
Actin Cytoskeletal Dynamics in Single-Cell Wound Repair

 , , and
, , and 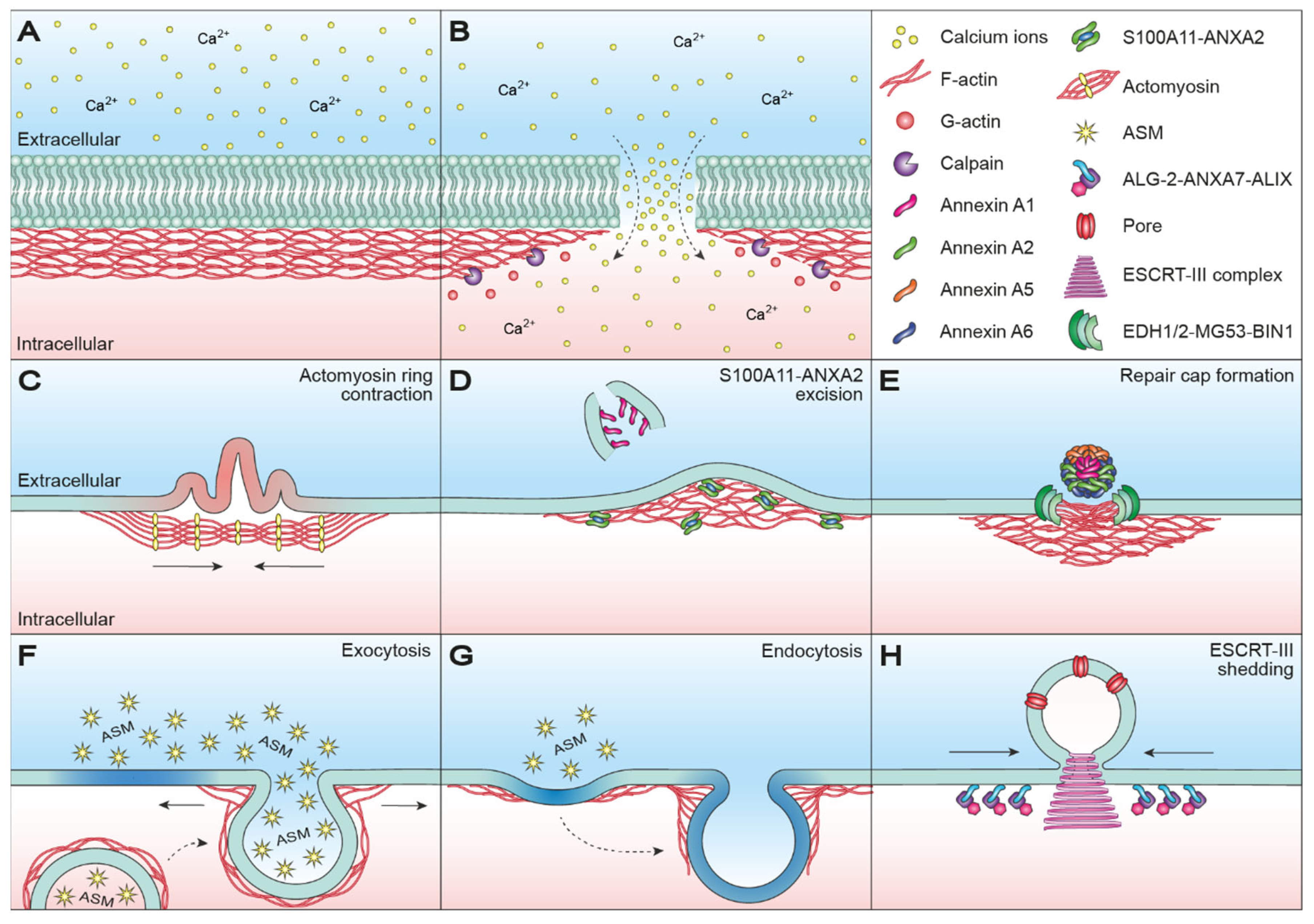{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Cortical Actin Dynamics in Membrane Resealing
2.1. Wound Healing in Oocytes
2.2. Would Healing in Somatic Cells
2.2.1. Protein-Mediated Plasma Membrane Repair
2.2.2. Exocytosis- and Endocytosis-Mediated Plasma Membrane Repair
2.2.3. Blebbing-Mediated Plasma Membrane Repair
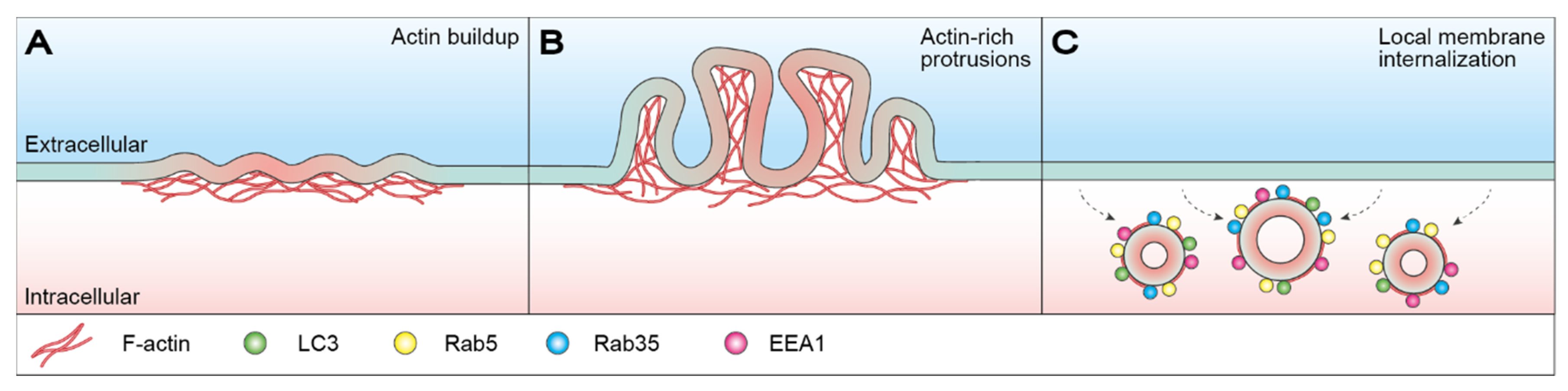
3. Cortical Actin Dynamics in Membrane Restructuring
3.1. Macropinocytosis-Mediated Membrane Restructuring
3.2. Plasma Membrane Repair Facilitated by Microparticle Shedding
4. Perinuclear Actin Dynamics
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Boye, T.L.; Nylandsted, J. Annexins in plasma membrane repair. Biol. Chem. 2016, 397, 961–969. [Google Scholar] [CrossRef] [PubMed]
- Cooper, S.T.; McNeil, P.L. Membrane Repair: Mechanisms and Pathophysiology. Physiol. Rev. 2015, 95, 1205–1240. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dias, C.; Nylandsted, J. Plasma membrane integrity in health and disease: Significance and therapeutic potential. Cell Discov. 2021, 7, 4. [Google Scholar] [CrossRef] [PubMed]
- Draeger, A.; Monastyrskaya, K.; Babiychuk, E.B. Plasma membrane repair and cellular damage control: The annexin survival kit. Biochem. Pharm. 2011, 81, 703–712. [Google Scholar] [CrossRef]
- Jimenez, A.J.; Perez, F. Physico-chemical and biological considerations for membrane wound evolution and repair in animal cells. Semin. Cell Dev. Biol. 2015, 45, 2–9. [Google Scholar] [CrossRef]
- Nicolson, G.L. Cell membrane fluid-mosaic structure and cancer metastasis. Cancer Res. 2015, 75, 1169–1176. [Google Scholar] [CrossRef] [Green Version]
- Clark, A.G.; Wartlick, O.; Salbreux, G.; Paluch, E.K. Stresses at the cell surface during animal cell morphogenesis. Curr. Biol. 2014, 24, R484–R494. [Google Scholar] [CrossRef] [Green Version]
- Pegoraro, A.F.; Janmey, P.; Weitz, D.A. Mechanical Properties of the Cytoskeleton and Cells. Cold Spring Harb. Perspect. Biol. 2017, 9, a022038. [Google Scholar] [CrossRef]
- Svitkina, T. The Actin Cytoskeleton and Actin-Based Motility. Cold Spring Harb. Perspect. Biol. 2018, 10, a018267. [Google Scholar] [CrossRef] [Green Version]
- Pollard, T.D. Actin and Actin-Binding Proteins. Cold Spring Harb. Perspect. Biol. 2016, 8, a018226. [Google Scholar] [CrossRef] [Green Version]
- Le Clainche, C.; Carlier, M.F. Regulation of actin assembly associated with protrusion and adhesion in cell migration. Physiol. Rev. 2008, 88, 489–513. [Google Scholar] [CrossRef] [Green Version]
- Heintzelman, M.B. Gliding motility in apicomplexan parasites. Semin. Cell Dev. Biol. 2015, 46, 135–142. [Google Scholar] [CrossRef]
- Lee, S.H.; Dominguez, R. Regulation of actin cytoskeleton dynamics in cells. Mol. Cells 2010, 29, 311–325. [Google Scholar] [CrossRef]
- Jimenez, A.J.; Perez, F. Plasma membrane repair: The adaptable cell life-insurance. Curr. Opin. Cell Biol. 2017, 47, 99–107. [Google Scholar] [CrossRef] [PubMed]
- Duan, D.; Goemans, N.; Takeda, S.; Mercuri, E.; Aartsma-Rus, A. Duchenne muscular dystrophy. Nat. Rev. Dis. Primers 2021, 7, 13. [Google Scholar] [CrossRef] [PubMed]
- Lauritzen, S.P.; Boye, T.L.; Nylandsted, J. Annexins are instrumental for efficient plasma membrane repair in cancer cells. Semin Cell Dev. Biol. 2015, 45, 32–38. [Google Scholar] [CrossRef] [PubMed]
- McElhanon, K.E.; Bhattacharya, S. Altered membrane integrity in the progression of muscle diseases. Life Sci. 2018, 192, 166–172. [Google Scholar] [CrossRef]
- Häger, S.C.; Nylandsted, J. Annexins: Players of single cell wound healing and regeneration. Commun. Integr. Biol. 2019, 12, 162–165. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sønder, S.L.; Boye, T.L.; Tölle, R.; Dengjel, J.; Maeda, K.; Jäättelä, M.; Simonsen, A.C.; Jaiswal, J.K.; Nylandsted, J. Annexin A7 is required for ESCRT III-mediated plasma membrane repair. Sci. Rep. 2019, 9, 6726. [Google Scholar] [CrossRef]
