
Application of a Novel Algorithm for Expanding Newborn Screening for Inherited Metabolic Disorders across Europe

 ,
,
Abstract
:1. Introduction
2. Methods
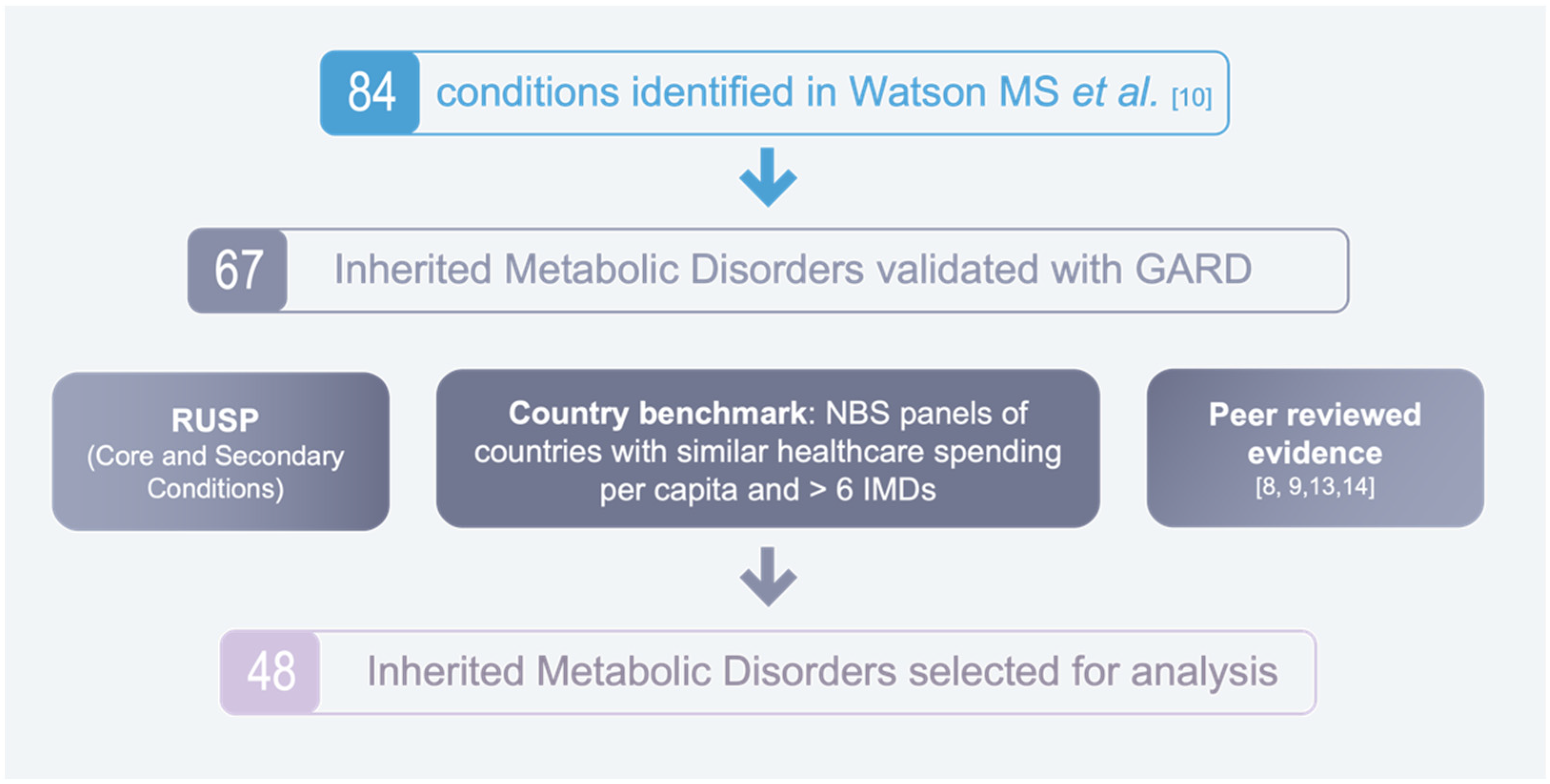
2.1. Identification of Disorders for Analysis
2.2. Assessment of Inherited Metabolic Disorders
3. Results
3.1. Characteristics of IMDs Identified for Analysis
- Twenty-one are lysosomal storage disorders (LSD), eight are disorders of organic acid metabolism (DOAM), seven are disorders of amino acid metabolism (DAAM), nine are disorders of fatty acid metabolism (DFAM), three disorders are classified as Other;
- Nine disorders had a frequency greater than or equal to 1 in 50,000; 10 disorders had a frequency between 1 in 50,000 and 1 in 100,000; seven disorders had a frequency between 1 in 100,000 and 1 in 150,000; eight disorders had a frequency between 1 in 150,000 and 1 in 250,000; 14 disorders had a frequency less than 1 in 250,000;
- Four disorders are screened for in over 20 European countries, 15 disorders are screened for in 11 to 20 European countries, nine disorders are screened for in at least one, but fewer than 10 European countries, and 17 disorders are not screened for in the European countries covered in Castineras DE et al. 2019 [7].
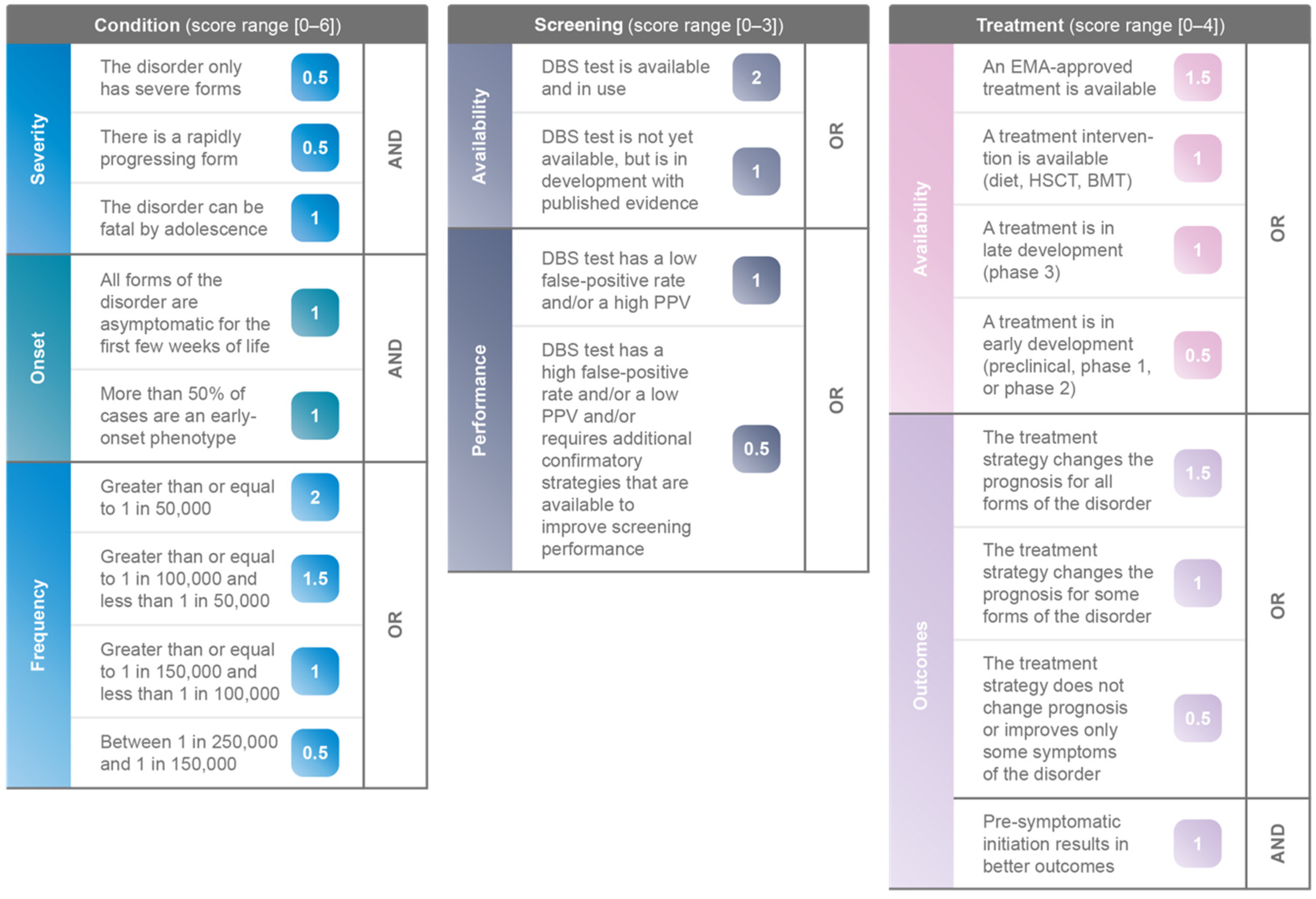
3.2. Scoring and Ranking of IMDs
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
Appendix A

| Disorder | Abbreviation | Type of Disorder | Frequency | Gene(s) Involved | European Countries with NBS Programme |
|---|---|---|---|---|---|
| 3-Hydroxy-3-methyglutaric aciduria | HMG/3HMG | DOAM | 1/125,000–1/1,000,000 | HMGCL (locus: 1p36.11) | Hungary, Iceland, Italy, Netherlands, Norway, Poland, Portugal, Slovakia, Slovenia |
| 3-Methylcrotonyl-CoA carboxylase deficiency | 3MCC | DOAM | 1/30,000–1/50,000 in Europe | MCCC1, MCCC2 (loci: 3q27.1, 5q13.2) | Austria, Hungary, Iceland, Italy, Macedonia, Netherlands, Poland, Portugal, Slovakia, Slovenia |
| Alpha-mannosidosis | α-mannosidosis | LSD | 1/500,000 | MAN2B1 (locus: 19p13.2-q12) | No screening programmes ° |
| Argininaemia | ARG | DAAM | 1/300,000–1/1,000,000 + | ARG1 (locus: 6q23.2) | Austria, Czech Republic, Estonia, Finland, Iceland, Italy, Macedonia, Poland, Portugal, Slovakia, Sweden |
| Argininosuccinic aciduria | ASA | DAAM | 1/70,000 [17] | ASL (locus: 7q11.21) | Austria, Denmark, Finland, Hungary, Iceland, Italy, Macedonia, Poland, Portugal, Sweden |
| Batten disease | CLN2 | LSD | Unknown, 1/25,000–1/50,000 for all NCLs [18] | CLN2 also known as TPP1 (locus: 11p15.4) | No screening programmes ° |
| Biotinidase deficiency | BIOT/BIO/BTD | Other | 1/60,000 | BTD (locus: 3p25.1) | Austria, Belgium (Flemish), Belgium (Walloon), Czech Republic, Denmark, Germany, Hungary, Italy, Latvia, Liechtenstein, Netherlands, Norway, Poland, San Marino, Spain, Sweden, Switzerland, Turkey |
| Carnitine acylcarnitine translocase deficiency | CACT | DFAM | Less than 60 cases worldwide | SLC25A20 (locus: 3p21.31) | Austria, Czech Republic, Estonia, Finland, Germany, Hungary, Iceland, Italy, Macedonia, Norway, Poland, Portugal, Slovakia, Slovenia, Sweden |
| Carnitine palmitoyltransferase, type I deficiency | CPT I/CPT1A | DFAM | 1/750,000–1/2,000,000 [19] | CPT1A (locus: 11q13.3) | Austria, Czech Republic, Estonia, Finland, Germany, Hungary, Iceland, Italy, Macedonia, Netherlands, Norway, Poland, Portugal, Slovakia, Slovenia, Sweden |
| Carnitine palmitoyltransferase, type II deficiency | CPT II | DFAM | 300 cases reported | CPT2 (locus: 1p32.3) | Austria, Czech Republic, Estonia, Finland, Germany, Hungary, Iceland, Italy, Macedonia, Norway, Poland, Portugal, Slovakia, Slovenia, Sweden |
| Carnitine uptake defect/carnitine transport defect | CUD | DFAM | 1/20,000–1/70,000 in Europe | SLC22A5 (locus: 5q23.3) | Austria, Croatia, Denmark, Estonia, Finland, Hungary, Iceland, Italy, Macedonia, Norway, Poland, Portugal, Slovakia, Slovenia, Sweden |
| Citrullinemia, type I | CIT/CTLN1 | DAAM | 1/250,000 [20] | ASS1 (locus: 9q34.11) | Austria, Czech Republic, Estonia, Finland, Hungary, Iceland, Italy, Macedonia, Poland, Portugal, Slovakia, Sweden |
| Classic galactosaemia | GALT/GAL | Other | 1/40,000–1/60,000 in Western countries | GALT (locus: 9p13.3) | Austria, Belgium (Walloon), Denmark, Germany, Greece, Hungary, Ireland, Italy, Latvia, Lithuania, Liechtenstein, Netherlands, Russia, San Marino, Spain, Sweden, Switzerland |
| Fabry disease | GLA | LSD | 1/80,000 | GLA (locus: Xq22) | No screening programmes ° |
