Discovery of Proteomic Code with mRNA Assisted Protein Folding

Abstract
:1. Introduction
2. Second Look at the Nirenberg Code
2.1. The 3D structure of mRNA
2.2. Physico-chemical definition of codon boundaries
2.3. The Common Periodic Table of Codons and Amino Acids
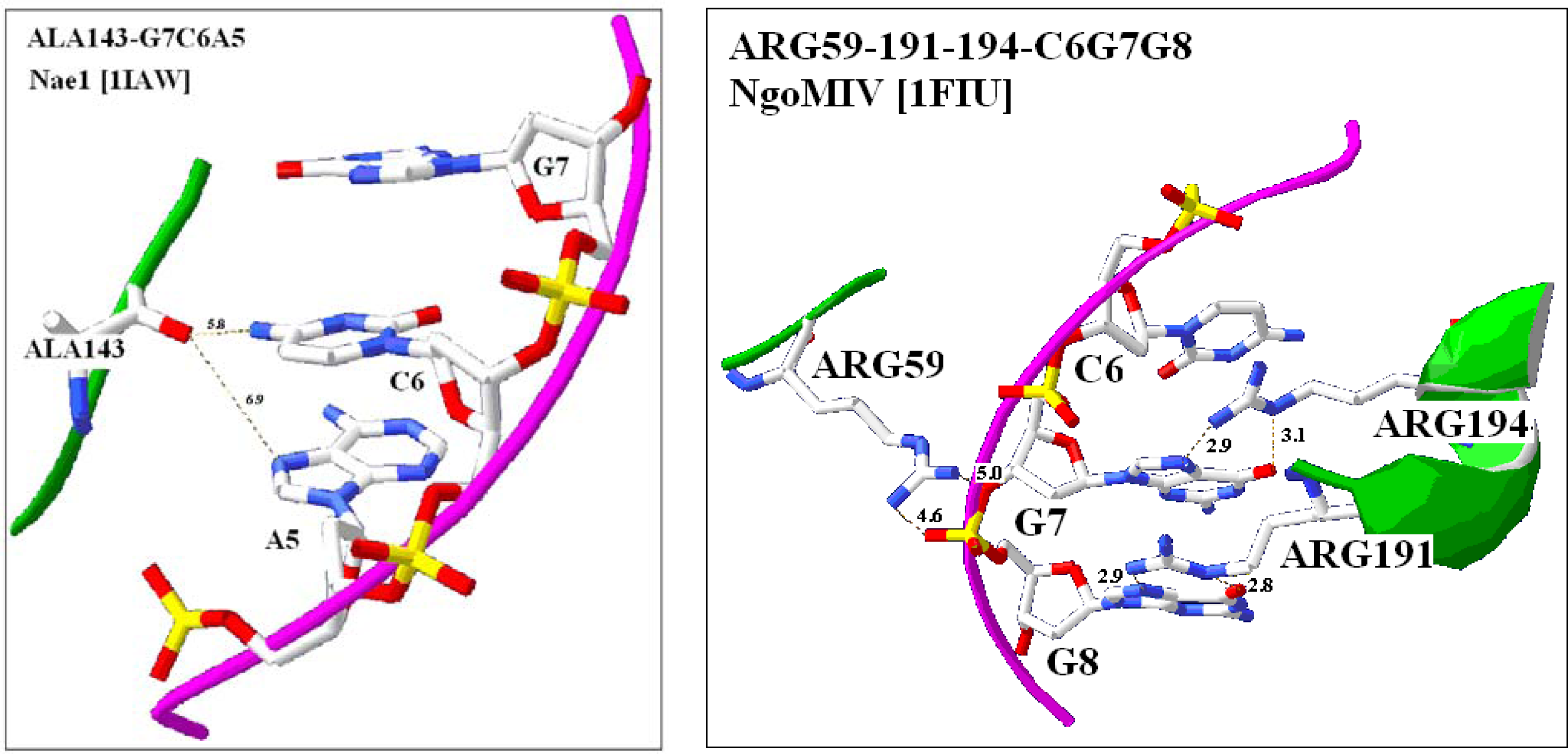
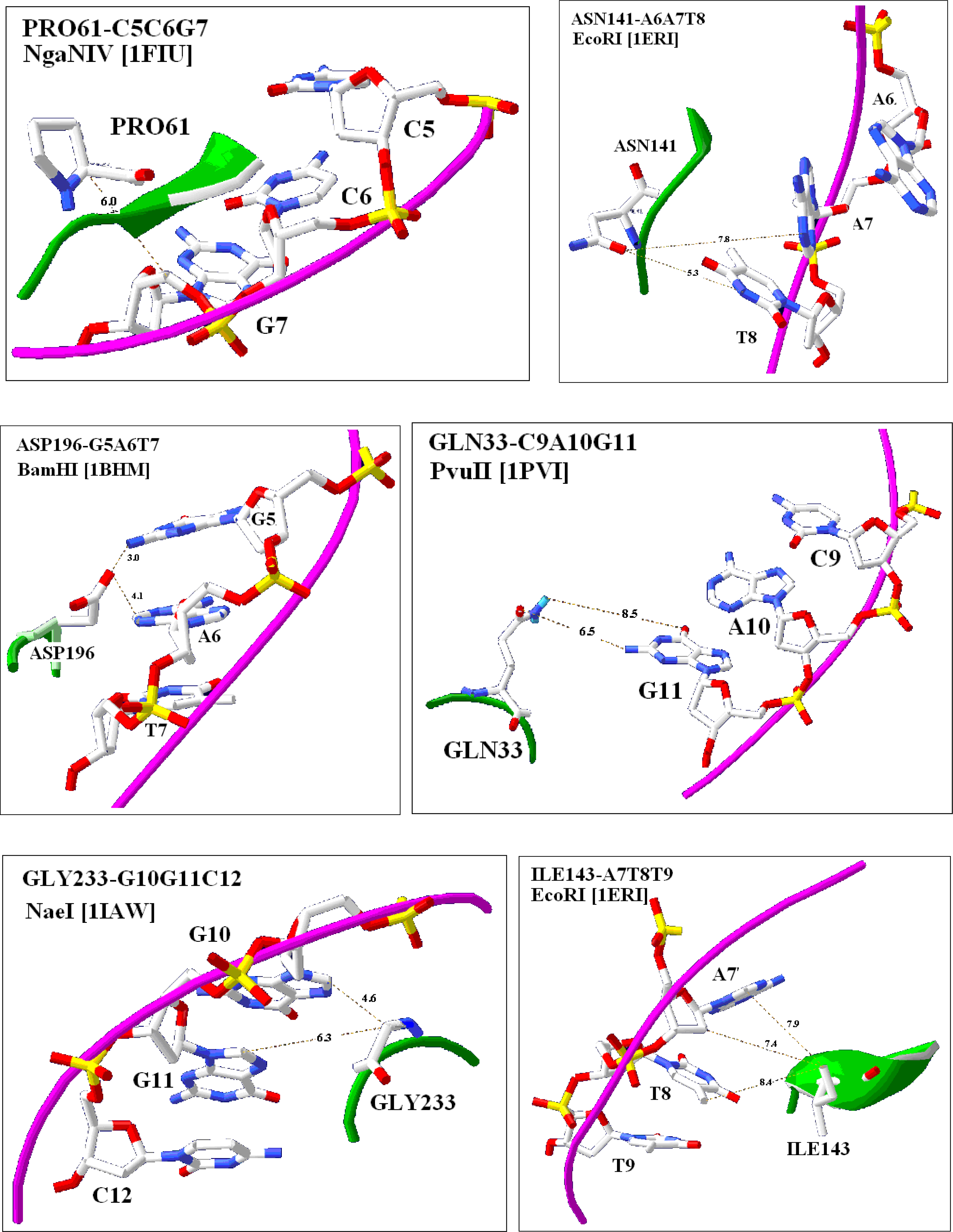
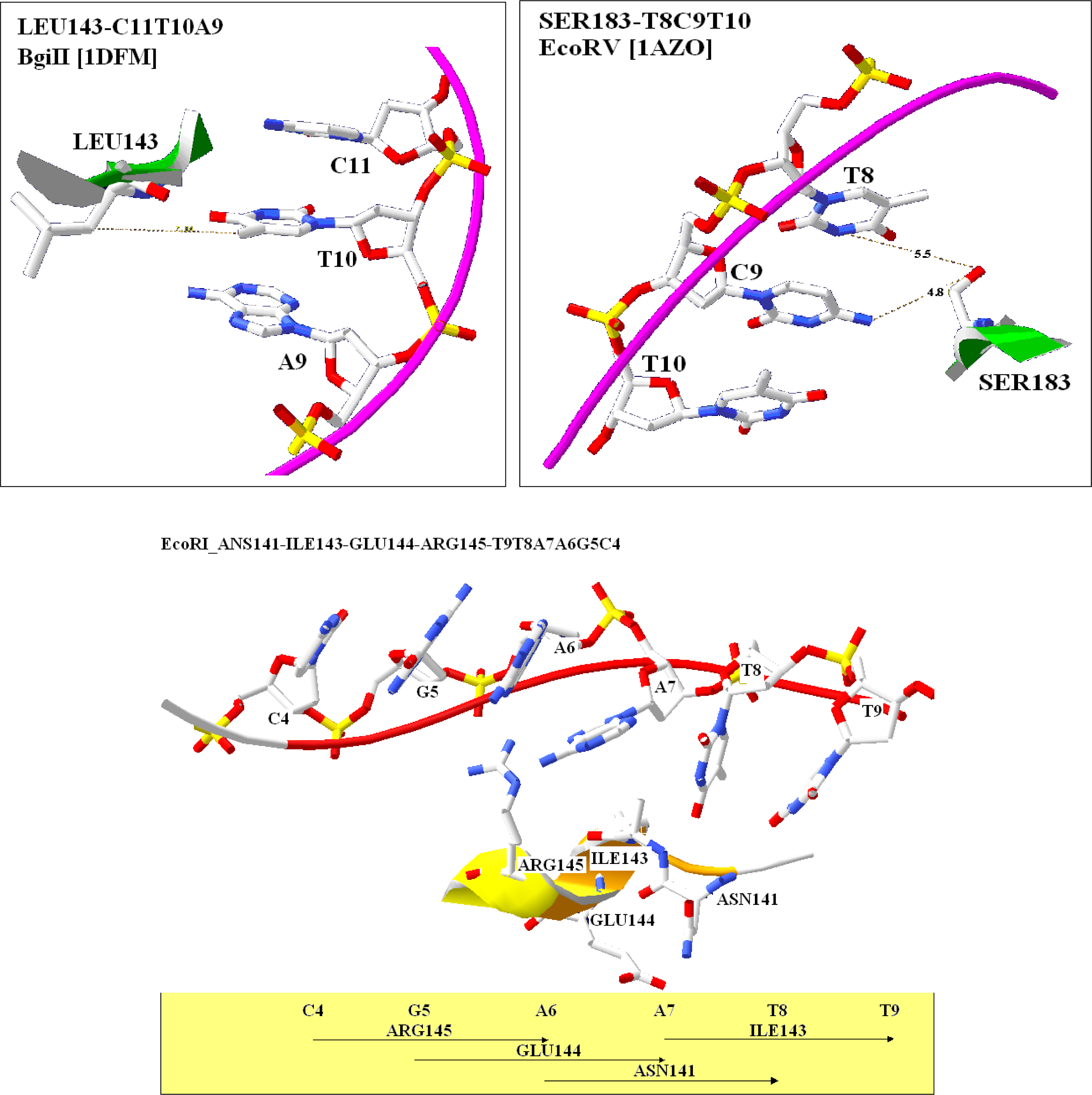
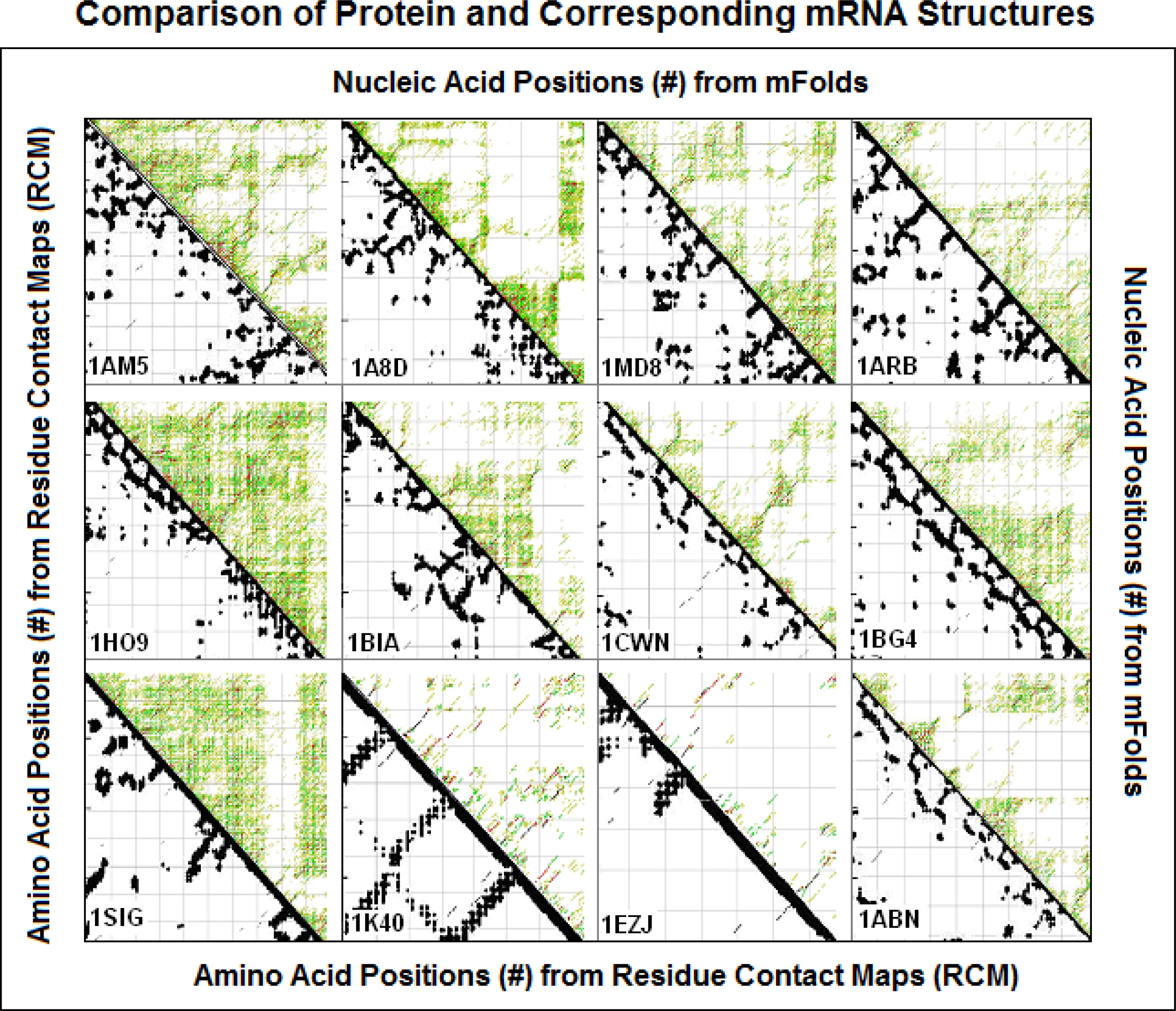
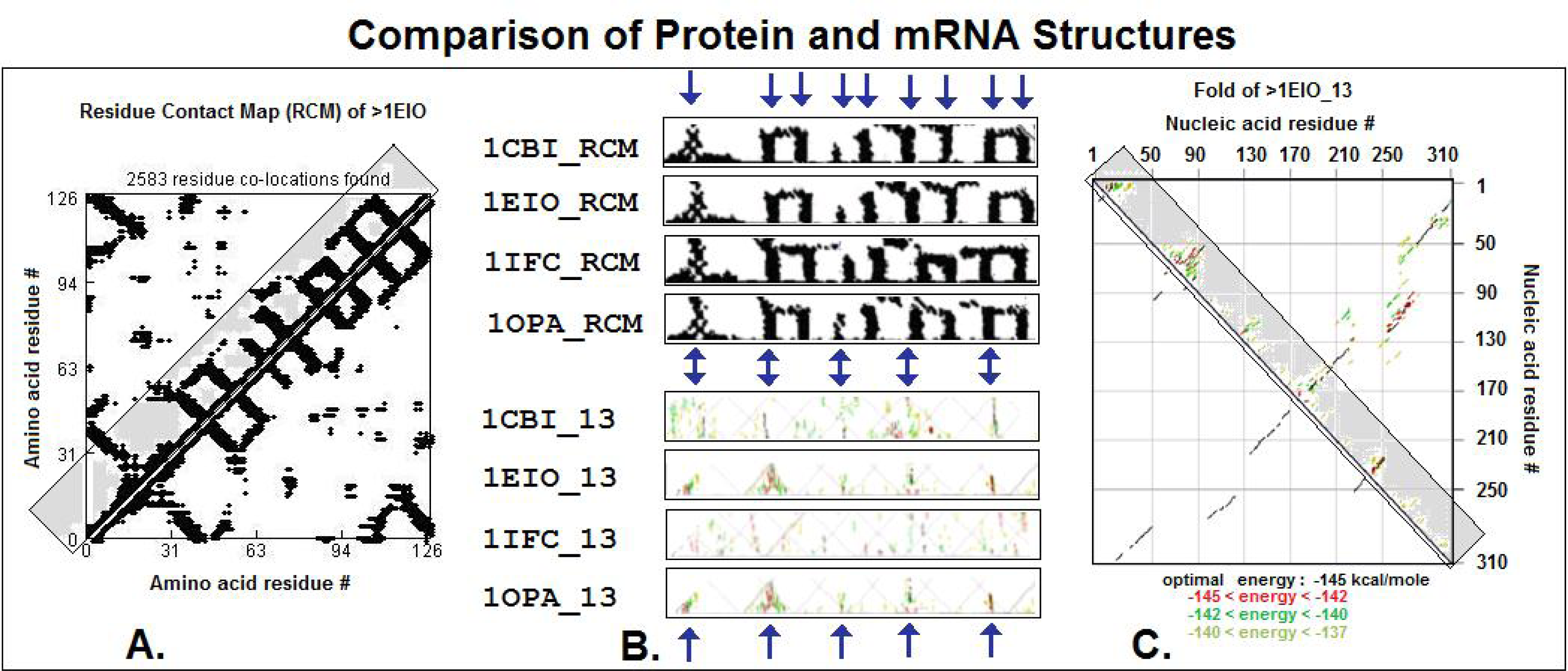
2.4. Visualization of specific nucleic acid – protein interactions
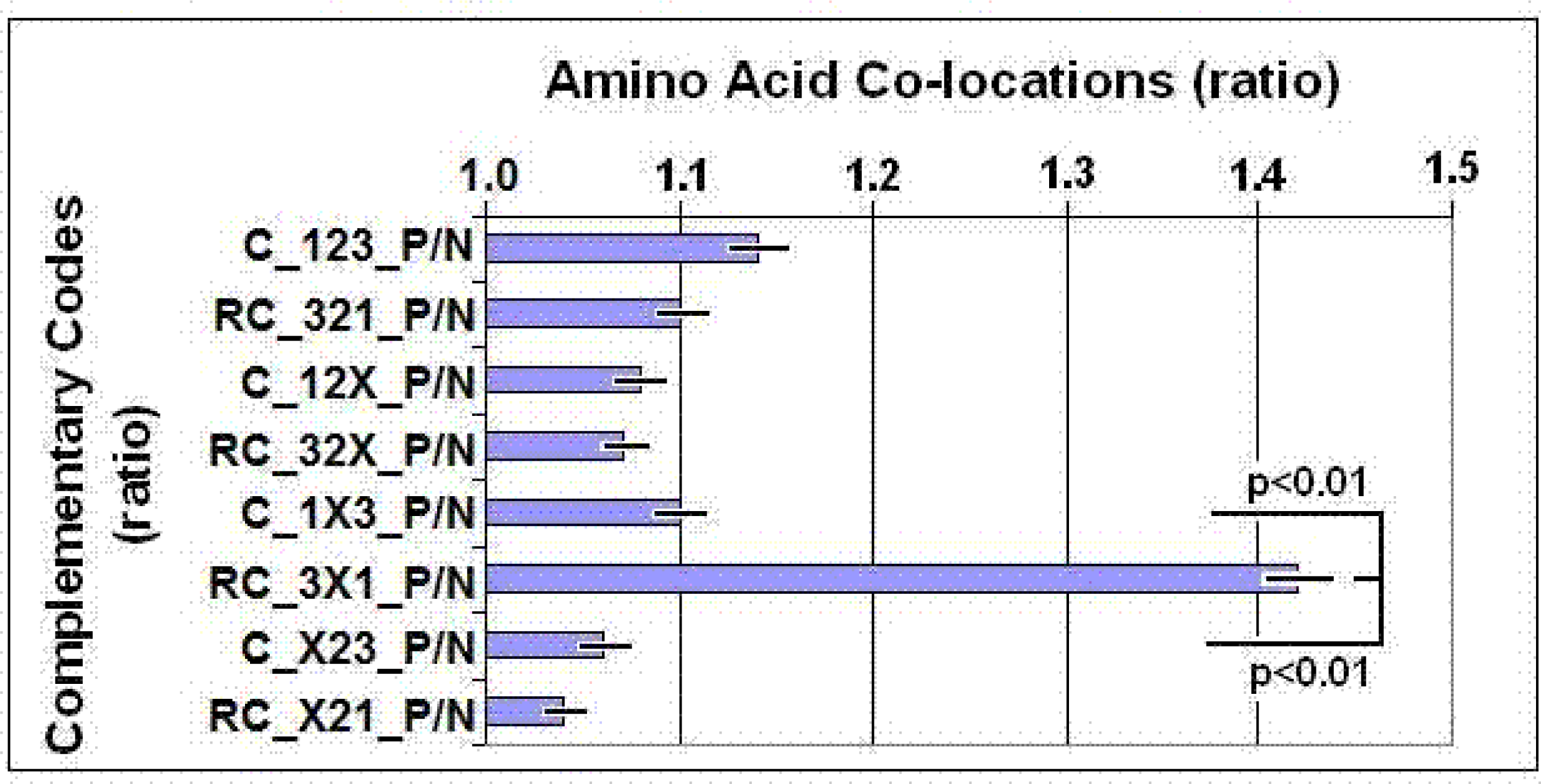
2.5. Partial Complementary Coding of Co-locating Amino Acids