- Moe, A.M.; Golding, A.E.; Bement, W.M. Cell healing: Calcium, repair and regeneration. Semin. Cell Dev. Biol. 2015, 45, 18–23. [Google Scholar] [CrossRef] [Green Version]
- Jaiswal, J.K. Calcium—How and why? J. Biosci. 2001, 26, 357–363. [Google Scholar] [CrossRef]
- Cai, C.; Masumiya, H.; Weisleder, N.; Matsuda, N.; Nishi, M.; Hwang, M.; Ko, J.K.; Lin, P.; Thornton, A.; Zhao, X.; et al. MG53 nucleates assembly of cell membrane repair machinery. Nat. Cell Biol. 2009, 11, 56–64. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bíró, E.N.; Venyaminov, S.Y. Depolymerization of actin in concentrated solutions of divalent metal chlorides. Acta Biochim. Biophys. Acad. Sci. Hung. 1979, 14, 31–42. [Google Scholar]
- Zechel, K. Stability differences of muscle F-actin in formamide in the presence of Mg2+ and Ca2+. Biochim. Biophys. Acta 1983, 742, 135–141. [Google Scholar] [CrossRef]
- Mellgren, R.L. A plasma membrane wound proteome: Reversible externalization of intracellular proteins following reparable mechanical damage. J. Biol. Chem. 2010, 285, 36597–36607. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mellgren, R.L.; Zhang, W.; Miyake, K.; McNeil, P.L. Calpain is required for the rapid, calcium-dependent repair of wounded plasma membrane. J. Biol. Chem. 2007, 282, 2567–2575. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McNeil, P.L.; Steinhardt, R.A. Plasma membrane disruption: Repair, prevention, adaptation. Annu. Rev. Cell Dev. Biol. 2003, 19, 697–731. [Google Scholar] [CrossRef]
- Godin, L.M.; Vergen, J.; Prakash, Y.S.; Pagano, R.E.; Hubmayr, R.D. Spatiotemporal dynamics of actin remodeling and endomembrane trafficking in alveolar epithelial type I cell wound healing. Am. J. Physiol. Lung Cell Mol. Physiol. 2011, 300, L615–L623. [Google Scholar] [CrossRef] [Green Version]
- Miyake, K.; McNeil, P.L.; Suzuki, K.; Tsunoda, R.; Sugai, N. An actin barrier to resealing. J. Cell Sci. 2001, 114, 3487–3494. [Google Scholar] [CrossRef]
- McNeil, P.L.; Vogel, S.S.; Miyake, K.; Terasaki, M. Patching plasma membrane disruptions with cytoplasmic membrane. J. Cell Sci. 2000, 113, 1891–1902. [Google Scholar] [CrossRef]
- Terasaki, M.; Miyake, K.; McNeil, P.L. Large plasma membrane disruptions are rapidly resealed by Ca2+-dependent vesicle-vesicle fusion events. J. Cell Biol. 1997, 139, 63–74. [Google Scholar] [CrossRef] [Green Version]
- Davenport, N.R.; Sonnemann, K.J.; Eliceiri, K.W.; Bement, W.M. Membrane dynamics during cellular wound repair. Mol. Biol. Cell 2016, 27, 2272–2285. [Google Scholar] [CrossRef]
- Reddy, A.; Caler, E.V.; Andrews, N.W. Plasma membrane repair is mediated by Ca(2+)-regulated exocytosis of lysosomes. Cell 2001, 106, 157–169. [Google Scholar] [CrossRef] [Green Version]
- McNeil, P.L. Repairing a torn cell surface: Make way, lysosomes to the rescue. J. Cell Sci. 2002, 115, 873–879. [Google Scholar] [CrossRef]
- Lorusso, A.; Covino, C.; Priori, G.; Bachi, A.; Meldolesi, J.; Chieregatti, E. Annexin2 coating the surface of enlargeosomes is needed for their regulated exocytosis. EMBO J. 2006, 25, 5443–5456. [Google Scholar] [CrossRef] [Green Version]
- Sharma, N.; Medikayala, S.; Defour, A.; Rayavarapu, S.; Brown, K.J.; Hathout, Y.; Jaiswal, J.K. Use of quantitative membrane proteomics identifies a novel role of mitochondria in healing injured muscles. J. Biol. Chem. 2012, 287, 30455–30467. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Atanassoff, A.P.; Wolfmeier, H.; Schoenauer, R.; Hostettler, A.; Ring, A.; Draeger, A.; Babiychuk, E.B. Microvesicle shedding and lysosomal repair fulfill divergent cellular needs during the repair of streptolysin O-induced plasmalemmal damage. PLoS ONE 2014, 9, e89743. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davenport, N.R.; Bement, W.M. Cell repair: Revisiting the patch hypothesis. Commun. Integr. Biol. 2016, 9, e1253643. [Google Scholar] [CrossRef] [PubMed]
- Cabukusta, B.; Neefjes, J. Mechanisms of lysosomal positioning and movement. Traffic 2018, 19, 761–769. [Google Scholar] [CrossRef] [PubMed]
- Bandyopadhyay, D.; Cyphersmith, A.; Zapata, J.A.; Kim, Y.J.; Payne, C.K. Lysosome transport as a function of lysosome diameter. PLoS ONE 2014, 9, e86847. [Google Scholar] [CrossRef] [Green Version]
