3.1.3. Synthesis of Compounds 9–11, 13, 14, 16–18, 22–24, 29–31, 36
To a solution of compound
5–
7, 15, 2,3-indolo-olenolic acid [
64] or
25–
27 (1 mmol) in CH
2Cl
2 (20 mL) (COCl)
2 (3 mmol; 0.26 mL) was added and stirred at room temperature for 2 h. The mixture was concentrated to dryness under reduced pressure and the resulting acid chloride was dissolved in CH
2Cl
2 (10 mL), 3 drops of Et
3N and 1.5 mmol of the corresponding amine were added: (a)
N-methylpiperazine (for synthesis of compounds
9–
11,
16,
22,
29–
31,
36); (b) morpholine (for synthesis of compounds
13,
17, and
24); (c) piperazine (for synthesis of compounds
14,
18, and
23). After completion of the reactions (TLC control) the organic layers were treated with 5% HCl (3 × 50 mL) until neutral pH, dried over CaCl
2, and evaporated under reduced pressure. The residue was purified by column chromatography on Al
2O
3 using petroleum ether–CHCl
3 (10:1 to 0:10) as eluent.
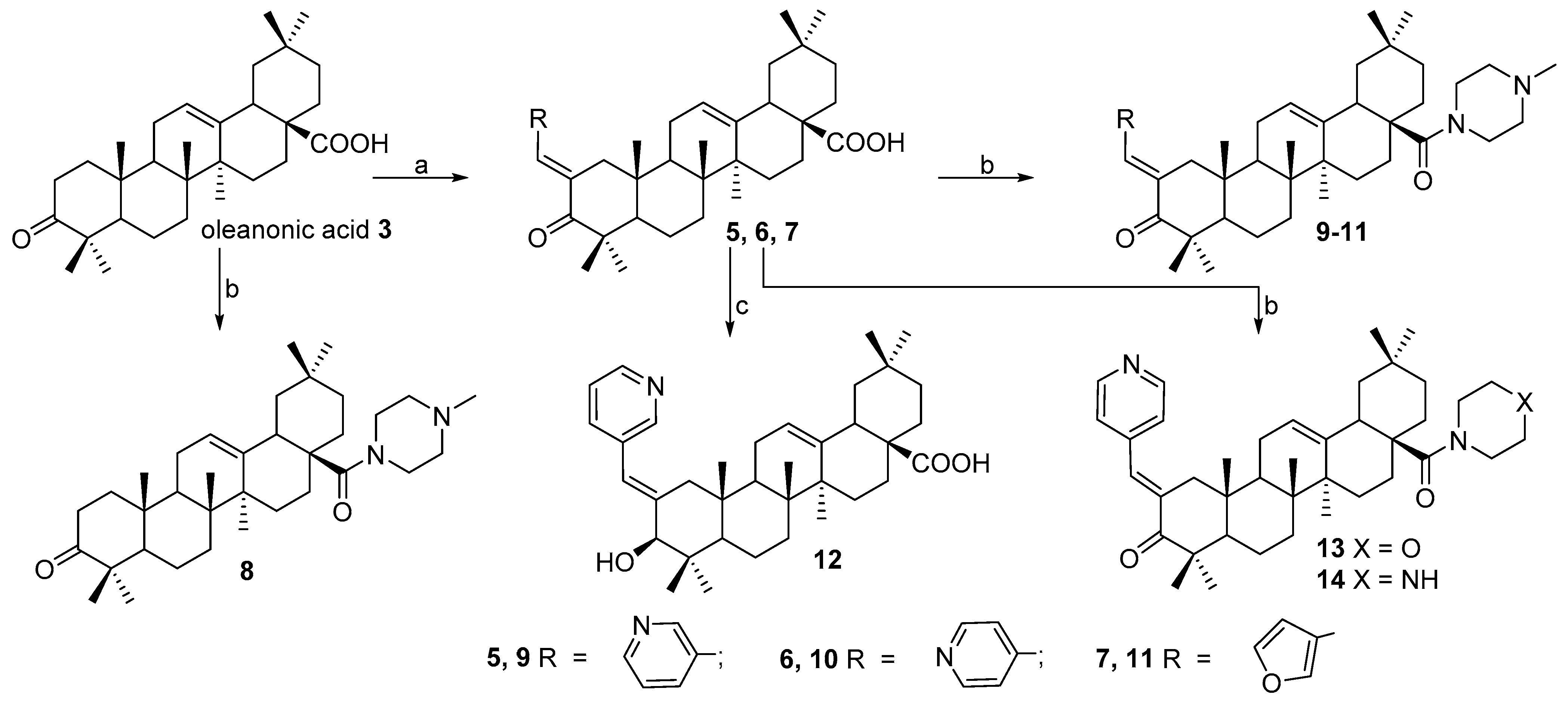
N-2-[3-(2E-Pyridinyl)-prop-2-en-1-one]-3-oxoolean-12-en-28-oyl)-methylpiperazine 9. Yield 0.54 g (87%). Rf 0.23; mp 177–178 °C. [α]D20 + 13.5 (c 0.1, CHCl3). 1H NMR (δ, ppm): 0.80, 0.90, 1.01, 1.11, 1.19, 1.35, 1.40 (7 s, 21H, 7CH3), 1.39–2.19 (m, 19H, CH and CH2), 2.30 (s, 3H, NCH3), 2.31–2.41 (m, 4H, 2CH2), 2.89–2.98 (m, 2H, H-1), 3.41–3.71 (m, 4H, 2CH2), 5.21 (s, 1H, H-12), 7.35 (m, 1H, Ar-CH), 7.41 (s, 1H, vinilic H), 7.73 (d, 1H, J 8 Hz), 8.52 (d, 1H, J 4.0 Hz, Ar-CH), 8.73 (s, 1H, Ar-CH). 13C NMR (δ, ppm): 15.5, 16.7, 17.5, 17.7, 18.5, 20.3, 21.3, 22.3, 22.7, 23.6, 24.5, 28.2, 29.7, 29.9, 30.6, 32.2, 34.2, 36.3, 38.7, 39.3, 39.6, 42.4, 44.2, 45.2, 45.3, 45.3, 46.0, 53.1, 55.1, 55.1, 123.4, 124.8 (C-12), 128.3, 131.8, 133.6, 137.1 (C-2), 144.4 (C-13), 149.1, 151.0, 175.1 (C-28), 207.4 (C-3). Analysis calculated for C41H59N3O2 (M 625.93): C 78.67, H 9.50, N 6.71; found: C 78.54, H 9.36, N 6.69. APCI (m/z): 626.73 (M + H)+ 100%.
