
Uncovering miRNA-mRNA Regulatory Modules in Developing Xylem of Pinus massoniana via Small RNA and Degradome Sequencing


Abstract
:1. Introduction
2. Results
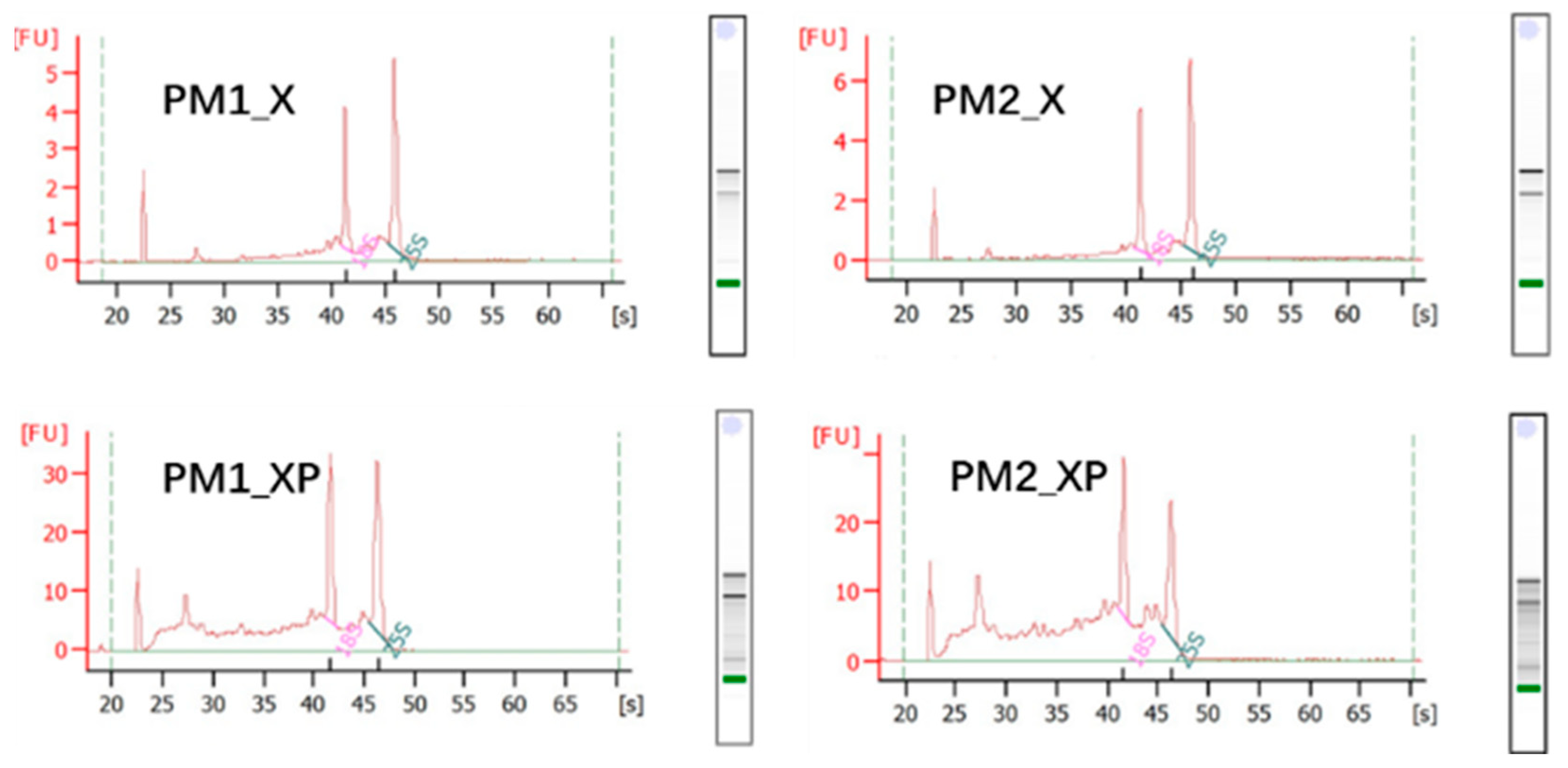
2.1. sRNAs Generated from Sequencing
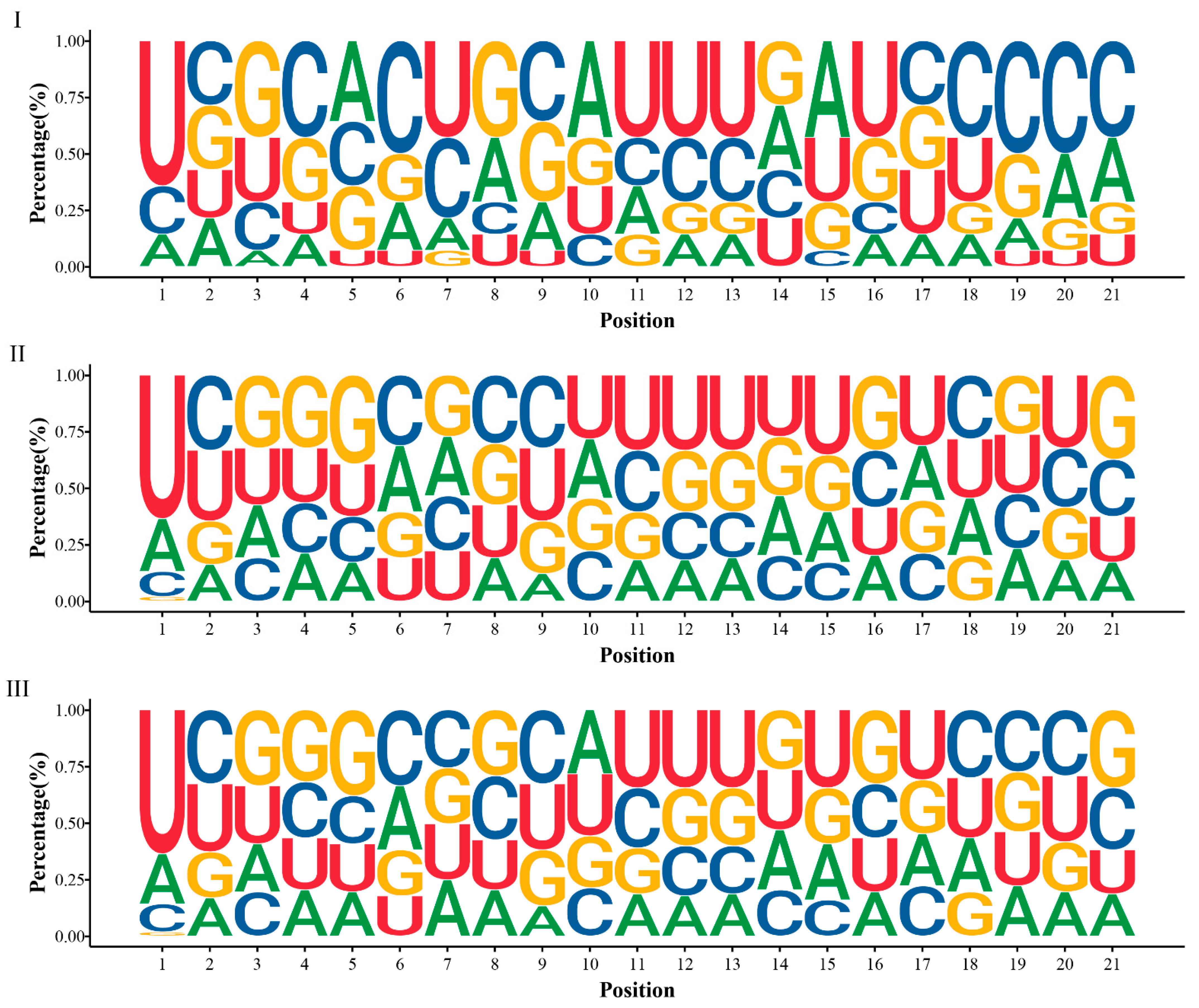
2.2. Identification of miRNAs Involved in Developing Xylem
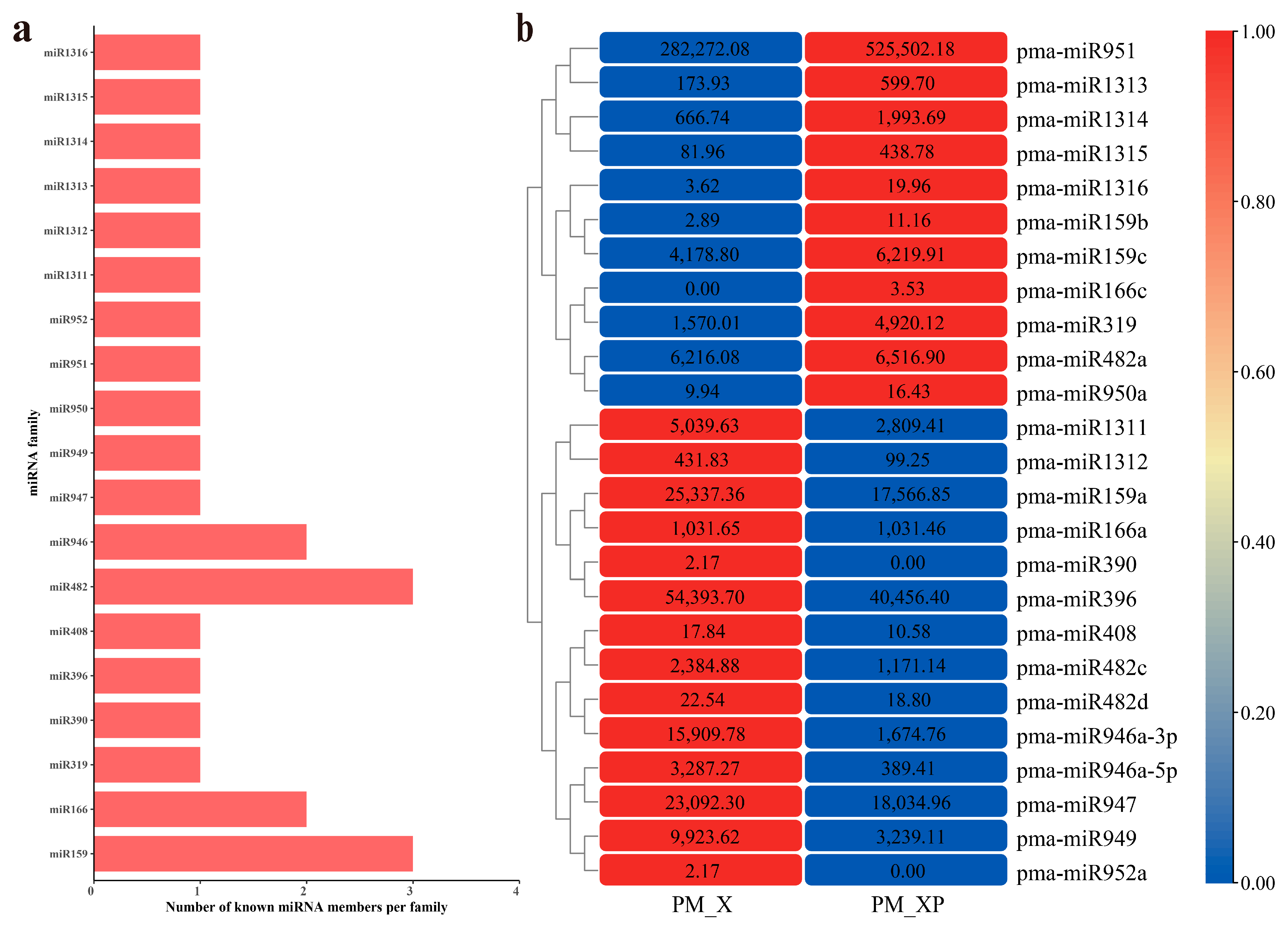
2.3. Differentially Expressed miRNAs (DEmiRNAs) between PM_XP and PM_X
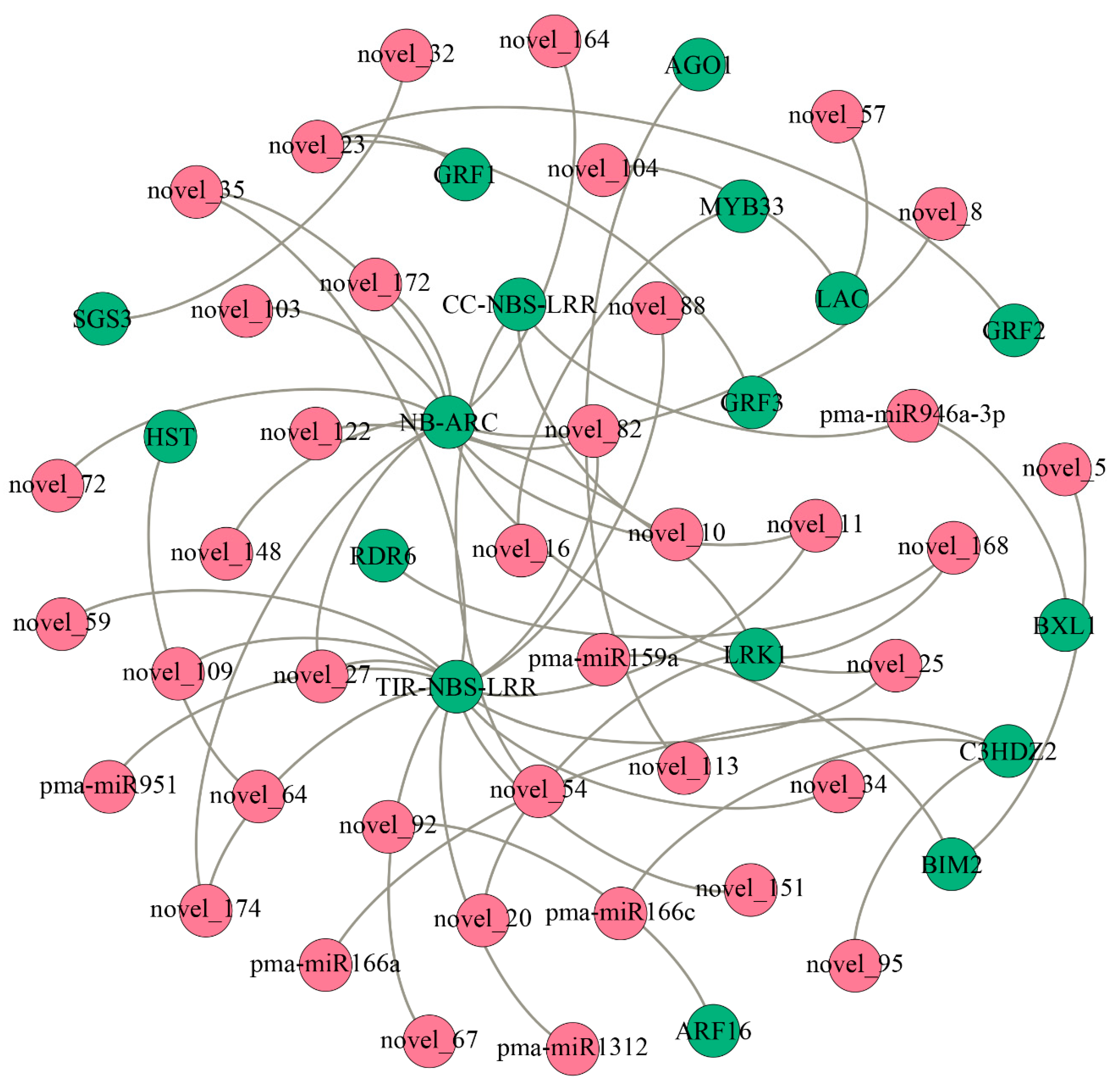
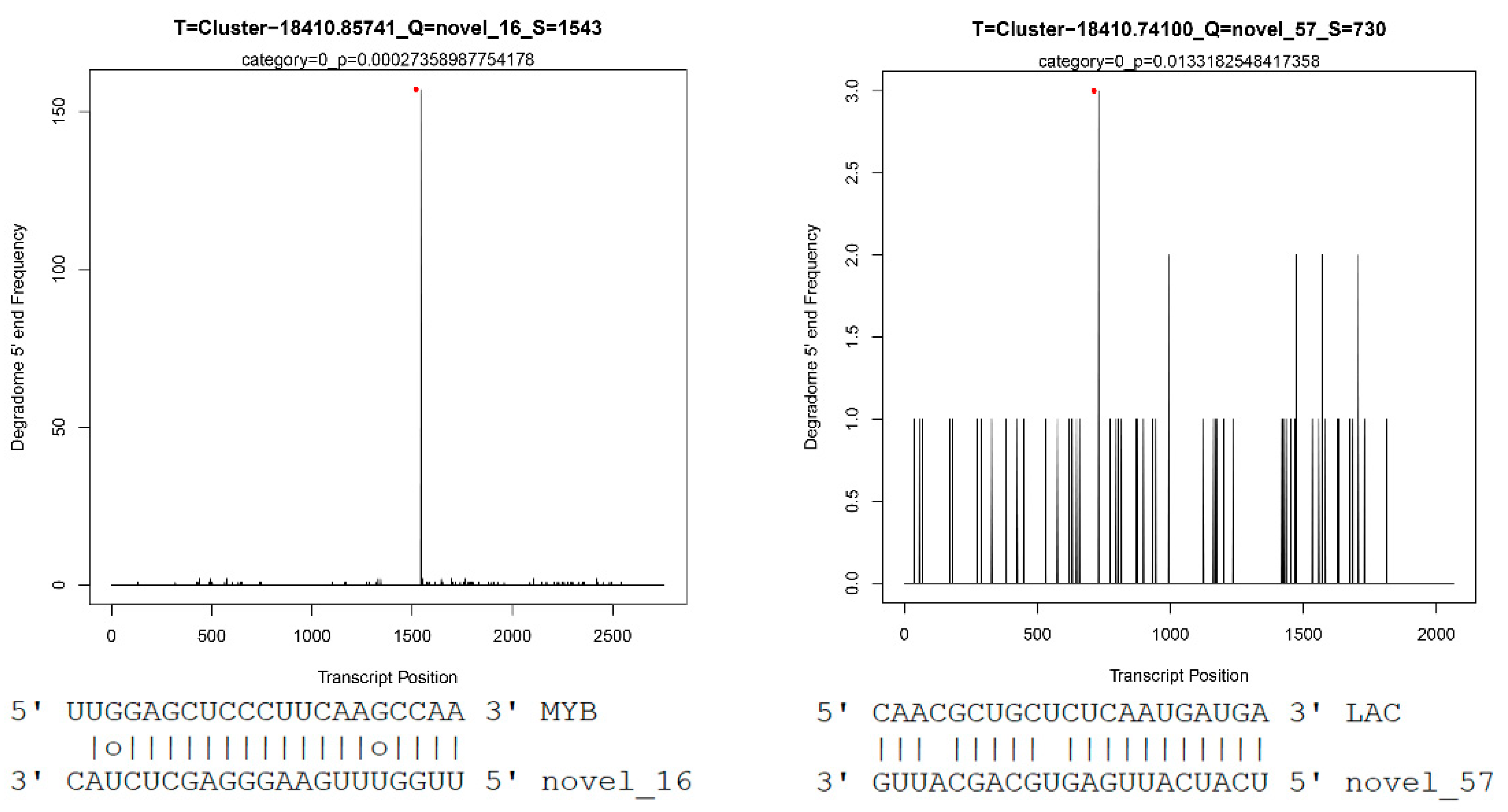
2.4. Gobal Analysis of miRNA Target Genes via Degradome Sequencing
2.5. Enrichment Statistics of miRNA Target Genes
3. Discussion
3.1. Mirna-Target Regulatory Pathways Related to the Stress Response in the Developing Xylem of Masson Pine
3.2. Conserved Mirna-Mrna Modules between Softwood and Hardwood Formation
4. Materials and Methods
4.1. Plant Materials and Sequencing
4.2. Identification of Conserved and Novel Mirnas
4.3. Identification of DEmiRNAs
4.4. Identification of miRNA Target Genes
4.5. Enrichment Analysis of miRNA Target Genes
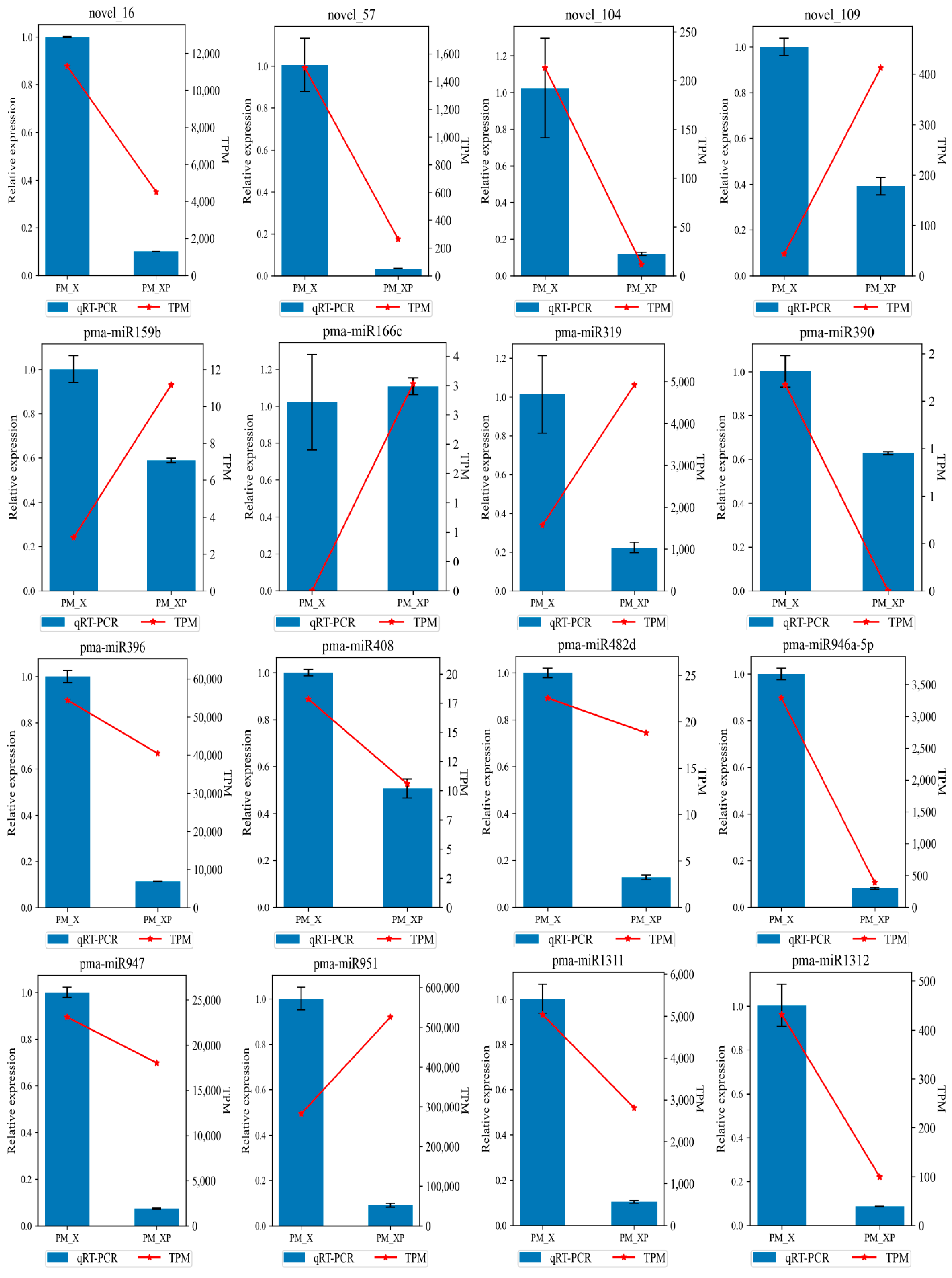
4.6. qRT-PCR Validation
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Fukuda, H.; Ohashi-Ito, K. Vascular tissue development in plants. Curr. Top. Dev. Biol. 2019, 131, 141–160. [Google Scholar]
- Venturas, M.D.; Sperry, J.S.; Hacke, U.G. Plant xylem hydraulics: What we understand, current research, and future challenges. J. Integr. Plant Biol. 2017, 59, 356–389. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mitsuda, N.; Seki, M.; Shinozaki, K.; Ohme-Takagi, M. The NAC Transcription Factors NST1 and NST2 of Arabidopsis Regulate Secondary Wall Thickenings and Are Required for Anther Dehiscence. Plant Cell 2005, 17, 2993–3006. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cosgrove, D.J. Loosening of plant cell walls by expansins. Nature 2000, 407, 321–326. [Google Scholar] [CrossRef]
- McFarlane, H.E.; Döring, A.; Persson, S. The cell biology of cellulose synthesis. Annu. Rev. Plant Biol. 2014, 65, 69–94. [Google Scholar] [CrossRef] [PubMed]
- Scheller, H.V.; Ulvskov, P. Hemicelluloses. Annu. Rev. Plant Biol. 2010, 61, 263–289. [Google Scholar] [CrossRef]
- Schädel, C.; Richter, A.; Blöchl, A.; Hoch, G. Hemicellulose concentration and composition in plant cell walls under extreme carbon source-sink imbalances. Physiol. Plant 2010, 139, 241–255. [Google Scholar] [CrossRef] [PubMed]
