
Insights into Mechanisms of Tumorigenesis in Neuroendocrine Neoplasms

 , and
, and
Abstract
:1. Introduction
2. Novel Tumor Suppressor Genes (TSGs)
3. Cooperative Tumorigenic Effects
4. Dysregulation of Splicing Machinery in PitNETs
5. Genomic Integrity
6. Permissive Chromatin Landscape
7. Aberrant Methylation
8. Cell Dedifferentiation
9. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Nuñez-Valdovinos, B.; Carmona-Bayonas, A.; Jimenez-Fonseca, P.; Capdevila, J.; Castaño-Pascual, Á.; Benavent, M.; Pi Barrio, J.J.; Teule, A.; Alonso, V.; Custodio, A.; et al. Neuroendocrine Tumor Heterogeneity Adds Uncertainty to the World Health Organization 2010 Classification: Real-World Data from the Spanish Tumor Registry (R-GETNE). Oncologist 2018, 23, 422–432. [Google Scholar] [CrossRef] [Green Version]
- Gill, A.J. Why did they change that? Practical implications of the evolving classification of neuroendocrine tumours of the gastrointestinal tract. Histopathology 2021, 78, 162–170. [Google Scholar] [CrossRef]
- Dasari, A.; Shen, C.; Halperin, D.; Zhao, B.; Zhou, S.; Xu, Y.; Shih, T.; Yao, J.C. Trends in the Incidence, Prevalence, and Survival Outcomes in Patients With Neuroendocrine Tumors in the United States. JAMA Oncol. 2017, 3, 1335–1342. [Google Scholar] [CrossRef]
- Patel, N.; Benipal, B. Incidence of Neuroendocrine Tumors in the United States from 2001-2015: A United States Cancer Statistics Analysis of 50 States. Cureus 2019, 11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Asa, S.L.; Casar-Borota, O.; Chanson, P.; Delgrange, E.; Earls, P.; Ezzat, S.; Grossman, A.; Ikeda, H.; Inoshita, N.; Karavitaki, N.; et al. From pituitary adenoma to pituitary neuroendocrine tumor (PitNET): An International Pituitary Pathology Club proposal. Endocr. Relat. Cancer 2017, 24, C5–C8. [Google Scholar] [CrossRef] [PubMed]
- La Rosa, S.; Uccella, S. Classification of neuroendocrine neoplasms: Lights and shadows. Rev. Endocr. Metab. Disord. 2021, 22, 527–538. [Google Scholar] [CrossRef]
- Milione, M.; Maisonneuve, P.; Spada, F.; Pellegrinelli, A.; Spaggiari, P.; Albarello, L.; Pisa, E.; Barberis, M.; Vanoli, A.; Buzzoni, R.; et al. The clinicopathologic heterogeneity of grade 3 gastroenteropancreatic neuroendocrine neoplasms: Morphological differentiation and proliferation identify different prognostic categories. Neuroendocrinology 2016, 104, 85–93. [Google Scholar] [CrossRef]
- Albertelli, M.; Grillo, F.; Calzo, F.L.; Puliani, G.; Rainone, C.; Colao, A.A.L.; Faggiano, A. Pathology Reporting in Neuroendocrine Neoplasms of the Digestive System: Everything You Always Wanted to Know but Were Too Afraid to Ask. Front. Endocrinol. 2021, 12, 680305. [Google Scholar] [CrossRef]
- Yang, Z.; Klimstra, D.S.; Hruban, R.H.; Tang, L.H. Immunohistochemical Characterization of the Origins of Metastatic Well-differentiated Neuroendocrine Tumors to the Liver. Am. J. Surg. Pathol. 2017, 41, 915–922. [Google Scholar] [CrossRef]
- Maxwell, J.E.; Sherman, S.K.; Stashek, K.M.; O’Dorisio, T.M.; Bellizzi, A.M.; Howe, J.R. A practical method to determine the site of unknown primary in metastatic neuroendocrine tumors. Surgery 2014, 156, 1359–1366. [Google Scholar] [CrossRef] [Green Version]
- Crona, J.; Skogseid, B. GEP- NETS UPDATE: Genetics of neuroendocrine tumors. Eur. J. Endocrinol. 2016, 174, R275–R290. [Google Scholar] [CrossRef] [Green Version]
- Scarpa, A.; Chang, D.K.; Nones, K.; Corbo, V.; Patch, A.M.; Bailey, P.; Lawlor, R.T.; Johns, A.L.; Miller, D.K.; Mafficini, A.; et al. Whole-genome landscape of pancreatic neuroendocrine tumours. Nature 2017, 543, 65–71. [Google Scholar] [CrossRef]
- Scarpa, A. The landscape of molecular alterations in pancreatic and small intestinal neuroendocrine tumours. Ann. Endocrinol. 2019, 80, 153–158. [Google Scholar] [CrossRef]
- Pipinikas, C.P.; Berner, A.M.; Sposito, T.; Thirlwell, C. The evolving (epi)genetic landscape of pancreatic neuroendocrine tumours. Endocr. Relat. Cancer 2019, 26, R519–R544. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brandi, M.L.; Agarwal, S.K.; Perrier, N.D.; Lines, K.E.; Valk, G.D.; Thakker, R. V Multiple Endocrine Neoplasia Type 1: Latest Insights. Endocr. Rev. 2021, 42, 133–170. [Google Scholar] [CrossRef]
