
Optical Fluorescence Imaging of Native Proteins Using a Fluorescent Probe with a Cell-Membrane-Permeable Carboxyl Group


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. Free Carboxyl Group-Containing Fluorescent Dye Ligates the Lysine (K) Residue of the AVI-Tag in Living Cells
2.2. Analysis of the Free Carboxylic Acid Dye Uptake Subcellular Localization and Its Fluorescent Labeling
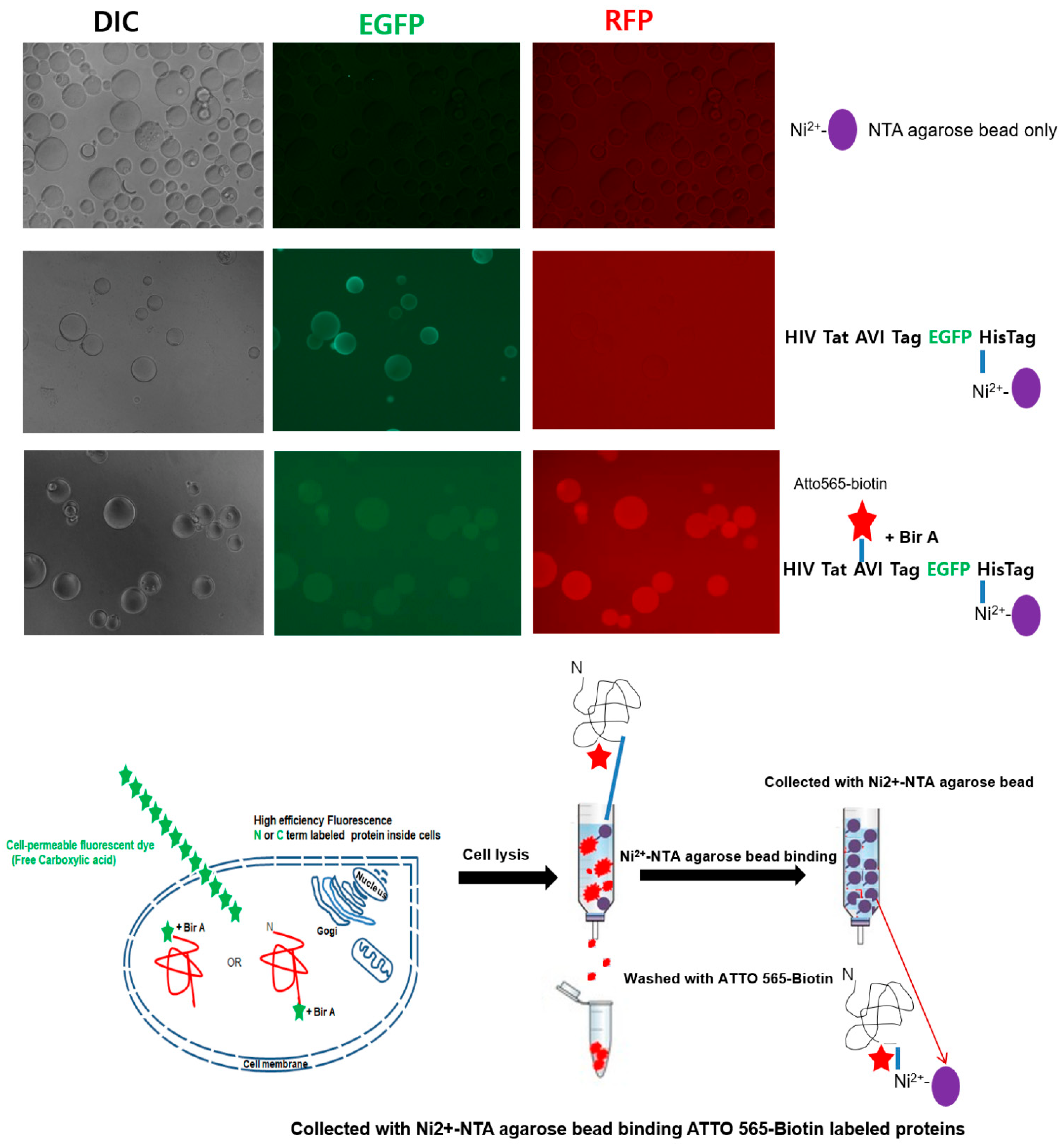
2.3. Detection of the Fluorescently Labeled NPI
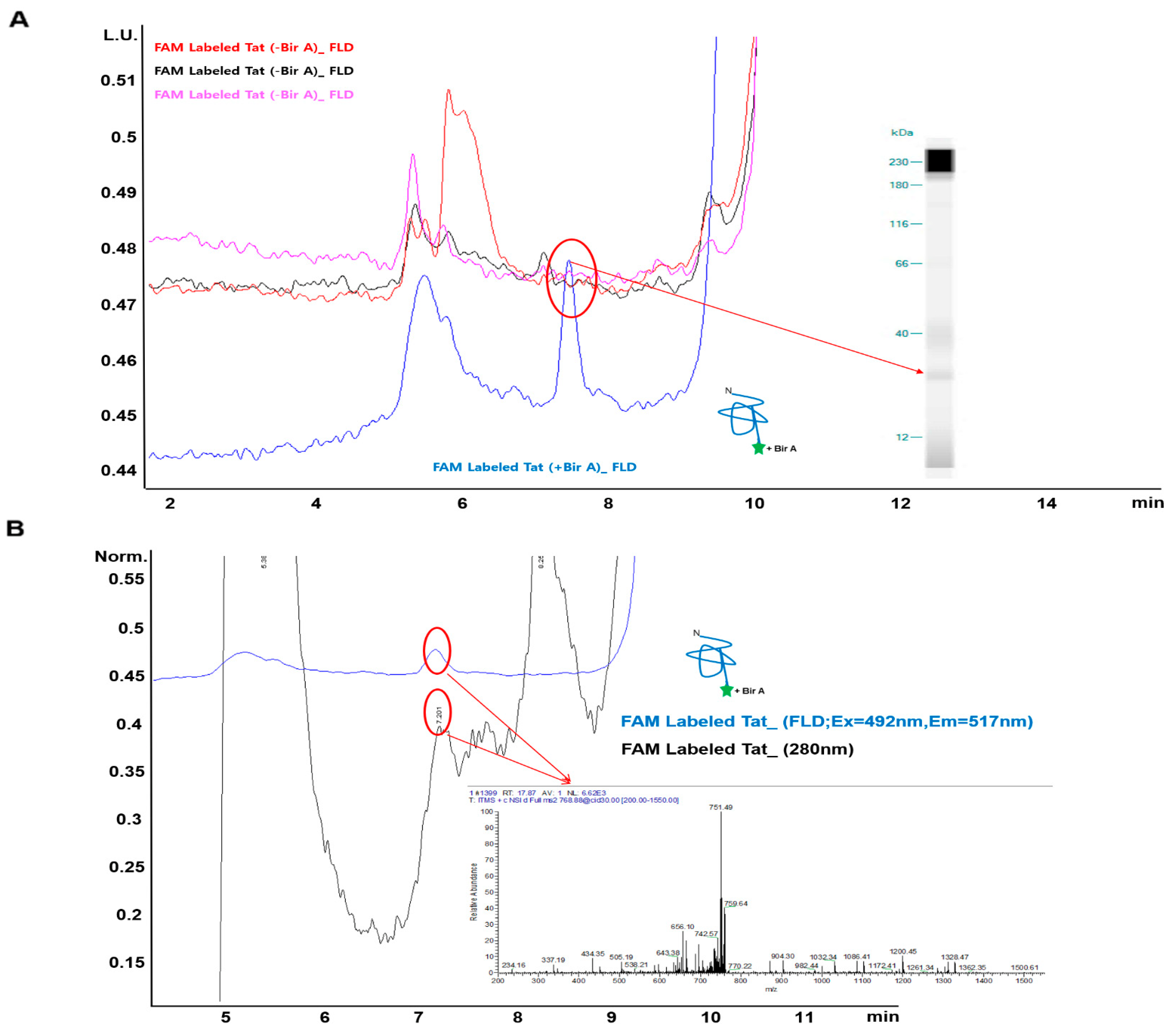
2.4. Identification of Fluorescence-Labeled NPIs Separated by SEC-HPLC According to LC–MS Spectra
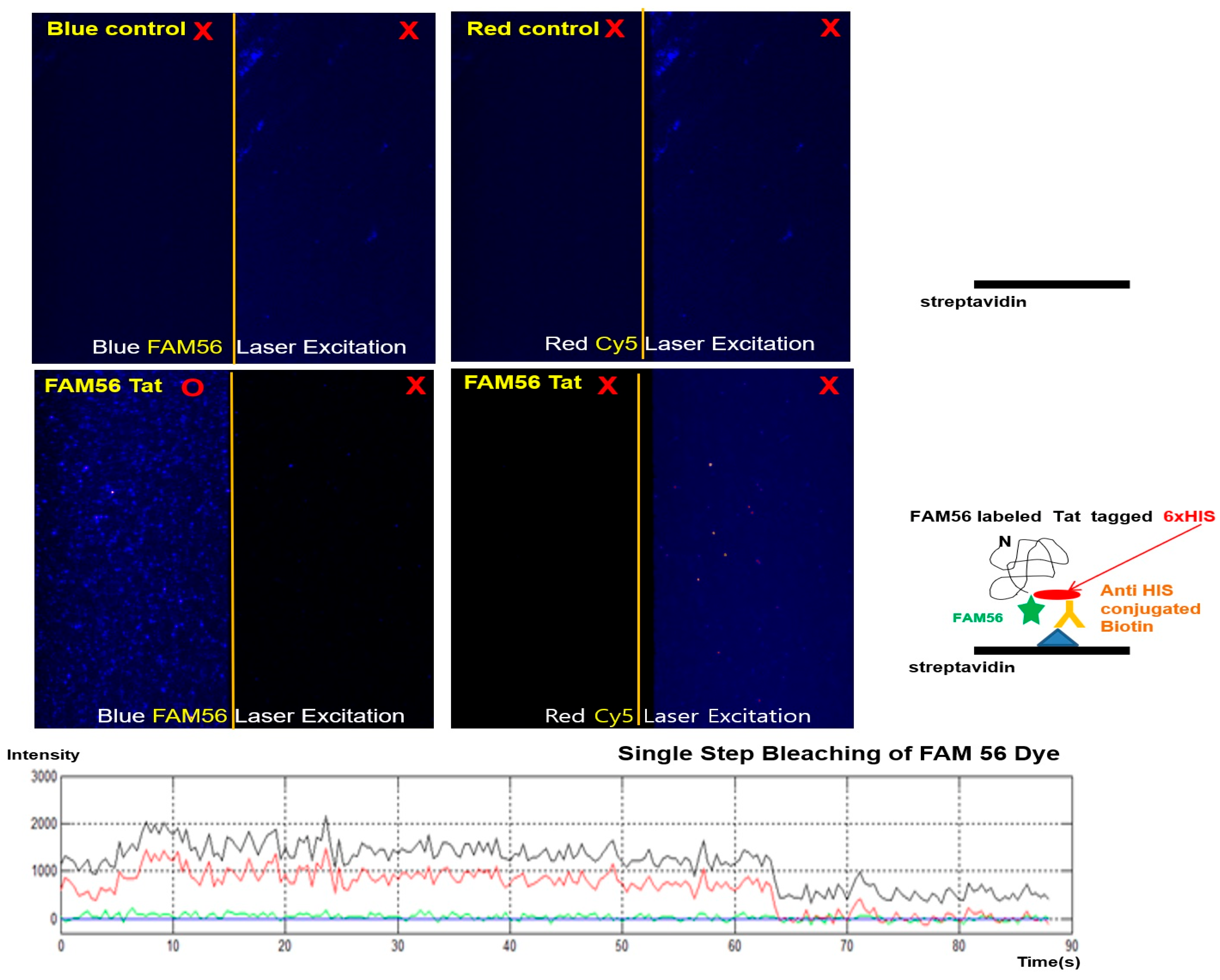
2.5. The Universal Carboxy Group Fluorescent Dye Mediated the Expression of Fluorescence-Labeled NPIs at the Single-Molecule Level
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Fluorescence Imaging of the Free Carboxyl Fluorescent Dyes in Living Cells
4.3. Construction of Expression Plasmids
4.4. Labeling of Living Cells for Co-Translational Tagging of Target Proteins and Free Carboxyl Fluorescent Dyes
4.5. Single Agarose Bead Assay for ATTO 565-Biotin-Labeled Tat-EGFP Protein Detection with Fluorescence Microscopy
4.6. Liquid Chromatography Analysis of Fluorescent Dyes Labeling of Target Proteins
4.7. LC–MS Analysis of Labeled Protein
4.8. Detection of Fluorescence and Biotin-Fused Tat
4.9. Statistical Analysis
5. Conclusions
6. Patents
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lafranchi, L.; Schlesinger, D.; Kimler, K.J.; Elsasser, S.J. Universal Single-Residue Terminal Labels for Fluorescent Live Cell Imaging of Microproteins. J. Am. Chem. Soc. 2020, 142, 20080–20087. [Google Scholar] [CrossRef] [PubMed]
- Mauriz, E.; Lechuga, L.M. Plasmonic Biosensors for Single-Molecule Biomedical Analysis. Biosensors 2021, 11, 123. [Google Scholar] [CrossRef] [PubMed]
- Beuwer, M.A.; Prins, M.W.; Zijlstra, P. Stochastic protein interactions monitored by hundreds of single-molecule plasmonic biosensors. Nano Lett. 2015, 15, 3507–3511. [Google Scholar] [CrossRef] [PubMed]
