Current Status of Idiopathic Nonspecific Interstitial Pneumonia. Semin. Respir. Crit. Care Med. 2012, 33, 440–449. [Google Scholar] [CrossRef]
- Tomassetti, S.; Ryu, J.H.; Piciucchi, S.; Chilosi, M.; Poletti, V. Nonspecific Interstitial Pneumonia: What Is the Optimal Approach to Management? Semin. Respir. Crit. Care Med. 2016, 37, 378–394. [Google Scholar] [CrossRef] [PubMed]
- Bergamasco, A.; Hartmann, N.; Wallace, L.; Verpillat, P. Epidemiology of Systemic Sclerosis and Systemic Sclerosis-Associated Interstitial Lung Disease. CLEP 2019, 11, 257–273. [Google Scholar] [CrossRef] [Green Version]
- Perelas, A.; Silver, R.M.; Arrossi, A.V.; Highland, K.B. Systemic Sclerosis-Associated Interstitial Lung Disease. Lancet Resp. Med. 2020, 8, 304–320. [Google Scholar] [CrossRef]
- Jee, A.S.; Sahhar, J.; Youssef, P.; Bleasel, J.; Adelstein, S.; Nguyen, M.; Corte, T.J. Review: Serum Biomarkers in Idiopathic Pulmonary Fibrosis and Systemic Sclerosis Associated Interstitial Lung Disease–Frontiers and Horizons. Pharmacol. Ther. 2019, 202, 40–52. [Google Scholar] [CrossRef] [PubMed]
- Valenzi, E.; Bulik, M.; Tabib, T.; Morse, C.; Sembrat, J.; Bittar, H.T.; Rojas, M.; Lafyatis, R. Single-Cell Analysis Reveals Fibroblast Heterogeneity and Myofibroblasts in Systemic Sclerosis-Associated Interstitial Lung Disease. Ann. Rheum. Dis. 2019, 78, 1379–1387. [Google Scholar] [CrossRef]
- Cottin, V.; Brown, K.K. Interstitial Lung Disease Associated with Systemic Sclerosis (SSc-ILD). Resp. Res. 2019, 20. [Google Scholar] [CrossRef] [PubMed]
- Distler, O.; Highland, K.B.; Gahlemann, M.; Azuma, A.; Fischer, A.; Mayes, M.D.; Raghu, G.; Sauter, W.; Girard, M.; Alves, M.; et al. Nintedanib for Systemic Sclerosis–Associated Interstitial Lung Disease. N. Engl. J. Med. 2019, 380, 2518–2528. [Google Scholar] [CrossRef] [PubMed]
- Bonhomme, O.; André, B.; Gester, F.; de Seny, D.; Moermans, C.; Struman, I.; Louis, R.; Malaise, M.; Guiot, J. Biomarkers in Systemic Sclerosis-Associated Interstitial Lung Disease: Review of the Literature. Rheumatology 2019, 58, 1534–1546. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elhai, M.; Hoffmann-Vold, A.M.; Avouac, J.; Pezet, S.; Cauvet, A.; Leblond, A.; Fretheim, H.; Garen, T.; Kuwana, M.; Molberg, Ø.; et al. Performance of Candidate Serum Biomarkers for Systemic Sclerosis–Associated Interstitial Lung Disease. Arthritis Rheumatol. 2019, 71, 972–982. [Google Scholar] [CrossRef]
- Shaw, M.; Collins, B.F.; Ho, L.A.; Raghu, G. Rheumatoid Arthritis-Associated Lung Disease. Eur. Respir. Rev. 2015, 24, 1–16. [Google Scholar] [CrossRef] [Green Version]
- Spagnolo, P.; Lee, J.S.; Sverzellati, N.; Rossi, G.; Cottin, V. The Lung in Rheumatoid Arthritis: Focus on Interstitial Lung Disease. Arthritis Rheumatol. 2018, 70, 1544–1554. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, D.; Zhang, J.; Lau, J.; Wang, S.; Taneja, V.; Matteson, E.L.; Vassallo, R. Mechanisms of Lung Disease Development in Rheumatoid Arthritis. Nat. Rev. Rheumatol. 2019, 15, 581–596. [Google Scholar] [CrossRef]