- Vaahtera, L.; Schulz, J.; Hamann, T. Cell wall integrity maintenance during plant development and interaction with the environment. Nat. Plants 2019, 5, 924–932. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Luo, L.; Zheng, L. Lignins: Biosynthesis and Biological Functions in Plants. Int. J. Mol. Sci. 2018, 19, 335. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhong, R.; Richardson, E.A.; Ye, Z.-H. Two NAC domain transcription factors, SND1 and NST1, function redundantly in regulation of secondary wall synthesis in fibers of Arabidopsis. Planta 2007, 225, 1603–1611. [Google Scholar] [CrossRef] [PubMed]
- Zhong, R.; Richardson, E.A.; Ye, Z.-H. The MYB46 transcription factor is a direct target of SND1 and regulates secondary wall biosynthesis in Arabidopsis. Plant Cell 2007, 19, 2776–2792. [Google Scholar] [CrossRef] [Green Version]
- Zhong, R.; Lee, C.; Zhou, J.; McCarthy, R.L.; Ye, Z.-H. A battery of transcription factors involved in the regulation of secondary cell wall biosynthesis in Arabidopsis. Plant Cell 2008, 20, 2763–2782. [Google Scholar] [CrossRef] [Green Version]
- Ko, J.-H.; Kim, W.-C.; Han, K.-H. Ectopic expression of MYB46 identifies transcriptional regulatory genes involved in secondary wall biosynthesis in Arabidopsis. Plant J. 2009, 60, 649–665. [Google Scholar] [CrossRef]
- Ko, J.-H.; Kim, W.-C.; Kim, J.-Y.; Ahn, S.-J.; Han, K.-H. MYB46-mediated transcriptional regulation of secondary wall biosynthesis. Mol. Plant 2012, 5, 961–963. [Google Scholar] [CrossRef] [Green Version]
- Ko, J.H.; Jeon, H.W.; Kim, W.C.; Kim, J.Y.; Han, K.H. The MYB46/MYB83-mediated transcriptional regulatory programme is a gatekeeper of secondary wall biosynthesis. Ann. Bot. 2014, 114, 1099–1107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakano, Y.; Nishikubo, N.; Goué, N.; Ohtani, M.; Yamaguchi, M.; Katayama, Y.; Demura, T. MYB transcription factors orchestrating the developmental program of xylem vessels in Arabidopsis roots. Plant Biotechnol. 2010, 27, 267–272. [Google Scholar] [CrossRef] [Green Version]
- McCarthy, R.L.; Zhong, R.; Ye, Z.-H. MYB83 Is a Direct Target of SND1 and Acts Redundantly with MYB46 in the Regulation of Secondary Cell Wall Biosynthesis in Arabidopsis. Plant Cell Physiol. 2009, 50, 1950–1964. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Axtell, M.J. Classification and Comparison of Small RNAs from Plants. Annu. Rev. Plant Biol. 2013, 64, 137–159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, Y.; Jia, T.; Chen, X. The ‘how’ and ‘where’ of plant microRNAs. New Phytol. 2017, 216, 1002–1017. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bartel, D.P. MicroRNAs: Genomics, Biogenesis, Mechanism, and Function. Cell 2004, 116, 281–297. [Google Scholar] [CrossRef] [Green Version]