2.6. The Role and Predictability of Wobble bases
2.7. Integrated Codon Systems
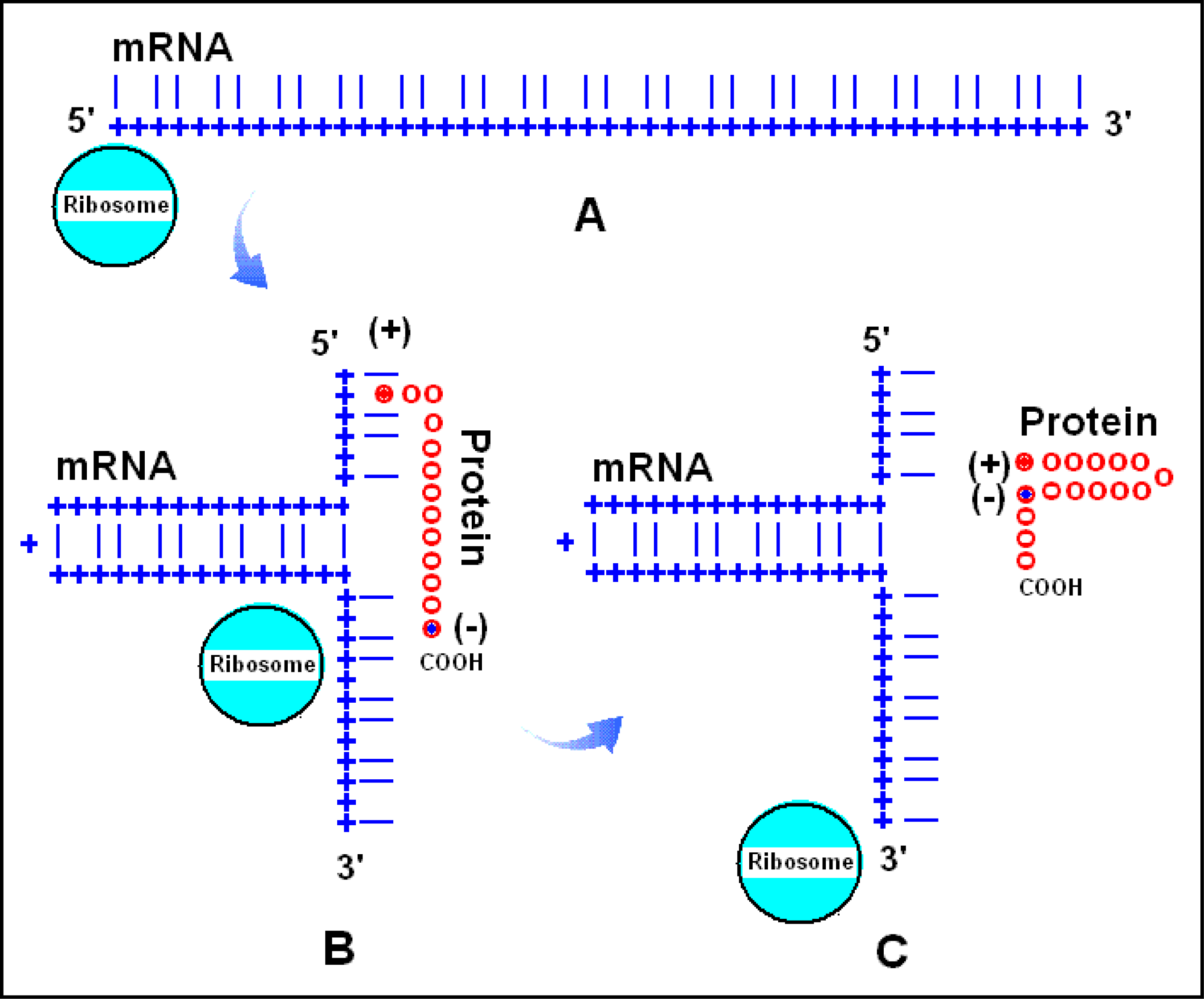
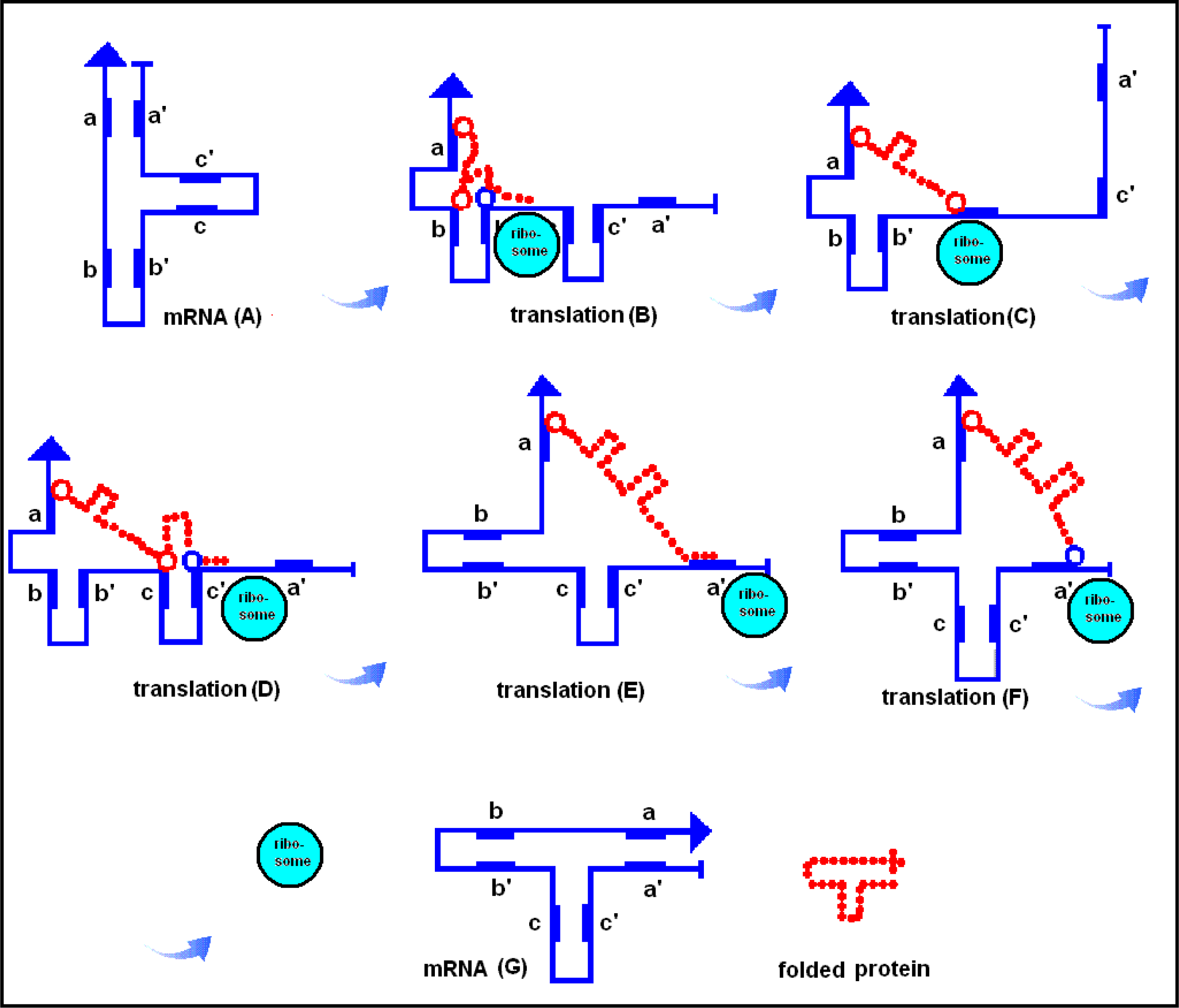
2.8. The RNA-assisted Protein Folding
2.9. Evolutionary Aspects of the Redundant Genetic and Proteomic Codes
3. Conclusions
Acknowledgments
References and Notes
- Hayes, B. The invention of the Genetic Code. Amer. Sci 1998, 1, 8–14. [Google Scholar]
- Franklin, R; Gosling, R. Molecular configuration in sodium thymonucleate. Nature 1953, 171, 740–741. [Google Scholar]
- Watson, JD; Crick, FFC. A structure for deoxyribose nucleic acids. Nature 1953, 171, 737–739. [Google Scholar]