- Goncalves, M.S. Fluorescent labeling of biomolecules with organic probes. Chem. Rev. 2009, 109, 190–212. [Google Scholar] [CrossRef] [PubMed]
- Lemieux, G.A.; De Graffenried, C.L.; Bertozzi, C.R. A fluorogenic dye activated by the staudinger ligation. J. Am. Chem. Soc. 2003, 125, 4708–4709. [Google Scholar] [CrossRef] [PubMed]
- Kowada, T.; Maeda, H.; Kikuchi, K. BODIPY-based probes for the fluorescence imaging of biomolecules in living cells. Chem. Soc. Rev. 2015, 44, 4953–4972. [Google Scholar] [CrossRef]
- Yin, X.; Low, J.Z.; Fallon, K.J.; Paley, D.W.; Campos, L.M. The butterfly effect in bisfluorenylidene-based dihydroacenes: Aggregation induced emission and spin switching. Chem. Sci. 2019, 10, 10733–10739. [Google Scholar] [CrossRef] [Green Version]
- Li, S.; Kimura, E.; Ng, R.; Fall, B.M.; Meuse, L.; Reyes, M.; Faulkner, J.A.; Chamberlain, J.S. A highly functional mini-dystrophin/GFP fusion gene for cell and gene therapy studies of Duchenne muscular dystrophy. Hum. Mol. Genet. 2006, 15, 1610–1622. [Google Scholar] [CrossRef]
- Rodriguez, E.A.; Campbell, R.E.; Lin, J.Y.; Lin, M.Z.; Miyawaki, A.; Palmer, A.E.; Shu, X.; Zhang, J.; Tsien, R.Y. The Growing and Glowing Toolbox of Fluorescent and Photoactive Proteins. Trends Biochem. Sci. 2017, 42, 111–129. [Google Scholar] [CrossRef] [Green Version]
- Shaner, N.C.; Steinbach, P.A.; Tsien, R.Y. A guide to choosing fluorescent proteins. Nat. Methods 2005, 2, 905–909. [Google Scholar] [CrossRef]
- Tsien, R.Y. The green fluorescent protein. Annu. Rev. Biochem. 1998, 67, 509–544. [Google Scholar] [CrossRef] [PubMed]
- Drobizhev, M.; Makarov, N.S.; Tillo, S.E.; Hughes, T.E.; Rebane, A. Two-photon absorption properties of fluorescent proteins. Nat. Methods 2011, 8, 393–399. [Google Scholar] [CrossRef] [PubMed]
- Ngo, J.T.; Champion, J.A.; Mahdavi, A.; Tanrikulu, I.C.; Beatty, K.E.; Connor, R.E.; Yoo, T.H.; Dieterich, D.C.; Schuman, E.M.; Tirrell, D.A. Cell-selective metabolic labeling of proteins. Nat. Chem. Biol. 2009, 5, 715–717. [Google Scholar] [CrossRef] [PubMed]