- Xiao, M.; Li, J.; Li, W.; Wang, Y.; Wu, F.; Xi, Y.; Zhang, L.; Ding, C.; Luo, H.; Li, Y.; et al. MicroRNAs activate gene transcription epigenetically as an enhancer trigger. RNA Biol. 2017, 14, 1326–1334. [Google Scholar] [CrossRef] [PubMed]
- Liang, Y.; Zou, Q.; Yu, W. Steering Against Wind: A New Network of NamiRNAs and Enhancers. Genom. Proteom. Bioinform. 2017, 15, 331–337. [Google Scholar] [CrossRef] [PubMed]
- Allen, E.; Xie, Z.; Gustafson, A.M.; Carrington, J.C. microRNA-Directed Phasing during Trans-Acting siRNA Biogenesis in Plants. Cell 2005, 121, 207–221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.; Teng, C.; Xia, R.; Meyers, B.C. PhasiRNAs in Plants: Their Biogenesis, Genic Sources, and Roles in Stress Responses, Development, and Reproduction. Plant Cell 2020, 32, 3059–3080. [Google Scholar] [CrossRef] [PubMed]
- Lauressergues, D.; Couzigou, J.-M.; Clemente, H.S.; Martinez, Y.; Dunand, C.; Bécard, G.; Combier, J.-P. Primary transcripts of microRNAs encode regulatory peptides. Nature 2015, 520, 90–93. [Google Scholar] [CrossRef]
- Chen, Q.-J.; Deng, B.-H.; Gao, J.; Zhao, Z.-Y.; Chen, Z.-L.; Song, S.-R.; Wang, L.; Zhao, L.-P.; Xu, W.-P.; Zhang, C.-X.; et al. A miRNA-Encoded Small Peptide, vvi-miPEP171d1, Regulates Adventitious Root Formation. Plant Physiol. 2020, 183, 656–670. [Google Scholar] [CrossRef] [Green Version]
- Ye, Y.; Wang, J.; Ni, Z.; Meng, X.; Feng, Y.; Yang, Z.; Xu, L.-A. Small RNA and degradome sequencing reveal roles of miRNAs in strobilus development in masson pine (Pinus massoniana). Ind. Crop. Prod. 2020, 154, 112724. [Google Scholar] [CrossRef]
- Lee, R.C.; Feinbaum, R.L.; Ambros, V. The C. elegans heterochronic gene lin-4 encodes small RNAs with antisense complementarity to lin-14. Cell 1993, 75, 843–854. [Google Scholar] [CrossRef]
- Griffiths-Jones, S.; Grocock, R.J.; van Dongen, S.; Bateman, A.; Enright, A.J. miRBase: microRNA sequences, targets and gene nomenclature. Nucleic Acids Res. 2006, 34, D140–D144. [Google Scholar] [CrossRef]
- He, L.; Tang, R.; Shi, X.; Wang, W.; Cao, Q.; Liu, X.; Wang, T.; Sun, Y.; Zhang, H.; Li, R.; et al. Uncovering anthocyanin biosynthesis related microRNAs and their target genes by small RNA and degradome sequencing in tuberous roots of sweetpotato. BMC Plant Biol. 2019, 19, 232. [Google Scholar] [CrossRef]
- Dai, X.; Zhuang, Z.; Zhao, P.X. psRNATarget: A plant small RNA target analysis server (2017 release). Nucleic Acids Res. 2018, 46, W49–W54. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Addo-Quaye, C.; Miller, W.; Axtell, M.J. CleaveLand: A pipeline for using degradome data to find cleaved small RNA targets. Bioinformatics 2009, 25, 130–131. [Google Scholar] [CrossRef]