| Farber disease | ACD | LSD | 200 cases reported worldwide | ASAH1 (locus: 8p22) | No screening programmes ° |
| Gaucher disease | GD | LSD | 1/50,000–1/100,000 + | GBA (locus: 1q22) | No screening programmes ° |
| Glutaric aciduria type 1 | GA1 | DOAM | 1/100,000 | GCDH (locus: 19p13.2) | Austria, Belgium (Flemish), Croatia, Czech Republic, Denmark, Estonia, Finland, Germany, Hungary, Iceland, Ireland, Italy, Macedonia, Netherlands, Norway, Poland, Portugal, Slovakia, Slovenia, Spain, Sweden, Switzerland, United Kingdom (UK) |
| Holocarboxylase synthetase deficiency | MCD/HCSD | DOAM | 1/200,000 | HLCS (locus: 21q22.1) | Austria, Denmark, Hungary, Iceland, Italy, Macedonia, Netherlands, Norway, Portugal, Slovenia |
| Homocystinuria | HCU/HCY | DAAM | 1/150,000 [21] | CBS also MTHFR, MTR, MTRR and MMADHC (loci: 21q22.3, 1p36.22, 1q43, 5p15.31, 2q23.2) * | Austria, Belgium (Walloon), Czech Republic, Estonia, Finland, Hungary, Iceland, Ireland, Italy, Macedonia, Netherlands, Norway, Poland, Portugal, Sweden, UK |
| Isovaleric acidaemia | IVA | DOAM | 1/120,000 [22] | IVD (locus: 15q15.1) | Austria, Belgium (Flemish), Croatia, Czech Republic, Denmark, Estonia, Finland, Germany, Hungary, Iceland, Italy, Macedonia, Netherlands, Norway, Poland, Portugal, Slovakia, Slovenia, Sweden, UK |
| Krabbe disease | GLD | LSD | 1/100,000 in Northern Europe | GALC (locus: 14q31.3) | Not studied in [4,7,23]. |
| Long-chain 3 hydroxyacyl-CoA dehydrogenase deficiency | LCHAD | DFAM | 1/110,000–1/150,000 [24] | HADHA (locus: 2p23.3) | Austria, Croatia, Czech Republic, Denmark, Estonia, Finland, Germany, Hungary, Iceland, Italy, Macedonia, Netherlands, Norway, Poland, Slovakia, Slovenia, Spain, Portugal, Sweden |
| Lysosomal acid lipase deficiency | LAL/LAL-D | LSD | 1/177,000 | LIPA (locus: 10q23.31) | Not studied in [4,7,23]. |
| Maple syrup urine disease | MSUD | DAAM | 1/135,000 [22] | BCKDHA, BCKDHB and DBT (loci: 19q13.2, 6q14.1, 1p21.2) | Austria, Belgium (Flemish), Belgium (Walloon), Czech Republic, Denmark, Estonia, Finland, Germany, Hungary, Iceland, Ireland, Italy, Netherlands, Macedonia, Norway, Poland, Portugal, Slovakia, Slovenia, Sweden, Switzerland, UK |
| Medium-chain acyl-CoA dehydrogenase deficiency | MCAD/MCADD | DFAM | 1/4900–1/27,000 in Caucasian population | ACADM (locus: 1p31,1) | Austria, Belgium (Flemish), Croatia, Czech Republic, Denmark, Estonia, Finland, France, Germany, Hungary, Iceland, Ireland, Italy, Luxembourg, Macedonia, Norway, Netherlands, Poland, Portugal, Slovakia, Slovenia, Spain, Sweden, Switzerland, UK |
| Metachromatic leukodystrophy | MLD | LSD | 1/40,000–1/160,000 ** | ARSA, rarely PSAP (loci: 22q13.33, 10q22.1) | Not studied in [4,7,23]. |
| Methylmalonic acidaemia (cobalamin disorders, Cbl A, B) | MMA/Cbl A,B | DOAM | Over 120 patients with cblA, 66 patients with cblB have been reported | MMAA, MMAB (loci: 4q31.21, 12q24.11) | Austria, Belgium (Flemish), Denmark, Estonia, Finland, Hungary, Iceland, Italy, Macedonia, Netherlands, Norway, Poland, Portugal, Slovakia, Slovenia, Sweden |
| Methylmalonic acidaemia (methylmalonyl-CoA mutase) | MUT | DOAM | 1/167,000 in Europe [25] | MMUT (locus: 6p12.3) | Austria, Belgium (Flemish), Denmark, Hungary, Iceland, Italy, Portugal, Sweden |
| Mucopolysaccharidosis, type II | MPS II | LSD | 1/166,000 in Europe | IDS (locus: Xq28) | No screening programmes ° |
| Mucopolysaccharidosis, type III | MPS III | LSD | 1/70,000 in Europe [26] | SGSH, NAGLU, HGSNAT, GNS (loci: 17q25.3, 17q21.2, 8p11.2-p11.1, 12q14.3) * | No screening programmes ° |
| Mucopolysaccharidosis, type IV | MPS IV | LSD | 1/77,000–1/1,400,000 in Europe [27] | GALNS for type IV A, GLB1 for type IV B (loci: 16q24.3, 3p22.3) | No screening programmes ° |
| Mucopolysaccharidosis, type IX | MPS IX | LSD | Only 4 known cases [28] | HYAL1 (locus: 3p21.31) | No screening programmes ° |
| Mucopolysaccharidosis, type VI | MPS VI | LSD | 1/43,000–1/1,505,000 [29] | ARSB (locus: 5q14.1) | No screening programmes ° |
| Mucopolysaccharidosis, type VII | MPS VII | LSD | 1/345,000–1/5,000,000 | GUSB (locus: 7q11.21) | No screening programmes ° |
| Mucopolysaccharidosis, type I | MPS I | LSD | 1/100,000 | IDUA (locus: 4p16.3) | No screening programmes ° |
| Multiple acyl-CoA dehydrogenase deficiency | MADD | DFAM | 1/200,000 | ETFA, ETFB, ETFDH (loci: 15q24.2-q24.3, 19q13.41, 4q32.1) | Austria, Belgium (Flemish), Finland, Hungary, Iceland, Italy, Macedonia, Poland, Portugal, Sweden |
