
Androgen Receptor Activation in Glioblastoma Can Be Achieved by Ligand-Independent Signaling through EGFR—A Potential Therapeutic Target


Abstract
:1. Introduction
2. Results
2.1. AR Expression in Glioblastoma Samples Correlates Significantly with EGFR Expression Levels
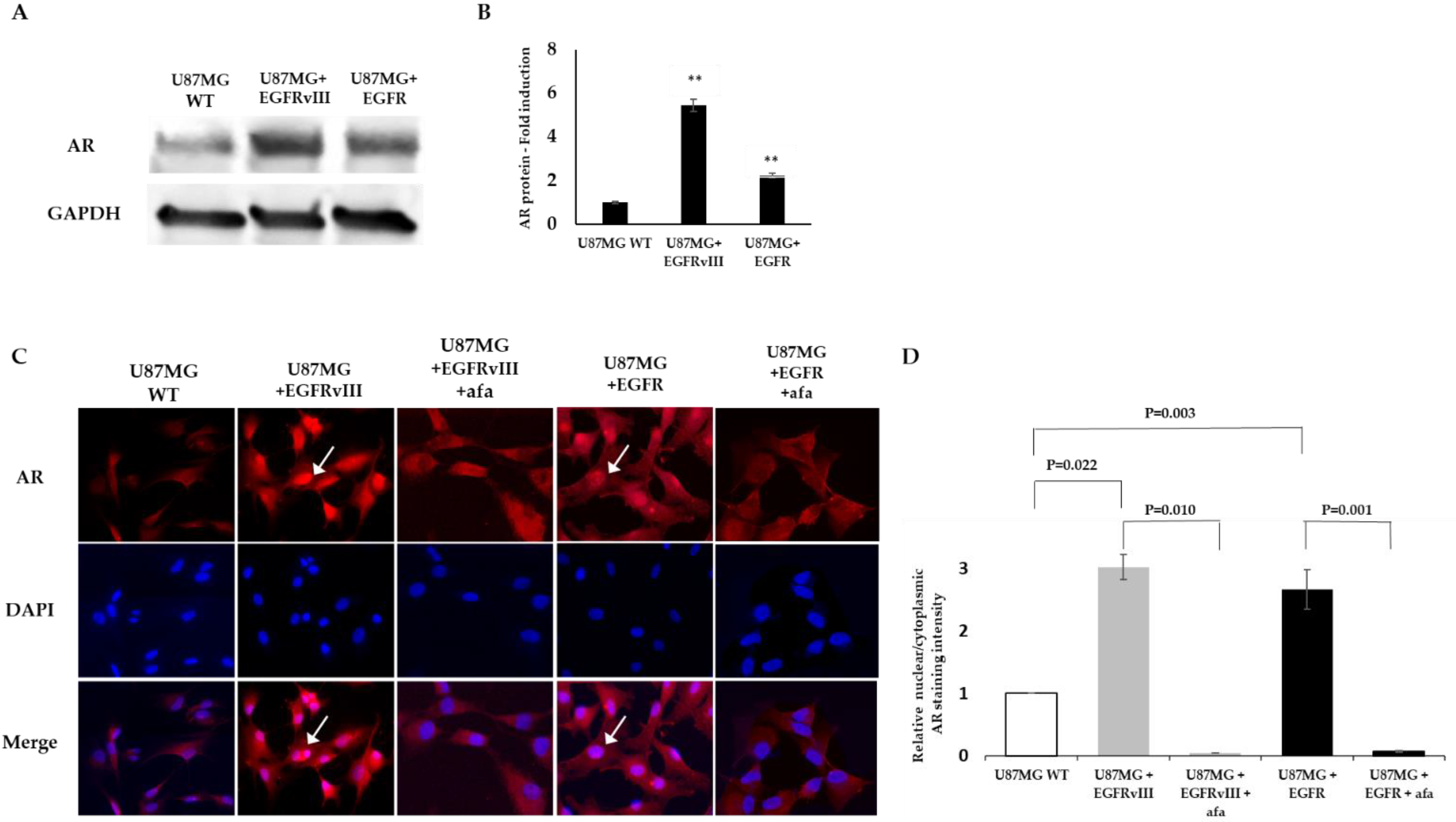
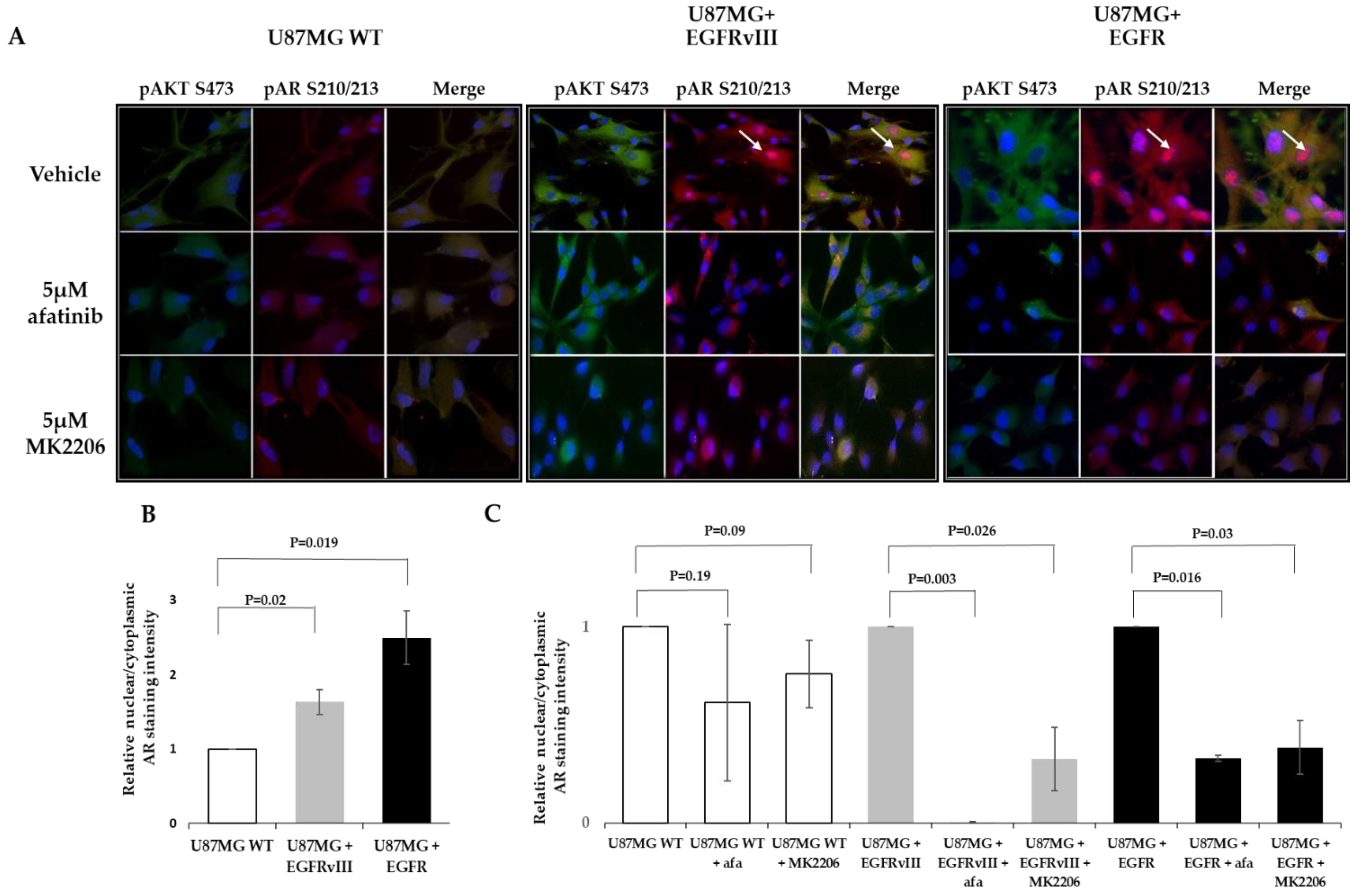
2.2. EGFR or EGFRvIII Overexpression Induces AR Overexpression and Nuclear Translocation, While EGFR and AKT Inhibitor Treatment Abrogates the AR Nuclear Translocation
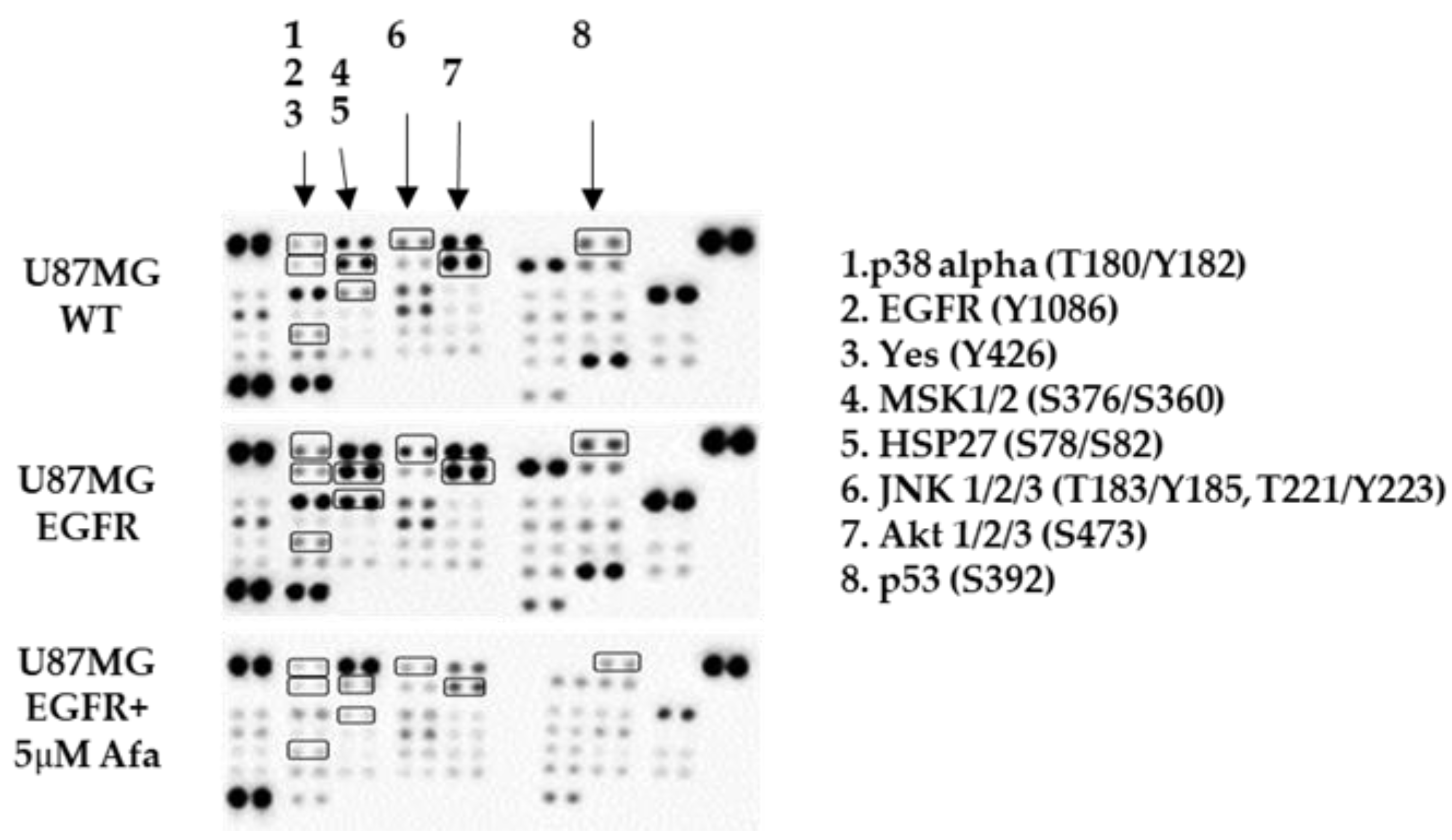
2.3. Phospho-Kinase Array Evaluation of EGFR Downstream Effectors
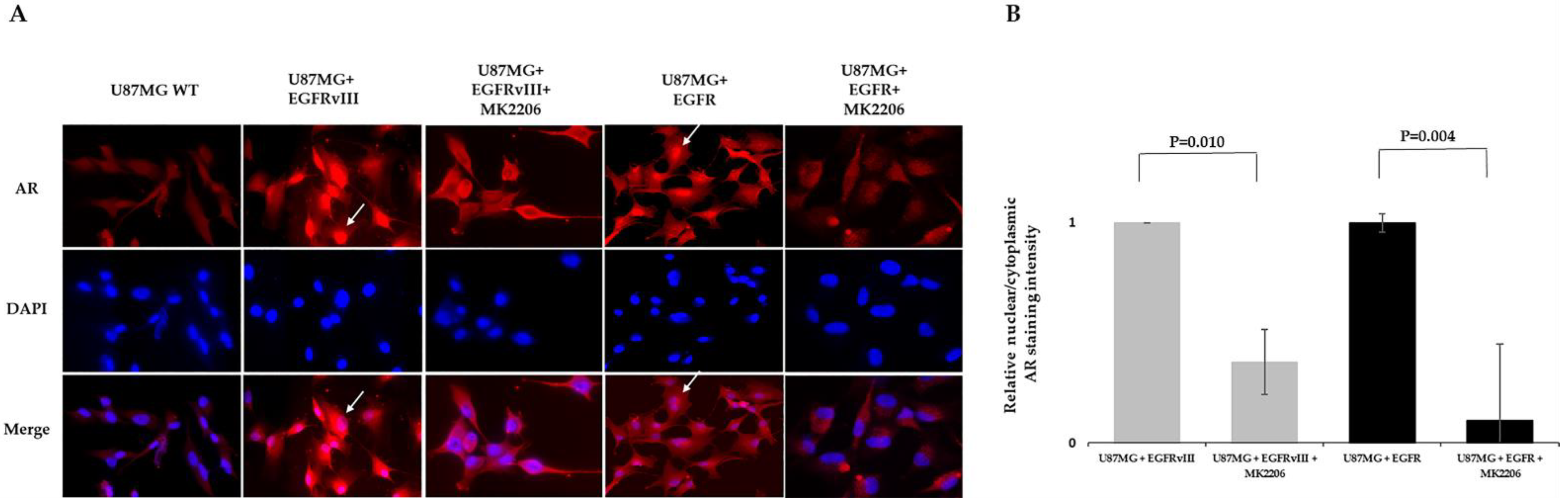
2.4. MK2206 Modulates EGFR-Induced AR Expression and Nuclear Translocation
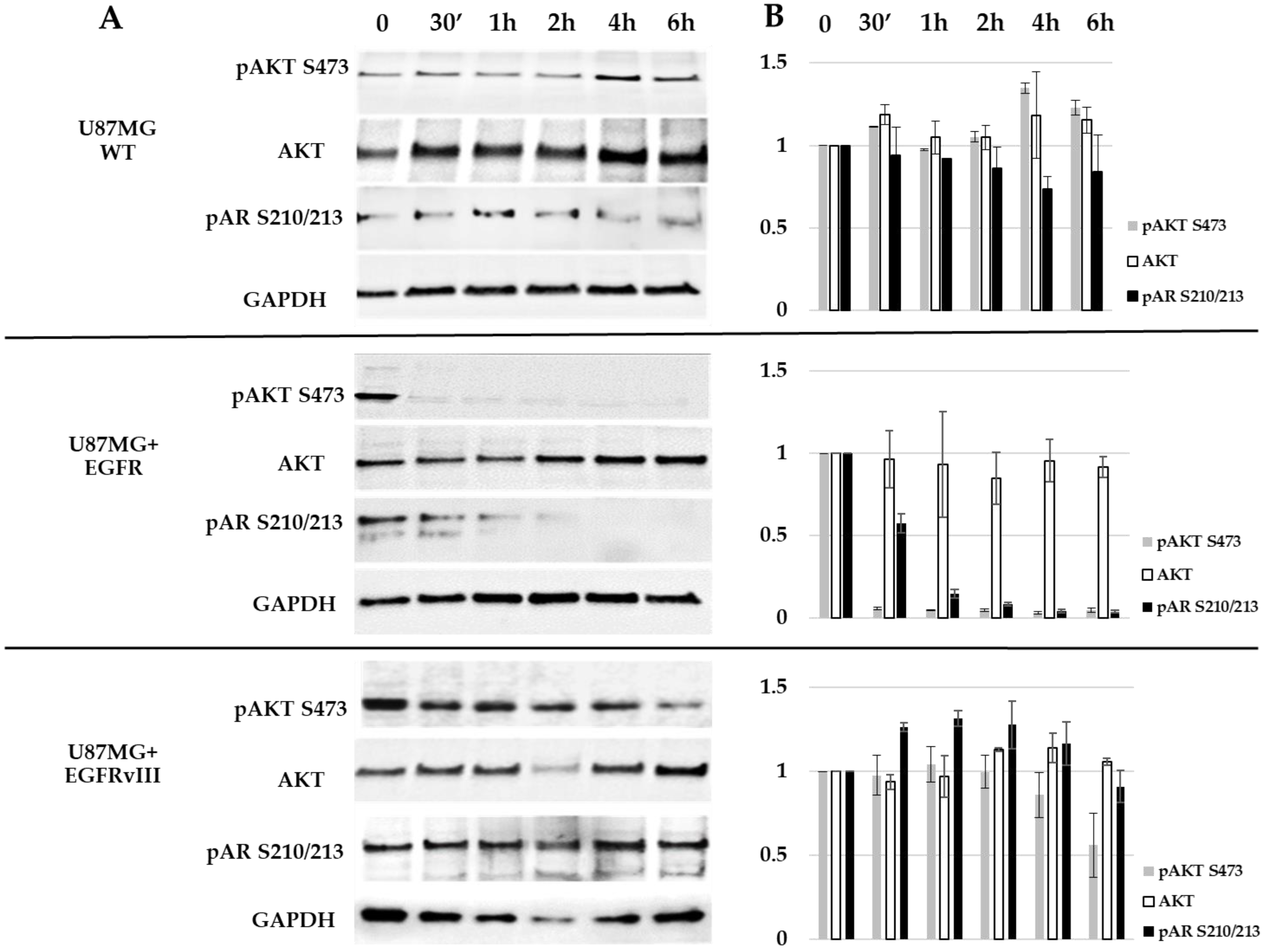
2.5. EGFR Probably Induces AR Phosphorylation on S210/213 through AKT S473
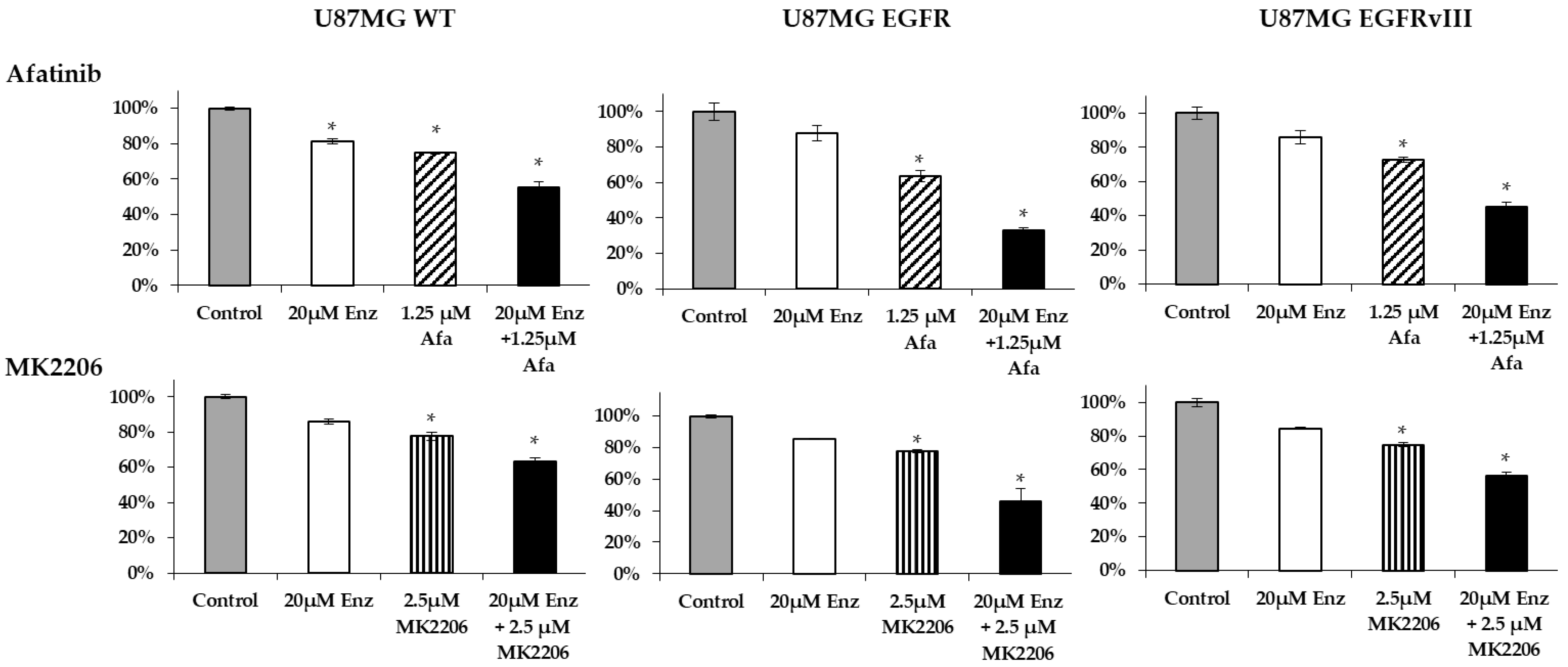
2.6. Afatinib and Enzalutamide Combination Therapy Yields Improved Efficacy in Glioblastoma Cell Lines
3. Discussion
4. Materials and Methods
4.1. Patients and Tumors
4.2. RNA Extraction, cDNA Preparation, and qPCR
4.3. Cell Culture and Treatments
4.4. Cell Survival and Drug Interaction Analysis
4.5. Immunofluorescence Staining, Western Blotting, and ELISA
4.6. Human Phospho-Kinase Antibody Array
4.7. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
Correction Statement
References
- Poisson, M.; Pertuiset, B.F.; Hauw, J.J.; Philippon, J.; Buge, A.; Moguilewsky, M.; Philibert, D. Steroid hormone receptors in human meningiomas, gliomas and brain metastases. J. Neuro-Oncol. 1983, 1, 179–189. [Google Scholar] [CrossRef] [PubMed]
- Felsberg, J.; Hentschel, B.; Kaulich, K.; Gramatzki, D.; Zacher, A.; Malzkorn, B.; Kamp, M.; Sabel, M.; Simon, M.; Westphal, M.; et al. Epidermal Growth Factor Receptor Variant III (EGFRvIII) Positivity in EGFR-Amplified Glioblastomas: Prognostic Role and Comparison between Primary and Recurrent Tumors. Clin. Cancer Res. 2017, 23, 6846–6855. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Wu, C.; Chen, N.; Gu, H.; Yen, A.; Cao, L.; Wang, E.; Wang, L. PI3K/Akt/mTOR signaling pathway and targeted therapy for glioblastoma. Oncotarget 2016, 7, 33440–33450. [Google Scholar] [CrossRef] [PubMed]