- Mullany, L.E.; Herrick, J.; Wolff, R.; Stevens, J.; Samowitz, W.; Slattery, M.L. MicroRNA transcription factor interactions and their combined effect on target gene expression in colon cancer cases. Genes Chromosomes Cancer 2018, 57, 192–202. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H.; Dai, X.; Huang, X.; Xu, M.; Wang, Q.; Yan, X.; Sederoff, R.R.; Li, Q. Single-cell RNA sequencing reveals a high-resolution cell atlas of xylem in Populus. J. Integr. Plant Biol. 2021. [Google Scholar] [CrossRef] [PubMed]
- Meyers, B.C.; Kozik, A.; Griego, A.; Kuang, H.; Michelmore, R.W. Genome-wide analysis of NBS-LRR-encoding genes in Arabidopsis. Plant Cell 2003, 15, 809–834. [Google Scholar] [CrossRef] [Green Version]
- Xia, R.; Xu, J.; Arikit, S.; Meyers, B.C. Extensive Families of miRNAs and PHAS Loci in Norway Spruce Demonstrate the Origins of Complex phasiRNA Networks in Seed Plants. Mol. Biol. Evol. 2015, 32, 2905–2918. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, F.; Pignatta, D.; Bendix, C.; Brunkard, J.O.; Cohn, M.M.; Tung, J.; Sun, H.; Kumar, P.; Baker, B. MicroRNA regulation of plant innate immune receptors. Proc. Natl. Acad. Sci. USA 2012, 109, 1790. [Google Scholar] [CrossRef] [Green Version]
- Arikit, S.; Xia, R.; Kakrana, A.; Huang, K.; Zhai, J.; Yan, Z.; Valdés-López, O.; Prince, S.; Musket, T.A.; Nguyen, H.T.; et al. An Atlas of Soybean Small RNAs Identifies Phased siRNAs from Hundreds of Coding Genes. Plant Cell 2014, 26, 4584–4601. [Google Scholar] [CrossRef] [Green Version]
- Zhai, J.; Jeong, D.H.; De Paoli, E.; Park, S.; Rosen, B.D.; Li, Y.; Gonzalez, A.J.; Yan, Z.; Kitto, S.L.; Grusak, M.A.; et al. MicroRNAs as master regulators of the plant NB-LRR defense gene family via the production of phased, trans-acting siRNAs. Genes Dev. 2011, 25, 2540–2553. [Google Scholar] [CrossRef] [Green Version]
- Fei, Q.; Xia, R.; Meyers, B.C. Phased, Secondary, Small Interfering RNAs in Posttranscriptional Regulatory Networks. Plant Cell 2013, 25, 2400–2415. [Google Scholar] [CrossRef] [Green Version]
- Zan, Y.; Ji, Y.; Zhang, Y.; Yang, S.; Song, Y.; Wang, J. Genome-wide identification, characterization and expression analysis of populusleucine-rich repeat receptor-like protein kinase genes. BMC Genom. 2013, 14, 318. [Google Scholar] [CrossRef] [Green Version]
- Torii, K.U. Leucine-rich repeat receptor kinases in plants: Structure, function, and signal transduction pathways. Int. Rev. Cytol. 2004, 234, 1–46. [Google Scholar]
- Hwang, S.-G.; Kim, D.S.; Jang, C.S. Comparative analysis of evolutionary dynamics of genes encoding leucine-rich repeat receptor-like kinase between rice and Arabidopsis. Genetica 2011, 139, 1023–1032. [Google Scholar] [CrossRef]