N-2-[4-(2E-pyridinyl)-prop-2-en-1-one]-3-oxoolean-12-en-28-oyl)-methylpiperazine 10. Yield 0.56 g (90%). Rf 0.25; mp 156–157 °C. [α]D20 + 7 (c 0.5, CHCl3). 1H NMR (δ, ppm): 0.80, 0.90, 1.01, 1.11, 1.19, 1.34, 1.41 (7 s, 21H, 7CH3), 1.39–2.19 (m, 19H, CH and CH2), 2.31 (s, 3H, NCH3), 2.31–2.42 (m, 4H, 2CH2), 2.90–2.98 (m, 2H, H-1), 3.41–3.71 (m, 4H, 2CH2), 5.23 (s, 1H, H-12), 6.85 (s, 1H, H-1′), 7.02 (d, 2H, J 5.84 Hz, Ar-CH), 8.36 (d, 2H, J 5.68 Hz, Ar-CH). 13C NMR (δ, ppm): 15.5, 16.7, 17.5, 17.7, 18.5, 20.3, 21.3, 22.3, 22.7, 23.6, 24.5, 28.2, 29.7, 29.9, 30.6, 31.5, 32.2, 34.2, 36.3, 38.7, 39.3, 39.6, 42.4, 44.2, 45.2, 45.3, 45.3, 46.0, 53.1, 55.1, 55.1, 122.0, 122.0, 125.1 (C-12), 134.0 (C-2), 138.0, 143.8 (C-13), 149.4, 149.4, 175.8 (C-28), 207.4 (C-3). Analysis calculated for C41H59N3O2 (M 625.93): C 78.67, H 9.50, N 6.71; found: C 78.54, H 9.36, N 6.69. APCI (m/z): 626.73 (M + H)+ 100%.
N-2-[2-(2E-furyl)-prop-2-en-1-one]-3-oxo-olean-12-en-28-oyl)-methylpiperazine 11. Yield 0.58 g (95%). Rf 0.35; mp 160–161 °C. [α]D20 + 24° (c 0.5, CHCl3). 1H NMR (δ, ppm): 0.79, 0.87, 0.91, 1.09, 1.10, 1.29, 1.41 (7 s, 21H, 7CH3), 1.39–2.19 (m, 19H, CH and CH2), 2.31 (s, 3H, NCH3), 2.31–2.51 (m, 4H, 2CH2), 3.02–3.11 (m, 2H, H-1), 3.61–3.79 (m, 4H, 2CH2), 5.31 (s, 1H, H-12), 6.45–6.48 (m, 1H), 6.60 (d, 1H, J 3.4 Hz), 7.28–7.30 (m, 1H), 7.56 (s, 1H, H-1′). 13C NMR (δ, ppm): 15.66, 16.44, 20.42, 22.46, 22.50, 22.67, 23.63, 24.04, 25.73, 27.87, 28.60, 29.90, 30.39, 31.98, 33.05, 33.99, 34.00, 35.73, 38.95, 42.11, 43.78, 44.36, 44.80, 45.51, 45.68, 46.41, 47.48, 52.70, 54.99, 54.99, 112.20, 115.43, 121.35, 124.12 (C-12), 130.92 (C-2), 144.36 (C-13), 144.91, 152.57, 174.97 (C-28), 207.33 (C-3). Analysis calculated for C40H58N2O3 (M 614.92): C 78.13, H 9.51, N 4.56; found: C 78.54, H 9.72, N 4.63. APCI (m/z): 615.71 (M + H)+ 100%.
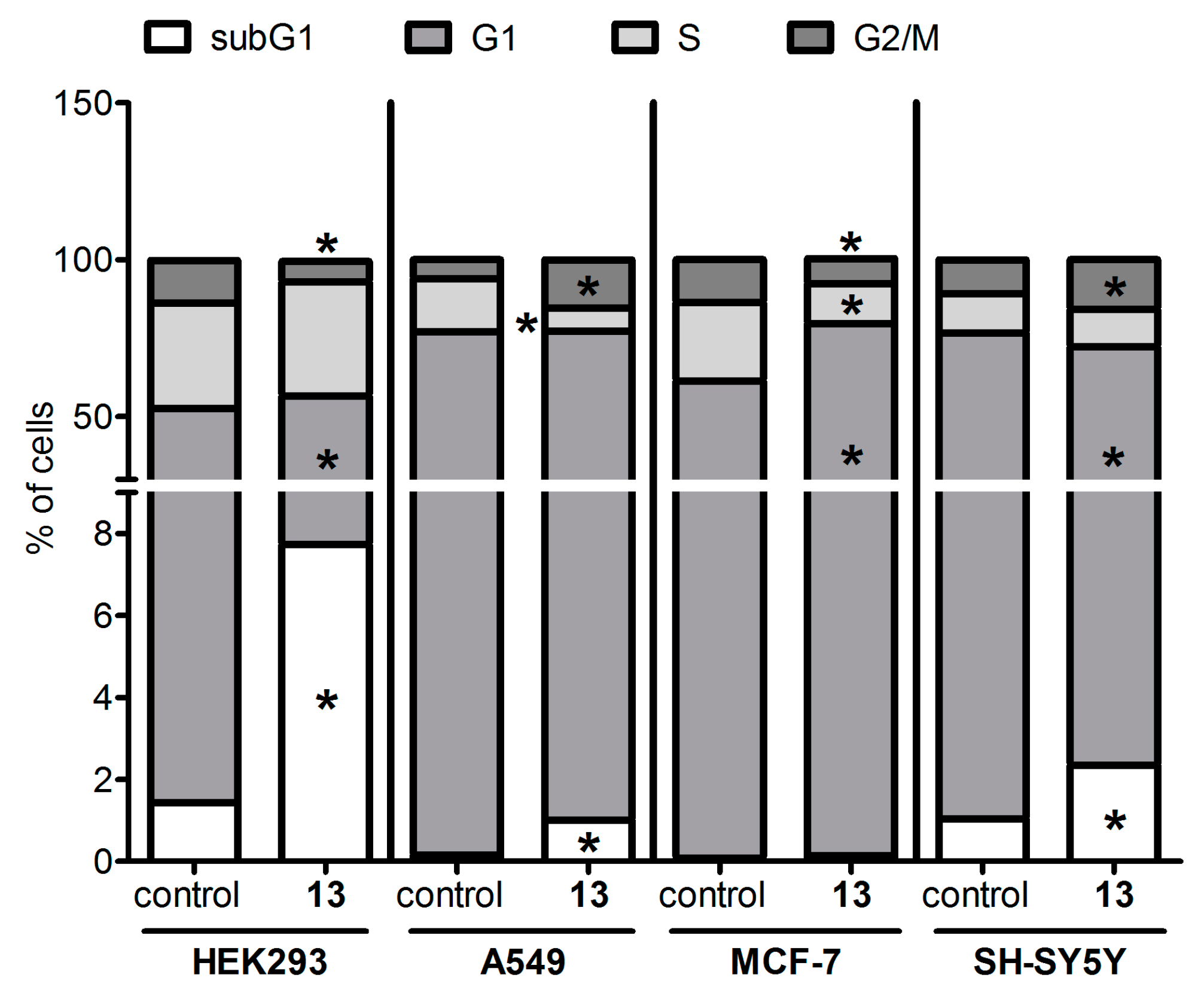
N-2-[4-(2E-pyridinyl)-prop-2-en-1-one]-3-oxo-olean-12-en-28-oyl)-morpholine 13. Yield 0.55 g (89%). Rf 0.30; mp 187–188 °C. [α]D20 + 34 (c 0.1, CHCl3). 1H NMR (δ, ppm): 0.79, 0.81, 0.85, 0.91, 1.09, 1.18, 1.21 (7 s, 21H, 7CH3), 1.39–3.12 (m, 19H, CH and CH2), 1.52–1.71 (m, 4H, 2CH2), 2.90–2.98 (m, 2H, H-1), 3.51–3.71 (m, 4H, 2CH2), 5.23 (s, 1H, H-12), 7.35 (d, 2H, J 5.6 Hz), 7.71 (s, 1H, vinilic H), 8.62 (d, 2H, J 7.2 Hz). 13C NMR (δ, ppm): 15.3, 16.5, 20.3, 22.6, 22.7, 23.2, 23.6, 24.0, 25.8, 27.8, 29.5, 29.6, 29.7, 29.8, 30.4, 31.9, 32.0, 33.0, 34.0, 36.4, 39.0, 42.1, 42.1, 43.7, 44.1, 45.2, 45.4, 45.5, 46.1, 46.3, 47.5, 53.0, 66.9, 66.9, 123 (C-12), 144.9 (C-13), 148.3 (C-1′), 150.7, 175.1 (C-28), 207.4 (C-3). Analysis calculated for C40H56N2O3 (M 612.88): C 78.39, H 9.21, N 4.57; found: C 78.38, H 9.19, N 4.56. APCI (m/z): 613.87 (M + H)+ 100%.