- Minkovsky, N.; Berezov, A. BIBW-2992, a dual receptor tyrosine kinase inhibitor for the treatment of solid tumors. Curr. Opin. Investig. Drugs 2008, 9, 1336–1346. [Google Scholar]
- Messersmith, W.A.; Ahnen, D.J. Targeting EGFR in colorectal cancer. N. Engl. J. Med. 2008, 359, 1834–1836. [Google Scholar] [CrossRef]
- Prados, M.D.; Byron, S.A.; Tran, N.L.; Phillips, J.J.; Molinaro, A.M.; Ligon, K.L.; Wen, P.Y.; Kuhn, J.G.; Mellinghoff, I.K.; de Groot, J.F.; et al. Toward precision medicine in glioblastoma: The promise and the challenges. Neuro. Oncol. 2015, 17, 1051–1063. [Google Scholar] [CrossRef]
- Von Achenbach, C.; Silginer, M.; Blot, V.; Weiss, W.A.; Weller, M. Depatuxizumab Mafodotin (ABT-414)-induced Glioblastoma Cell Death Requires EGFR Overexpression, but not EGFR(Y1068) Phosphorylation. Mol. Cancer 2020, 19, 1328–1339. [Google Scholar] [CrossRef]
- Tan, M.H.; Li, J.; Xu, H.E.; Melcher, K.; Yong, E.L. Androgen receptor: Structure, role in prostate cancer and drug discovery. Acta Pharmacol. Sin. 2015, 36, 3–23. [Google Scholar] [CrossRef]
- Feldman, B.J.; Feldman, D. The development of androgen-independent prostate cancer. Nat. Rev. Cancer 2001, 1, 34–45. [Google Scholar] [CrossRef]
- Ricke, E.A.; Williams, K.; Lee, Y.F.; Couto, S.; Wang, Y.; Hayward, S.W.; Cunha, G.R.; Ricke, W.A. Androgen hormone action in prostatic carcinogenesis: Stromal androgen receptors mediate prostate cancer progression, malignant transformation and metastasis. Carcinogenesis 2012, 33, 1391–1398. [Google Scholar] [CrossRef]
- Lin, C.Y.; Jan, Y.J.; Kuo, L.K.; Wang, B.J.; Huo, C.; Jiang, S.S.; Chen, S.C.; Kuo, Y.Y.; Chang, C.R.; Chuu, C.P. Elevation of androgen receptor promotes prostate cancer metastasis by induction of epithelial-mesenchymal transition and reduction of KAT5. Cancer Sci. 2018, 109, 3564–3574. [Google Scholar] [CrossRef]