| Niemann-Pick disease, type A/B | ASMD | LSD | 1/250,000 + | SMPD1 (locus: 11p15.4) | No screening programmes ° |
| Niemann-Pick disease, type C | NPC1 and NPC2 | LSD | 1/150,000 + | NPC1, NPC2 (loci: 18q11.2, 14q24.3) | No screening programmes ° |
| Phenylketonuria | PKU/HPA | DAAM | 1/10,000 in Europe | PAH (locus: 12q23.2) | Andorra, Austria, Belarus, Belgium (Flemish), Belgium (Walloon), Bosnia-Herzegovina, Bulgaria, Croatia, Cyprus, Czech Republic, Denmark, Estonia, Finland, France, Georgia, Germany, Greece, Hungary, Iceland, Ireland, Italy, Latvia, Liechtenstein, Lithuania, Luxembourg, Macedonia, Malta, Moldova, Monaco, Netherlands, Norway, Poland, Portugal, Romania, Russia, San Marino, Serbia, Slovakia, Slovenia, Spain, Sweden, Switzerland, Turkey, Ukraine, UK |
| Pompe disease | GSD 2 | LSD | 1/40,000 [17] | GAA (locus: 17q25.3) | No screening programmes ° |
| Propionic acidaemia | PROP/PA | DOAM | 1/45,000–1/313,000 in Europe [30] | PCCA, PCCB (loci: 13q32.3, 3q22.3) | Austria, Belgium (Flemish), Denmark, Estonia, Finland, Hungary, Iceland, Italy, Macedonia, Netherlands, Norway, Poland, Portugal, Serbia, Slovakia, Slovenia, Sweden |
| Sandhoff disease (GM2 gangliosidosis, type II) | SD | LSD | 1/130,000 in Europe | HEXB (locus: 5q13.3) | No screening programmes ° |
| Severe Combined Immunodeficiency | SCID | Other | 1/50,000 | must common ADA, also DCLRE1C, IL2RG, IL7R, JAK3, NHEJ1, PTPRC (loci: 20q13.12, 10p13, Xq13, 5p13.2, 19p13.11, 2q35, 1q31.3-q32.1) * | Denmark, Germany, Iceland, Norway, Sweden, Switzerland |
| Tay-Sachs disease (GM2 gangliosidosis, type I) | TSD | LSD | 1/320,000 | HEXA (locus: 15q23) | No screening programmes ° |
| Trifunctional protein deficiency | TFP | DFAM | Less than 100 cases reported | HADHA and HADHB (loci: 2p23.3, 2p23.3) | Austria, Denmark, Germany, Hungary, Iceland, Italy, Portugal, Sweden |
| Tyrosinemia, type 1 | TYR 1 | DAAM | 1/100,000 | FAH (locus: 15q25.1) | Austria, Belgium (Walloon), Denmark, Estonia, Finland, Germany, Hungary, Italy, Netherlands, Macedonia, Norway, Poland, Portugal, Romania, Slovakia, Slovenia, Sweden |
| Very long-chain acyl-CoA dehydrogenase deficiency | VLCAD | DFAM | 1/25,000 in the European Union [31] | ACADVL (locus: 17p13.1) | Austria, Belgium, Croatia, Czech Republic, Denmark, Estonia, Finland, Germany, Hungary, Iceland, Italy, Macedonia, Netherlands, Poland, Slovakia, Slovenia, Portugal, Sweden |
| X-linked adrenoleukodystrophy | X-ALD | Other | 1/14,700 [32] | ABCD1 (locus: Xq28) | Netherlands |
| Disorder | Score (0–6) | Condition | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Severity | Onset | Frequency | ||||||||
| The Condition Only Has Severe Forms | There Is a Rapidly Progressing Form | The Condition Can Be Fatal by Adolescence | All Forms of the Condition Are Asymptomatic for the First Few Weeks of Life | More than 50% of Cases Are an Early-Onset Phenotype | Greater than or Equal to 1 in 50,000 | Greater than or Equal to 1 in 100,000 and Less than 1 in 50,000 | Greater than or Equal to 1 in 150,000 and Less than 1 in 100,000 | Between 1 in 250,000 and 1 in 150,000 | ||
| AND | AND | OR | ||||||||
| 0.5 | 0.5 | 1 | 1 | 1 | 2 | 1.5 | 1 | 0.5 | ||
| Carnitine uptake defect/carnitine transport defect (CUD) | 5.5 | 0 | 0.5 | 1 | 1 | 1 | 2 | 0 | 0 | 0 |
| Severe combined immunodeficiency (SCID) | 6 | 0.5 | 0.5 | 1 | 1 | 1 | 2 | 0 | 0 | 0 |
| Glutaric aciduria type 1 (GA1) | 5.5 | 0.5 | 0.5 | 1 | 1 | 1 | 0 | 1.5 | 0 | 0 |
| Homocystinuria (HCU) | 4.5 | 0 | 0.5 | 1 | 1 | 1 | 0 | 0 | 1 | 0 |
| Phenylketonuria (PKU) | 4.5 | 0 | 0.5 | 0 | 1 | 1 | 2 | 0 | 0 | 0 |
| Tyrosinemia, type 1 (TYR 1) | 5 | 0 | 0.5 | 1 | 1 | 1 | 0 | 1.5 | 0 | 0 |
| Classic galactosaemia (GALT) | 5 | 0.5 | 0.5 | 1 | 0 | 1 | 2 | 0 | 0 | 0 |
| 3-Hydroxy-3-methyglutaric aciduria (HMG) | 4.5 | 0 | 0.5 | 1 | 1 | 1 | 0 | 0 | 1 | 0 |
| Pompe disease | 4.5 | 0 | 0.5 | 1 | 1 | 0 | 2 | 0 | 0 | 0 |
| X-linked adrenoleukodystrophy (X-ALD) | 4.5 | 0 | 0.5 | 1 | 1 | 0 | 2 | 0 | 0 | 0 |
| Argininosuccinic aciduria (ASA) | 4.5 | 0.5 | 0.5 | 1 | 0 | 1 | 0 | 1.5 | 0 | 0 |
| Carnitine palmitoyltransferase, type I deficiency (CPT I) | 4 | 0.5 | 0.5 | 1 | 1 | 1 | 0 | 0 | 0 | 0 |
| Long-chain 3 hydroxyacyl-CoA dehydrogenase deficiency (LCHAD) | 5 | 0.5 | 0.5 | 1 | 1 | 1 | 0 | 0 | 1 | 0 |
| Methylmalonic acidaemia (cobalamin disorders, Cbl A, B) | 4 | 0.5 | 0.5 | 1 | 1 | 1 | 0 | 0 | 0 | 0 |
| Metachromatic leukodystrophy (MLD) | 5.5 | 0.5 | 0.5 | 1 | 1 | 1 | 0 | 1.5 | 0 | 0 |