- Schuh, M. An actin-dependent mechanism for long-range vesicle transport. Nat. Cell Biol. 2011, 13, 1431–1436. [Google Scholar] [CrossRef] [PubMed]
- Talukder, M.S.U.; Pervin, M.S.; Tanvir, M.I.O.; Fujimoto, K.; Tanaka, M.; Itoh, G.; Yumura, S. Ca(2+)-Calmodulin Dependent Wound Repair in Dictyostelium Cell Membrane. Cells 2020, 9, 1058. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mandato, C.A.; Bement, W.M. Actomyosin transports microtubules and microtubules control actomyosin recruitment during Xenopus oocyte wound healing. Curr. Biol. 2003, 13, 1096–1105. [Google Scholar] [CrossRef] [Green Version]
- Bement, W.M.; Benink, H.A.; von Dassow, G. A microtubule-dependent zone of active RhoA during cleavage plane specification. J. Cell Biol. 2005, 170, 91–101. [Google Scholar] [CrossRef] [PubMed]
- Abreu-Blanco, M.T.; Verboon, J.M.; Parkhurst, S.M. Cell wound repair in Drosophila occurs through three distinct phases of membrane and cytoskeletal remodeling. J. Cell Biol. 2011, 193, 455–464. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bement, W.M.; Mandato, C.A.; Kirsch, M.N. Wound-induced assembly and closure of an actomyosin purse string in Xenopus oocytes. Curr. Biol. 1999, 9, 579–587. [Google Scholar] [CrossRef] [Green Version]
- Mandato, C.A.; Bement, W.M. Contraction and polymerization cooperate to assemble and close actomyosin rings around Xenopus oocyte wounds. J. Cell Biol. 2001, 154, 785–797. [Google Scholar] [CrossRef]
- Benink, H.A.; Bement, W.M. Concentric zones of active RhoA and Cdc42 around single cell wounds. J. Cell Biol. 2005, 168, 429–439. [Google Scholar] [CrossRef] [Green Version]
- Abreu-Blanco, M.T.; Watts, J.J.; Verboon, J.M.; Parkhurst, S.M. Cytoskeleton responses in wound repair. Cell Mol. Life Sci. 2012, 69, 2469–2483. [Google Scholar] [CrossRef] [Green Version]
- Burkel, B.M.; Benink, H.A.; Vaughan, E.M.; von Dassow, G.; Bement, W.M. A Rho GTPase signal treadmill backs a contractile array. Dev. Cell 2012, 23, 384–396. [Google Scholar] [CrossRef] [Green Version]
- Abreu-Blanco, M.T.; Verboon, J.M.; Parkhurst, S.M. Coordination of Rho family GTPase activities to orchestrate cytoskeleton responses during cell wound repair. Curr. Biol. 2014, 24, 144–155. [Google Scholar] [CrossRef] [Green Version]
- Rohatgi, R.; Ma, L.; Miki, H.; Lopez, M.; Kirchhausen, T.; Takenawa, T.; Kirschner, M.W. The interaction between N-WASP and the Arp2/3 complex links Cdc42-dependent signals to actin assembly. Cell 1999, 97, 221–231. [Google Scholar] [CrossRef] [Green Version]
- Benink, H.A.; Mandato, C.A.; Bement, W.M. Analysis of cortical flow models in vivo. Mol. Biol. Cell 2000, 11, 2553–2563. [Google Scholar] [CrossRef] [Green Version]
- Kawano, Y.; Fukata, Y.; Oshiro, N.; Amano, M.; Nakamura, T.; Ito, M.; Matsumura, F.; Inagaki, M.; Kaibuchi, K. Phosphorylation of myosin-binding subunit (MBS) of myosin phosphatase by Rho-kinase in vivo. J. Cell Biol. 1999, 147, 1023–1038. [Google Scholar] [CrossRef] [Green Version]
- Bishop, A.L.; Hall, A. Rho GTPases and their effector proteins. Biochem. J. 2000, 348, 241–255. [Google Scholar] [CrossRef]
- Vaughan, E.M.; You, J.S.; Elsie Yu, H.Y.; Lasek, A.; Vitale, N.; Hornberger, T.A.; Bement, W.M. Lipid domain-dependent regulation of single-cell wound repair. Mol. Biol. Cell 2014, 25, 1867–1876. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schwayer, C.; Sikora, M.; Slováková, J.; Kardos, R.; Heisenberg, C.P. Actin Rings of Power. Dev. Cell 2016, 37, 493–506. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DeKraker, C.; Goldin-Blais, L.; Boucher, E.; Mandato, C.A. Dynamics of actin polymerisation during the mammalian single-cell wound healing response. BMC Res. Notes 2019, 12, 420. [Google Scholar] [CrossRef] [PubMed]