- Matkar, S.; Thiel, A.; Hua, X. Menin: A scaffold protein that controls gene expression and cell signaling. Trends Biochem. Sci. 2013, 38, 394. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bouwmeester, W.; Zuithoff, N.P.A.; Mallett, S.; Geerlings, M.I.; Vergouwe, Y.; Steyerberg, E.W.; Altman, D.G.; Moons, K.G.M. Reporting and Methods in Clinical Prediction Research: A Systematic Review. PLoS Med. 2012, 9, e1001221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Greek, R.; Menache, A. Systematic Reviews of Animal Models: Methodology versus Epistemology. Int. J. Med. Sci. 2013, 10, 206. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Ohki, R. P53-phlda3-akt network: The key regulators of neuroendocrine tumorigenesis. Int. J. Mol. Sci. 2020, 21, 4098. [Google Scholar] [CrossRef]
- Xu, E.Y.; Vosburgh, E.; Wong, C.; Tang, L.H.; Notterman, D.A. Genetic analysis of the cooperative tumorigenic effects of targeted deletions of tumor suppressors Rb1, Trp53, Men1, and Pten in neuroendocrine tumors in mice. Oncotarget 2020, 11, 2718–2739. [Google Scholar] [CrossRef]
- Vázquez-Borrego, M.C.; Fuentes-Fayos, A.C.; Venegas-Moreno, E.; Rivero-Cortés, E.; Dios, E.; Moreno-Moreno, P.; Madrazo-Atutxa, A.; Remón, P.; Solivera, J.; Wildemberg, L.E.; et al. Splicing machinery is dysregulated in pituitary neuroendocrine tumors and is associated with aggressiveness features. Cancers 2019, 11, 1439. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, X.; Chen, W.; Chen, P.; Zhou, W.; Rong, Y.; Lv, Y.; Li, J.A.; Ji, Y.; Chen, W.; Lou, W.; et al. Aberration of ARID1A Is Associated with the Tumorigenesis and Prognosis of Sporadic Nonfunctional Pancreatic Neuroendocrine Tumors. Pancreas 2020, 49, 514–523. [Google Scholar] [CrossRef] [PubMed]
- Wasylishen, A.R.; Sun, C.; Moyer, S.M.; Qi, Y.; Chau, G.P.; Aryal, N.K.; McAllister, F.; Kim, M.P.; Barton, M.C.; Estrella, J.S.; et al. Daxx maintains endogenous retroviral silencing and restricts cellular plasticity in vivo. Sci. Adv. 2020, 6, eaba8415. [Google Scholar] [CrossRef] [PubMed]
- Lakis, V.; Lawlor, R.T.; Newell, F.; Patch, A.M.; Mafficini, A.; Sadanandam, A.; Koufariotis, L.T.; Johnston, R.L.; Leonard, C.; Wood, S.; et al. DNA methylation patterns identify subgroups of pancreatic neuroendocrine tumors with clinical association. Commun. Biol. 2021, 4, 155. [Google Scholar] [CrossRef] [PubMed]
- Saghafinia, S.; Homicsko, K.; Di Domenico, A.; Wullschleger, S.; Perren, A.; Marinoni, I.; Ciriello, G.; Michael, I.P.; Hanahan, D. Cancer cells retrace a stepwise differentiation program during malignant progression. Cancer Discov. 2021. [Google Scholar] [CrossRef] [PubMed]
- Ohki, R.; Saito, K.; Chen, Y.; Kawase, T.; Hiraoka, N.; Saigawa, R.; Minegishi, M.; Aita, Y.; Yanai, G.; Shimizu, H.; et al. PHLDA3 is a novel tumor suppressor of pancreatic neuroendocrine tumors. Proc. Natl. Acad. Sci. USA 2014, 111. [Google Scholar] [CrossRef] [Green Version]
- Manning, B.D.; Toker, A. AKT/PKB Signaling: Navigating the Network. Cell 2017, 169, 381–405. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiao, Y.; Shi, C.; Edil, B.H.; De Wilde, R.F.; Klimstra, D.S.; Maitra, A.; Schulick, R.D.; Tang, L.H.; Wolfgang, C.L.; Choti, M.A.; et al. DAXX/ATRX, MEN1, and mTOR pathway genes are frequently altered in pancreatic neuroendocrine tumors. Science 2011, 331, 1199–1203. [Google Scholar] [CrossRef] [Green Version]
- Tena-Suck, M.L.; Ortiz-Plata, A.; de la Vega, H.A. Phosphatase and tensin homologue and pituitary tumor-transforming gene in pituitary adenomas. Clinical-pathologic and immunohistochemical analysis. Ann. Diagn. Pathol. 2008, 12, 275–282. [Google Scholar] [CrossRef]
- Cakir, M.; Grossman, A.B. Targeting MAPK (Ras/ERK) and PI3K/Akt pathways in pituitary tumorigenesis. Expert Opin. Ther. Targets 2009, 13, 1121–1134. [Google Scholar] [CrossRef]
- Kawase, T.; Ohki, R.; Shibata, T.; Tsutsumi, S.; Kamimura, N.; Inazawa, J.; Ohta, T.; Ichikawa, H.; Aburatani, H.; Tashiro, F.; et al. PH Domain-Only Protein PHLDA3 Is a p53-Regulated Repressor of Akt. Cell 2009, 136, 535–550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clewemar Antonodimitrakis, P.; Sundin, A.; Wassberg, C.; Granberg, D.; Skogseid, B.; Eriksson, B. Streptozocin and 5-Fluorouracil for the Treatment of Pancreatic Neuroendocrine Tumors: Efficacy, Prognostic Factors and Toxicity. Neuroendocrinology 2016, 103, 345–353. [Google Scholar] [CrossRef]