- England, B.R.; Hershberger, D. Management Issues in Rheumatoid Arthritis-Associated Interstitial Lung Disease. Curr. Opin. Rheumatol. 2020, 32, 255–263. [Google Scholar] [CrossRef] [PubMed]
- Bendstrup, E.; Møller, J.; Kronborg-White, S.; Prior, T.S.; Hyldgaard, C. Interstitial Lung Disease in Rheumatoid Arthritis Remains a Challenge for Clinicians. JCM 2019, 8, 2038. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Juge, P.-A.; Lee, J.S.; Ebstein, E.; Furukawa, H.; Dobrinskikh, E.; Gazal, S.; Kannengiesser, C.; Ottaviani, S.; Oka, S.; Tohma, S.; et al. MUC5B Promoter Variant and Rheumatoid Arthritis with Interstitial Lung Disease. N. Engl. J. Med. 2018, 379, 2209–2219. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.C.; Choi, K.H.; Jacob, J.; Song, J.W. Prognostic Role of Blood KL-6 in Rheumatoid Arthritis–Associated Interstitial Lung Disease. PLoS ONE 2020, 15, e0229997. [Google Scholar] [CrossRef] [PubMed]
- Farid, S.S.; Azizi, G.; Mirshafiey, A. Anti-Citrullinated Protein Antibodies and Their Clinical Utility in Rheumatoid Arthritis. Int. J. Rheum. Dis. 2013, 16, 379–386. [Google Scholar] [CrossRef]
- Harlow, L.; Gochuico, B.R.; Rosas, I.O.; Doyle, T.J.; Osorio, J.C.; Travers, T.S.; Camacho, C.C.; Oddis, C.V.; Ascherman, D.P. Anti-Citrullinated Heat Shock Protein 90 Antibodies Identified in Bronchoalveolar Lavage Fluid Are a Marker of Lung-Specific Immune Responses. Clin. Immunol. 2014, 155, 60–70. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.; Song, S.; Liu, Y.; Liu, D.; Lin, Y.; Ge, S.; Ascherman, D.P. Autoreactive T Cells to Citrullinated HSP90 Are Associated with Interstitial Lung Disease in Rheumatoid Arthritis. Int. J. Rheum. Dis. 2018, 21, 1398–1405. [Google Scholar] [CrossRef]
- Tukaj, S.; Mantej, J.; Sobala, M.; Potrykus, K.; Sitko, K. Autologous Extracellular Hsp70 Exerts a Dual Role in Rheumatoid Arthritis. Cell Stress Chaperon. 2020, 25, 1105–1110. [Google Scholar] [CrossRef]
- Spagnolo, P.; Rossi, G.; Trisolini, R.; Sverzellati, N.; Baughman, R.P.; Wells, A.U. Pulmonary Sarcoidosis. Lancet Resp. Med. 2018, 6, 389–402. [Google Scholar] [CrossRef]
- Bonham, C.A.; Strek, M.E.; Patterson, K.C. From Granuloma to Fibrosis: Sarcoidosis Associated Pulmonary Fibrosis. Curr. Opin. Pulm. Med. 2016, 22, 484–491. [Google Scholar] [CrossRef] [Green Version]
- Patterson, K.C.; Chen, E.S. The Pathogenesis of Pulmonary Sarcoidosis and Implications for Treatment. Chest 2018, 153, 1432–1442. [Google Scholar] [CrossRef] [PubMed]
- Dubaniewicz, A. Mycobacterium Tuberculosis Heat Shock Proteins and Autoimmunity in Sarcoidosis. Autoimmun. Rev. 2010, 9, 419–424. [Google Scholar] [CrossRef] [PubMed]