- Gamow, G. Possible relation between deoxyribonucleic acid and protein structures. Nature 1954, 173, 318–318. [Google Scholar]
- Gamow, G. Possible mathematical relation between deoxyribonucleic acid and proteins. Det Kongelige Danske Videnskabernes Selskab, Biologiske Meddelelser 1954, 22, 1–13. [Google Scholar]
- Gamow, G; Rich, A; Ycas, M. The problem of information transfer from nucleic acids to proteins. Adv. Bio. Med. Phys 1956, 4, 23–68. [Google Scholar]
- Brenner, S. On the impossibility of all overlapping triplet codes in information transfer from nucleic acid to proteins. Proc. Nat. Acad. Sci. USA 1957, 43, 687–694. [Google Scholar]
- Crick, FHC; Griffith, JS; Orgel, LE. Codes without commas. Proc. Nat. Acad. Sci. USA 1957, 43, 416–421. [Google Scholar]
- Nirenberg, MW; Matthaei, JH. The dependence of cell-free protein synthesis in E. coli upon naturally occurring or synthetic polyribonucleotides. Proc. Nat. Acad. Sci. USA 1961, 47, 1588–1602. [Google Scholar]
- Leder, P; Nirenberg, M. RNA code words and protein synthesis, II. Nucleotide sequence of a valine RNA codeword. Proc. Nat. Acad. Sci. USA 1964, 52, 420–427. [Google Scholar]
- Workman, C; Krogh, A. No evidence that mRNAs have lower folding free energies than random sequences with the same dinucleotide distribution. Nucleic Acids Res 2002, 24, 4816–4822. [Google Scholar]