- Bogdanova, Y.A.; Schultz, C.; Belousov, V.V. Local Generation and Imaging of Hydrogen Peroxide in Living Cells. Curr. Protoc. Chem. Biol. 2017, 9, 117–127. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.L.; Wang, Z.G.; Xie, H.Y.; Liu, A.A.; Lamb, D.C.; Pang, D.W. Single-Virus Tracking: From Imaging Methodologies to Virological Applications. Chem. Rev. 2020, 120, 1936–1979. [Google Scholar] [CrossRef] [Green Version]
- Ooi, S.L.; Henikoff, J.G.; Henikoff, S. A native chromatin purification system for epigenomic profiling in Caenorhabditis elegans. Nucleic Acids Res. 2010, 38, e26. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.; Seo, M.H.; Lee, S.; Cho, K.; Yang, A.; Woo, K.; Kim, H.S.; Park, H.S. Simple and efficient strategy for site-specific dual labeling of proteins for single-molecule fluorescence resonance energy transfer analysis. Anal. Chem. 2013, 85, 1468–1474. [Google Scholar] [CrossRef]
- Elliott, G.; O’Hare, P. Live-cell analysis of a green fluorescent protein-tagged herpes simplex virus infection. J. Virol. 1999, 73, 4110–4119. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.M.; Seong, B.L.; Lim, D.K. Bead based facile assay for sensitive quantification of native state green fluorescent protein. RSC Adv. 2020, 10, 13095–13099. [Google Scholar] [CrossRef] [Green Version]
- Spriestersbach, A.; Kubicek, J.; Schafer, F.; Block, H.; Maertens, B. Purification of His-Tagged Proteins. Methods Enzymol. 2015, 559, 1–15. [Google Scholar]
- Crowe, J.; Dobeli, H.; Gentz, R.; Hochuli, E.; Stuber, D.; Henco, K. 6xHis-Ni-NTA chromatography as a superior technique in recombinant protein expression/purification. Methods Mol. Biol. 1994, 31, 371–387. [Google Scholar] [PubMed]
- Howarth, M.; Takao, K.; Hayashi, Y.; Ting, A.Y. Targeting quantum dots to surface proteins in living cells with biotin ligase. Proc. Natl. Acad. Sci. USA 2005, 102, 7583–7588. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bagautdinov, B.; Matsuura, Y.; Bagautdinova, S.; Kunishima, N. Protein biotinylation visualized by a complex structure of biotin protein ligase with a substrate. J. Biol. Chem. 2008, 283, 14739–14750. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heiss, T.K.; Dorn, R.S.; Prescher, J.A. Bioorthogonal Reactions of Triarylphosphines and Related Analogues. Chem. Rev. 2021, 121, 6802–6849. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, T.; Kamiya, N.; Nagamune, T. N-terminal glycine-specific protein conjugation catalyzed by microbial transglutaminase. FEBS Lett. 2005, 579, 2092–2096. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Slavoff, S.A.; Chen, I.; Choi, Y.A.; Ting, A.Y. Expanding the substrate tolerance of biotin ligase through exploration of enzymes from diverse species. J. Am. Chem. Soc. 2008, 130, 1160–1162. [Google Scholar] [CrossRef] [Green Version]
- Fairhead, M.; Howarth, M. Site-specific biotinylation of purified proteins using BirA. Methods Mol. Biol. 2015, 1266, 171–184. [Google Scholar]
- Cronan, J.E., Jr. Biotination of proteins in vivo. A post-translational modification to label, purify, and study proteins. J. Biol. Chem. 1990, 265, 10327–10333. [Google Scholar] [CrossRef]
- Beckett, D.; Kovaleva, E.; Schatz, P.J. A minimal peptide substrate in biotin holoenzyme synthetase-catalyzed biotinylation. Protein Sci. 1999, 8, 921–929. [Google Scholar] [CrossRef] [Green Version]
- Guo, W.; Yang, W.; Wang, Y.; Sun, X.; Liu, Z.; Zhang, B.; Chang, J.; Chen, X. Color Tunable Gd-Zn-Cu-In-S/ZnS Quantum Dots for Dual Modality Magnetic Resonance and Fluorescence Imaging. Nano Res. 2014, 7, 1581–1591. [Google Scholar] [CrossRef] [Green Version]
- Faridi, A.; Sun, Y.; Mortimer, M.; Aranha, R.R.; Nandakumar, A.; Li, Y.; Javed, I.; Kakinen, A.; Fan, Q.; Purcell, A.W.; et al. Graphene quantum dots rescue protein dysregulation of pancreatic beta-cells exposed to human islet amyloid polypeptide. Nano Res. 2019, 12, 2827–2834. [Google Scholar] [CrossRef] [PubMed]
- Brown, K.; Thurn, T.; Xin, L.; Liu, W.; Bazak, R.; Chen, S.; Lai, B.; Vogt, S.; Jacobsen, C.; Paunesku, T.; et al. Intracellular in situ labeling of TiO2 nanoparticles for fluorescence microscopy detection. Nano Res. 2018, 11, 464–476. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Juillerat, A.; Gronemeyer, T.; Keppler, A.; Gendreizig, S.; Pick, H.; Vogel, H.; Johnsson, K. Directed evolution of O6-alkylguanine-DNA alkyltransferase for efficient labeling of fusion proteins with small molecules in vivo. Chem. Biol. 2003, 10, 313–317. [Google Scholar] [CrossRef] [Green Version]