- Yu, X.; Jiang, Y.; Wei, W.; Cong, P.; Ding, Y.; Xiang, L.; Wu, K. Androgen receptor signaling regulates growth of glioblastoma multiforme in men. Tumour Biol. J. Int. Soc. Oncodev. Biol. Med. 2015, 36, 967–972. [Google Scholar] [CrossRef] [PubMed]
- Zalcman, N.; Canello, T.; Ovadia, H.; Charbit, H.; Zelikovitch, B.; Mordechai, A.; Fellig, Y.; Rabani, S.; Shahar, T.; Lossos, A.; et al. Androgen receptor: A potential therapeutic target for glioblastoma. Oncotarget 2018, 9, 19980–19993. [Google Scholar] [CrossRef] [PubMed]
- Orevi, M.; Shamni, O.; Zalcman, N.; Chicheportiche, A.; Mordechai, A.; Moscovici, S.; Shoshan, Y.; Shahar, T.; Charbit, H.; Gutreiman, M.; et al. [18F]-FDHT PET/CT as a Tool for Imaging Androgen Receptor Expression in High-Grade Glioma. Neuro-Oncol. Adv. 2021, 3, vdab019. [Google Scholar] [CrossRef] [PubMed]
- Mellinghoff, I.K.; Vivanco, I.; Kwon, A.; Tran, C.; Wongvipat, J.; Sawyers, C.L. HER2/neu kinase-dependent modulation of androgen receptor function through effects on DNA binding and stability. Cancer Cell 2004, 6, 517–527. [Google Scholar] [CrossRef] [PubMed]
- Gregory, C.W.; Whang, Y.E.; McCall, W.; Fei, X.; Liu, Y.; Ponguta, L.A.; French, F.S.; Wilson, E.M.; Earp, H.S. Heregulin-induced activation of HER2 and HER3 increases androgen receptor transactivation and CWR-R1 human recurrent prostate cancer cell growth. Clin. Cancer Res. 2005, 11, 1704–1712. [Google Scholar] [CrossRef] [PubMed]
- Culig, Z.; Hobisch, A.; Cronauer, M.V.; Radmayr, C.; Trapman, J.; Hittmair, A.; Bartsch, G.; Klocker, H. Androgen receptor activation in prostatic tumor cell lines by insulin-like growth factor-I, keratinocyte growth factor, and epidermal growth factor. Cancer Res. 1994, 54, 5474–5478. [Google Scholar] [CrossRef] [PubMed]
- Hu, R.; Dunn, T.A.; Wei, S.; Isharwal, S.; Veltri, R.W.; Humphreys, E.; Han, M.; Partin, A.W.; Vessella, R.L.; Isaacs, W.B. Ligand-independent androgen receptor variants derived from splicing of cryptic exons signify hormone-refractory prostate cancer. Cancer Res. 2009, 69, 16–22. [Google Scholar] [CrossRef]