- Zanini, S.; Renzi, S.; Giovinazzo, F.; Bermano, G. mTOR Pathway in Gastroenteropancreatic Neuroendocrine Tumor (GEP-NETs). Front. Endocrinol. 2020, 11, 562505. [Google Scholar] [CrossRef] [PubMed]
- Nitulescu, G.M.; Van De Venter, M.; Nitulescu, G.; Ungurianu, A.; Juzenas, P.; Peng, Q.; Olaru, O.T.; Grǎdinaru, D.; Tsatsakis, A.; Tsoukalas, D.; et al. The Akt pathway in oncology therapy and beyond (Review). Int. J. Oncol. 2018, 53, 2319–2331. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sigel, C.S.; Krauss Silva, V.W.; Reid, M.D.; Chhieng, D.; Basturk, O.; Sigel, K.M.; Daniel, T.D.; Klimstra, D.S.; Tang, L.H. Assessment of cytologic differentiation in high-grade pancreatic neuroendocrine neoplasms: A multi-institutional study. Cancer Cytopathol. 2018, 126, 44–53. [Google Scholar] [CrossRef]
- Ogino, A.; Yoshino, A.; Katayama, Y.; Watanabe, T.; Ota, T.; Komine, C.; Yokoyama, T.; Fukushima, T. The p15INK4b/p16INK4a/RB1 pathway is frequently deregulated in human pituitary adenomas. J. Neuropathol. Exp. Neurol. 2005, 64, 398–403. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simpson, D.J.; Hibberts, N.A.; McNicol, A.M.; Clayton, R.N.; Farrell, W.E. Loss of pRb expression in pituitary adenomas is associated with methylation of the RB1 CpG island. Cancer Res. 2000, 60, 1211–1216. [Google Scholar] [PubMed]
- Hu, N.; Gutsmann, A.; Herbert, D.C.; Bradley, A.; Lee, W.H.; Lee, E.Y.H. Heterozygous Rb-1(Δ20)/+ mice are predisposed to tumors of the pituitary gland with a nearly complete penetrance. Oncogene 1994, 9, 1021–1027. [Google Scholar]
- Glenn, S.T.; Jones, C.A.; Sexton, S.; LeVea, C.M.; Caraker, S.M.; Hajduczok, G.; Gross, K.W. Conditional deletion of p53 and Rb in the renin-expressing compartment of the pancreas leads to a highly penetrant metastatic pancreatic neuroendocrine carcinoma. Oncogene 2014, 33, 5706–5715. [Google Scholar] [CrossRef] [Green Version]
- Crabtree, J.S.; Scacheri, P.C.; Ward, J.M.; McNally, S.R.; Swain, G.P.; Montagna, C.; Hager, J.H.; Hanahan, D.; Edlund, H.; Magnuson, M.A.; et al. Of Mice and MEN1: Insulinomas in a Conditional Mouse Knockout. Mol. Cell. Biol. 2003, 23, 6075–6085. [Google Scholar] [CrossRef] [Green Version]
- Bertolino, P.; Tong, W.M.; Galendo, D.; Wang, Z.Q.; Zhang, C.X. Heterozygous Men1 mutant mice develop a range of endocrine tumors mimicking multiple endocrine neoplasia type 1. Mol. Endocrinol. 2003, 17, 1880–1892. [Google Scholar] [CrossRef] [Green Version]
- Biondi, C.A.; Gartside, M.G.; Waring, P.; Loffler, K.A.; Stark, M.S.; Magnuson, M.A.; Kay, G.F.; Hayward, N.K. Conditional Inactivation of the Men1 Gene Leads to Pancreatic and Pituitary Tumorigenesis but Does Not Affect Normal Development of These Tissues. Mol. Cell. Biol. 2004, 24, 3125–3131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bai, F.; Pei, X.-H.; Pandolfi, P.P.; Xiong, Y. p18Ink4c and Pten Constrain a Positive Regulatory Loop between Cell Growth and Cell Cycle Control. Mol. Cell. Biol. 2006, 26, 4564–4576. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wong, C.; Tang, L.H.; Davidson, C.; Vosburgh, E.; Chen, W.; Foran, D.J.; Notterman, D.A.; Levine, A.J.; Xu, E.Y. Two well-differentiated pancreatic neuroendocrine tumor mouse models. Cell Death Differ. 2020, 27, 269–283. [Google Scholar] [CrossRef] [Green Version]
- McLellan, M.A.; Rosenthal, N.A.; Pinto, A.R. Cre-loxP-Mediated Recombination: General Principles and Experimental Considerations. Curr. Protoc. Mouse Biol. 2017, 7, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; Kim, M.; Im, S.-K.; Fang, S. Mouse Cre-LoxP system: General principles to determine tissue-specific roles of target genes. Lab. Anim. Res. 2018, 34, 147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quintanal-Villalonga, Á.; Chan, J.M.; Yu, H.A.; Pe’er, D.; Sawyers, C.L.; Sen, T.; Rudin, C.M. Lineage plasticity in cancer: A shared pathway of therapeutic resistance. Nat. Rev. Clin. Oncol. 2020, 17, 360–371. [Google Scholar] [CrossRef]