N-2-[4-(2E-pyridinyl)-prop-2-en-1-one]-3-oxo-olean-12-en-28-oyl)-piperazine 14. Yield 0.46 g (75%). Rf 0.15; mp 148–149 °C. [α]D20 + 14 (c 0.5, CHCl3). 1H NMR (δ, ppm): 0.78, 0.81, 0.85, 0.90, 1.09, 1.18, 1.21 (7 s, 21H, 7CH3), 1.22–2.15 (m, 19H, CH and CH2), 3.10–3.21 (m, 4H, 2CH2), 2.90–2.98 (m, 2H, H-1), 3.55–3.69 (m, 4H, 2CH2), 4.21 (br. s, 1H, NH), 5.23 (s, 1H, H-12), 7.35 (d, 2H, J 5.6 Hz), 7.70 (s, 1H, vinilic H), 8.63 (d, 2H, J 7.2 Hz). 13C NMR (δ, ppm): 15.3, 16.5, 20.3, 22.7, 22.8, 23.1, 23.6, 24.1, 25.8, 27.8, 29.5, 29.6, 29.7, 29.9, 30.5, 31.9, 32.0, 33.0, 34.0, 36.4, 39.0, 42.1, 42.1, 43.8, 44.1, 45.2, 45.4, 45.5, 46.1, 46.3, 47.5, 53.0, 58.5, 58.8, 123 (C-12), 144.9 (C-13), 148.3 (C-1′), 150.7, 175.6 (C-28), 207.5 (C-3). Analysis calculated for C40H57N3O2 (M 611.90): C 78.51, H 9.39, N 6.87; found: C 78.50, H 9.38, N 6.86. APCI (m/z): 612.87 (M + H)+ 100%.
N-(3β-Nicotinoyloxy-olean-12-en-28-oyl)-methylpiperazine 16. Yield 0.50 g (78%). Rf 0.35; mp 201–202 °C. [α]D20 + 7 (c 0.1, CHCl3). 1H NMR (δ, ppm): 0.76, 0.80, 0.84, 0.90, 1.09, 1.13, 1.17 (7 s, 21H, 7CH3), 1.31–2.20 (m, 24H, CH and CH2), 2.15–2.38 (m, 4H, 2CH2), 2.31 (s, 3H, NCH3), 3.61–3.78 (m, 4H, 2CH2), 5.31 (s, 1H, H-12), 7.42 (1 H, dd, J 4.9 Hz, 4.7, Harom), 8.23 (1 H, ddd, J 5.1 Hz, 2.1 Hz, 1.8 Hz, Harom), 8.76 (1 H, t, J 4.6 Hz, Harom), 9.22 (1 H, dd, J 1.9 Hz, 7.0 Hz, Harom). 13C NMR (125.5 MHz, CDCl3): 15.7, 16.9, 17.5, 19.3, 21.3, 23.3, 23.4, 23.7, 23.8, 28.3, 30.6, 31.0, 32.6, 34.0, 34.3, 35.6, 37.0, 37.4, 37.7, 38.8, 39.6, 41.5, 42.3, 45.3, 45.9, 46.1, 46.4, 48.6, 53.4, 55.2, 55.2, 83.0 (C-3), 121.4, 123.9 (C-12), 127.4, 138.5, 144.7 (C-13), 149.5, 151.8, 164.3, 176.0 (C-28). Analysis calculated for C41H61N3O3 (M 643.94): C 76.47, H 9.55, N 6.53; found: C 76.43, H 9.54, N 6.52. APCI (m/z): 644.92 (M + H)+ 100%.
N-(3β-Nicotinoyloxy-olean-12-en-28-oyl)-morpholine 17. Yield 0.54 g (85%). Rf 0.30; mp 197–198 °C. [α]D20 + 18 (c 0.5, CHCl3). 1H NMR (δ, ppm): 0.75, 0.81, 0.89, 0.91, 0.99, 1.02, 1.12 (7 s, 21H, 7CH3), 1.21–2.15 (m, 23H, CH and CH2), 3.01–3.11 (m, 4H, 2CH2), 3.52–3.69 (m, 4H, 2CH2), 4.75 (t, 1H, J = 8.4, H-3), 5.23 (s, 1H, H-12), 7.41 (1 H, dd, J 4.9 Hz, 4.7 Hz, Harom), 8.24 (1 H, ddd, J 5.1 Hz, 2.1 Hz, 1.8 Hz, Harom), 8.75 (1 H, t, J 4.6 Hz, Harom), 9.21 (1 H, dd, J 1.9 Hz, 7.0 Hz, Harom). 13C NMR (δ, ppm): 15.4, 16.7, 17.0, 18.2, 22.7, 23.4, 23.6, 24.1, 26.0, 27.9, 28.2, 30.0, 30.4, 32.7, 33.1, 34.0, 37.0, 37.8, 38.0, 38.1, 39.1, 41.9, 43.5, 46.0, 46.3, 47.4, 47.7, 55.4, 67.0, 67.0, 83.0 (C-3), 121.4, 123.9 (C-12), 127.4, 138.5, 144.7 (C-13), 149.5, 151.8, 164.3, 175.2 (C-28). Analysis calculated for C40H58N2O4 (M 630.90): C 76.15, H 9.27, N 4.44; found: C 76.13, H 9.26, N 4.44. APCI (m/z): 631.89 (M + H)+ 100%.