- Ekstrand, A.J.; James, C.D.; Cavenee, W.K.; Seliger, B.; Pettersson, R.F.; Collins, V.P. Genes for epidermal growth factor receptor, transforming growth factor alpha, and epidermal growth factor and their expression in human gliomas in vivo. Cancer Res. 1991, 51, 2164–2172. [Google Scholar]
- Libermann, T.A.; Nusbaum, H.R.; Razon, N.; Kris, R.; Lax, I.; Soreq, H.; Whittle, N.; Waterfield, M.D.; Ullrich, A.; Schlessinger, J. Amplification, enhanced expression and possible rearrangement of EGF receptor gene in primary human brain tumours of glial origin. Nature 1985, 313, 144–147. [Google Scholar] [CrossRef]
- Nishikawa, R.; Ji, X.D.; Harmon, R.C.; Lazar, C.S.; Gill, G.N.; Cavenee, W.K.; Huang, H.J. A mutant epidermal growth factor receptor common in human glioma confers enhanced tumorigenicity. Proc. Natl. Acad. Sci. USA 1994, 91, 7727–7731. [Google Scholar] [CrossRef]
- Feldman, H.A.; Longcope, C.; Derby, C.A.; Johannes, C.B.; Araujo, A.B.; Coviello, A.D.; Bremner, W.J.; McKinlay, J.B. Age trends in the level of serum testosterone and other hormones in middle-aged men: Longitudinal results from the Massachusetts male aging study. J. Clin. Endocrinol. Metab. 2002, 87, 589–598. [Google Scholar] [CrossRef]
- Narayan, R.S.; Fedrigo, C.A.; Brands, E.; Dik, R.; Stalpers, L.J.; Baumert, B.G.; Slotman, B.J.; Westerman, B.A.; Peters, G.J.; Sminia, P. The allosteric AKT inhibitor MK2206 shows a synergistic interaction with chemotherapy and radiotherapy in glioblastoma spheroid cultures. BMC Cancer 2017, 17, 204. [Google Scholar] [CrossRef] [PubMed]
- Wen, Y.; Hu, M.C.; Makino, K.; Spohn, B.; Bartholomeusz, G.; Yan, D.H.; Hung, M.C. HER-2/neu promotes androgen-independent survival and growth of prostate cancer cells through the Akt pathway. Cancer Res. 2000, 60, 6841–6845. [Google Scholar]
- Lin, H.K.; Hu, Y.C.; Yang, L.; Altuwaijri, S.; Chen, Y.T.; Kang, H.Y.; Chang, C. Suppression versus induction of androgen receptor functions by the phosphatidylinositol 3-kinase/Akt pathway in prostate cancer LNCaP cells with different passage numbers. J. Biol. Chem. 2003, 278, 50902–50907. [Google Scholar] [CrossRef] [PubMed]