- The Nucleic Acid Database Project. The State University of New Jersey: Rutgers, 2008.
- Zuker, M. Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res 2003, 31, 3406–3415. [Google Scholar]
- Biro, JC. Correlation between nucleotide composition and folding energy of coding sequences with special attention to wobble bases. Theor. Biol. Med. Model 2008, 5, 14. [Google Scholar]
- Meyer, IM; Miklós, I. Statistical evidence for conserved, local secondary structure in the coding regions of eukaryotic mRNAs and pre-mRNAs. Nucleic Acids Res 2005, 33, 6338–6348. [Google Scholar]
- Katz, L; Burge, CB. Widespread selection for local RNA secondary structure in coding regions of bacterial genes. Genome Res 2003, 13, 2042–2051. [Google Scholar]
- Hatfield, GW; Roth, DA. Optimizing scaleup yield for protein production: Computationally Optimized DNA Assembly (CODA) and translation engineeringtrade mark. Biotechnol. Annu. Rev 2007, 13, 27–42. [Google Scholar]
- Zhang, W; Xiao, W; Wei, H; Zhang, J; Tian, Z. mRNA secondary structure at start AUG codon is a key limiting factor for human protein expression in Escherichia coli. Biochem. Biophys. Res. Commun 2006, 349, 69–78. [Google Scholar]
- Codon Usage Database, NCBI-GenBank Flat File Release 160.0.