- Rickman, D.S.; Beltran, H.; Demichelis, F.; Rubin, M.A. Biology and evolution of poorly differentiated neuroendocrine tumors. Nat. Med. 2017, 23, 664–673. [Google Scholar] [CrossRef]
- Peculis, R.; Niedra, H.; Rovite, V. Large scale molecular studies of pituitary neuroendocrine tumors: Novel markers, mechanisms and translational perspectives. Cancers 2021, 13, 1395. [Google Scholar] [CrossRef]
- Ronchi, C.L.; Peverelli, E.; Herterich, S.; Weigand, I.; Mantovani, G.; Schwarzmayr, T.; Sbiera, S.; Allolio, B.; Honegger, J.; Appenzeller, S.; et al. Landscape of somatic mutations in sporadic GH-secreting pituitary adenomas. Eur. J. Endocrinol. 2016, 174, 363–372. [Google Scholar] [CrossRef] [Green Version]
- Li, C.; Xie, W.; Rosenblum, J.S.; Zhou, J.; Guo, J.; Miao, Y.; Shen, Y.; Wang, H.; Gong, L.; Li, M.; et al. Somatic SF3B1 hotspot mutation in prolactinomas. Nat. Commun. 2020, 11, 2506. [Google Scholar] [CrossRef] [PubMed]
- Salomon, M.P.; Wang, X.; Marzese, D.M.; Hsu, S.C.; Nelson, N.; Zhang, X.; Matsuba, C.; Takasumi, Y.; Ballesteros-Merino, C.; Fox, B.A.; et al. The epigenomic landscape of pituitary adenomas reveals specific alterations and differentiates among acromegaly, Cushing’s disease and endocrine-inactive subtypes. Clin. Cancer Res. 2018, 24, 4126–4136. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zatelli, M.C. Pathogenesis of non-functioning pituitary adenomas. Pituitary 2018, 21, 130–137. [Google Scholar] [CrossRef] [PubMed]
- Sveen, A.; Kilpinen, S.; Ruusulehto, A.; Lothe, R.A.; Skotheim, R.I. Aberrant RNA splicing in cancer; Expression changes and driver mutations of splicing factor genes. Oncogene 2016, 35, 2413–2427. [Google Scholar] [CrossRef]
- Luque, R.M.; Ibáñez-Costa, A.; Neto, L.V.; Taboada, G.F.; Hormaechea-Agulla, D.; Kasuki, L.; Venegas-Moreno, E.; Moreno-Carazo, A.; Gálvez, M.Á.; Soto-Moreno, A.; et al. Truncated somatostatin receptor variant sst5TMD4 confers aggressive features (proliferation, invasion and reduced octreotide response) to somatotropinomas. Cancer Lett. 2015, 359, 299–306. [Google Scholar] [CrossRef] [Green Version]
- Ibáñez-Costa, A.; Gahete, M.D.; Rivero-Cortés, E.; Rincón-Fernández, D.; Nelson, R.; Beltrán, M.; De La Riva, A.; Japón, M.A.; Venegas-Moreno, E.; Gálvez, M.Á.; et al. In1-ghrelin splicing variant is overexpressed in pituitary adenomas and increases their aggressive features. Sci. Rep. 2015, 5, 8714. [Google Scholar] [CrossRef] [Green Version]
- Barabutis, N.; Siejka, A.; Schally, A.V.; Block, N.L.; Cai, R.; Varga, J.L. Activation of mitogen-activated protein kinases by a splice variant of GHRH receptor. J. Mol. Endocrinol. 2010, 44, 127–134. [Google Scholar] [CrossRef] [PubMed]
- Altenberger, T.; Bilban, M.; Auer, M.; Knosp, E.; Wolfsberger, S.; Gartner, W.; Mineva, I.; Zielinski, C.; Wagner, L.; Luger, A. Identification of DLK1 variants in pituitary- and neuroendocrine tumors. Biochem. Biophys. Res. Commun. 2006, 340, 995–1005. [Google Scholar] [CrossRef]
- Vitale, L.; Lenzi, L.; Huntsman, S.A.; Canaider, S.; Frabetti, F.; Casadei, R.; Facchin, F.; Carinci, P.; Zannotti, M.; Coppola, D.; et al. Differential expression of alternatively spliced mRNA forms of the insulin-like growth factor 1 receptor in human neuroendocrine tumors. Oncol. Rep. 2006, 15, 1249–1256. [Google Scholar] [CrossRef] [Green Version]
- Lekva, T.; Berg, J.P.; Lyle, R.; Heck, A.; Ringstad, G.; Olstad, O.K.; Michelsen, A.E.; Casar-Borota, O.; Bollerslev, J.; Ueland, T. Epithelial Splicing Regulator Protein 1 and Alternative Splicing in Somatotroph Adenomas. Endocrinology 2013, 154, 3331–3343. [Google Scholar] [CrossRef] [Green Version]
- Ríos, Y.; Melmed, S.; Lin, S.; Liu, N.A. Zebrafish usp39 mutation leads to rb1 mRNA splicing defect and pituitary lineage expansion. PLoS Genet. 2011, 7, e1001271. [Google Scholar] [CrossRef] [Green Version]
- Kotake, Y.; Sagane, K.; Owa, T.; Mimori-Kiyosue, Y.; Shimizu, H.; Uesugi, M.; Ishihama, Y.; Iwata, M.; Mizui, Y. Splicing factor SF3b as a target of the antitumor natural product pladienolide. Nat. Chem. Biol. 2007, 3, 570–575. [Google Scholar] [CrossRef]