N-(3β-Nicotinoyloxy-olean-12-en-28-oyl)-piperazine 18. Yield 0.44 g (70%). Rf 0.30; mp 131–132 °C. [α]D20 + 17 (c 0.5, CHCl3). 1H NMR (δ, ppm): 0.76, 0.82, 0.89, 0.92, 0.99, 1.03, 1.15 (7 s, 21H, 7CH3), 1.23–2.16 (m, 23H, CH and CH2), 3.10–3.21 (m, 4H, 2CH2), 3.55–3.69 (m, 4H, 2CH2), 4.21 (br. s, 1H, NH), 4.76 (t, 1H, J 8.4 Hz, H-3), 5.24 (s, 1H, H-12), 7.40 (1 H, dd, J 4.9 Hz, 4.7 Hz, Harom), 8.25 (1 H, ddd, J 5.1 Hz, 2.1 Hz, 1.8 Hz, Harom), 8.76 (1 H, t, J 4.6 Hz, Harom), 9.22 (1 H, dd, J 1.9 Hz, 7.0 Hz, Harom). 13C NMR (125.5 MHz, CDCl3): 15.4, 16.7, 17.1, 18.2, 22.8, 23.4, 23.7, 24.1, 26.1, 27.9, 28.3, 30.1, 30.4, 31.0, 32.8, 33.1, 34.1, 37.0, 37.8, 38.0, 38.2, 39.1, 41.9, 43.6, 46.0, 46.3, 47.4, 47.8, 58.6, 58.6, 83.0 (C-3), 121.4, 124.0 (C-12), 127.5, 138.5, 144.5 (C-13), 149.5, 151.8, 164.3, 176.5 (C-28). Analysis calculated for C40H59N3O3 (M 629.92): C 76.27, H 9.44, N 6.67; found: C 76.26, H 9.42, N 6.65. APCI (m/z): 630.91 (M + H)+ 100%.
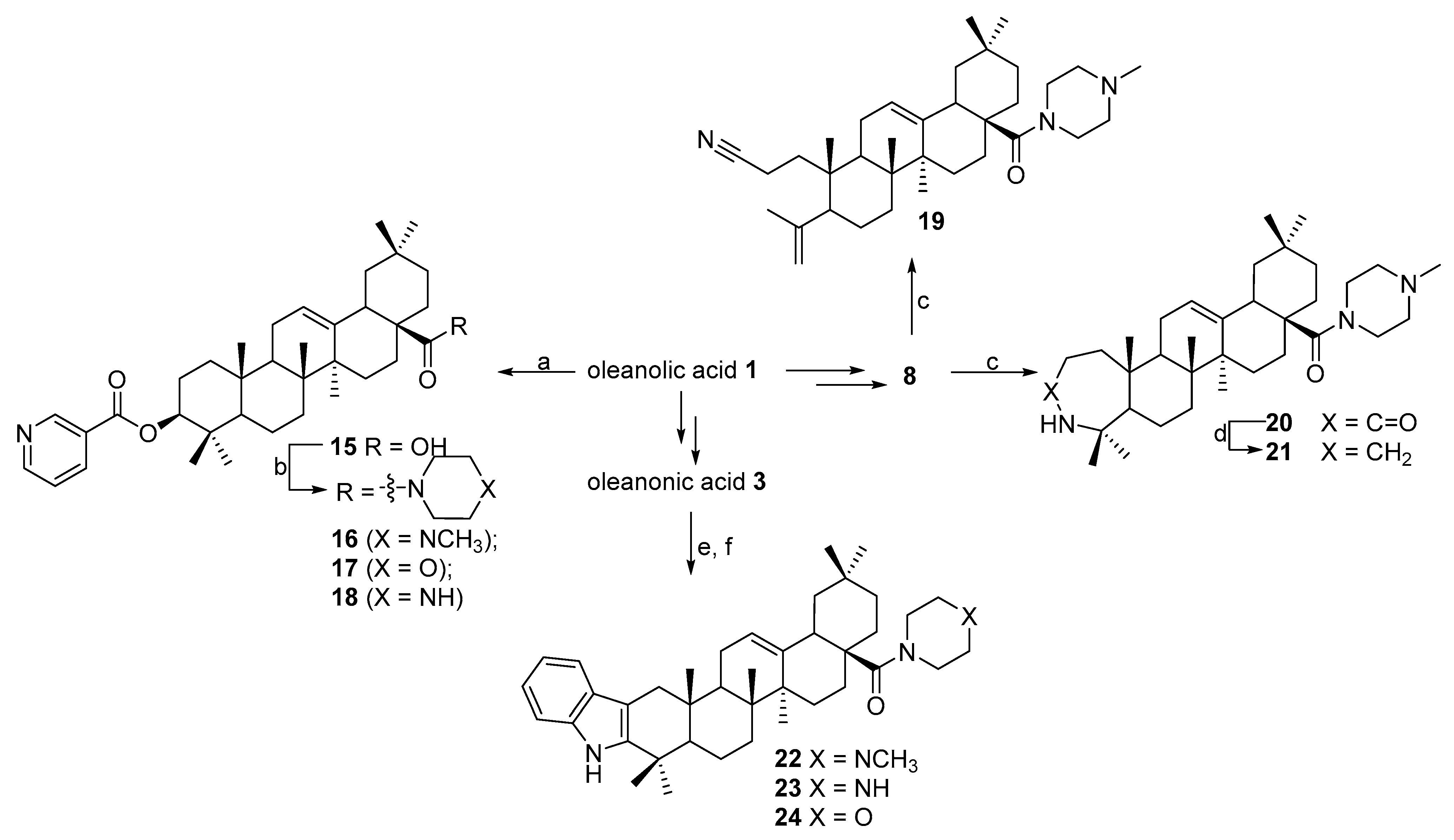
N-([3,2b]-Indolo-olean-12-en-28-oyl)-N-methylpiperazine 22. Yield 0.54 g (90%). Rf 0.35; mp 174–175 °C. [α]D20 + 63 (c 0.1, CHCl3). 1H NMR (500 MHz, CDCl3): 0.83, 0.91, 0.99, 1.01, 1.10, 1.15, 1.32 (7 s, 21H, 7CH3), 1.31–2.20 (m, 19H, CH and CH2), 2.15–2.38 (m, 4H, 2CH2), 2.31 (s, 3H, NCH3), 2.71–2.95 (m, 2H, H-1), 3.61–3.78 (m, 4H, 2CH2), 5.31 (s, 1H, H-12), 7.07–7.44 (m, 4Harom, 4CH), 8.10 (br. s, 1H, NH). 13C NMR (125.5 MHz, CDCl3): 15.7, 16.9, 17.5, 19.3, 21.3, 23.3, 23.4, 23.7, 23.8, 28.3, 30.6, 31.0, 32.6, 34.0, 34.3, 35.6, 37.0, 37.4, 38.8, 39.6, 41.5, 42.3, 45.3, 45.9, 46.1, 46.4, 48.6, 53.4, 55.2, 55.2, 106.8, 110.4 (C-2), 118.0, 118.8, 120.8, 121.9 (C-12), 128.3, 136.2, 141.0 (C-3), 144.6 (C-13), 175.1 (C-28). Analysis calculated for C41H59N3O (M 609.47): C 80.74, H 9.75, N 6.89; found: C 80.72, H 9.73, N 6.85. APCI (m/z): 610.47 (M + H)+ 100%.