- Biro, JC. Indications that “codon boundaries” are physico-chemically defined and that protein-folding information is contained in the redundant exon bases. Theor. Biol. Med. Model 2006, 3, 28. [Google Scholar]
- Shabalina, SA; Ogurtsov, AY; Spiridonov, NA. A periodic pattern of mRNA secondary structure created by the genetic code. Nucleic Acids Res 2006, 34, 82428–2437. [Google Scholar]
- Stoletzki, N; Eyre-Walker, A. Synonymous codon usage in Escherichia coli: selection for translational accuracy. Mol. Biol. Evol 2007, 24, 374–381. [Google Scholar]
- Smith, NG; Eyre-Walker, A. Why are translationally sub-optimal synonymous codons used in Escherichia coli? J. Mol. Evol 2001, 53, 225–236. [Google Scholar]
- Akashi, H. Synonymous Codon Usage in Drosophila Melanogaster: Natural Selection and Translational Accuracy. Genetics 1994, 136, 927–935. [Google Scholar]
- Zhang, J; Long, M; Li, L. Translational effects of differential codon usage among intragenic domains of new genes in Drosophila. Biochim. Biophys. Acta (BBA) - Gene Struct. Express 2005, 1728, 135–142. [Google Scholar]
- Pelc, SR; Welton, MGE. Stereochemical relationship between coding triplets and amino-acids. Nature 1966, 209, 868–870. [Google Scholar]
- Welton, MGE; Pelc, SR. Specificity of the Stereochemical relationship between ribonucleic acid-triplets and amino-acids. Nature 1966, 209, 870–872. [Google Scholar]
- Crick, FHC. An Error in Model Building. Nature 1967, 213, 798–798. [Google Scholar]
- Woese, CR. The molecular basis for gene expression. In The Genetic Code; Harper & Row: New York, 1967; Chapters 6–7pp. 156–160. [Google Scholar]
- Biro, JC; Benyo, B; Sansom, C; Szlavecz, A; Fordos, G; Micsik, T; Benyo, Z. A common periodic table of codons and amino acids. Biochem. Biophys. Res. Commun 2003, 306, 408–415. [Google Scholar]
- Biro, JC; Biro, JMK. Frequent occurrence of recognition Site-like sequences in the restriction endonucleases. BMC Bioinformat 2004, 5, 30. [Google Scholar]
- Biro, J. Comparative analysis of specificity in protein-protein interactions. Part I: A theoretical and mathematical approach to specificity in protein-protein interactions. Med. Hypotheses 1981, 7, 969–979. [Google Scholar]
- Biro, J. Comparative analysis of specificity in protein-protein interactions. Part II: The complementary coding of some proteins as the possible source of specificity in protein-protein interactions. Med. Hypotheses 1981, 7, 981–993. [Google Scholar]
- Biro, J. Comparative analysis of specificity in protein-protein interactions. Part III: Models of the gene expression based on the sequential complementary coding of some pituitary proteins. Med. Hypotheses 1981, 7, 995–1007. [Google Scholar]
- Mekler, LB. Specific selective interaction between amino acid groups of polypeptide chains. Biofizika 1969, 14, 581–584. [Google Scholar]
- Mekler, LB; Idlis, RG. VINITI Deposited Doc, 1981; 1476–1481.
- Blalock, JE; Bost, KL. Binding of peptides that are specified by complementary RNAs. Biochem. J 1986, 234, 679–683. [Google Scholar]
- Segerstéen, U; Nordgren, H; Biro, JC. Frequent occurrence of short complementary sequences in nucleic acids. Biochem. Biophys. Res. Commun 1986, 139, 94–101. [Google Scholar]
- Blalock, JE; Smith, EM. Hydropathic anti-complementarity of amino acids based on the genetic code. Biochem. Biophys. Res. Commun 1984, 121, 203–207. [Google Scholar]
- Root-Bernstein, RS. Amino acid pairing. J. Theor. Biol 1982, 94, 885–894. [Google Scholar]
- Siemion, IZ; Stefanowicz, P. Periodical changes of amino acid reactivity within the genetic code. Biosystems 1992, 27, 77–84. [Google Scholar]
- Heal, JR; Roberts, GW; Raynes, JG; Bhakoo, A; Miller, AD. Specific interactions between sense and complementary peptides: the basis for the proteomic code. Chembiochemistry 2002, 3, 136–151. [Google Scholar]
- Baranyi, L; Campbell, W; Ohshima, K; Fujimoto, S; Boros, M; Okada, H. The antisense homology box: A new motif within proteins that encodes biologically active peptides. Nat. Med 1995, 1, 894–901. [Google Scholar]
- Biro, JC; Fördös, G. SeqX a tool to detect, analyze and visualize residue co-locations in protein and nucleic acid structures. BMC Bioinformat 2005, 6, 170. [Google Scholar]
- Biro, JC. Amino acid size, charge, hydropathy indices and matrices for protein structure analysis. Theor. Biol. Med. Model 2006, 3, 15. [Google Scholar]
- Biro, JC. The Proteomic Code: A molecular recognition code for proteins. Review. Theor. Biol. Med. Model 2007, 4, 45. [Google Scholar]
- Biro, JC. Protein folding information in nucleic acids which is not present in the genetic code. Ann. NY Acad. Sci 2006, 1091, 399–411. [Google Scholar]
- Anfinsen, CB; Redfield, RR; Choate, WI; Page, J; Carroll, WR. Studies on the gross structure, cross-linkages, and terminal sequences in ribonuclease. J. Biol. Chem 1954, 207, 201–210. [Google Scholar]
- Makhatadze, GI; Privalov, PL. Energetics of Protein Structure. In Advances in Protein Chemistry; Anfinsen, CB, Edsall, JT, Richards, FM, Eds.; Academic Press: New York, 1995; Volume 47, pp. 308–405. [Google Scholar]
- Thirumalai, D; Hyeon, C. RNA and protein folding: common themes and variations. Biochemistry 2005, 44, 4957–4970. [Google Scholar]
- Thirumalai, D; Lorimer, GH. Chaperonin-mediated protein folding. Annu. Rev. Biophys. Biomol. Struct 2001, 30, 245–69. [Google Scholar]
- Levinthal, C. How to fold graciously in Mossbauer spectroscopy in biological systems. In Proceedings of a Meeting held at Allerton House; Debrunner, P, Tsibris, JCM, Munck, E, Urbana, IL, Eds.; University of Illinois Press: Monticello, 1969; pp. 22–24. [Google Scholar]