- Dubaniewicz, A.; Dubaniewicz-Wybieralska, M.; Sternau, A.; Zwolska, Z.; Iżycka-Świeszewska, E.; Augustynowicz-Kopeć, E.; Skokowski, J.; Singh, M.; Zimnoch, L. Mycobacterium Tuberculosis Complex and Mycobacterial Heat Shock Proteins in Lymph Node Tissue from Patients with Pulmonary Sarcoidosis. J. Clin. Microbiol. 2006, 44, 3448–3451. [Google Scholar] [CrossRef] [Green Version]
- Massart, A.; Hunt, D.P. Pulmonary Manifestations of Inflammatory Bowel Disease. Am. J. Med. 2020, 133, 39–43. [Google Scholar] [CrossRef] [PubMed]
- Bewig, B.; Manske, I.; Böttcher, H.; Bastian, A.; Nitsche, R.; Fölsch, U.R. Crohn’s Disease Mimicking Sarcoidosis in Bronchoalveolar Lavage. Respiration 1999, 66, 467–469. [Google Scholar] [CrossRef] [PubMed]
- Grimstad, T.; Kvivik, I.; Kvaløy, J.T.; Aabakken, L.; Omdal, R. Heat Shock Protein 90 and Inflammatory Activity in Newly Onset Crohn’s Disease. Scand. J. Gastroenterol. 2018, 53, 1453–1458. [Google Scholar] [CrossRef]
- Cullinan, P.; Reid, P. Pneumoconiosis. Prim. Care Resp. J. 2013, 22, 249–252. [Google Scholar] [CrossRef] [Green Version]
- Fujimura, N. Pathology and Pathophysiology of Pneumoconiosis. Curr. Opini. Pulmon. Med. 2000, 6, 140–144. [Google Scholar] [CrossRef] [PubMed]
- Qi, X.-M.; Luo, Y.; Song, M.-Y.; Liu, Y.; Shu, T.; Liu, Y.; Pang, J.-L.; Wang, J.; Wang, C. Pneumoconiosis: Current Status and Future Prospects. Chin. Med. J. 2021, 134, 898–907. [Google Scholar] [CrossRef] [PubMed]
- McGrath, E.E.; Bardsley, P. An Association between Mycobacterium Malmoense and Coal Workers’ Pneumoconiosis. Lung 2009, 187, 51–54. [Google Scholar] [CrossRef] [PubMed]
- Raghu, G.; Remy-Jardin, M.; Ryerson, C.J.; Myers, J.L.; Kreuter, M.; Vasakova, M.; Bargagli, E.; Chung, J.H.; Collins, B.F.; Bendstrup, E.; et al. Diagnosis of Hypersensitivity Pneumonitis in Adults. An Official ATS/JRS/ALAT Clinical Practice Guideline. Am. J. Respir. Crit. Care Med. 2020, 202, e36–e69. [Google Scholar] [CrossRef]
- Nogueira, R. Hypersensitivity Pneumonitis: Antigen Diversity and Disease Implications. Hypersensitivity Pneumonitis 2019, 25, 97–108. [Google Scholar] [CrossRef]
- Pereira, C.A. Carlos AC Pereira1 Andréa Gimenez2 Lilian Kuranishi2 Karin Storrer2. J. Asthma Allergy. 2016, 9, 171. [Google Scholar] [CrossRef]
- Miyazaki, Y. Treatment and Monitoring of Hypersensitivity Pneumonitis. Expert Rev. Clin. Immunol. 2016, 12, 953–962. [Google Scholar] [CrossRef]
- Okamoto, T. The Usefulness of KL-6 and SP-D for the Diagnosis and Management of Chronic Hypersensitivity Pneumonitis. Respir. Med. 2015, 109, 1576–1581. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Classification | Nomenclature | Weight Discrimination | Localisation | ATP Depending |
|---|---|---|---|---|
| HSP27 | HSPB1 | Small molecular weight | Cytoplasm/nucleus | ATP-independant |
| αB-Crystallin | HSPB5 | Cytoplasm/nucleus | ATP-independant | |
| HSP90 | HSPC | High molecular weight | Cytoplasm/Endoplasmic reticulum/Membrane | ATP- dependent |