- Rymond, B. Targeting the spliceosome. Nat. Chem. Biol. 2007, 3, 533–535. [Google Scholar] [CrossRef]
- Ribeiro-Silva, C.; Vermeulen, W.; Lans, H. SWI/SNF: Complex complexes in genome stability and cancer. DNA Repair 2019, 77, 87–95. [Google Scholar] [CrossRef]
- Khursheed, M.; Kolla, J.N.; Kotapalli, V.; Gupta, N.; Gowrishankar, S.; Uppin, S.G.; Sastry, R.A.; Koganti, S.; Sundaram, C.; Pollack, J.R.; et al. ARID1B, a member of the human SWI/SNF chromatin remodeling complex, exhibits tumour-suppressor activities in pancreatic cancer cell lines. Br. J. Cancer 2013, 108, 2056–2062. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Watanabe, R.; Ui, A.; Kanno, S.I.; Ogiwara, H.; Nagase, T.; Kohno, T.; Yasui, A. SWI/SNF factors required for cellular resistance to dna damage include arid1a and arid1b and show interdependent protein stability. Cancer Res. 2014, 74, 2465–2475. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, S.C.; Nassour, I.; Xiao, S.; Zhang, S.; Luo, X.; Lee, J.; Li, L.; Sun, X.; Nguyen, L.H.; Chuang, J.C.; et al. SWI/SNF component ARID1A restrains pancreatic neoplasia formation. Gut 2019, 68, 1259–1270. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, W.; Friedland, S.C.; Guo, B.; O’Dell, M.R.; Alexander, W.B.; Whitney-Miller, C.L.; Agostini-Vulaj, D.; Huber, A.R.; Myers, J.R.; Ashton, J.M.; et al. ARID1A, a SWI/SNF subunit, is critical to acinar cell homeostasis and regeneration and is a barrier to transformation and epithelial-mesenchymal transition in the pancreas. Gut 2019, 68, 1245–1258. [Google Scholar] [CrossRef] [PubMed]
- You, J.S.; Jones, P.A. Cancer Genetics and Epigenetics: Two Sides of the Same Coin? Cancer Cell 2012, 22, 9–20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shen, H.; Laird, P.W. Interplay between the cancer genome and epigenome. Cell 2013, 153, 38–55. [Google Scholar] [CrossRef] [Green Version]
- Dyer, M.A.; Qadeer, Z.A.; Valle-Garcia, D.; Bernstein, E. ATRX and DAXX: Mechanisms and Mutations. Cold Spring Harb. Perspect. Med. 2017, 7, a026567. [Google Scholar] [CrossRef]
- Voon, H.P.J.; Collas, P.; Wong, L.H. Compromised telomeric heterochromatin promotes ALTernative lengthening of telomeres. Trends Cancer 2016, 2, 114–116. [Google Scholar] [CrossRef]
- Voon, H.P.J.; Wong, L.H. New players in heterochromatin silencing: Histone variant H3.3 and the ATRX/DAXX chaperone. Nucleic Acids Res. 2015, 44, 1496–1501. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elsässer, S.J.; Noh, K.M.; Diaz, N.; Allis, C.D.; Banaszynski, L.A. Histone H3.3 is required for endogenous retroviral element silencing in embryonic stem cells. Nature 2015, 522, 240–244. [Google Scholar] [CrossRef] [Green Version]
- Wolf, G.; Rebollo, R.; Karimi, M.M.; Ewing, A.D.; Kamada, R.; Wu, W.; Wu, B.; Bachu, M.; Ozato, K.; Faulkner, G.J.; et al. On the role of H3.3 in retroviral silencing. Nature 2017, 548, E1–E3. [Google Scholar] [CrossRef] [PubMed]
- Geis, F.K.; Goff, S.P. Silencing and transcriptional regulation of endogenous retroviruses: An overview. Viruses 2020, 12, 884. [Google Scholar] [CrossRef] [PubMed]
- Jansz, N.; Faulkner, G.J. Endogenous retroviruses in the origins and treatment of cancer. Genome Biol. 2021, 22, 147. [Google Scholar] [CrossRef] [PubMed]
- Desboiss, C.; Hogue, D.A.; Karsentyt, G. The Mouse Osteocalcin Gene Cluster Contains Three Genes with Two Separate Spatial and Temporal Patterns of Expression. J. Biol. Chem. 1994, 269, 1183–1190. [Google Scholar] [CrossRef]
- Jiao, J.; Feng, G.; Wu, M.; Wang, Y.; Li, R.; Liu, J. MiR-140-5p promotes osteogenic differentiation of mouse embryonic bone marrow mesenchymal stem cells and post-fracture healing of mice. Cell Biochem. Funct. 2020, 38, 1152–1160. [Google Scholar] [CrossRef]
- Zhou, Y.; Sun, B.; Li, W.; Zhou, J.; Gao, F.; Wang, X.; Cai, M.; Sun, Z. Pancreatic stellate cells: A rising translational physiology star as a potential stem cell type for beta cell neogenesis. Front. Physiol. 2019, 10, 218. [Google Scholar] [CrossRef]
- Haqq, J.; Howells, L.M.; Garcea, G.; Metcalfe, M.S.; Steward, W.P.; Dennison, A.R. Pancreatic stellate cells and pancreas cancer: Current perspectives and future strategies. Eur. J. Cancer 2014, 50, 2570–2582. [Google Scholar] [CrossRef]