N-([3,2b]-Indolo-olean-12-en-28-oyl)-piperazine 23. Yield 0.51 g (85%). Rf 0.20; mp 205–206 °C. [α]D20 + 34 (c 0.5, CHCl3). 1H NMR (500 MHz, CDCl3): 0.84, 0.91, 0.99, 1.02, 1.11, 1.15, 1.31 (7 s, 21H, 7CH3), 1.31–2.20 (m, 19H, CH and CH2), 3.10–3.21 (m, 4H, 2CH2), 2.80–2.95 (m, 2H, H-1), 3.55–3.69 (m, 4H, 2CH2), 4.21 (br. s, 1H, NH), 5.31 (s, 1H, H-12), 7.07–7.44 (m, 4Harom, 4CH), 8.10 (br. s, 1H, NH). 13C NMR (125.5 MHz, CDCl3): 15.7, 16.9, 17.5, 19.3, 21.3, 23.3, 23.4, 23.7, 23.8, 28.3, 30.6, 31.0, 32.6, 34.0, 34.3, 35.6, 37.0, 37.4, 38.8, 39.6, 41.5, 42.3, 45.3, 45.9, 46.4, 48.6, 53.4, 55.2, 55.2, 106.8, 110.4 (C-2), 118.0, 118.8, 120.8, 121.9 (C-12), 128.3, 136.2, 141.0 (C-3), 143.9 (C-13), 176.2 (C-28). Analysis calculated for C40H57N3O (M 595.90): C 80.62, H 9.64, N 7.05; found: C 80.60, H 9.63, N 7.04. APCI (m/z): 596.89 (M + H)+ 100%.
N-([3,2b]-Indolo-olean-12-en-28-oyl)-morpholine 24. Yield 0.52 g (87%). Rf 0.25; mp 190–191 °C. [α]D20 + 56 (c 0.1, CHCl3). 1H NMR (500 MHz, CDCl3): 0.84, 0.92, 0.99, 1.02, 1.11, 1.15, 1.31 (7 s, 21H, 7CH3), 1.31–2.20 (m, 19H, CH and CH2), 3.01–3.11 (m, 4H, 2CH2), 2.71–2.95 (m, 2H, H-1), 3.52–3.69 (m, 4H, 2CH2), 5.26 (s, 1H, H-12), 7.08–7.45 (m, 4Harom, 4CH), 8.09 (br. s, 1H, NH). 13C NMR (125.5 MHz, CDCl3): 15.8, 16.9, 17.6, 19.3, 21.3, 23.3, 23.4, 23.8, 28.3, 30.7, 31.0, 32.6, 34.0, 34.3, 35.6, 37.0, 37.4, 38.8, 39.6, 41.5, 42.3, 45.3, 45.9, 46.1, 46.4, 48.6, 55.4, 67.0, 67.0, 106.8, 111.0 (C-2), 118.0, 118.6, 120.8, 121.9 (C-12), 128.3, 136.2, 141.0 (C-3), 144.6 (C-13), 176.2 (C-28). Analysis calculated for C40H56N2O2 (M 596.88): C 80.50, H 9.46, N 4.69; found: C 80.49, H 9.45, N 4.68. APCI (m/z): 597.87 (M + H)+ 100%.
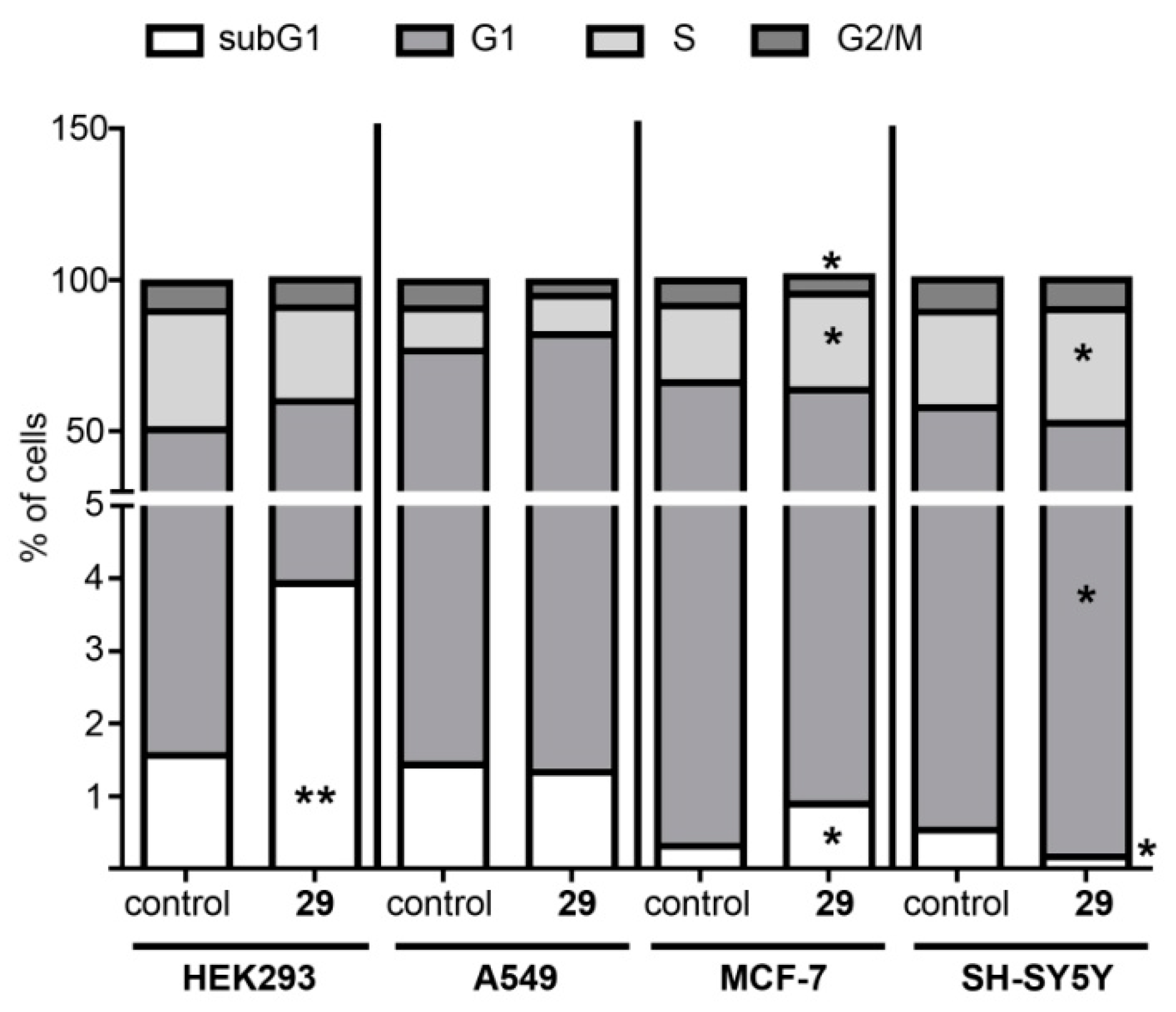
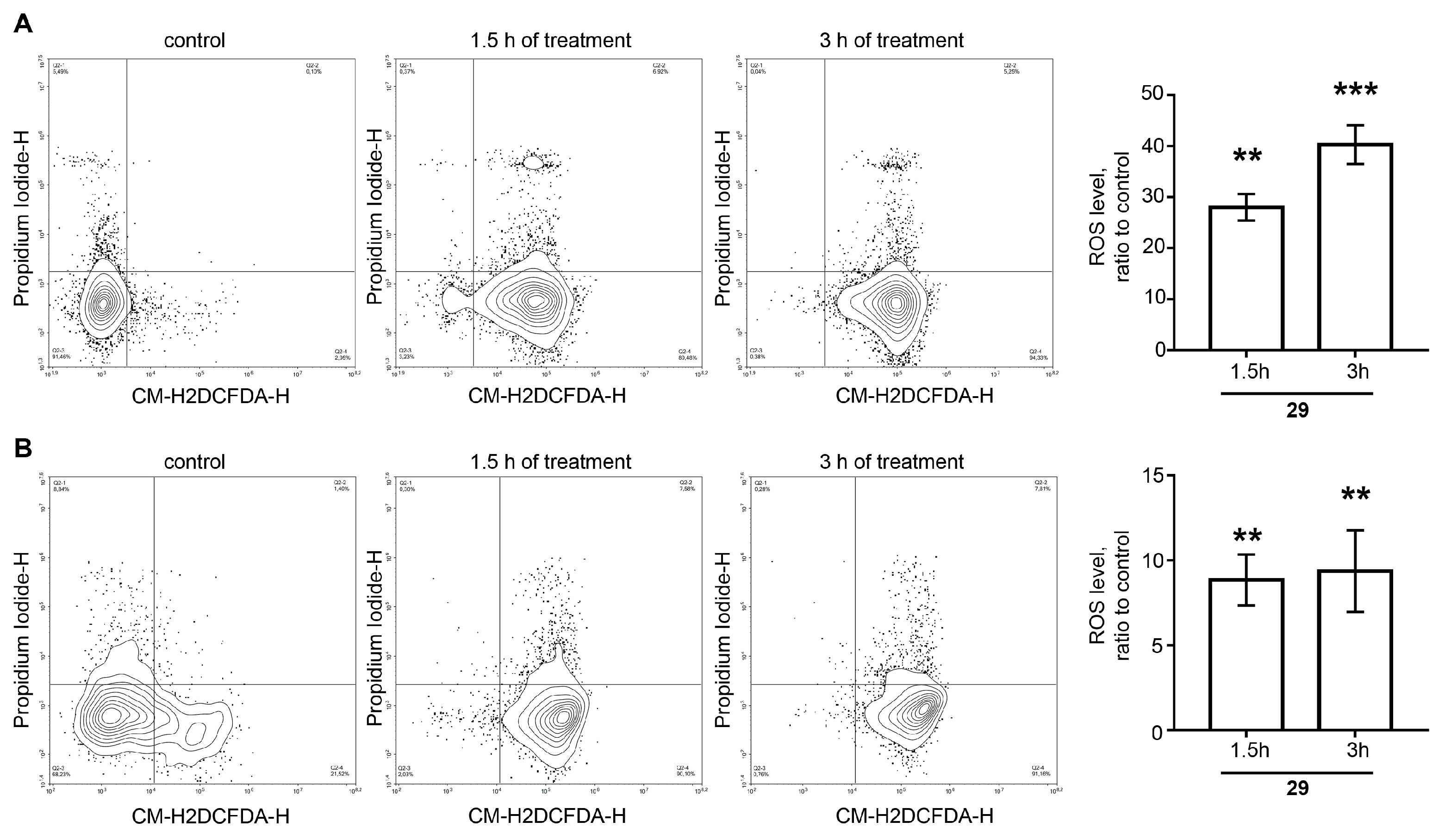
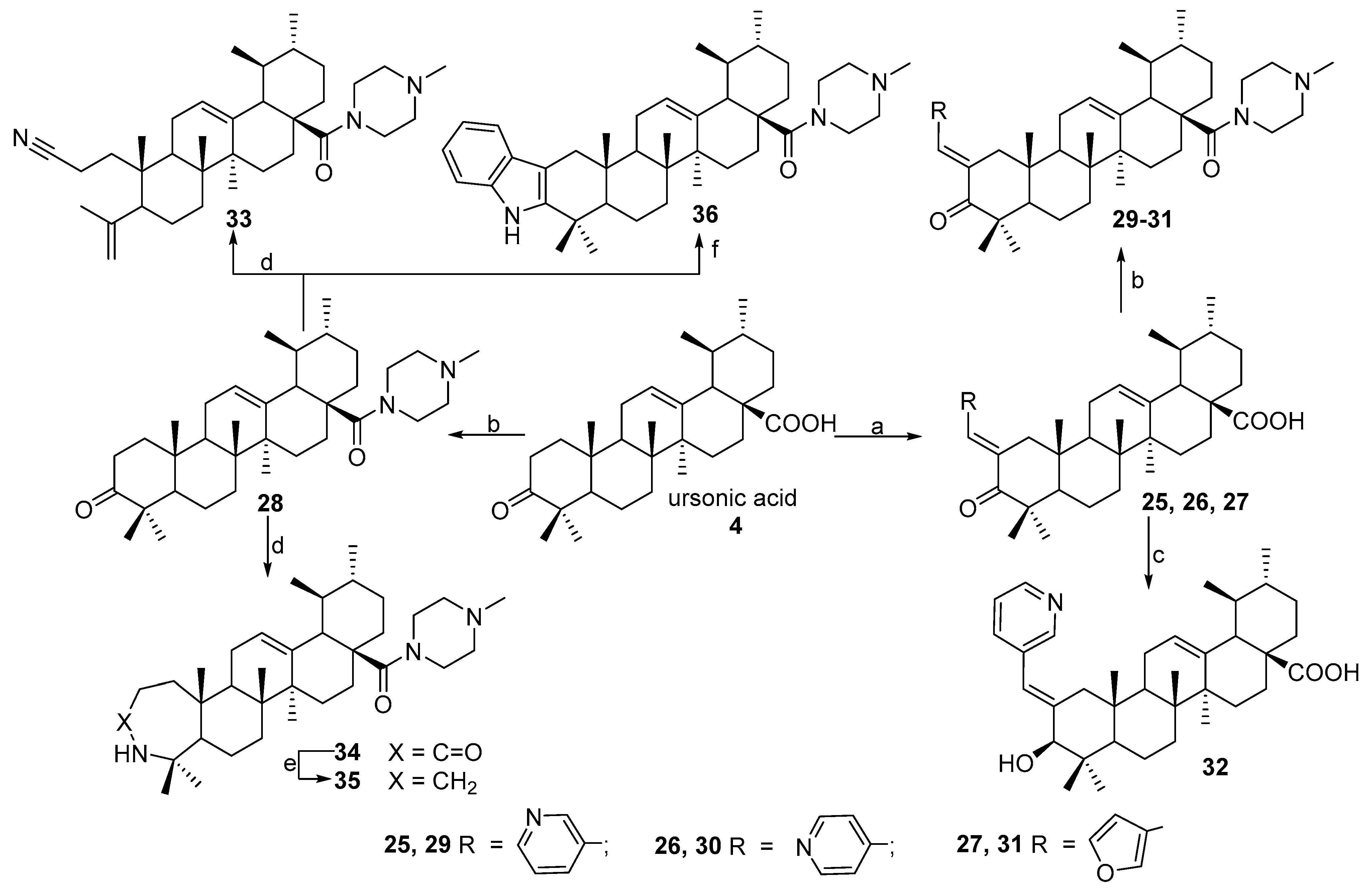