- Boye, T.L.; Maeda, K.; Pezeshkian, W.; Sønder, S.L.; Haeger, S.C.; Gerke, V.; Simonsen, A.C.; Nylandsted, J. Annexin A4 and A6 induce membrane curvature and constriction during cell membrane repair. Nat. Commun. 2017, 8, 1623. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jaiswal, J.K.; Lauritzen, S.P.; Scheffer, L.; Sakaguchi, M.; Bunkenborg, J.; Simon, S.M.; Kallunki, T.; Jäättelä, M.; Nylandsted, J. S100A11 is required for efficient plasma membrane repair and survival of invasive cancer cells. Nat. Commun. 2014, 5, 3795. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hayes, M.J.; Rescher, U.; Gerke, V.; Moss, S.E. Annexin-actin interactions. Traffic 2004, 5, 571–576. [Google Scholar] [CrossRef]
- Hayes, M.J.; Shao, D.; Bailly, M.; Moss, S.E. Regulation of actin dynamics by annexin 2. EMBO J. 2006, 25, 1816–1826. [Google Scholar] [CrossRef] [Green Version]
- Morel, E.; Parton, R.G.; Gruenberg, J. Annexin A2-dependent polymerization of actin mediates endosome biogenesis. Dev. Cell 2009, 16, 445–457. [Google Scholar] [CrossRef]
- Bouter, A.; Gounou, C.; Bérat, R.; Tan, S.; Gallois, B.; Granier, T.; d’Estaintot, B.L.; Pöschl, E.; Brachvogel, B.; Brisson, A.R. Annexin-A5 assembled into two-dimensional arrays promotes cell membrane repair. Nat. Commun. 2011, 2, 270. [Google Scholar] [CrossRef] [Green Version]
- Melle, C.; Ernst, G.; Schimmel, B.; Bleul, A.; Mothes, H.; Kaufmann, R.; Settmacher, U.; Von Eggeling, F. Different expression of calgizzarin (S100A11) in normal colonic epithelium, adenoma and colorectal carcinoma. Int. J. Oncol. 2006, 28, 195–200. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cross, S.S.; Hamdy, F.C.; Deloulme, J.C.; Rehman, I. Expression of S100 proteins in normal human tissues and common cancers using tissue microarrays: S100A6, S100A8, S100A9 and S100A11 are all overexpressed in common cancers. Histopathology 2005, 46, 256–269. [Google Scholar] [CrossRef]
- Rehman, I.; Azzouzi, A.R.; Cross, S.S.; Deloulme, J.C.; Catto, J.W.; Wylde, N.; Larre, S.; Champigneuille, J.; Hamdy, F.C. Dysregulated expression of S100A11 (calgizzarin) in prostate cancer and precursor lesions. Hum. Pathol. 2004, 35, 1385–1391. [Google Scholar] [CrossRef] [PubMed]
- Xiao, M.B.; Jiang, F.; Ni, W.K.; Chen, B.Y.; Lu, C.H.; Li, X.Y.; Ni, R.Z. High expression of S100A11 in pancreatic adenocarcinoma is an unfavorable prognostic marker. Med. Oncol. 2012, 29, 1886–1891. [Google Scholar] [CrossRef]
- Tian, T.; Hao, J.; Xu, A.; Hao, J.; Luo, C.; Liu, C.; Huang, L.; Xiao, X.; He, D. Determination of metastasis-associated proteins in non-small cell lung cancer by comparative proteomic analysis. Cancer Sci. 2007, 98, 1265–1274. [Google Scholar] [CrossRef]
- Melle, C.; Ernst, G.; Schimmel, B.; Bleul, A.; von Eggeling, F. Colon-derived liver metastasis, colorectal carcinoma, and hepatocellular carcinoma can be discriminated by the Ca(2+)-binding proteins S100A6 and S100A11. PLoS ONE 2008, 3, e3767. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Christensen, M.V.; Høgdall, C.K.; Jochumsen, K.M.; Høgdall, E.V.S. Annexin A2 and cancer: A systematic review. Int. J. Oncol. 2018, 52, 5–18. [Google Scholar] [CrossRef]
- Wang, T.; Wang, Z.; Niu, R.; Wang, L. Crucial role of Anxa2 in cancer progression: Highlights on its novel regulatory mechanism. Cancer Biol. Med. 2019, 16, 671–687. [Google Scholar] [CrossRef] [PubMed]
- Rocha, M.R.; Barcellos-de-Souza, P.; Sousa-Squiavinato, A.C.M.; Fernandes, P.V.; de Oliveira, I.M.; Boroni, M.; Morgado-Diaz, J.A. Annexin A2 overexpression associates with colorectal cancer invasiveness and TGF-ß induced epithelial mesenchymal transition via Src/ANXA2/STAT3. Sci. Rep. 2018, 8, 11285. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, C.Y.; Lin, Y.S.; Chen, C.H.; Chen, Y.J. Annexin A2-mediated cancer progression and therapeutic resistance in nasopharyngeal carcinoma. J. Biomed. Sci. 2018, 25, 30. [Google Scholar] [CrossRef] [PubMed]
- Sharma, M.C. Annexin A2 (ANX A2): An emerging biomarker and potential therapeutic target for aggressive cancers. Int. J. Cancer 2019, 144, 2074–2081. [Google Scholar] [CrossRef] [PubMed]
- Emoto, K.; Sawada, H.; Yamada, Y.; Fujimoto, H.; Takahama, Y.; Ueno, M.; Takayama, T.; Uchida, H.; Kamada, K.; Naito, A.; et al. Annexin II overexpression is correlated with poor prognosis in human gastric carcinoma. Anticancer Res. 2001, 21, 1339–1345. [Google Scholar] [PubMed]