| Mucopolysaccharidosis, type I (MPS I) | 5 | 0 | 0.5 | 1 | 1 | 1 | 0 | 1.5 | 0 | 0 |
| Propionic acidaemia (PROP) | 3.5 | 0.5 | 0.5 | 1 | 0 | 1 | 0 | 0 | 0 | 0.5 |
| Biotinidase deficiency (BIOT) | 4.5 | 0.5 | 0.5 | 1 | 1 | 0 | 0 | 1.5 | 0 | 0 |
| Medium-chain acyl-CoA dehydrogenase deficiency (MCADD) | 4.5 | 0 | 0.5 | 1 | 1 | 0 | 2 | 0 | 0 | 0 |
| 3-Methylcrotonyl-CoA carboxylase deficiency (3MCC) | 4.5 | 0 | 0.5 | 1 | 1 | 0 | 2 | 0 | 0 | 0 |
| Citrullinemia, type I (CIT) | 3 | 0 | 0.5 | 1 | 1 | 0 | 0 | 0 | 0 | 0.5 |
| Holocarboxylase synthetase deficiency (MCD) | 3.5 | 0.5 | 0.5 | 1 | 0 | 1 | 0 | 0 | 0 | 0.5 |
| Krabbe disease | 5 | 0 | 0.5 | 1 | 1 | 1 | 0 | 1.5 | 0 | 0 |
| Argininaemia (ARG) | 3.5 | 0 | 0.5 | 1 | 1 | 1 | 0 | 0 | 0 | 0 |
| Carnitine acylcarnitine translocase deficiency (CACT) | 3 | 0.5 | 0.5 | 1 | 0 | 1 | 0 | 0 | 0 | 0 |
| Very long-chain acyl-CoA dehydrogenase deficiency (VLCAD) | 4.5 | 0 | 0.5 | 1 | 1 | 0 | 2 | 0 | 0 | 0 |
| Isovaleric acidaemia (IVA) | 2.5 | 0 | 0.5 | 1 | 0 | 0 | 0 | 0 | 1 | 0 |
| Maple syrup urine disease (MSUD) | 2.5 | 0 | 0.5 | 1 | 0 | 0 | 0 | 0 | 1 | 0 |
| Methylmalonic acidaemia (methylmalonyl-CoA mutase) (MUT) | 3 | 0 | 0.5 | 1 | 1 | 0 | 0 | 0 | 0 | 0.5 |
| Carnitine palmitoyltransferase, type II deficiency (CPT II) | 2.5 | 0 | 0.5 | 1 | 1 | 0 | 0 | 0 | 0 | 0 |
| Batten disease (CLN2) | 4 | 0.5 | 0.5 | 1 | 1 | 1 | 0 | 0 | 0 | 0 |
| Niemann Pick A/B (ASM deficiency) | 4.5 | 0.5 | 0.5 | 1 | 1 | 1 | 0 | 0 | 0 | 0.5 |
| Trifunctional protein deficiency (TFP) | 2.5 | 0 | 0.5 | 1 | 0 | 1 | 0 | 0 | 0 | 0 |
| Gaucher disease | 4 | 0 | 0.5 | 1 | 1 | 0 | 0 | 1.5 | 0 | 0 |
| Lysosomal acid lipase deficiency (LAL-D/Wolman/CESD) | 3 | 0 | 0.5 | 1 | 1 | 0 | 0 | 0 | 0 | 0.5 |
| Multiple acyl-CoA dehydrogenase deficiency (MADD) | 2 | 0 | 0.5 | 1 | 0 | 0 | 0 | 0 | 0 | 0.5 |
| MPS VI (Maroteaux-Lamy syndrome) | 3.5 | 0 | 0.5 | 1 | 1 | 1 | 0 | 0 | 0 | 0 |
| Alpha-mannosidosis | 2.5 | 0 | 0.5 | 1 | 1 | 0 | 0 | 0 | 0 | 0 |
| Fabry disease | 2.5 | 0 | 0 | 0 | 1 | 0 | 0 | 1.5 | 0 | 0 |
| MPS II (Hunter syndrome) | 2.5 | 0 | 0 | 0 | 1 | 1 | 0 | 0 | 0 | 0.5 |
| MPS III (Sanfilippo syndrome) | 3.5 | 0 | 0 | 0 | 1 | 1 | 0 | 1.5 | 0 | 0 |
| Niemann-Pick type C disease | 3.5 | 0 | 0.5 | 1 | 1 | 0 | 0 | 0 | 1 | 0 |
| MPS IV (Morquio syndrome) | 2 | 0 | 0 | 0 | 1 | 1 | 0 | 0 | 0 | 0 |
| Sandhoff disease (GM2 gangliosidosis, type II) | 4.5 | 0 | 0.5 | 1 | 1 | 1 | 0 | 0 | 1 | 0 |
| Farber disease | 3 | 0.5 | 0.5 | 1 | 0 | 1 | 0 | 0 | 0 | 0 |
| Tay-Sachs disease (GM2 gangliosidosis, type I) | 3.5 | 0 | 0.5 | 1 | 1 | 1 | 0 | 0 | 0 | 0 |
| MPS VII (Sly syndrome) | 1.5 | 0 | 0.5 | 1 | 0 | 0 | 0 | 0 | 0 | 0 |
| MPS IX (hyaluronidase deficiency) | 1 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 |
| Disorder | Score (0–3) | Screening | |||
|---|---|---|---|---|---|
| Availability | Performance | ||||
| DBS Test Is Available and in Use | DBS Test Is Not Yet Available, but Is in Development with Published Evidence | DBS Test Has a Low False-Positive Rate or a High Positive Predictive Value | DBS Test Has a High False-Positive Rate or a Low PPV, or Additional Confirmatory Strategies Are Required That Are Available to Improve Screening Performance | ||
| OR | OR | ||||
| 2 | 1 | 1 | 0.5 | ||
| Carnitine uptake defect/carnitine transport defect (CUD) | 3 | 2 | 0 | 1 | 0 |
| Severe combined immunodeficiency (SCID) | 2.5 | 2 | 0 | 0 | 0.5 |
| Glutaric aciduria type 1 (GA1) | 3 | 2 | 0 | 1 | 0 |
| Homocystinuria (HCU) | 3 | 2 | 0 | 1 | 0 |
| Phenylketonuria (PKU) | 3 | 2 | 0 | 1 | 0 |
| Tyrosinemia, type 1 (TYR 1) | 2.5 | 2 | 0 | 0 | 0.5 |
| Classic galactosaemia (GALT) | 3 | 2 | 0 | 1 | 0 |
| 3-Hydroxy-3-methyglutaric aciduria (HMG) | 3 | 2 | 0 | 1 | 0 |
| Pompe disease | 2.5 | 2 | 0 | 0 | 0.5 |
| X-linked adrenoleukodystrophy (X-ALD) | 3 | 2 | 0 | 1 | 0 |
| Argininosuccinic aciduria (ASA) | 3 | 2 | 0 | 1 | 0 |
| Carnitine palmitoyltransferase, type I deficiency (CPT I) | 3 | 2 | 0 | 1 | 0 |
| Long-chain 3 hydroxyacyl-CoA dehydrogenase deficiency (LCHAD) | 3 | 2 | 0 | 1 | 0 |
| Methylmalonic acidaemia (cobalamin disorders, Cbl A, B) | 3 | 2 | 0 | 1 | 0 |
| Metachromatic leukodystrophy (MLD) | 1.5 | 0 | 1 | 0 | 0.5 |
| Mucopolysaccharidosis, type I (MPS I) | 2.5 | 2 | 0 | 0 | 0.5 |
| Propionic acidaemia (PROP) | 3 | 2 | 0 | 1 | 0 |
| Biotinidase deficiency (BIOT) | 2.5 | 2 | 0 | 0 | 0.5 |
| Medium-chain acyl-CoA dehydrogenase deficiency (MCADD) | 3 | 2 | 0 | 1 | 0 |