- Atack, T.C.; Raymond, D.D.; Blomquist, C.A.; Pasaje, C.F.; McCarren, P.R.; Moroco, J.; Befekadu, H.B.; Robinson, F.P.; Pal, D.; Esherick, L.Y.; et al. Targeted Covalent Inhibition of Plasmodium FK506 Binding Protein 35. ACS Med. Chem. Lett. 2020, 11, 2131–2138. [Google Scholar] [CrossRef]
- Macchia, E.; Manoli, K.; Di Franco, C.; Scamarcio, G.; Torsi, L. New trends in single-molecule bioanalytical detection. Anal. Bioanal. Chem. 2020, 412, 5005–5014. [Google Scholar] [CrossRef] [Green Version]
- Huang, M.; Yu, L.; Zhang, M.; Wang, Z.; Xiao, B.; Liu, Y.; He, J.; Chang, S. Developing Longer-Lived Single Molecule Junctions with a Functional Flexible Electrode. Small 2021, 17, e2101911. [Google Scholar] [CrossRef]
- Xie, X.S.; Choi, P.J.; Li, G.W.; Lee, N.K.; Lia, G. Single-molecule approach to molecular biology in living bacterial cells. Annu. Rev. Biophys. 2008, 37, 417–444. [Google Scholar] [CrossRef] [Green Version]
- Li, L.; Jiang, N. Proximity and single-molecule energetics. Science 2021, 373, 392–393. [Google Scholar] [CrossRef]
- Akkilic, N.; Geschwindner, S.; Hook, F. Single-molecule biosensors: Recent advances and applications. Biosens. Bioelectron. 2020, 151, 111944. [Google Scholar] [CrossRef]
- Varnaite, R.; MacNeill, S.A. Meet the neighbors: Mapping local protein interactomes by proximity-dependent labeling with BioID. Proteomics 2016, 16, 2503–2518. [Google Scholar] [CrossRef] [Green Version]
- Cheah, J.S.; Jacobs, K.A.; Lai, T.W.; Caballelo, R.; Yee, J.L.; Ueda, S.; Heinrich, V.; Yamada, S. Spatial proximity of proteins surrounding zyxin under force-bearing conditions. Mol. Biol. Cell 2021, 32, 1221–1228. [Google Scholar] [CrossRef] [PubMed]
- Habel, J.E. Biotin Proximity Labeling for Protein-Protein Interaction Discovery: The BioID Method. Methods Mol. Biol. 2021, 2261, 357–379. [Google Scholar] [PubMed]