- Luo, S.; Xie, C.; Wu, P.; He, J.; Tang, Y.; Xu, J.; Zhao, S. Annexin A2 is an independent prognostic biomarker for evaluating the malignant progression of laryngeal cancer. Exp. Med. 2017, 14, 6113–6118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rezvanpour, A.; Santamaria-Kisiel, L.; Shaw, G.S. The S100A10-annexin A2 complex provides a novel asymmetric platform for membrane repair. J. Biol. Chem. 2011, 286, 40174–40183. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dempsey, B.R.; Rezvanpour, A.; Lee, T.W.; Barber, K.R.; Junop, M.S.; Shaw, G.S. Structure of an asymmetric ternary protein complex provides insight for membrane interaction. Structure 2012, 20, 1737–1745. [Google Scholar] [CrossRef] [Green Version]
- Ozorowski, G.; Milton, S.; Luecke, H. Structure of a C-terminal AHNAK peptide in a 1:2:2 complex with S100A10 and an acetylated N-terminal peptide of annexin A2. Acta Cryst. D Biol. Cryst. 2013, 69, 92–104. [Google Scholar] [CrossRef]
- Hohaus, A.; Person, V.; Behlke, J.; Schaper, J.; Morano, I.; Haase, H. The carboxyl-terminal region of ahnak provides a link between cardiac L-type Ca2+ channels and the actin-based cytoskeleton. FASEB J. 2002, 16, 1205–1216. [Google Scholar] [CrossRef]
- Benaud, C.; Gentil, B.J.; Assard, N.; Court, M.; Garin, J.; Delphin, C.; Baudier, J. AHNAK interaction with the annexin 2/S100A10 complex regulates cell membrane cytoarchitecture. J. Cell Biol. 2004, 164, 133–144. [Google Scholar] [CrossRef]
- Cocucci, E.; Racchetti, G.; Podini, P.; Rupnik, M.; Meldolesi, J. Enlargeosome, an exocytic vesicle resistant to nonionic detergents, undergoes endocytosis via a nonacidic route. Mol. Biol. Cell 2004, 15, 5356–5368. [Google Scholar] [CrossRef] [Green Version]
- Cocucci, E.; Racchetti, G.; Podini, P.; Meldolesi, J. Enlargeosome traffic: Exocytosis triggered by various signals is followed by endocytosis, membrane shedding or both. Traffic 2007, 8, 742–757. [Google Scholar] [CrossRef] [PubMed]
- Cerny, J.; Feng, Y.; Yu, A.; Miyake, K.; Borgonovo, B.; Klumperman, J.; Meldolesi, J.; McNeil, P.L.; Kirchhausen, T. The small chemical vacuolin-1 inhibits Ca(2+)-dependent lysosomal exocytosis but not cell resealing. EMBO Rep. 2004, 5, 883–888. [Google Scholar] [CrossRef] [PubMed]
- Swaggart, K.A.; Demonbreun, A.R.; Vo, A.H.; Swanson, K.E.; Kim, E.Y.; Fahrenbach, J.P.; Holley-Cuthrell, J.; Eskin, A.; Chen, Z.; Squire, K.; et al. Annexin A6 modifies muscular dystrophy by mediating sarcolemmal repair. Proc. Natl. Acad. Sci. USA 2014, 111, 6004–6009. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Demonbreun, A.R.; Quattrocelli, M.; Barefield, D.Y.; Allen, M.V.; Swanson, K.E.; McNally, E.M. An actin-dependent annexin complex mediates plasma membrane repair in muscle. J. Cell Biol. 2016, 213, 705–718. [Google Scholar] [CrossRef] [PubMed]
- Keefe, D.; Shi, L.; Feske, S.; Massol, R.; Navarro, F.; Kirchhausen, T.; Lieberman, J. Perforin triggers a plasma membrane-repair response that facilitates CTL induction of apoptosis. Immunity 2005, 23, 249–262. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Borgonovo, B.; Cocucci, E.; Racchetti, G.; Podini, P.; Bachi, A.; Meldolesi, J. Regulated exocytosis: A novel, widely expressed system. Nat. Cell Biol. 2002, 4, 955–962. [Google Scholar] [CrossRef]
- Tam, C.; Idone, V.; Devlin, C.; Fernandes, M.C.; Flannery, A.; He, X.; Schuchman, E.; Tabas, I.; Andrews, N.W. Exocytosis of acid sphingomyelinase by wounded cells promotes endocytosis and plasma membrane repair. J. Cell Biol. 2010, 189, 1027–1038. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodríguez, A.; Webster, P.; Ortego, J.; Andrews, N.W. Lysosomes behave as Ca2+-regulated exocytic vesicles in fibroblasts and epithelial cells. J. Cell Biol. 1997, 137, 93–104. [Google Scholar] [CrossRef] [PubMed]
- Andrews, N.W.; Corrotte, M.; Castro-Gomes, T. Above the fray: Surface remodeling by secreted lysosomal enzymes leads to endocytosis-mediated plasma membrane repair. Semin Cell Dev. Biol. 2015, 45, 10–17. [Google Scholar] [CrossRef] [Green Version]