| 3-Methylcrotonyl-CoA carboxylase deficiency (3MCC) | 3 | 2 | 0 | 1 | 0 |
| Citrullinemia, type I (CIT) | 3 | 2 | 0 | 1 | 0 |
| Holocarboxylase synthetase deficiency (MCD) | 3 | 2 | 0 | 1 | 0 |
| Krabbe disease | 2.5 | 2 | 0 | 0 | 0.5 |
| Argininaemia (ARG) | 2.5 | 2 | 0 | 0 | 0.5 |
| Carnitine acylcarnitine translocase deficiency (CACT) | 3 | 2 | 0 | 1 | 0 |
| Very long-chain acyl-CoA dehydrogenase deficiency (VLCAD) | 3 | 2 | 0 | 1 | 0 |
| Isovaleric acidaemia (IVA) | 3 | 2 | 0 | 1 | 0 |
| Maple syrup urine disease (MSUD) | 3 | 2 | 0 | 1 | 0 |
| Methylmalonic acidaemia (methylmalonyl-CoA mutase) (MUT) | 3 | 2 | 0 | 1 | 0 |
| Carnitine palmitoyltransferase, type II deficiency (CPT II) | 3 | 2 | 0 | 1 | 0 |
| Batten disease (CLN2) | 2 | 0 | 1 | 1 | 0 |
| Niemann Pick A/B (ASM deficiency) | 3 | 2 | 0 | 1 | 0 |
| Trifunctional protein deficiency (TFP) | 3 | 2 | 0 | 1 | 0 |
| Gaucher disease | 2.5 | 2 | 0 | 0 | 0.5 |
| Lysosomal acid lipase deficiency (LAL-D/Wolman/CESD) | 3 | 2 | 0 | 1 | 0 |
| Multiple acyl-CoA dehydrogenase deficiency (MADD) | 3 | 2 | 0 | 1 | 0 |
| MPS VI (Maroteaux-Lamy syndrome) | 1.5 | 0 | 1 | 0 | 0.5 |
| Alpha-mannosidosis | 2 | 0 | 1 | 1 | 0 |
| Fabry disease | 2.5 | 2 | 0 | 0 | 0.5 |
| MPS II (Hunter syndrome) | 1.5 | 0 | 1 | 0 | 0.5 |
| MPS III (Sanfilippo syndrome) | 2 | 0 | 1 | 1 | 0 |
| Niemann-Pick type C disease | 1 | 0 | 1 | 0 | 0 |
| MPS IV (Morquio syndrome) | 1.5 | 0 | 1 | 0 | 0.5 |
| Sandhoff disease (GM2 gangliosidosis, type II) | 0 | 0 | 0 | 0 | 0 |
| Farber disease | 0 | 0 | 0 | 0 | 0 |
| Tay-Sachs disease (GM2 gangliosidosis, type I) | 0 | 0 | 0 | 0 | 0 |
| MPS VII (Sly syndrome) | 0 | 0 | 0 | 0 | 0 |
| MPS IX (hyaluronidase deficiency) | 0 | 0 | 0 | 0 | 0 |
| Disorder | Score (0–4) | Treatment | |||||||
|---|---|---|---|---|---|---|---|---|---|
| Availability | Outcomes | ||||||||
| An EMA-Approved Therapy Is Available | A Therapeutic Strategy Is Available (Diet, HSCT, BMT) | A Therapy Is in Late Development (Phase 3) | A Therapy Is in Early Development (Preclinical, Phase 1, or Phase 2) | The Therapeutic Strategy Changes the Prognosis for All Forms of the Condition | The Therapeutic Strategy Changes the Prognosis Only for Some Forms of the Condition | The Therapeutic Strategy Does Not Change Prognosis or Improves Only Some Symptoms | Pre-Symptomatic Initiation Results in Better Outcomes | ||
| OR | OR | AND | |||||||
| 1.5 | 1 | 1 | 0.5 | 1.5 | 1 | 0.5 | 1 | ||
| Carnitine uptake defect/carnitine transport defect (CUD) | 4 | 1.5 | 0 | 0 | 0 | 1.5 | 0 | 0 | 1 |
| Severe combined immunodeficiency (SCID) | 3.5 | 1.5 | 0 | 0 | 0 | 0 | 1 | 0 | 1 |
| Glutaric aciduria type 1 (GA1) | 3 | 0 | 1 | 0 | 0 | 0 | 1 | 0 | 1 |
| Homocystinuria (HCU) | 4 | 1.5 | 0 | 0 | 0 | 1.5 | 0 | 0 | 1 |
| Phenylketonuria (PKU) | 4 | 1.5 | 0 | 0 | 0 | 1.5 | 0 | 0 | 1 |
| Tyrosinemia, type 1 (TYR 1) | 4 | 1.5 | 0 | 0 | 0 | 1.5 | 0 | 0 | 1 |
| Classic galactosaemia (GALT) | 3 | 0 | 1 | 0 | 0 | 0 | 1 | 0 | 1 |
| 3-Hydroxy-3-methyglutaric aciduria (HMG) | 3.5 | 0 | 1 | 0 | 0 | 1.5 | 0 | 0 | 1 |
| Pompe disease | 4 | 1.5 | 0 | 0 | 0 | 1.5 | 0 | 0 | 1 |
| X-linked adrenoleukodystrophy (X-ALD) | 3 | 0 | 1 | 0 | 0 | 0 | 1 | 0 | 1 |
| Argininosuccinic aciduria (ASA) | 3 | 1.5 | 0 | 0 | 0 | 1.5 | 0 | 0 | 0 |
| Carnitine palmitoyltransferase, type I deficiency (CPT I) | 3.5 | 0 | 1 | 0 | 0 | 1.5 | 0 | 0 | 1 |
| Long-chain 3 hydroxyacyl-CoA dehydrogenase deficiency (LCHAD) | 2.5 | 0 | 1 | 0 | 0 | 0 | 0 | 0.5 | 1 |
| Methylmalonic acidaemia (cobalamin disorders, Cbl A, B) | 3.5 | 0 | 1 | 0 | 0 | 1.5 | 0 | 0 | 1 |
| Metachromatic leukodystrophy (MLD) | 3.5 | 1.5 | 0 | 0 | 0 | 0 | 1 | 0 | 1 |
| Mucopolysaccharidosis, type I (MPS I) | 3 | 1.5 | 0 | 0 | 0 | 0 | 0 | 0.5 | 1 |
| Propionic acidaemia (PROP) | 4 | 1.5 | 0 | 0 | 0 | 1.5 | 0 | 0 | 1 |
| Biotinidase deficiency (BIOT) | 3.5 | 0 | 1 | 0 | 0 | 1.5 | 0 | 0 | 1 |
| Medium-chain acyl-CoA dehydrogenase deficiency (MCADD) | 2.5 | 0 | 1 | 0 | 0 | 1.5 | 0 | 0 | 0 |
| 3-Methylcrotonyl-CoA carboxylase deficiency (3MCC) | 2.5 | 0 | 1 | 0 | 0 | 1.5 | 0 | 0 | 0 |
| Citrullinemia, type I (CIT) | 4 | 1.5 | 0 | 0 | 0 | 1.5 | 0 | 0 | 1 |
| Holocarboxylase synthetase deficiency (MCD) | 3.5 | 0 | 1 | 0 | 0 | 1.5 | 0 | 0 | 1 |
| Krabbe disease | 2.5 | 0 | 1 | 0 | 0 | 0 | 0 | 0.5 | 1 |
| Argininaemia (ARG) | 3.5 | 0 | 1 | 0 | 0 | 1.5 | 0 | 0 | 1 |
| Carnitine acylcarnitine translocase deficiency (CACT) | 3.5 | 0 | 1 | 0 | 0 | 1.5 | 0 | 0 | 1 |