- Zhong, R.; McCarthy, R.L.; Lee, C.; Ye, Z.-H. Dissection of the transcriptional program regulating secondary wall biosynthesis during wood formation in poplar. Plant Physiol. 2011, 157, 1452–1468. [Google Scholar] [CrossRef] [Green Version]
- Chai, G.; Qi, G.; Cao, Y.; Wang, Z.; Yu, L.; Tang, X.; Yu, Y.; Wang, D.; Kong, Y.; Zhou, G. Poplar PdC3H17 and PdC3H18 are direct targets of PdMYB3 and PdMYB21, and positively regulate secondary wall formation in Arabidopsis and poplar. New Phytol. 2014, 203, 520–534. [Google Scholar] [CrossRef]
- Yang, L.; Zhao, X.; Ran, L.; Li, C.; Fan, D.; Luo, K. PtoMYB156 is involved in negative regulation of phenylpropanoid metabolism and secondary cell wall biosynthesis during wood formation in poplar. Sci. Rep. 2017, 7, 41209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Woodward, A.W.; Bartel, B. A Receptor for Auxin. Plant Cell 2005, 17, 2425–2429. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, B.; Li, Y.-H.; Wu, J.-Y.; Chen, Q.-Z.; Huang, X.; Chen, Y.-F.; Huang, X.-L. Over-expression of mango (Mangifera indica L.) MiARF2 inhibits root and hypocotyl growth of Arabidopsis. Mol. Biol. Rep. 2011, 38, 3189–3194. [Google Scholar] [CrossRef]
- Liu, P.-P.; Montgomery, T.A.; Fahlgren, N.; Kasschau, K.D.; Nonogaki, H.; Carrington, J.C. Repression of AUXIN RESPONSE FACTOR10 by microRNA160 is critical for seed germination and post-germination stages. Plant J. 2007, 52, 133–146. [Google Scholar] [CrossRef] [PubMed]
- Fu, C.; Sunkar, R.; Zhou, C.; Shen, H.; Zhang, J.-Y.; Matts, J.; Wolf, J.; Mann, D.G.J.; Stewart, C.N.; Tang, Y.; et al. Overexpression of miR156 in switchgrass (Panicum virgatum L.) results in various morphological alterations and leads to improved biomass production. Plant Biotechnol. J. 2012, 10, 443–452. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Z.-B.; Geng, X.; He, C.; Zhang, F.; Wang, R.; Horst, W.J.; Ding, Z. TAA1-regulated local auxin biosynthesis in the root-apex transition zone mediates the aluminum-induced inhibition of root growth in Arabidopsis. Plant Cell 2014, 26, 2889–2904. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peng, C.; Wang, X.; Feng, T.; He, R.; Zhang, M.; Li, Z.; Zhou, Y.; Duan, L. System Analysis of in Maize Internode Elongation. Biomolecules 2019, 9, 417. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, F.; Dixon, R.A. Lignin modification improves fermentable sugar yields for biofuel production. Nat. Biotechnol. 2007, 25, 759–761. [Google Scholar] [CrossRef]