- Bakker, A.C.; Webster, P.; Jacob, W.A.; Andrews, N.W. Homotypic fusion between aggregated lysosomes triggered by elevated [Ca2+]i in fibroblasts. J. Cell Sci. 1997, 110, 2227–2238. [Google Scholar] [CrossRef]
- Li, D.; Ropert, N.; Koulakoff, A.; Giaume, C.; Oheim, M. Lysosomes are the major vesicular compartment undergoing Ca2+-regulated exocytosis from cortical astrocytes. J. Neurosci. 2008, 28, 7648–7658. [Google Scholar] [CrossRef]
- Cheng, X.; Zhang, X.; Gao, Q.; Ali Samie, M.; Azar, M.; Tsang, W.L.; Dong, L.; Sahoo, N.; Li, X.; Zhuo, Y.; et al. The intracellular Ca²⁺ channel MCOLN1 is required for sarcolemma repair to prevent muscular dystrophy. Nat. Med. 2014, 20, 1187–1192. [Google Scholar] [CrossRef] [Green Version]
- Jaiswal, J.K.; Andrews, N.W.; Simon, S.M. Membrane proximal lysosomes are the major vesicles responsible for calcium-dependent exocytosis in nonsecretory cells. J. Cell Biol. 2002, 159, 625–635. [Google Scholar] [CrossRef] [Green Version]
- Andrews, N.W.; Corrotte, M. Plasma membrane repair. Curr. Biol. 2018, 28, R392–R397. [Google Scholar] [CrossRef] [Green Version]
- Andrews, N.W.; Almeida, P.E.; Corrotte, M. Damage control: Cellular mechanisms of plasma membrane repair. Trends Cell Biol. 2014, 24, 734–742. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Togo, T.; Krasieva, T.B.; Steinhardt, R.A. A decrease in membrane tension precedes successful cell-membrane repair. Mol. Biol. Cell 2000, 11, 4339–4346. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boucher, E.; Mandato, C.A. Plasma membrane and cytoskeleton dynamics during single-cell wound healing. Biochim. Biophys. Acta 2015, 1853, 2649–2661. [Google Scholar] [CrossRef] [Green Version]
- Barlan, K.; Gelfand, V.I. Microtubule-Based Transport and the Distribution, Tethering, and Organization of Organelles. Cold Spring Harb. Perspect. Biol. 2017, 9, a025817. [Google Scholar] [CrossRef] [PubMed]
- Noordstra, I.; Akhmanova, A. Linking cortical microtubule attachment and exocytosis. F1000Research 2017, 6, 469. [Google Scholar] [CrossRef] [PubMed]
- Li, P.; Bademosi, A.T.; Luo, J.; Meunier, F.A. Actin Remodeling in Regulated Exocytosis: Toward a Mesoscopic View. Trends Cell Biol. 2018, 28, 685–697. [Google Scholar] [CrossRef] [Green Version]
- Miklavc, P.; Frick, M. Actin and Myosin in Non-Neuronal Exocytosis. Cells 2020, 9, 1455. [Google Scholar] [CrossRef] [PubMed]
- Wollman, R.; Meyer, T. Coordinated oscillations in cortical actin and Ca2+ correlate with cycles of vesicle secretion. Nat. Cell Biol. 2012, 14, 1261–1269. [Google Scholar] [CrossRef] [Green Version]
- Holopainen, J.M.; Angelova, M.I.; Kinnunen, P.K. Vectorial budding of vesicles by asymmetrical enzymatic formation of ceramide in giant liposomes. Biophys. J. 2000, 78, 830–838. [Google Scholar] [CrossRef] [Green Version]
- Schissel, S.L.; Schuchman, E.H.; Williams, K.J.; Tabas, I. Zn2+-stimulated sphingomyelinase is secreted by many cell types and is a product of the acid sphingomyelinase gene. J. Biol. Chem. 1996, 271, 18431–18436. [Google Scholar] [CrossRef] [Green Version]
- Jenkins, R.W.; Canals, D.; Hannun, Y.A. Roles and regulation of secretory and lysosomal acid sphingomyelinase. Cell Signal. 2009, 21, 836–846. [Google Scholar] [CrossRef] [Green Version]
- Corrotte, M.; Almeida, P.E.; Tam, C.; Castro-Gomes, T.; Fernandes, M.C.; Millis, B.A.; Cortez, M.; Miller, H.; Song, W.; Maugel, T.K.; et al. Caveolae internalization repairs wounded cells and muscle fibers. Elife 2013, 2, e00926. [Google Scholar] [CrossRef]
- Kelly, B.T.; Graham, S.C.; Liska, N.; Dannhauser, P.N.; Höning, S.; Ungewickell, E.J.; Owen, D.J. Clathrin adaptors. AP2 controls clathrin polymerization with a membrane-activated switch. Science 2014, 345, 459–463. [Google Scholar] [CrossRef] [Green Version]
- Mooren, O.L.; Galletta, B.J.; Cooper, J.A. Roles for actin assembly in endocytosis. Annu Rev. Biochem. 2012, 81, 661–686. [Google Scholar] [CrossRef]
- Akamatsu, M.; Vasan, R.; Serwas, D.; Ferrin, M.A.; Rangamani, P.; Drubin, D.G. Principles of self-organization and load adaptation by the actin cytoskeleton during clathrin-mediated endocytosis. Elife 2020, 9, e49840. [Google Scholar] [CrossRef] [PubMed]