- Wasylishen, A.R.; Sun, C.; Chau, G.P.; Qi, Y.; Su, X.; Kim, M.P.; Estrella, J.S.; Lozano, G. Men1 maintains exocrine pancreas homeostasis in response to inflammation and oncogenic stress. Proc. Natl. Acad. Sci. USA 2020, 117, 6622–6629. [Google Scholar] [CrossRef]
- Agarwal, S.K.; Guru, S.C.; Heppner, C.; Erdos, M.R.; Collins, R.M.; Park, S.Y.; Saggar, S.; Chandrasekharappa, S.C.; Collins, F.S.; Spiegel, A.M.; et al. Menin interacts with the AP1 transcription factor JunD and represses JunD-activated transcription. Cell 1999, 96, 143–152. [Google Scholar] [CrossRef] [Green Version]
- Yokoyama, A.; Cleary, M.L. Menin Critically Links MLL Proteins with LEDGF on Cancer-Associated Target Genes. Cancer Cell 2008, 14, 36–46. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Milne, T.A.; Hughes, C.M.; Lloyd, R.; Yang, Z.; Rozenblatt-Rosen, O.; Dou, Y.; Schnepp, R.W.; Krankel, C.; LiVolsi, V.A.; Gibbs, D.; et al. Menin and MLL cooperatively regulate expression of cyclin-dependent kinase inhibitors. Proc. Natl. Acad. Sci. USA 2005, 102, 749–754. [Google Scholar] [CrossRef] [Green Version]
- Arroyo, M.; Larrosa, R.; Claros, M.G.; Bautista, R. Expression Change Correlations Between Transposons and Their Adjacent Genes in Lung Cancers Reveal a Genomic Location Dependence and Highlights Cancer-Significant Genes. In Lecture Notes in Computer Science (Including Subseries Lecture Notes in Artificial Intelligence and Lecture Notes in Bioinformatics); Springer: Cham, Switzerland, 2019; Volume 11465, pp. 84–92. [Google Scholar]
- Girot, P.; Dumars, C.; Mosnier, J.F.; Muzellec, L.; Senellart, H.; Foubert, F.; Caroli-Bosc, F.X.; Cauchin, E.; Regenet, N.; Matysiak-Budnik, T.; et al. Evaluation of O6-methylguanine-DNA methyltransferase as a predicting factor of response to temozolomide-based chemotherapy in well-differentiated metastatic pancreatic neuroendocrine tumors. Eur. J. Gastroenterol. Hepatol. 2017, 29, 826–830. [Google Scholar] [CrossRef] [PubMed]
- Chan, C.S.; Laddha, S.V.; Lewis, P.W.; Koletsky, M.S.; Robzyk, K.; Da Silva, E.; Torres, P.J.; Untch, B.R.; Li, J.; Bose, P.; et al. ATRX, DAXX or MEN1 mutant pancreatic neuroendocrine tumors are a distinct alpha-cell signature subgroup. Nat. Commun. 2018, 9, 4158. [Google Scholar] [CrossRef] [Green Version]
- Conemans, E.B.; Lodewijk, L.; Moelans, C.B.; Offerhaus, G.J.A.; Pieterman, C.R.C.; Morsink, F.H.; Dekkers, O.M.; De Herder, W.W.; Hermus, A.R.; Van Der Horst-Schrivers, A.N.; et al. DNA methylation profiling in MEN1-related pancreatic neuroendocrine tumors reveals a potential epigenetic target for treatment. Eur. J. Endocrinol. 2018, 179, 153–160. [Google Scholar] [CrossRef] [PubMed]
- Stefanoli, M.; La Rosa, S.; Sahnane, N.; Romualdi, C.; Pastorino, R.; Marando, A.; Capella, C.; Sessa, F.; Furlan, D. Prognostic relevance of aberrant DNA methylation in G1 and G2 pancreatic neuroendocrine tumors. Neuroendocrinology 2014, 100, 26–34. [Google Scholar] [CrossRef]
- Olson, P.; Lu, J.; Zhang, H.; Shai, A.; Chun, M.G.; Wang, Y.; Libutti, S.K.; Nakakura, E.K.; Golub, T.R.; Hanahan, D. MicroRNA dynamics in the stages of tumorigenesis correlate with hallmark capabilities of cancer. Genes Dev. 2009, 23, 2152–2165. [Google Scholar] [CrossRef] [Green Version]
- Sadanandam, A.; Wullschleger, S.; Lyssiotis, C.A.; Grötzinger, C.; Barbi, S.; Bersani, S.; Körner, J.; Wafy, I.; Mafficini, A.; Lawlo, R.T.; et al. A cross-species analysis in pancreatic neuroendocrine tumors reveals molecular subtypes with distinctive clinical, metastatic, developmental, and metabolic characteristics. Cancer Discov. 2015, 5, 1296–1313. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cejas, P.; Drier, Y.; Dreijerink, K.M.A.; Brosens, L.A.A.; Deshpande, V.; Epstein, C.B.; Conemans, E.B.; Morsink, F.H.M.; Graham, M.K.; Valk, G.D.; et al. Enhancer signatures stratify and predict outcomes of non-functional pancreatic neuroendocrine tumors. Nat. Med. 2019, 25, 1264–1269. [Google Scholar] [CrossRef] [PubMed]
- Peri, A.K.; Wilgenbus, P.; Dahl, U.; Semb, H.; Christofori, G. A causal role for E-cadherin in the transition from adenoma to carcinoma. Nature 1998, 392, 190–193. [Google Scholar] [CrossRef]