| Very long-chain acyl-CoA dehydrogenase deficiency (VLCAD) | 2 | 0 | 1 | 0 | 0 | 0 | 1 | 0 | 0 |
| Isovaleric acidaemia (IVA) | 3 | 0 | 1 | 0 | 0 | 0 | 1 | 0 | 1 |
| Maple syrup urine disease (MSUD) | 3.5 | 0 | 1 | 0 | 0 | 1.5 | 0 | 0 | 1 |
| Methylmalonic acidaemia (methylmalonyl-CoA mutase) (MUT) | 3 | 0 | 1 | 0 | 0 | 0 | 1 | 0 | 1 |
| Carnitine palmitoyltransferase, type II deficiency (CPT II) | 3.5 | 0 | 1 | 0 | 0 | 1.5 | 0 | 0 | 1 |
| Batten disease (CLN2) | 3 | 1.5 | 0 | 0 | 0 | 1.5 | 0 | 0 | 0 |
| Niemann Pick A/B (ASM deficiency) | 1.5 | 0 | 0 | 1 | 0 | 0 | 0 | 0.5 | 0 |
| Trifunctional protein deficiency (TFP) | 3 | 0 | 1 | 0 | 0 | 0 | 1 | 0 | 1 |
| Gaucher disease | 2 | 1.5 | 0 | 0 | 0 | 0 | 0 | 0.5 | 0 |
| Lysosomal acid lipase deficiency (LAL-D/Wolman/CESD) | 2.5 | 1.5 | 0 | 0 | 0 | 0 | 1 | 0 | 0 |
| Multiple acyl-CoA dehydrogenase deficiency (MADD) | 3 | 0 | 1 | 0 | 0 | 0 | 1 | 0 | 1 |
| MPS VI (Maroteaux-Lamy syndrome) | 3 | 1.5 | 0 | 0 | 0 | 0 | 0 | 0.5 | 1 |
| Alpha-mannosidosis | 3 | 1.5 | 0 | 0 | 0 | 0 | 0 | 0.5 | 1 |
| Fabry disease | 2.5 | 1.5 | 0 | 0 | 0 | 0 | 1 | 0 | 0 |
| MPS II (Hunter syndrome) | 3 | 1.5 | 0 | 0 | 0 | 0 | 0 | 0.5 | 1 |
| MPS III (Sanfilippo syndrome) | 1 | 0 | 0 | 0 | 0.5 | 0 | 0 | 0.5 | 0 |
| Niemann-Pick type C disease | 2 | 1.5 | 0 | 0 | 0 | 0 | 0 | 0.5 | 0 |
| MPS IV (Morquio syndrome) | 2 | 1.5 | 0 | 0 | 0 | 0 | 0 | 0.5 | 0 |
| Sandhoff disease (GM2 gangliosidosis, type II) | 1 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 |
| Farber disease | 2 | 0 | 1 | 0 | 0 | 0 | 1 | 0 | 0 |
| Tay-Sachs disease (GM2 gangliosidosis, type I) | 1 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 |
| MPS VII (Sly syndrome) | 2 | 1.5 | 0 | 0 | 0 | 0 | 0 | 0.5 | 0 |
| MPS IX (hyaluronidase deficiency) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
References
- Ferreira, C.R.; Rahman, S.; Keller, M.; Zschocke, J.; ICIMD Advisory Group. An International Classification of Inherited Metabolic Disorders (ICIMD). J. Inherit. Metab. Dis. 2021, 44, 164–177. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, C.R.; van Karnebeek, C.D.M.; Vockley, J.; Blau, N. A Proposed Nosology of Inborn Errors of Metabolism. Genet. Med. 2019, 21, 102–106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- George, R.S.; Moat, S.J. Effect of Dried Blood Spot Quality on Newborn Screening Analyte Concentrations and Recommendations for Minimum Acceptance Criteria for Sample Analysis. Clin. Chem. 2016, 62, 466–475. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Loeber, J.G.; Platis, D.; Zetterström, R.H.; Almashanu, S.; Boemer, F.; Bonham, J.R.; Borde, P.; Brincat, I.; Cheillan, D.; Dekkers, E.; et al. Neonatal Screening in Europe Revisited: An ISNS Perspective on the Current State and Developments Since 2010. Int. J. Neonatal Screen. 2021, 7, 15. [Google Scholar] [CrossRef] [PubMed]
- la Marca, G. La organización del cribado neonatal en Italia: Comparación con Europa y el resto del mundo. Rev. Esp. Salud Pública 2021, 95, 26. [Google Scholar]
- Cornel, M.; Rigter, T.; Weinreich, S.; Burgard, P.; Hoffmann, G.F.; Lindner, M.; Loeber, J.G.; Rupp, K.; Taruscio, D.; Vittozzi, L. Evaluation of Population Newborn Screening Practices for Rare Disorders in Member States of the European Union. Newborn Screening in Europe; Expert Opinion Document. EU Network of Experts on Newborn Screening. 2011. Available online: https://isns-neoscreening.org/wp-content/uploads/2016/06/Expert-opinion-document-on-NBS-FINAL.pdf (accessed on 21 June 2021).
- Castiñeras, D.E.; Couce, M.-L.; Marin, J.L.; González-Lamuño, D.; Rocha, H. Newborn screening for metabolic disorders in Spain and worldwide. An. Pediatr. 2019, 91, 128.e1–128.e14. [Google Scholar] [CrossRef]
- Burlina, A.; Jones, S.A.; Chakrapani, A.; Church, H.J.; Heales, S.; Wu, T.H.Y.; Morton, G.; Roberts, P.; Sluys, E.F.; Cheillan, D. A new approach to objectively evaluate inherited metabolic diseases for inclusion on newborn screening programmes. Int. J. Neonatal Screen. 2022, 8, 25. [Google Scholar]
- Wilson, J.M.; Jungner, Y.G. Principles and practice of mass screening for disease. Bol. Oficina Sanit. Panam. 1968, 65, 281–393. [Google Scholar]
- Watson, M.S.; Mann, M.Y.; Lloyd-Puryear, M.A.; Rinaldo, P.; Howell, R.R. Newborn Screening: Toward a Uniform Screening Panel and System. Genet Med. 2006, 8 (Suppl. 1), 1S–252S. [Google Scholar] [CrossRef] [Green Version]
- OECD iLibrary. Available online: https://0-www-oecd--ilibrary-org.brum.beds.ac.uk/ (accessed on 1 July 2021).