- Lin, H.K.; Yeh, S.; Kang, H.Y.; Chang, C. Akt suppresses androgen-induced apoptosis by phosphorylating and inhibiting androgen receptor. Proc. Natl. Acad. Sci. USA 2001, 98, 7200–7205. [Google Scholar] [CrossRef] [PubMed]
- Taneja, S.S.; Ha, S.; Swenson, N.K.; Huang, H.Y.; Lee, P.; Melamed, J.; Shapiro, E.; Garabedian, M.J.; Logan, S.K. Cell-specific regulation of androgen receptor phosphorylation in vivo. J. Biol. Chem. 2005, 280, 40916–40924. [Google Scholar] [CrossRef] [PubMed]
- Palazzolo, I.; Burnett, B.G.; Young, J.E.; Brenne, P.L.; La Spada, A.R.; Fischbeck, K.H.; Howell, B.W.; Pennuto, M. Akt blocks ligand binding and protects against expanded polyglutamine androgen receptor toxicity. Hum. Mol. Genet. 2007, 16, 1593–1603. [Google Scholar] [CrossRef] [PubMed]
- Kasina, S.; Macoska, J.A. The CXCL12/CXCR4 axis promotes ligand-independent activation of the androgen receptor. Mol. Cell Endocrinol. 2012, 351, 249–263. [Google Scholar] [CrossRef] [PubMed]
- Linn, D.E.; Yang, X.; Xie, Y.; Alfano, A.; Deshmukh, D.; Wang, X.; Shimelis, H.; Chen, H.; Li, W.; Xu, K.; et al. Differential regulation of androgen receptor by PIM-1 kinases via phosphorylation-dependent recruitment of distinct ubiquitin E3 ligases. J. Biol. Chem. 2012, 287, 22959–22968. [Google Scholar] [CrossRef]
- Varisli, L.; Gonen-Korkmaz, C.; Syed, H.M.; Bogurcu, N.; Debelec-Butuner, B.; Erbaykent-Tepedelen, B.; Korkmaz, K.S. Androgen regulated HN1 leads proteosomal degradation of androgen receptor (AR) and negatively influences AR mediated transactivation in prostate cells. Mol. Cell Endocrinol. 2012, 350, 107–117. [Google Scholar] [CrossRef]
- Craft, N.; Shostak, Y.; Carey, M.; Sawyers, C.L. A mechanism for hormone-independent prostate cancer through modulation of androgen receptor signaling by the HER-2/neu tyrosine kinase. Nat. Med. 1999, 5, 280–285. [Google Scholar] [CrossRef] [PubMed]
- Yeh, S.; Lin, H.K.; Kang, H.Y.; Thin, T.H.; Lin, M.F.; Chang, C. From HER2/Neu signal cascade to androgen receptor and its coactivators: A novel pathway by induction of androgen target genes through MAP kinase in prostate cancer cells. Proc. Natl. Acad. Sci. USA 1999, 96, 5458–5463. [Google Scholar] [CrossRef] [PubMed]