- Title, A.C.; Hong, S.J.; Pires, N.D.; Hasenöhrl, L.; Godbersen, S.; Stokar-Regenscheit, N.; Bartel, D.P.; Stoffel, M. Genetic dissection of the miR-200–Zeb1 axis reveals its importance in tumor differentiation and invasion. Nat. Commun. 2018, 9, 4671. [Google Scholar] [CrossRef] [PubMed]
- Mujica-Mota, R.; Varley-Campbell, J.; Tikhonova, I.; Cooper, C.; Griffin, E.; Haasova, M.; Peters, J.; Lucherini, S.; Talens-Bou, J.; Long, L.; et al. Everolimus, lutetium-177 DOTATATE and sunitinib for advanced, unresectable or metastatic neuroendocrine tumours with disease progression: A systematic review and cost-effectiveness analysis. Health Technol. Assess. 2018, 22, 1–325. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bartel, D.P. Metazoan MicroRNAs. Cell 2018, 173, 20–51. [Google Scholar] [CrossRef] [Green Version]
- Vidigal, J.A.; Ventura, A. The biological functions of miRNAs: Lessons from in vivo studies. Trends Cell Biol. 2015, 25, 137–147. [Google Scholar] [CrossRef] [Green Version]
- Basso, K.; Margolin, A.A.; Stolovitzky, G.; Klein, U.; Dalla-Favera, R.; Califano, A. Reverse engineering of regulatory networks in human B cells. Nat. Genet. 2005, 37, 382–390. [Google Scholar] [CrossRef]
- Margolin, A.A.; Nemenman, I.; Basso, K.; Wiggins, C.; Stolovitzky, G.; Favera, R.D.; Califano, A. ARACNE: An algorithm for the reconstruction of gene regulatory networks in a mammalian cellular context. BMC Bioinform. 2006, 7, S7. [Google Scholar] [CrossRef] [Green Version]
- Alvarez, M.J.; Shen, Y.; Giorgi, F.M.; Lachmann, A.; Ding, B.B.; Hilda Ye, B.; Califano, A. Functional characterization of somatic mutations in cancer using network-based inference of protein activity. Nat. Genet. 2016, 48, 838–847. [Google Scholar] [CrossRef]
- Song, N.; Liu, B.; Wu, J.L.; Zhang, R.F.; Duan, L.; He, W.S.; Zhang, C.M. Prognostic value of HMGB3 expression in patients with non-small cell lung cancer. Tumor Biol. 2013, 34, 2599–2603. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Wang, H.; Wen, Y.; Chen, X.; Liu, X.; Gao, J.; Su, P.; Xu, Y.; Zhou, W.; Shi, L.; et al. MEIS2 regulates endothelial to hematopoietic transition of human embryonic stem cells by targeting TAL1 06 Biological Sciences 0604 Genetics. Stem Cell Res. Ther. 2018, 9, 340. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Ghareeb, W.M.; Zhang, Y.; Yu, Q.; Lu, X.; Huang, Y.; Huang, S.; Sun, Y.; Chi, P. Hypermethylated and downregulated MEIS2 are involved in stemness properties and oxaliplatin-based chemotherapy resistance of colorectal cancer. J. Cell. Physiol. 2019, 234, 18180–18191. [Google Scholar] [CrossRef]
- Wan, Z.; Chai, R.; Yuan, H.; Chen, B.; Dong, Q.; Zheng, B.; Mou, X.; Pan, W.; Tu, Y.; Yang, Q.; et al. MEIS2 promotes cell migration and invasion in colorectal cancer. Oncol. Rep. 2019, 42, 213–223. [Google Scholar] [CrossRef]
- Gaudenzi, G.; Carra, S.; Dicitore, A.; Cantone, M.C.; Persani, L.; Vitale, G. Fishing for neuroendocrine tumors. Endocr. Relat. Cancer 2020, 27, R163–R176. [Google Scholar] [CrossRef]
- Gaudenzi, G.; Albertelli, M.; Dicitore, A.; Würth, R.; Gatto, F.; Barbieri, F.; Cotelli, F.; Florio, T.; Ferone, D.; Persani, L.; et al. Patient-derived xenograft in zebrafish embryos: A new platform for translational research in neuroendocrine tumors. Endocrine 2017, 57, 214–219. [Google Scholar] [CrossRef] [PubMed]


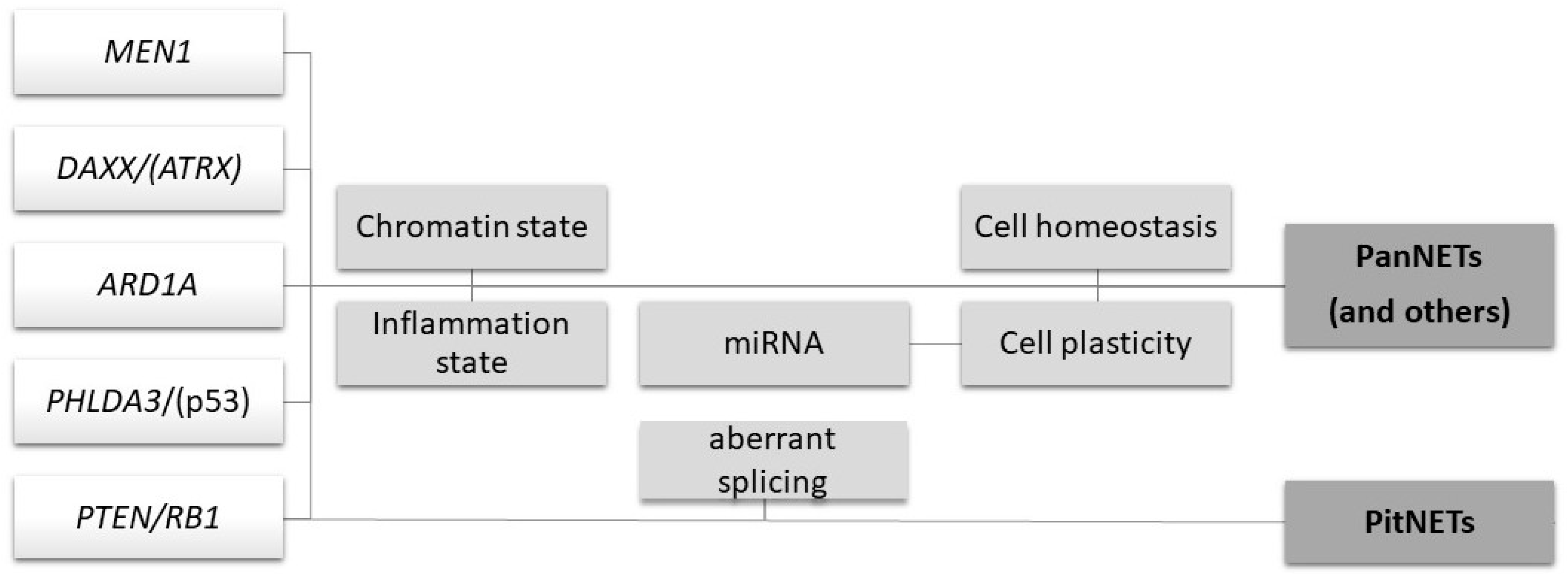{kind=link}
| Topic and Main Reference | Methods | Results |
|---|---|---|
| Novel Tumor Suppressor Genes [19] | PHLDA3 Studies in Tissues from Human PanNETs, MIN6 Cells (β-Cells Derived), Transgenic PHLDA3-Deficient Mice. | Loss of PHLDA3, a p53 Target and Inhibitor of Akt, Disrupts the Balance of p53-PHLDA3-Akt Axis and Promotes NETs’ Tumorigenesis. |
| Cooperative Tumorigenic Effects [20] | Pairwise and Single Homozygous Deletions of RB1, PTEN, MEN1, p53 in Insulin II Gene Expressing Cells (Cre-LoxP System). Histopathology of the Pituitary and Pancreas in the Mice. Scoring of the Cooperative Role of the Aforementioned Genes in PitNETs and PanNETs. | In PitNETs, the Order of Relevance in Initiation and/or Progression was Established as RB1, PTEN, MEN1, and p53, while, as Expected, in Islet Tumorigenesis it was MEN1, PTEN, RB1, and Lastly p53. |