N-2-[3-(2E-pyridinyl)-prop-2-en-1-one]-3-oxoursan-12-en-28-oyl)-methylpiperazine 29. Yield 0.53 g (85%). Rf 0.32; mp 167–168°C. [α]D20 + 24 (c 0.1, CHCl3). 1H NMR (δ, ppm): 0.80, 0.90, 1.01, 1.11, 1.19, 1.35, 1.40 (7 s, 21H, 7CH3), 1.39–2.19 (m, 19H, CH and CH2), 2.30 (s, 3H, NCH3), 2.31–2.41 (m, 4H, 2CH2), 2.89–2.98 (m, 2H, H-1), 3.41–3.71 (m, 4H, 2CH2), 5.21 (s, 1H, H-12), 7.35 (m, 1H, Ar-CH), 7.41 (s, 1H, vinilic H), 7.73 (d, 1H, J 8 Hz), 8.52 (d, 1H, J 4.0 Hz, Ar-CH), 8.73 (s, 1H, Ar-CH). 13C NMR (δ, ppm): 15.5, 16.7, 17.5, 17.7, 18.5, 20.3, 21.3, 22.3, 22.7, 23.6, 24.5, 28.2, 29.7, 29.9, 30.6, 32.2, 34.2, 36.3, 38.7, 39.3, 39.6, 42.4, 44.2, 45.2, 45.3, 45.3, 46.0, 53.1, 55.1, 55.1, 123.4, 124.8 (C-12), 128.3, 131.8, 133.6, 135.9 (C-13), 137.1 (C-2), 149.1, 151.0, 175.1 (CON), 207.4 (C-3). Analysis calculated for C41H59N3O2 (M 625.93): C 78.67, H 9.50, N 6.71; found: C 78.54, H 9.36, N 6.69. APCI (m/z): 626.73 (M + H)+ 100%.
N-2-[4-(2E-pyridinyl)-prop-2-en-1-one]-3-oxoursan-12-en-28-oyl)-methylpiperazine 30. Yield 0.53 g (85%). Rf 0.30; mp 150–151 °C. [α]D20 + 8 (c 0.5, CHCl3). 1H NMR (δ, ppm): 0.80, 0.90, 1.01, 1.11, 1.19, 1.34, 1.41 (7 s, 21H, 7CH3), 1.39–2.19 (m, 19H, CH and CH2), 2.31 (s, 3H, NCH3), 2.31–2.42 (m, 4H, 2CH2), 2.90–2.98 (m, 2H, H-1), 3.41–3.71 (m, 4H, 2CH2), 5.23 (s, 1H, H-12), 6.85 (s, 1H, vinilic H), 7.02 (d, 2H, J 5.84 Hz, Ar-CH), 8.36 (d, 2H, J 5.68 Hz, Ar-CH). 13C NMR (δ, ppm): 15.5, 16.7, 17.5, 17.7, 18.5, 20.3, 21.3, 22.3, 22.7, 23.6, 24.5, 28.2, 29.7, 29.9, 30.6, 31.5, 32.2, 34.2, 36.3, 38.7, 39.3, 39.6, 42.4, 44.2, 45.2, 45.3, 45.3, 46.0, 53.1, 55.1, 55.1, 122.0, 122.0, 125.1 (C-12), 134.0 (C-2), 138.0 (C-13), 143.8, 149.4, 149.4, 175.8 (CON), 207.4 (C-3). Analysis calculated for C41H59N3O2 (M 625.93): C 78.67, H 9.50, N 6.71; found: C 78.54, H 9.36, N 6.69. APCI (m/z): 626.73 (M + H)+ 100%.