- Fard, S.S.; Saliminejad, K.; Sotoudeh, M.; Soleimanifard, N.; Kouchaki, S.; Yazdanbod, M.; Mahmoodzadeh, H.; Ghavamzadeh, A.; Malekzadeh, R.; Chahardouli, B.; et al. The Correlation between EGFR and Androgen Receptor Pathways: A Novel Potential Prognostic Marker in Gastric Cancer. Anti-Cancer Agents Med. Chem. 2019, 19, 2097–2107. [Google Scholar] [CrossRef]
- Ji, Z.; Yang, L.; Ruan, Q. Correlation of epidermal growth factor receptor (EGFR), androgen receptor (AR) and 14-3-3 sigma expression in breast cancer. Int. J. Clin. Exp. Pathol. 2017, 10, 10419–10430. [Google Scholar] [PubMed]
- Baek, K.H.; Hong, M.E.; Jung, Y.Y.; Lee, C.H.; Lee, T.J.; Park, E.S.; Kim, M.K.; Yoo, J.H.; Lee, S.W. Correlation of AR, EGFR, and HER2 Expression Levels in Prostate Cancer: Immunohistochemical Analysis and Chromogenic In Situ Hybridization. Cancer Res. Treat. 2012, 44, 50–56. [Google Scholar] [CrossRef] [PubMed]
- Mandel, A.; Larsson, P.; Sarwar, M.; Semenas, J.; Syed Khaja, A.S.; Persson, J.L. The interplay between AR, EGF receptor and MMP-9 signaling pathways in invasive prostate cancer. Mol. Med. 2018, 24, 34. [Google Scholar] [CrossRef]
- Huang, P.H.; Xu, A.M.; White, F.M. Oncogenic EGFR Signaling Networks in Glioma. Sci. Signal. 2009, 2, re6. [Google Scholar] [CrossRef]
- Vengoji, R.; Macha, M.A.; Nimmakayala, R.K.; Rachagani, S.; Siddiqui, J.A.; Mallya, K.; Gorantla, S.; Jain, M.; Ponnusamy, M.P.; Batra, S.K.; et al. Afatinib and Temozolomide combination inhibits tumorigenesis by targeting EGFRvIII-cMet signaling in glioblastoma cells. J. Exp. Clin. Cancer Res. 2019, 38, 266. [Google Scholar] [CrossRef] [PubMed]
- Górowska-Wójtowicz, E.; Hejmej, A.; Kamińska, A.; Pardyak, L.; Kotula-Balak, M.; Dulińska-Litewka, J.; Laidler, P.; Bilińska, B. Anti-androgen 2-hydroxyflutamide modulates cadherin, catenin and androgen receptor phosphorylation in androgen-sensitive LNCaP and androgen-independent PC3 prostate cancer cell lines acting via PI3K/Akt and MAPK/ERK1/2 pathways. Toxicol. Vitr. 2017, 40, 324–335. [Google Scholar] [CrossRef] [PubMed]