| Dysregulation of Splicing Machinery in Pitnets [21] | Analysis of the Expression Levels of Spliceosome Core Components by Dynamic qRT-PCR Microfluidic array in the Main PitNETs’ Subtypes. Scoring of mRNA Expression Levels. Evaluation of the mRNA and Protein Expression Levels of SF3B1 in Cell Lines Under Administration of Pladienolide-B. | Dysregulation of Splicing Machinery is a Unique Fingerprint in PitNETs and a Potential Therapeutic Target. Pladienolide-B Reduces Cell Proliferation and Hormone Secretion. |
| Genomic Integrity [22] | Exome Sequencing of Tissues from Sporadic PanNETs. The qRT-PCR and Immunohistochemistry to Evaluate mRNA Level and Protein Expression of ARID1A. | Loss of ARID1A Contributes to Tumorigenesis and Metastatic Behavior in Sporadic PanNETs. |
| Permissive Chromatin Landscape [23] | Conditional DAXX Allele in Mice. Whole-Transcriptome Analysis. Evidence of Dysregulation of Heterochromatin. Combination of DAXX Loss with MEN1 Loss and/or Inflammatory Stress. Comprehensive Transcriptome and Chromatin Accessibility Profiling. Evidence of Dysregulation of ERVs, Gene Expression Changes, Altered Cell State, and Impaired Pancreas Recovery. RNA Sequencing on Human PanNETs and Evidence of Dysregulation of ERVs and Genes Downstream of DAXX Alteration. | DAXX Loss Leads to a Permissive Transcriptional State that, in Association with Environmental Stress and Men1 Loss, Alters Gene Expression and Cell State. DAXX Loss-Associated Transcriptional Changes Dysregulate ERVs and Nearby Genes also in Human PanNETs. |
| Aberrant Methylation [24] | Genome-Wide Scan of DNA Methylation in PanNETs. Identification of Methylation Subgroups with Correlation to Clinical and Genomic Features. | Methylation Drives Tumorigenesis Together with Somatic LOH/Copy Number Changes and Contributes to the NETs’ Heterogeneity. Potential Role in Stratifying Prognosis and Supporting Therapeutic Choices for PanNETs. |
| Cell Dedifferentiation [25] | Profiling for mRNA and miRNa and Proteomic Analysis of Samples from Primary Tumors and Metastases from RT2 Genetically Engineered Mouse Models. Isolation of Two Clusters with Different Profiles and also Expression of Mature β–Cell or Progenitor Markers, i.e., Islet Tumors (IT) and Metastasis-Like Primary Tumors (MLP). Development of mRNA and miRNA Signature for the MLP Cluster. Identification of miRNAs Responsible for the Activation of the MLP Program in IT, i.e., miR-181c and miR-181d, Demonstrated by the Overexpression of this miRNA Cluster in the βTC3 Cell Line (IT-Like) with piggyBac Transposon System and the Application of the MLP mRNA Signature onto the Transcriptome Profiles of the Samples in Order to Evaluate the Activation of the Progenitor-like Program. Identification of the Transcription Factors Influenced by the mi-RNA Cluster and Regulating the Dedifferentiation from IT to MLP subtype. | Description of a Novel Mechanism that Modulates Cancer Cell Plasticity. MLP Tumors Arise from IT via Dedifferentiation and Acquisition of β-Cells’ Progenitor-Like Phenotype. The microRNA-181cd Cluster Induces the IT-to-MLP Transition by Suppressing Expression of Meis2 and Consequent Upregulation of Hmgb3. IT-to-MLP Transition is a Discrete Step Preceding the Proliferation of Cancer Cells in Tumorigenesis. |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pastorino, L.; Grillo, F.; Albertelli, M.; Ghiorzo, P.; Bruno, W. Insights into Mechanisms of Tumorigenesis in Neuroendocrine Neoplasms. Int. J. Mol. Sci. 2021, 22, 10328. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms221910328
Pastorino L, Grillo F, Albertelli M, Ghiorzo P, Bruno W. Insights into Mechanisms of Tumorigenesis in Neuroendocrine Neoplasms. International Journal of Molecular Sciences. 2021; 22(19):10328. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms221910328
Chicago/Turabian StylePastorino, Lorenza, Federica Grillo, Manuela Albertelli, Paola Ghiorzo, and William Bruno. 2021. "Insights into Mechanisms of Tumorigenesis in Neuroendocrine Neoplasms" International Journal of Molecular Sciences 22, no. 19: 10328. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms221910328