- Loeber, J.G. Neonatal Screening in Europe; the Situation in 2004. J. Inherit. Metab. Dis. 2007, 30, 430–438. [Google Scholar] [CrossRef]
- Loeber, J.G.; Burgard, P.; Cornel, M.C.; Rigter, T.; Weinreich, S.S.; Rupp, K.; Hoffmann, G.F.; Vittozzi, L. Newborn Screening Programmes in Europe; Arguments and Efforts Regarding Harmonization. Part 1. From Blood Spot to Screening Result. J. Inherit. Metab. Dis. 2012, 35, 603–611. [Google Scholar] [CrossRef]
- United Kingdom National Screening Committee. Screening in the UK: Making Effective Recommendations 1 April 2018 to 31 March 2019. Page Last Updated on 30 July 2021. Available online: https://www.gov.uk/government/publications/uk-national-screening-committee-recommendations-annual-report/screening-in-the-uk-making-effective-recommendations-1-april-2018-to-31-march-2019 (accessed on 4 September 2021).
- Sonnet Study. Heel Nederland Screent Straks Op SCID! The Whole of The Netherlands Will Soon Be Screening for SCID! Available online: http://sonnetstudie.nl/heel-nederland-screent-straks-op-scid (accessed on 5 January 2021).
- Burlina, A.B.; Polo, G.; Salviati, L.; Duro, G.; Zizzo, C.; Dardis, A.; Bembi, B.; Cazzorla, C.; Rubert, L.; Zordan, R.; et al. Newborn Screening for Lysosomal Storage Disorders by Tandem Mass Spectrometry in North East Italy. J. Inherit. Metab. Dis. 2018, 41, 209–219. [Google Scholar] [CrossRef] [PubMed]
- Martínez-Morillo, E.; Prieto García, B.; Álvarez Menéndez, F.V. Challenges for Worldwide Harmonization of Newborn Screening Programs. Clin. Chem. 2016, 62, 689–698. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- National Institute of Neurological Disorders and Stroke. Batten Disease Fact Sheet. Page Last Updated on 13 March 2020. Available online: https://www.ninds.nih.gov/Disorders/Patient-Caregiver-Education/Fact-Sheets/Batten-Disease-Fact-Sheet (accessed on 1 July 2021).
- Almannai, M.; Alfadhel, M.; El-Hattab, A.W. Carnitine Inborn Errors of Metabolism. Molecules 2019, 24, 3251. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zielonka, M.; Kölker, S.; Gleich, F.; Stützenberger, N.; Nagamani, S.C.S.; Gropman, A.L.; Hoffmann, G.F.; Garbade, S.F.; Posset, R.; Urea Cycle Disorders Consortium (UCDC) and the European Registry and Network for Intoxication type Metabolic Diseases (E-IMD) Consortia Study Group. Early Prediction of Phenotypic Severity in Citrullinemia Type 1. Ann. Clin. Transl. Neurol. 2019, 6, 1858–1871. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wasim, M.; Awan, F.R.; Khan, H.N.; Tawab, A.; Iqbal, M.; Ayesha, H. Aminoacidopathies: Prevalence, Etiology, Screening, and Treatment Options. Biochem. Genet. 2018, 56, 7–21. [Google Scholar] [CrossRef] [PubMed]
- Bessey, A.; Chilcott, J.; Pandor, A.; Paisley, S. The Cost-Effectiveness of Expanding the UK Newborn Bloodspot Screening Programme to Include Five Additional Inborn Errors of Metabolism. Int. J. Neonatal Screen. 2020, 6, 93. [Google Scholar] [CrossRef] [PubMed]
- Therrell, B.L.; Padilla, C.D.; Loeber, J.G.; Kneisser, I.; Saadallah, A.; Borrajo, G.J.C.; Adams, J. Current Status of Newborn Screening Worldwide: 2015. Semin. Perinatol. 2015, 39, 171–187. [Google Scholar] [CrossRef] [Green Version]
- Merritt, J.L.; Norris, M.; Kanungo, S. Fatty Acid Oxidation Disorders. Ann. Transl. Med. 2018, 6, 473. [Google Scholar] [CrossRef]
- Almási, T.; Guey, L.T.; Lukacs, C.; Csetneki, K.; Vokó, Z.; Zelei, T. Systematic Literature Review and Meta-Analysis on the Epidemiology of Methylmalonic Acidemia (MMA) with a Focus on MMA Caused by Methylmalonyl-CoA Mutase (Mut) Deficiency. Orphanet J. Rare Dis. 2019, 14, 84. [Google Scholar] [CrossRef]
- Zelei, T.; Csetneki, K.; Vokó, Z.; Siffel, C. Epidemiology of Sanfilippo Syndrome: Results of a Systematic Literature Review. Orphanet J. Rare Dis. 2018, 13, 53. [Google Scholar] [CrossRef] [Green Version]
- Khan, S.A.; Peracha, H.; Ballhausen, D.; Wiesbauer, A.; Rohrbach, M.; Gautschi, M.; Mason, R.W.; Giugliani, R.; Suzuki, Y.; Orii, K.E.; et al. Epidemiology of Mucopolysaccharidoses. Mol. Genet. Metab. 2017, 121, 227–240. [Google Scholar] [CrossRef] [PubMed]
- Imundo, L.; Leduc, C.A.; Guha, S.; Brown, M.; Perino, G.; Gushulak, L.; Triggs-Raine, B.; Chung, W.K. A Complete Deficiency of Hyaluronoglucosaminidase 1 (HYAL1) Presenting as Familial Juvenile Idiopathic Arthritis. J. Inherit. Metab. Dis. 2011, 34, 1013–1022. [Google Scholar] [CrossRef] [PubMed]
- Valayannopoulos, V.; Nicely, H.; Harmatz, P.; Turbeville, S. Mucopolysaccharidosis VI. Orphanet J. Rare Dis. 2010, 5, 5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Almási, T.; Guey, L.T.; Lukacs, C.; Csetneki, K.; Vokó, Z.; Zelei, T. Systematic Literature Review and Meta-Analysis on the Epidemiology of Propionic Acidemia. Orphanet J. Rare Dis. 2019, 14, 40. [Google Scholar] [CrossRef] [PubMed]
- EMA. EU/3/15/1508: Orphan Designation for the Treatment of Very Long-Chain Acyl-CoA Dehydrogenase Deficiency. Available online: https://www.ema.europa.eu/en/medicines/human/orphan-designations/eu3151508 (accessed on 1 July 2021).