- Deep, G.; Oberlies, N.H.; Kroll, D.J.; Agarwal, R. Isosilybin B causes androgen receptor degradation in human prostate carcinoma cells via PI3K-Akt-Mdm2-mediated pathway. Oncogene 2008, 27, 3986–3998. [Google Scholar] [CrossRef] [PubMed]
- Koul, D. PTEN signaling pathways in glioblastoma. Cancer Biol. Ther. 2008, 7, 1321–1325. [Google Scholar] [CrossRef]
- Wen, S.; Niu, Y.; Huang, H. Posttranslational regulation of androgen dependent and independent androgen receptor activities in prostate cancer. Asian J. Urol. 2020, 7, 203–218. [Google Scholar] [CrossRef] [PubMed]
- Cuenca-Lopez, M.D.; Montero, J.C.; Morales, J.C.; Prat, A.; Pandiella, A.; Ocana, A. Phospho-kinase profile of triple negative breast cancer and androgen receptor signaling. BMC Cancer 2014, 14, 302. [Google Scholar] [CrossRef] [PubMed]
- Fan, Q.W.; Cheng, C.K.; Gustafson, W.C.; Charron, E.; Zipper, P.; Wong, R.A.; Chen, J.; Lau, J.; Knobbe-Thomsen, C.; Weller, M.; et al. EGFR phosphorylates tumor-derived EGFRvIII driving STAT3/5 and progression in glioblastoma. Cancer Cell 2013, 24, 438–449. [Google Scholar] [CrossRef] [PubMed]
- Eliceiri, K.T.P.; Jug, F.; Carpenter, A.; Berthold, M.; Swedlow, J.; Rasband, W.; Rueden, C.; Dietz, C.; Northan, B.; Hiner, M. Colocalization Analysis with ImageJ. 2020. Available online: https://imagej.net/Colocalization_Analysis (accessed on 1 May 2020).
- Schneider, C.A.; Rasband, W.S.; Eliceiri, K.W. NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 2012, 9, 671–675. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Kinase | U87MG Control Pixels Densitometry | U87MG EGFR Pixels Densitometry | EGFR-Derived Fold Induction | U87MG EGFR + Afatinib Pixels Densitometry | Afatinib-Derived Fold Reduction |
|---|---|---|---|---|---|
| EGFR (Y1086) | 14 | 203 | 14.5 | 1 | 203 |
| Yes (Y426) | 117 | 397 | 3.4 | 1 | 397 |
| p53 (S392) | 1406 | 2412 | 1.7 | 682 | 3.5 |
| p38 alpha (T180/Y182) | 7 | 904 | 129 | 174 | 5.2 |
| HSP27 (S78/S82) | 381 | 6336 | 17 | 1 | 6336 |