- Hemalatha, A.; Mayor, S. Recent advances in clathrin-independent endocytosis. F1000Research 2019, 8. [Google Scholar] [CrossRef]
- Idone, V.; Tam, C.; Goss, J.W.; Toomre, D.; Pypaert, M.; Andrews, N.W. Repair of injured plasma membrane by rapid Ca2+-dependent endocytosis. J. Cell Biol. 2008, 180, 905–914. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nygård Skalman, L.; Holst, M.R.; Larsson, E.; Lundmark, R. Plasma membrane damage caused by listeriolysin O is not repaired through endocytosis of the membrane pore. Biol. Open 2018, 7, bio035287. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dai, J.; Sheetz, M.P. Membrane tether formation from blebbing cells. Biophys. J. 1999, 77, 3363–3370. [Google Scholar] [CrossRef] [Green Version]
- Charras, G.T.; Hu, C.K.; Coughlin, M.; Mitchison, T.J. Reassembly of contractile actin cortex in cell blebs. J. Cell Biol. 2006, 175, 477–490. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Babiychuk, E.B.; Monastyrskaya, K.; Potez, S.; Draeger, A. Blebbing confers resistance against cell lysis. Cell Death Differ. 2011, 18, 80–89. [Google Scholar] [CrossRef] [Green Version]
- Skočaj, M.; Yu, Y.; Grundner, M.; Resnik, N.; Bedina Zavec, A.; Leonardi, A.; Križaj, I.; Guella, G.; Maček, P.; Kreft, M.E.; et al. Characterisation of plasmalemmal shedding of vesicles induced by the cholesterol/sphingomyelin binding protein, ostreolysin A-mCherry. Biochim. Biophys. Acta 2016, 1858, 2882–2893. [Google Scholar] [CrossRef]
- Keyel, P.A.; Loultcheva, L.; Roth, R.; Salter, R.D.; Watkins, S.C.; Yokoyama, W.M.; Heuser, J.E. Streptolysin O clearance through sequestration into blebs that bud passively from the plasma membrane. J. Cell Sci. 2011, 124, 2414–2423. [Google Scholar] [CrossRef] [Green Version]
- Brito, C.; Cabanes, D.; Sarmento Mesquita, F.; Sousa, S. Mechanisms protecting host cells against bacterial pore-forming toxins. Cell Mol. Life Sci. 2019, 76, 1319–1339. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Romero, M.; Keyel, M.; Shi, G.; Bhattacharjee, P.; Roth, R.; Heuser, J.E.; Keyel, P.A. Intrinsic repair protects cells from pore-forming toxins by microvesicle shedding. Cell Death Differ. 2017, 24, 798–808. [Google Scholar] [CrossRef] [PubMed]
- Babiychuk, E.B.; Monastyrskaya, K.; Potez, S.; Draeger, A. Intracellular Ca(2+) operates a switch between repair and lysis of streptolysin O-perforated cells. Cell Death Differ. 2009, 16, 1126–1134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gerke, V.; Creutz, C.E.; Moss, S.E. Annexins: Linking Ca2+ signalling to membrane dynamics. Nat. Rev. Mol. Cell Biol. 2005, 6, 449–461. [Google Scholar] [CrossRef]
- Wolfmeier, H.; Radecke, J.; Schoenauer, R.; Koeffel, R.; Babiychuk, V.S.; Drücker, P.; Hathaway, L.J.; Mitchell, T.J.; Zuber, B.; Draeger, A.; et al. Active release of pneumolysin prepores and pores by mammalian cells undergoing a Streptococcus pneumoniae attack. Biochim. Biophys. Acta 2016, 1860, 2498–2509. [Google Scholar] [CrossRef] [Green Version]
- Jiao, M.; Wu, D.; Wei, Q. Myosin II-interacting guanine nucleotide exchange factor promotes bleb retraction via stimulating cortex reassembly at the bleb membrane. Mol. Biol. Cell 2018, 29, 643–656. [Google Scholar] [CrossRef] [Green Version]
- Aoki, K.; Maeda, F.; Nagasako, T.; Mochizuki, Y.; Uchida, S.; Ikenouchi, J. A RhoA and Rnd3 cycle regulates actin reassembly during membrane blebbing. Proc. Natl. Acad. Sci. USA 2016, 113, E1863–E1871. [Google Scholar] [CrossRef] [Green Version]
- Chikina, A.S.; Svitkina, T.M.; Alexandrova, A.Y. Time-resolved ultrastructure of the cortical actin cytoskeleton in dynamic membrane blebs. J. Cell Biol. 2019, 218, 445–454. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sønder, S.L.; Häger, S.C.; Heitmann, A.S.B.; Frankel, L.B.; Dias, C.; Simonsen, A.C.; Nylandsted, J. Restructuring of the plasma membrane upon damage by LC3-associated macropinocytosis. Sci. Adv. 2021, 7, eabg1969. [Google Scholar] [CrossRef]