- Novaes, E.; Kirst, M.; Chiang, V.; Winter-Sederoff, H.; Sederoff, R. Lignin and biomass: A negative correlation for wood formation and lignin content in trees. Plant Physiol. 2010, 154, 555–561. [Google Scholar] [CrossRef] [Green Version]
- Pedersen, J.F.; Vogel, K.P.; Funnell, D.L. Impact of Reduced Lignin on Plant Fitness. Crop Sci. 2005, 45, 812–819. [Google Scholar] [CrossRef] [Green Version]
- Xu, M.X. Establishment and Application of Transient Expression System in Protoplast of Pinus massoniana Lamb. Master’s Thesis, Nanjing Forestry University, Nanjing, China, 2019. [Google Scholar]
- Chen, C.; Zhong, Y.; Yu, F.; Xu, M. Deep sequencing identifies miRNAs and their target genes involved in the biosynthesis of terpenoids in Cinnamomum camphora. Ind. Crop. Prod. 2020, 145, 111853. [Google Scholar] [CrossRef]
- Tsuji, J.; Weng, Z. DNApi: A De Novo Adapter Prediction Algorithm for Small RNA Sequencing Data. PLoS ONE 2016, 11, e0164228. [Google Scholar] [CrossRef]
- Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet. J. 2011, 17, 10–12. [Google Scholar] [CrossRef]
- Wen, M.; Shen, Y.; Shi, S.; Tang, T. miREvo: An integrative microRNA evolutionary analysis platform for next-generation sequencing experiments. BMC Bioinform. 2012, 13, 140. [Google Scholar] [CrossRef] [Green Version]
- Kuang, Z.; Wang, Y.; Li, L.; Yang, X. miRDeep-P2: Accurate and fast analysis of the microRNA transcriptome in plants. Bioinformatics 2019, 35, 2521–2522. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, C.; Chen, H.; Zhang, Y.; Thomas, H.R.; Frank, M.H.; He, Y.; Xia, R. TBtools: An Integrative Toolkit Developed for Interactive Analyses of Big Biological Data. Mol. Plant 2020, 13, 1194–1202. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. fastp: An ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 2018, 34, i884–i890. [Google Scholar] [CrossRef] [PubMed]
- Götz, S.; García-Gómez, J.M.; Terol, J.; Williams, T.D.; Nagaraj, S.H.; Nueda, M.J.; Robles, M.; Talón, M.; Dopazo, J.; Conesa, A. High-throughput functional annotation and data mining with the Blast2GO suite. Nucleic Acids Res. 2008, 36, 3420–3435. [Google Scholar] [CrossRef]
- Wu, T.; Hu, E.; Xu, S.; Chen, M.; Guo, P.; Dai, Z.; Feng, T.; Zhou, L.; Tang, W.; Zhan, L.; et al. clusterProfiler 4.0: A universal enrichment tool for interpreting omics data. Innovation 2021, 2, 100141. [Google Scholar]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Category | PM1_X | PM2_X | PM1_XP | PM2_XP | Average |
|---|---|---|---|---|---|
| Raw Reads | 19,122,111 | 21,352,761 | 17,656,738 | 17,053,919 | - |
| Q20 | 97.67% | 97.70% | 97.67% | 97.67% | 97.68% |
| Q30 | 94.80% | 94.83% | 94.72% | 94.74% | 94.77% |