N-2-[3-(2E-furyl)-prop-2-en-1-one]-3-oxoursan-12-en-28-oyl)-methylpiperazine 31. Yield 0.55 g (90%). Rf 0.28; mp 147–148 °C. [α]D20 + 73 (c 0.5, CHCl3). 1H NMR (δ, ppm): 0.79, 0.87, 0.91, 1.09, 1.15, 1.28, 1.41 (7 s, 21H, 7CH3), 1.39–2.19 (m, 19H, CH and CH2), 2.31 (s, 3H, NCH3), 2.36–2.49 (m, 4H, 2CH2), 3.08–3.12 (m, 2H, H-1), 3.61–3.72 (m, 4H, 2CH2), 5.29 (s, 1H, H-12), 6.45–6.48 (m, 1H, furf-CH), 6.58 (d, 1H, J 3.4 Hz, furf-CH), 7.27–7.29 (m, 1H, furf-CH), 7.55 (s, 1H, vinilic H). 13C NMR (δ, ppm): 15.8, 16.6, 17.5, 20.4, 21.3, 22.5, 22.6, 23.6, 23.6, 28.2, 29.9, 30.5, 31.4, 32.1, 34.3, 34.4, 35.7, 38.7, 39.3, 39.5, 39.6, 42.3, 44.3, 44.8, 44.9, 45.3, 45.7, 52.8, 54.9, 54.9, 112.2, 115.5, 124.2, 125.1 (C-12), 130.9 (C-2), 138.1 (C-13), 144.4, 152.6, 175.3 (C-28), 207.2 (C-3). Analysis calculated for C40H58N2O3 (M 614.92): C 78.13, H 9.51, N 4.56; found: C 78.10, H 9.48, N 4.50. APCI (m/z): 615.85 (M + H)+ 100%.
N-([3,2b]-Indolo-ursan-12-en-28-oyl)-methylpiperazine 36. Yield 0.53 g (88%). Rf 0.30; mp 195–196 °C. [α]D20 + 36 (c 0.1, CHCl3). 1H NMR (500 MHz, CDCl3): 0.80, 0.91, 0.99, 1.10, 1.20, 1.32, 1.41 (7 s, 21H, 7CH3), 1.35–1.86 (m, 19H, CH and CH2), 2.15–2.38 (m, 4H, 2CH2), 2.35 (s, 3H, NCH3), 2.75–2.98 (m, 2H, H-1), 3.61–3.78 (m, 4H, 2CH2), 5.35 (s, 1H, H-12), 7.07–7.44 (m, 4Harom, 4CH), 7.85 (br. s, 1H, NH). 13C NMR (125.5 MHz, CDCl3): 15.7, 16.9, 17.5, 19.3, 21.3, 23.3, 23.4, 23.7, 23.8, 28.3, 30.6, 31.0, 32.6, 34.0, 34.3, 35.6, 37.0, 37.4, 38.1, 38.8, 39.6, 41.5, 42.3, 45.3, 45.9, 46.1, 46.4, 48.6, 53.3, 55.1, 55.1, 106.9, 110.3 (C-2), 117.9, 118.9, 120.9, 125.5 (C-12), 128.3, 135.6 (C-13), 136.1, 140.9 (C-3), 175.4 (C-28). Analysis calculated for C41H59N3O (M 609.47): C 80.74, H 9.75, N 6.89; found: C 80.73, H 9.74, N 6.85. APCI (m/z): 610.47 (M + H)+ 100%.