- Turk, B.R.; Theda, C.; Fatemi, A.; Moser, A.B. X-Linked Adrenoleukodystrophy: Pathology, Pathophysiology, Diagnostic Testing, Newborn Screening and Therapies. Int. J. Dev. Neurosci. 2020, 80, 52–72. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferreira, C.R.; Gahl, W.A. Lysosomal Storage Diseases. Transl. Sci. Rare Dis. 2017, 2, 1–71. [Google Scholar] [CrossRef] [Green Version]

{kind=link}
{kind=link}

| Disorder | Score (0–13) | Condition | Screening | Treatment | ||||
|---|---|---|---|---|---|---|---|---|
| Severity | Onset | Frequency | Availability | Performance | Availability | Outcomes | ||
| Carnitine uptake defect/carnitine transport defect (CUD) | 12.5 | 1.5 | 2 | 2 | 2 | 1 | 1.5 | 2.5 |
| Severe combined immunodeficiency (SCID) | 12 | 2 | 2 | 2 | 2 | 0.5 | 1.5 | 2 |
| Glutaric aciduria type 1 (GA1) | 11.5 | 2 | 2 | 1.5 | 2 | 1 | 1 | 2 |
| Homocystinuria (HCU) | 11.5 | 1.5 | 2 | 1 | 2 | 1 | 1.5 | 2.5 |
| Phenylketonuria (PKU) | 11.5 | 0.5 | 2 | 2 | 2 | 1 | 1.5 | 2.5 |
| Tyrosinemia, type 1 (TYR 1) | 11.5 | 1.5 | 2 | 1.5 | 2 | 0.5 | 1.5 | 2.5 |
| Classic galactosaemia (GALT) | 11 | 2 | 1 | 2 | 2 | 1 | 1 | 2 |
| 3-Hydroxy-3-methyglutaric aciduria (HMG) | 11 | 1.5 | 2 | 1 | 2 | 1 | 1 | 2.5 |
| Pompe disease | 11 | 1.5 | 1 | 2 | 2 | 0.5 | 1.5 | 2.5 |
| X-linked adrenoleukodystrophy (X-ALD) | 10.5 | 1.5 | 1 | 2 | 2 | 1 | 1 | 2 |
| Argininosuccinic aciduria (ASA) | 10.5 | 2 | 1 | 1.5 | 2 | 1 | 1.5 | 1.5 |
| Carnitine palmitoyltransferase, type I deficiency (CPT I) | 10.5 | 2 | 2 | 0 | 2 | 1 | 1 | 2.5 |
| Long-chain 3 hydroxyacyl-CoA dehydrogenase deficiency (LCHAD) | 10.5 | 2 | 2 | 1 | 2 | 1 | 1 | 1.5 |
| Methylmalonic acidaemia (cobalamin disorders, Cbl A, B) | 10.5 | 2 | 2 | 0 | 2 | 1 | 1 | 2.5 |
| Metachromatic leukodystrophy (MLD) | 10.5 | 2 | 2 | 1.5 | 1 | 0.5 | 1.5 | 2 |
| Mucopolysaccharidosis, type I (MPS I) | 10.5 | 1.5 | 2 | 1.5 | 2 | 0.5 | 1.5 | 1.5 |
| Propionic acidaemia (PROP) | 10.5 | 2 | 1 | 0.5 | 2 | 1 | 1.5 | 2.5 |
| Biotinidase deficiency (BIOT) | 10.5 | 2 | 1 | 1.5 | 2 | 0.5 | 1 | 2.5 |
| Medium-chain acyl-CoA dehydrogenase deficiency (MCADD) | 10 | 1.5 | 1 | 2 | 2 | 1 | 1 | 1.5 |
| 3-Methylcrotonyl-CoA carboxylase deficiency (3MCC) | 10 | 1.5 | 1 | 2 | 2 | 1 | 1 | 1.5 |
| Citrullinemia, type I (CIT) | 10 | 1.5 | 1 | 0.5 | 2 | 1 | 1.5 | 2.5 |
| Holocarboxylase synthetase deficiency (MCD) | 10 | 2 | 1 | 0.5 | 2 | 1 | 1 | 2.5 |
| Krabbe disease | 10 | 1.5 | 2 | 1.5 | 2 | 0.5 | 1 | 1.5 |
| Argininaemia (ARG) | 9.5 | 1.5 | 2 | 0 | 2 | 0.5 | 1 | 2.5 |
| Carnitine acylcarnitine translocase deficiency (CACT) | 9.5 | 2 | 1 | 0 | 2 | 1 | 1 | 2.5 |
| Very long-chain acyl-CoA dehydrogenase deficiency (VLCAD) | 9.5 | 1.5 | 1 | 2 | 2 | 1 | 1 | 1 |
| Maple syrup urine disease (MSUD) | 9 | 1.5 | 0 | 1 | 2 | 1 | 1 | 2.5 |
| Methylmalonic acidaemia (methylmalonyl-CoA mutase) (MUT) | 9 | 1.5 | 1 | 0.5 | 2 | 1 | 1 | 2 |
| Carnitine palmitoyltransferase, type II deficiency (CPT II) | 9 | 1.5 | 1 | 0 | 2 | 1 | 1 | 2.5 |
| Batten disease (CLN2) | 9 | 2 | 2 | 0 | 1 | 1 | 1.5 | 1.5 |
| Niemann Pick A/B (ASM deficiency) | 9 | 2 | 2 | 0.5 | 2 | 1 | 1 | 0.5 |
| Isovaleric acidaemia (IVA) | 8.5 | 1.5 | 0 | 1 | 2 | 1 | 1 | 2 |
| Trifunctional protein deficiency (TFP) | 8.5 | 1.5 | 1 | 0 | 2 | 1 | 1 | 2 |
| Gaucher disease | 8.5 | 1.5 | 1 | 1.5 | 2 | 0.5 | 1.5 | 0.5 |
| Lysosomal acid lipase deficiency (LAL-D/Wolman/CESD) | 8.5 | 1.5 | 1 | 0.5 | 2 | 1 | 1.5 | 1 |
| Multiple acyl-CoA dehydrogenase deficiency (MADD) | 8 | 1.5 | 0 | 0.5 | 2 | 1 | 1 | 2 |
| MPS VI (Maroteaux-Lamy syndrome) | 8 | 1.5 | 2 | 0 | 1 | 0.5 | 1.5 | 1.5 |
| Alpha-mannosidosis | 7.5 | 1.5 | 1 | 0 | 1 | 1 | 1.5 | 1.5 |
| Fabry disease | 7.5 | 0 | 1 | 1.5 | 2 | 0.5 | 1.5 | 1 |
| MPS II (Hunter syndrome) | 7 | 0 | 2 | 0.5 | 1 | 0.5 | 1.5 | 1.5 |
| MPS III (Sanfilippo syndrome) | 6.5 | 0 | 2 | 1.5 | 1 | 1 | 0.5 | 0.5 |
| Niemann-Pick type C disease | 6.5 | 1.5 | 1 | 1 | 1 | 0 | 1.5 | 0.5 |
| MPS IV (Morquio syndrome) | 5.5 | 0 | 2 | 0 | 1 | 0.5 | 1.5 | 0.5 |
| Sandhoff disease (GM2 gangliosidosis, type II) | 5.5 | 1.5 | 2 | 1 | 0 | 0 | 1 | 0 |
| Farber disease | 5 | 2 | 1 | 0 | 0 | 0 | 1 | 1 |
| Tay-Sachs disease (GM2 gangliosidosis, type I) | 4.5 | 1.5 | 2 | 0 | 0 | 0 | 1 | 0 |
| MPS VII (Sly syndrome) | 3.5 | 1.5 | 0 | 0 | 0 | 0 | 1.5 | 0.5 |
| MPS IX (hyaluronidase deficiency) | 1 | 0 | 1 | 0 | 0 | 0 | 0 | 0 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jones, S.A.; Cheillan, D.; Chakrapani, A.; Church, H.J.; Heales, S.; Wu, T.H.Y.; Morton, G.; Roberts, P.; Sluys, E.F.; Burlina, A. Application of a Novel Algorithm for Expanding Newborn Screening for Inherited Metabolic Disorders across Europe. Int. J. Neonatal Screen. 2022, 8, 20. https://0-doi-org.brum.beds.ac.uk/10.3390/ijns8010020
Jones SA, Cheillan D, Chakrapani A, Church HJ, Heales S, Wu THY, Morton G, Roberts P, Sluys EF, Burlina A. Application of a Novel Algorithm for Expanding Newborn Screening for Inherited Metabolic Disorders across Europe. International Journal of Neonatal Screening. 2022; 8(1):20. https://0-doi-org.brum.beds.ac.uk/10.3390/ijns8010020
Chicago/Turabian StyleJones, Simon A., David Cheillan, Anupam Chakrapani, Heather J. Church, Simon Heales, Teresa H. Y. Wu, Georgina Morton, Patricia Roberts, Erica F. Sluys, and Alberto Burlina. 2022. "Application of a Novel Algorithm for Expanding Newborn Screening for Inherited Metabolic Disorders across Europe" International Journal of Neonatal Screening 8, no. 1: 20. https://0-doi-org.brum.beds.ac.uk/10.3390/ijns8010020