| Clean Reads | 18,456,504 | 20,654,598 | 17,068,608 | 16,485,018 | - |
| Clean Reads of sRNA | 14,681,663 | 17,072,786 | 9,997,932 | 10,846,339 | - |
| Mapped sRNA Reads | 11,619,254 | 12,936,717 | 7,439,780 | 7,686,301 | - |
| Mapped Rate | 79.14% | 75.77% | 74.41% | 70.87% | |
| Conserved miRNA Reads | 341,974 | 108,456 | 93,012 | 76,618 | - |
| Uniq Conserved miRNA Reads | 464 | 316 | 332 | 307 | - |
| Conserved miRNA Hairpin | 28 | 24 | 26 | 26 | - |
| Conserved miRNA Mature | 24 | 20 | 22 | 21 | - |
| Novel miRNA Reads | 371,500 | 182,024 | 52,850 | 50,364 | - |
| Uniq Novel miRNA Reads | 2454 | 1481 | 1556 | 1442 | - |
| Novel miRNA Mature | 165 | 162 | 152 | 153 | - |
| Novel miRNA Hairpin | 173 | 171 | 163 | 161 | - |
| miRNA | Target Transcript | Transcript Annotation | Degradome Category | DegradomePval |
|---|---|---|---|---|
| pma-miR951 | Cluster-18410.91163 | TIR-NBS-LRR | 0 | 0.022455112 |
| pma-miR946a-3p | Cluster-17513.1 | CC-NBS-LRR | 0 | 0.006001718 |
| pma-miR946a-3p | Cluster-18410.68882 | CC-NBS-LRR | 0 | 0.053772939 |
| pma-miR946a-3p | Cluster-6417.0 | CC-NBS-LRR | 0 | 0.003278143 |
| pma-miR946a-3p | Cluster-6417.1 | CC-NBS-LRR | 0 | 0.003550836 |
| pma-miR946a-3p | Cluster-6417.2 | CC-NBS-LRR | 0 | 0.006545539 |
| pma-miR946a-3p | Cluster-18410.69983 | BXL1 | 0 | 0.025393001 |
| pma-miR482a | Cluster-18410.4683 | Zinc Ion-binding protein | 0 | 0.009260186 |
| pma-miR396 | Cluster-18410.26942 | CYCT1-3 | 2 | 0.045402037 |
| pma-miR166c | Cluster-18410.59035 | C3HDZ2 | 0 | 0.00027359 |
| pma-miR166a | Cluster-18410.60044 | C3HDZ2 | 2 | 0.011549044 |
| pma-miR166a | Cluster-18410.65256 | C3HDZ2 | 2 | 0.022964707 |
| pma-miR159a | Cluster-18410.64102 | BIM3 | 1 | 0.000935432 |
| pma-miR1314 | Cluster-22970.0 | BAM3 | 0 | 0.00027359 |
| pma-miR1312 | Cluster-18410.35178 | TIR-NBS-LRR | 1 | 0.018543312 |
| novel_95 | Cluster-18410.58110 | C3HDZ2 | 0 | 0.000820545 |
| novel_92 | Cluster-18410.48111 | ARF16 | 0 | 0.00109391 |
| novel_92 | Cluster-18410.55513 | ARF16 | 0 | 0.002186624 |
| novel_92 | Cluster-18410.62143 | ARF16 | 0 | 0.000547105 |
| novel_92 | Cluster-18410.71517 | ARF16 | 0 | 0.001367201 |
| novel_92 | Cluster-18410.73418 | ARF16 | 0 | 0.005185428 |
| novel_92 | Cluster-18410.73420 | ARF16 | 0 | 0.003823454 |
| novel_92 | Cluster-18410.73422 | ARF16 | 0 | 0.004913182 |
| novel_82 | Cluster-18410.37093 | TIR-NBS-LRR | 1 | 0.022210506 |
| novel_82 | Cluster-18410.114635 | TIR-NBS-LRR | 1 | 0.031318655 |
| novel_82 | Cluster-18410.19951 | TIR-NBS-LRR | 0 | 0.011156025 |
| novel_67 | Cluster-18410.75829 | TIR-NBS-LRR | 2 | 0.034248531 |
| novel_59 | Cluster-18410.68783 | TIR-NBS-LRR | 2 | 0.034248531 |