3.1.5. Synthesis of Compounds 19, 20, 33 and 34
To a solution of 3-oximino-derivatives (1 mmol), obtained from 8 or 28, in dry dioxane (15 mL) SOCl2 (0.4 mL) was added and the mixture was stirred for 30 min, then poured into H2O (50 mL); the precipitate was filtered, washed with water and dried. The residue was purified by column chromatography on Al2O3 eluting using petroleum ether—ethyl acetate (10:1 to 0:10) as eluent.
N-(3,4-Seco-2-cyano-olean-4(23),12(13)-diene-28-oyl)-methylpiperaine 19. Yield 0.16 g (30%). Rf 0.82; mp 200–201 °C. [α]D20 + 10 (c 0.1, CHCl3). 1H NMR (δ, ppm): 0.72, 0.81, 0.83, 0.84, 1.12, 1.71 (6 s, 18H, 6CH3), 1.12–2.41 (m, 23H, CH and CH2), 2.26 (s, 3H, NCH3), 2.52–2.48 (m, 4H, 2CH2), 3.52–3.70 (m, 4H, 2CH2), 4.61 and 4.87 (both d, 2J = 0.8 Hz, 2H, H-24), 5.25 (s, 1H, H-12). 13C NMR (δ, ppm): 11.5, 16.9, 19.1, 20.5, 22.6, 23.0, 23.6, 24.0, 24.2, 25.8, 26.4, 27.9, 29.8, 30.4, 31.5, 33.0, 34.0, 34.4, 38.0, 38.8, 39.5, 42.3, 43.6, 45.2, 45.9, 46.3, 47.3, 50.7, 55.1, 114.1, 120.3, 120.7 (C-12), 145.0 (C-13), 146.9, 174.9 (C-28). Analysis calculated for C35H55N3O (M 533.83): C 78.75, H 10.39, N 7.87; found: C 78.74, H 10.36, N 7.85. APCI (m/z): 534.43 (M + H)+ 100%.
N-(3-Oxo-3a-homo-3a-aza-olean-12-en-28-oyl)-methylpiperazine 20. Yield 0.38 g (68%). Rf 0.20; mp 156–157 °C. [α]D20 + 8 (c 0.1, CHCl3). 1H NMR (δ, ppm): 0.78, 0.82, 0.89, 0.99, 1.01, 1.15, 1.31 (7 s, 21H, 7CH3), 1.21–2.21 (m, 23H, CH and CH2), 2.31 (s, 3H, NCH3), 2.31–2.52 (m, 4H, 2CH2), 3.41–3.71 (m, 4H, 2CH2), 5.28 (s, 1H, H-12), 5.62 (br. s, 1H, NH). 13C NMR (δ, ppm): 13.9, 14.8, 19.2, 21.5, 23.5, 24.1, 25.5, 25.7, 27.7, 28.2, 29.3, 30.4, 30.9, 31.4, 31.8, 32.4, 34.7, 36.9, 37.0, 37.6, 37.8, 38.3, 38.8, 39.3, 39.8, 40.1, 41.3, 43.0, 43.3, 43.8, 43.8, 43.9, 44.1, 45.2, 51.3, 52.0, 52.3, 52.5, 53.0, 53.2, 53.7, 54.2, 119.3, 142.4, 172.6, 174.9 (C-28). Analysis calculated for C35H57N3O2 (M 551.73): C 76.18, H 10.41, N 7.61; found: C 76.16, H 10.40, N 7.60. APCI (m/z): 552.45 (M + H)+ 100%.
N-(3,4-Seco-2-cyano-ursan-4(23),12(13)-dien-28-oyl)-methylpiperazine 33. Yield 0.17 g (32%). Rf 0.82; mp 197–198 °C. [α]D20 + 17 (c 0.1, CHCl3). 1H NMR (δ, ppm): 0.72, 0.81, 0.88, 0.99, 1.12, 1.76 (6 s, 18H, 6CH3), 1.12–2.61 (m, 23H, CH and CH2), 2.21 (s, 3H, NCH3), 2.21–2.65 (m, 4H, 2CH2), 3.31–3.62 (m, 4H, 2CH2), 4.41 and 4.71 (both d, J 0.8 Hz, 2H, H-24), 5.32 (s, 1H, H-12). 13C NMR (δ, ppm): 13.5, 17.0, 19.1, 20.4, 23.0, 23.5, 23.6, 23.7, 24.0, 25.6, 27.2, 29.3, 30.7, 32.3, 33.0, 34.0, 34.3, 37.8, 39.1, 39.4, 42.0, 42.4, 45.7, 46.2, 46.4, 50.5, 52.4, 53.8, 54.4, 55.5, 114.2, 120.1, 123.0 (C-12), 139.0 (C-13), 146.6, 177.7 (C-28). Analysis calculated for C35H55N3O (M 533.80): C 78.75, H 10.39, N 7.87; found: C 78.74, H 10.36, N 7.85. APCI (m/z): 534.43 (M + H)+ 100%.
N-(3-Oxo-3a-homo-3a-aza-ursan-12-en-28-oyl)-methylpiperazine 34. Yield 0.34 g (62%). Rf 0.28; mp 144–145 °C. [α]D20 + 4 (c 0.1, CHCl3). 1H NMR (δ, ppm): 0.72, 0.81, 0.89, 0.99, 1.11, 1.39, 1.71 (7 s, 21H, 7CH3), 1.12–2.41 (m, 23H, CH and CH2), 2.21 (s, 3H, NCH3), 2.31–2.52 (m, 4H, 2CH2), 3.29–3.61 (m, 4H, 2CH2), 5.15 (s, 1H, H-12), 5.63 (br. s, 1H, NH). 13C NMR (δ, ppm): 17.5, 17.7, 18.9, 20.8, 21.2, 23.0, 23.3, 24.5, 27.8, 28.2, 30.4, 30.6, 32.7, 33.2, 33.6, 33.7, 34.1, 35.7, 37.6, 38.6, 39.4, 39.9, 40.3, 41.5, 42.5, 45.3, 46.0, 53.4, 54.1, 54.5, 55.0, 124.7 (C-12), 138.9 (C-13), 175.1, 177.6 (C-28). Analysis calculated for C35H57N3O2 (M 551.73): C 76.18, H 10.41, N 7.61; found: C 76.16, H 10.40, N 7.60. APCI (m/z): 552.45 (M + H)+ 100%.


 ,
, 












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}