| MSK1/2 (S376/S360) | 4191 | 8184 | 1.95 | 525 | 15.6 |
| AKT 1/2/3 (S473) | 7060 | 10,753 | 1.5 | 1015 | 10.6 |
| JNK 1/2/3 (T183/Y185, T221/Y223) | 774 | 2305 | 3 | 341 | 6.8 |
| Treatment | U87MG WT | U87MG EGFRvIII | U87MG EGFR | |
|---|---|---|---|---|
| Vehicle | Pearson correlation R values | 0.98 | 0.94 | 0.87 |
| Pearson Correlation p-value | <0.00001 | <0.00001 | 0.010899 | |
| 5 μM Afatinib | Pearson correlation R values | 0.99 | 0.464 | 0.86 |
| Pearson Correlation p-value | 0.00015 | 0.000037 | 0.017 | |
| 5 μm MK2206 | Pearson correlation R value | 0.834 | 0.62 | 0.836 |
| Pearson Correlation p-value | 0.009546 | <0.00001 | 0.000026 |
| Gene | Primer Sequence |
|---|---|
| AR | F: ACCGAGGAGCTTTCCAGAATC |
| R: AGGCTCTGGGACGCAACCT | |
| EGFR | F: GCGTCTCTTGCCGGAATGT |
| R: CTTGGCTCACCCTCCAGAAG | |
| TATA-binding protein (TBP) | F: CCACTCACAGACTCTCACAAC |
| R: CTGCGGTACAATCCCAGAACT | |
| Hypoxanthine phosphoribosyltransferase 1 (HPRT) | F: GATGGTCAAGGTCGCAAGC |
| R: ATATCCTACAACAAACTTGTCTGGAA |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zalcman, N.; Gutreiman, M.; Shahar, T.; Weller, M.; Lavon, I. Androgen Receptor Activation in Glioblastoma Can Be Achieved by Ligand-Independent Signaling through EGFR—A Potential Therapeutic Target. Int. J. Mol. Sci. 2021, 22, 10954. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms222010954
Zalcman N, Gutreiman M, Shahar T, Weller M, Lavon I. Androgen Receptor Activation in Glioblastoma Can Be Achieved by Ligand-Independent Signaling through EGFR—A Potential Therapeutic Target. International Journal of Molecular Sciences. 2021; 22(20):10954. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms222010954
Chicago/Turabian StyleZalcman, Nomi, Mijal Gutreiman, Tal Shahar, Michael Weller, and Iris Lavon. 2021. "Androgen Receptor Activation in Glioblastoma Can Be Achieved by Ligand-Independent Signaling through EGFR—A Potential Therapeutic Target" International Journal of Molecular Sciences 22, no. 20: 10954. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms222010954