- Grantcharova, V; Alm, EJ; Baker, D; Horwich, AL. Mechanisms of protein folding. Curr. Opin. Struct. Biol 2001, 11, 70–82. [Google Scholar]
- Hartl, FU; Hayer-Hartl, M. Molecular chaperones in the cytosol: from nascent chain to folded protein. Science 2002, 295, 1852–1858. [Google Scholar]
- Genevaux, P; Georgopoulos, C; Kelley, WL. The Hsp70 chaperone machines of Escherichia coli: a paradigm for the repartition of chaperone functions. Mol. Microbiol 2007, 66, 840–857. [Google Scholar]
- Thanaraj, TA; Argos, P. Protein secondary structural types are differentially coded on messenger RNA. Protein Sci 1996, 5, 1973–1983. [Google Scholar]
- Brunak, S; Engelbrecht, J. Protein structure and the sequential structure of mRNA: alpha-Helix and beta-sheet signals at the nucleotide level. Proteins 1996, 25, 237–252. [Google Scholar]
- Gupta, SK; Majumdar, S; Bhattacharya, TK; Ghosh, TC. Studies on the relationships between the synonymous codon usage and protein secondary structural units. Biochem. Biophys. Res. Commun 2000, 269, 692–696. [Google Scholar]
- Chiusano, ML; Alvarez-Valin, F; DI Giulio, M. Second codon positions of genes and the secondary structures of proteins. Relationships and implications for the origin of the genetic code. Gene 2000, 261, 63–69. [Google Scholar]
- Gu, W; Zhoa, T; Ma, J; Sun, X; Lu, Z. The relationship between synonymous codon usage and protein structure in Escherichia coli and Homo sapiens. Biosystems 2004, 73, 89–97. [Google Scholar]
- Ermolaeva, O. Synonymous codon usage in bacteria. Curr. Issues Mol. Biol 2001, 3, 91–97. [Google Scholar]
- Adzhubei, IA; Adzhubei, AA. ISSD Version 2.0: Taxonomic range extended. Nucleic Acids Res 1999, 27, 268–271. [Google Scholar]
- Biro, JC. Does codon bias have an evolutionary origin? Theor. Biol. Med. Model 2008, 5, 16. [Google Scholar]
- Kimchi-Sarfaty, C; Oh, JM; Kim, IW; Sauna, ZE; Calcagno, AM; Ambudkar, SV; Gottesman, MM. A “silent” polymorphism in the MDR1 gene changes substrate specificity. Science 2007, 315, 525–528. [Google Scholar]
- Sauna, ZE; Kimchi-Sarfaty, C; Ambudkar, SV; Gottesman, MM. Silent polymorphisms speak: how they affect pharmacogenomics and the treatment of cancer. Cancer Res 2007, 67, 9609–9612. [Google Scholar]
- Duan, J; Wainwright, MS; Comeron, JM; Saitou, N; Sanders, AR; Gelernter, J; Gejman, PV. Synonymous mutations in the human dopamine receptor D2 (DRD2) affect mRNA stability and synthesis of the receptor. Hum. Mol. Genet 2003, 12, 205–216. [Google Scholar]
- Pagani, F; Raponi, M; Baralle, FE. Synonymous mutations in CFTR exon 12 affect splicing and are not neutral in evolution. Proc. Nat. Acad. Sci. USA 2005, 102, 6368–6372. [Google Scholar]
- Nielsen, KB; Sorensen, S; Cartegni, L; Corydon, TJ; Doktor, TK; Schroeder, LD; Reinert, LS; Elpeleg, O; Krainer, AR; Gregersen, N; Kjems, J; Andresen, BS. Seemingly neutral polymorphic variants may confer immunity to splicing inactivating mutations: A synonymous SNP in exon 5 of MCAD protects from deleterious mutations in a flanking exonic splicing enhancer. Am. J. Hum. Genet 2007, 80, 416–432. [Google Scholar]
- Sauna, ZE; Kimchi-Sarfaty, C; Ambudkar, SV; Gottesman, MM. The sounds of silence: Synonymous mutations affect function. Pharmacogenomics 2007, 8, 527–532. [Google Scholar]
- Komar, AA. Genetics. SNPs, silent but not invisible. Science 2007, 315, 466–467. [Google Scholar]
- Soares, C. Codon spell check. Silent mutations are not so silent after. Sci. Am 2007, 296, 23–24. [Google Scholar]
- Drake, JW. Too many mutants with multiple mutations. Crit. Rev. Biochem. Mol. Biol 2007, 42, 247–258. [Google Scholar]
- Drake, JW; Bebenek, A; Kissling, GE; Peddada, S. Clusters of mutations from transient hypermutability. Proc. Nat. Acad. Sci. USA 2005, 102, 12849–12854. [Google Scholar]
- Poon, A; Chao, L. The rate of compensatory mutation in the DNA bacteriophage ϕX174. Genetics 2005, 170, 989–999. [Google Scholar]
- Plotnikova, OV; Kondrashov, FA; Vlasov, PK; Grigorenko, AP; Ginter, EK; Rogaev, EI. Conversion and compensatory evolution of the gamma-crystallin genes and identification of a cataractogenic mutation that reverses the sequence of the human CRYGD gene to an ancestral state. Am. J. Hum. Genet 2007, 81, 32–43. [Google Scholar]
- Kim, H; Shen, T; Sun, DP; Ho, NT; Madrid, M; Tam, MF; Zou, M; Cottam, PF; Ho, C. Restoring allosterism with compensatory mutations in hemoglobin. Proc. Nat. Acad. Sci. USA 1994, 91, 11547–11551. [Google Scholar]
- Biro, JC. Nucleic acid chaperons: a theory of an RNA-assisted protein folding. Theor. Biol. Med. Model 2005, 2, 35. [Google Scholar]
- Ikehara, K. Origins of gene, genetic code, protein and life: Comprehensive view of life systems from a GNC-SNS primitive genetic code hypothesis. J. Biosci 2002, 27, 165–186. [Google Scholar]
- Ikehara, K; Omori, Y; Arai, R; Hirose, A. A novel theory on the origin of the genetic code: A GNC-SNS hypothesis. J. Mol. Evol 2002, 54, 530–538. [Google Scholar]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}


© 2008 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/). This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Biro, J.C. Discovery of Proteomic Code with mRNA Assisted Protein Folding. Int. J. Mol. Sci. 2008, 9, 2424-2446. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms9122424
Biro JC. Discovery of Proteomic Code with mRNA Assisted Protein Folding. International Journal of Molecular Sciences. 2008; 9(12):2424-2446. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms9122424
Chicago/Turabian StyleBiro, Jan C. 2008. "Discovery of Proteomic Code with mRNA Assisted Protein Folding" International Journal of Molecular Sciences 9, no. 12: 2424-2446. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms9122424