- Samavarchi-Tehrani, P.; Samson, R.; Gingras, A.C. Proximity Dependent Biotinylation: Key Enzymes and Adaptation to Proteomics Approaches. Mol. Cell Proteom. 2020, 19, 757–773. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, D.I.; Jensen, S.C.; Noble, K.A.; Kc, B.; Roux, K.H.; Motamedchaboki, K.; Roux, K.J. An improved smaller biotin ligase for BioID proximity labeling. Mol. Biol. Cell 2016, 27, 1188–1196. [Google Scholar] [CrossRef] [PubMed]
- Peng, T.; Hang, H.C. Site-Specific Bioorthogonal Labeling for Fluorescence Imaging of Intracellular Proteins in Living Cells. J. Am. Chem. Soc. 2016, 138, 14423–14433. [Google Scholar] [CrossRef] [Green Version]
- Chudakov, D.M.; Matz, M.V.; Lukyanov, S.; Lukyanov, K.A. Fluorescent proteins and their applications in imaging living cells and tissues. Physiol. Rev. 2010, 90, 1103–1163. [Google Scholar] [CrossRef]
- Sedlak, S.M.; Schendel, L.C.; Melo, M.C.R.; Pippig, D.A.; Luthey-Schulten, Z.; Gaub, H.E.; Bernardi, R.C. Direction Matters: Monovalent Streptavidin/Biotin Complex under Load. Nano Lett. 2019, 19, 3415–3421. [Google Scholar] [CrossRef]
- Howarth, M.; Ting, A.Y. Imaging proteins in live mammalian cells with biotin ligase and monovalent streptavidin. Nat. Protoc. 2008, 3, 534–545. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.M.; Seong, B.L. Highly chromophoric Cy5-methionine for N-terminal fluorescent tagging of proteins in eukaryotic translation systems. Sci. Rep. 2017, 7, 11642. [Google Scholar] [CrossRef] [Green Version]
- Wu, N.; Bao, L.; Ding, L.; Ju, H. A Single Excitation-Duplexed Imaging Strategy for Profiling Cell Surface Protein-Specific Glycoforms. Angew. Chem. Int. Ed. Engl. 2016, 55, 5220–5224. [Google Scholar] [CrossRef]
- Bjerneld, E.J.; Johansson, J.D.; Laurin, Y.; Hagner-McWhirter, A.; Ronn, O.; Karlsson, R. Pre-labeling of diverse protein samples with a fixed amount of Cy5 for sodium dodecyl sulfate-polyacrylamide gel electrophoresis analysis. Anal. Biochem. 2015, 484, 51–57. [Google Scholar] [CrossRef] [PubMed]