- Scheffer, L.L.; Sreetama, S.C.; Sharma, N.; Medikayala, S.; Brown, K.J.; Defour, A.; Jaiswal, J.K. Mechanism of Ca²⁺-triggered ESCRT assembly and regulation of cell membrane repair. Nat. Commun 2014, 5, 5646. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jimenez, A.J.; Maiuri, P.; Lafaurie-Janvore, J.; Divoux, S.; Piel, M.; Perez, F. ESCRT machinery is required for plasma membrane repair. Science 2014, 343, 1247136. [Google Scholar] [CrossRef] [PubMed]
- Potez, S.; Luginbühl, M.; Monastyrskaya, K.; Hostettler, A.; Draeger, A.; Babiychuk, E.B. Tailored protection against plasmalemmal injury by annexins with different Ca2+ sensitivities. J. Biol. Chem. 2011, 286, 17982–17991. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wolfmeier, H.; Schoenauer, R.; Atanassoff, A.P.; Neill, D.R.; Kadioglu, A.; Draeger, A.; Babiychuk, E.B. Ca²⁺-dependent repair of pneumolysin pores: A new paradigm for host cellular defense against bacterial pore-forming toxins. Biochim. Biophys. Acta 2015, 1853, 2045–2054. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mageswaran, S.K.; Yang, W.Y.; Chakrabarty, Y.; Oikonomou, C.M.; Jensen, G.J. Plasma membrane damage removal by F-actin-mediated shedding from repurposed filopodia. bioRxiv 2019. [Google Scholar] [CrossRef]
- Cauwenberghs, S.; Feijge, M.A.; Harper, A.G.; Sage, S.O.; Curvers, J.; Heemskerk, J.W. Shedding of procoagulant microparticles from unstimulated platelets by integrin-mediated destabilization of actin cytoskeleton. FEBS Lett. 2006, 580, 5313–5320. [Google Scholar] [CrossRef] [Green Version]
- Fox, J.E.; Austin, C.D.; Boyles, J.K.; Steffen, P.K. Role of the membrane skeleton in preventing the shedding of procoagulant-rich microvesicles from the platelet plasma membrane. J. Cell Biol. 1990, 111, 483–493. [Google Scholar] [CrossRef] [Green Version]
- Middelhoven, P.J.; van Buul, J.D.; Kleijer, M.; Roos, D.; Hordijk, P.L. Actin polymerization induces shedding of FcgammaRIIIb (CD16) from human neutrophils. Biochem. Biophys. Res. Commun. 1999, 255, 568–574. [Google Scholar] [CrossRef]
- Meng, X.; Yang, Q.; Yu, X.; Zhou, J.; Ren, X.; Zhou, Y.; Xu, S. Actin Polymerization and ESCRT Trigger Recruitment of the Fusogens Syntaxin-2 and EFF-1 to Promote Membrane Repair in C. elegans. Dev. Cell 2020, 54, 624–638.e625. [Google Scholar] [CrossRef]
- Ohashi, K.; Fujiwara, S.; Mizuno, K. Roles of the cytoskeleton, cell adhesion and rho signalling in mechanosensing and mechanotransduction. J. Biochem. 2017, 161, 245–254. [Google Scholar] [CrossRef] [Green Version]
- Shao, X.; Li, Q.; Mogilner, A.; Bershadsky, A.D.; Shivashankar, G.V. Mechanical stimulation induces formin-dependent assembly of a perinuclear actin rim. Proc. Natl. Acad. Sci. USA 2015, 112, E2595–E2601. [Google Scholar] [CrossRef] [Green Version]
- Wales, P.; Schuberth, C.E.; Aufschnaiter, R.; Fels, J.; García-Aguilar, I.; Janning, A.; Dlugos, C.P.; Schäfer-Herte, M.; Klingner, C.; Wälte, M.; et al. Calcium-mediated actin reset (CaAR) mediates acute cell adaptations. Elife 2016, 5, e19850. [Google Scholar] [CrossRef] [PubMed]
- Raab, M.; Gentili, M.; de Belly, H.; Thiam, H.R.; Vargas, P.; Jimenez, A.J.; Lautenschlaeger, F.; Voituriez, R.; Lennon-Duménil, A.M.; Manel, N.; et al. ESCRT III repairs nuclear envelope ruptures during cell migration to limit DNA damage and cell death. Science 2016, 352, 359–362. [Google Scholar] [CrossRef] [PubMed]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ebstrup, M.L.; Dias, C.; Heitmann, A.S.B.; Sønder, S.L.; Nylandsted, J. Actin Cytoskeletal Dynamics in Single-Cell Wound Repair. Int. J. Mol. Sci. 2021, 22, 10886. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms221910886
Ebstrup ML, Dias C, Heitmann ASB, Sønder SL, Nylandsted J. Actin Cytoskeletal Dynamics in Single-Cell Wound Repair. International Journal of Molecular Sciences. 2021; 22(19):10886. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms221910886
Chicago/Turabian StyleEbstrup, Malene Laage, Catarina Dias, Anne Sofie Busk Heitmann, Stine Lauritzen Sønder, and Jesper Nylandsted. 2021. "Actin Cytoskeletal Dynamics in Single-Cell Wound Repair" International Journal of Molecular Sciences 22, no. 19: 10886. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms221910886