| HSP70 | HSPA1A | Cytoplasm/nucleus/mitochondrie/endoplasmic reticulum | ATP- dependent | |
| HSP47 | DNAJ | Endoplasmic reticulum | ATP-independant | |
| HSP60 | HSPD1 | Mitochondrie | ATP- dependent | |
| HSP65 Mtb HSP16 Mtb HSP70 Mtb | DNAK | Membrane/cytosol/envelope | ATP- dependent | |
| Alpha-2-HS glycoprotein | Fetuin-A | Cytosol | ATP-independant |
| Pathology | eHSP | Protein | Auto-Antibody | Biological Fluids | Prognosis/Diagnosis | Authors |
|---|---|---|---|---|---|---|
| IPF | HSP90 | + | − | Serum | Prognosis | [16] |
| HSP70 | − | + | Serum, BALF | Prognosis/Diagnosis | [38,39,40] | |
| HSP47 | + | + | Serum | Prognosis | [41,42,43] | |
| SSc | HSP70 | + | − | Serum, BALF | Prognostic | [44,45] |
| HSP90 | + | − | Blood | Prognostic | [46] | |
| HSP27 | + | − | Blood | Diagnosis | [47] | |
| α2 HSP | + | − | BALF | Diagnosis | [48] | |
| RA | HSP90 citrunillated | + | + | Blood | Prognosis/Diagnosis | [49] |
| HSP70 | + | − | Blood | Under investigation | [50] | |
| SA | HSP70 | − | + | Serum | Diagnosis | [51] |
| HSPMtb 16 | + | − | Serum | Diagnosis | [52] | |
| HSPMtb 65 | + | − | Serum, BALF | Diagnosis | [53] | |
| HSPMtb 70 | + | − | Serum | Diagnosis | [54] | |
| HSP70 | + | − | BALF | Diagnosis | [53] | |
| HSP90 | Deserve further investigated in fibrosing ILD context | |||||
| HSP60 | ||||||
| NSIP | HSP47 | + | + | Serum, BALF | Diagnosis/Prognosis | [55] |
| [42] | ||||||
| [56] | ||||||
| PNC | HSP70 | + | − | Serum | Diagnosis | [57] |
| HSP27 | + | − | Serum | Diagnosis | [58] | |
| HSP90 | + | − | Blood | Under investigation | [59] | |
| HSPMtb | Deserve further investigated in fibrosing ILD context | |||||
| HP | HSP70 | + | − | BALF | Diagnosis | [60] |
| α2 HSP | + | − | BALF | Diagnosis | [61] | |
| HSPMtb 65 | Deserve further investigated in fibrosing ILD context | |||||
| DILD | HSP47 | + | − | Serum | Diagnosis | [62] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tanguy, J.; Pommerolle, L.; Garrido, C.; Kolb, M.; Bonniaud, P.; Goirand, F.; Bellaye, P.-S. Extracellular Heat Shock Proteins as Therapeutic Targets and Biomarkers in Fibrosing Interstitial Lung Diseases. Int. J. Mol. Sci. 2021, 22, 9316. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22179316
Tanguy J, Pommerolle L, Garrido C, Kolb M, Bonniaud P, Goirand F, Bellaye P-S. Extracellular Heat Shock Proteins as Therapeutic Targets and Biomarkers in Fibrosing Interstitial Lung Diseases. International Journal of Molecular Sciences. 2021; 22(17):9316. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22179316
Chicago/Turabian StyleTanguy, Julie, Lenny Pommerolle, Carmen Garrido, Martin Kolb, Philippe Bonniaud, Françoise Goirand, and Pierre-Simon Bellaye. 2021. "Extracellular Heat Shock Proteins as Therapeutic Targets and Biomarkers in Fibrosing Interstitial Lung Diseases" International Journal of Molecular Sciences 22, no. 17: 9316. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22179316