- Seo, M.H.; Lee, T.S.; Kim, E.; Cho, Y.L.; Park, H.S.; Yoon, T.Y.; Kim, H.S. Efficient single-molecule fluorescence resonance energy transfer analysis by site-specific dual-labeling of protein using an unnatural amino acid. Anal. Chem. 2011, 83, 8849–8854. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Park, K.Y.; Suazo, K.F.; Distefano, M.D. Recent progress in enzymatic protein labelling techniques and their applications. Chem. Soc. Rev. 2018, 47, 9106–9136. [Google Scholar] [CrossRef] [PubMed]
- Divine, R.; Dang, H.V.; Ueda, G.; Fallas, J.A.; Vulovic, I.; Sheffler, W.; Saini, S.; Zhao, Y.T.; Raj, I.X.; Morawski, P.A.; et al. Designed proteins assemble antibodies into modular nanocages. Science 2021, 372, eabd9994. [Google Scholar] [CrossRef]
- Brinkley, M. A brief survey of methods for preparing protein conjugates with dyes, haptens, and cross-linking reagents. Bioconjug. Chem. 1992, 3, 2–13. [Google Scholar] [CrossRef]
- Penalva, L.O.; Keene, J.D. Biotinylated tags for recovery and characterization of ribonucleoprotein complexes. Bio. Tech. 2004, 37, 608–610. [Google Scholar] [CrossRef] [Green Version]
- Rudolph, C.; Adam, G.; Simm, A. Determination of copy number of c-Myc protein per cell by quantitative Western blotting. Anal. Biochem. 1999, 269, 66–71. [Google Scholar] [CrossRef]
- Jo, M.H.; Shin, S.; Jung, S.R.; Kim, E.; Song, J.J.; Hohng, S. Human Argonaute 2 Has Diverse Reaction Pathways on Target RNAs. Mol. Cell 2015, 59, 117–124. [Google Scholar] [CrossRef] [Green Version]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, J.M.; Kang, Y.-M. Optical Fluorescence Imaging of Native Proteins Using a Fluorescent Probe with a Cell-Membrane-Permeable Carboxyl Group. Int. J. Mol. Sci. 2022, 23, 5841. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23105841
Kim JM, Kang Y-M. Optical Fluorescence Imaging of Native Proteins Using a Fluorescent Probe with a Cell-Membrane-Permeable Carboxyl Group. International Journal of Molecular Sciences. 2022; 23(10):5841. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23105841
Chicago/Turabian StyleKim, Jung Min, and Young-Mi Kang. 2022. "Optical Fluorescence Imaging of Native Proteins Using a Fluorescent Probe with a Cell-Membrane-Permeable Carboxyl Group" International Journal of Molecular Sciences 23, no. 10: 5841. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23105841