| novel_59 | Cluster-18410.68790 | TIR-NBS-LRR | 0 | 0.000547105 |
| novel_57 | Cluster-18410.74100 | LAC | 0 | 0.013318255 |
| novel_54 | Cluster-18410.61870 | CC-NBS-LRR | 0 | 0.027257968 |
| novel_5 | Cluster-18410.33788 | BIM2 | 2 | 0.05642673 |
| novel_35 | Cluster-18410.30641 | TIR-NBS-LRR | 0 | 0.097521346 |
| novel_34 | Cluster-18410.90022 | TIR-NBS-LRR | 0 | 0.017359701 |
| novel_32 | Cluster-18410.57154 | SGS3 | 0 | 0.076790575 |
| novel_27 | Cluster-18410.37995 | TIR-NBS-LRR | 2 | 0.078095614 |
| novel_25 | Cluster-18410.82637 | TIR-NBS-LRR | 2 | 0.011549044 |
| novel_20 | Cluster-18410.84714 | LRK1 | 0 | 0.003550836 |
| novel_174 | Cluster-18410.33579 | TIR-NBS-LRR | 0 | 0.035210347 |
| novel_174 | Cluster-18410.101866 | TIR-NBS-LRR | 0 | 0.009531243 |
| novel_168 | Cluster-18410.19527 | RDR6 | 0 | 0.082080284 |
| novel_168 | Cluster-18410.92321 | LRK1 | 0 | 0.020044798 |
| novel_16 | Cluster-18410.85741 | MYB33 | 0 | 0.00027359 |
| novel_16 | Cluster-18410.85743 | MYB33 | 0 | 0.000547105 |
| novel_16 | Cluster-18410.27112 | MYB33 | 0 | 0.00109391 |
| novel_151 | Cluster-18410.19951 | TIR-NBS-LRR | 2 | 0.078095614 |
| novel_146 | Cluster-18410.109049 | TIR-NBS-LRR | 0 | 0.002186624 |
| novel_122 | Cluster-18410.49874 | LRK1 | 1 | 0.010241759 |
| novel_113 | Cluster-18410.63994 | AGO1 | 0 | 0.004913182 |
| novel_113 | Cluster-18410.63996 | AGO1 | 0 | 0.004640862 |
| novel_11 | Cluster-18410.33711 | TIR-NBS-LRR | 2 | 0.0673241 |
| novel_11 | Cluster-18410.33065 | TIR-NBS-LRR | 0 | 0.019776619 |
| novel_109 | Cluster-18410.27403 | TIR-NBS-LRR | 2 | 0.034248531 |
| novel_10 | Cluster-18410.113971 | CC-NBS-LRR | 0 | 0.000547105 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shen, T.; Xu, M.; Qi, H.; Feng, Y.; Yang, Z.; Xu, M. Uncovering miRNA-mRNA Regulatory Modules in Developing Xylem of Pinus massoniana via Small RNA and Degradome Sequencing. Int. J. Mol. Sci. 2021, 22, 10154. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms221810154
Shen T, Xu M, Qi H, Feng Y, Yang Z, Xu M. Uncovering miRNA-mRNA Regulatory Modules in Developing Xylem of Pinus massoniana via Small RNA and Degradome Sequencing. International Journal of Molecular Sciences. 2021; 22(18):10154. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms221810154
Chicago/Turabian StyleShen, Tengfei, Mengxuan Xu, Haoran Qi, Yuanheng Feng, Zhangqi Yang, and Meng Xu. 2021. "Uncovering miRNA-mRNA Regulatory Modules in Developing Xylem of Pinus massoniana via Small RNA and Degradome Sequencing" International Journal of Molecular Sciences 22, no. 18: 10154. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms221810154