
Bidimensional Polyiodide Netting Stabilized by a Cu(II) Macrocyclic Complex


Department of Chemistry “Ugo Schiff”, University of Florence, Via della Lastruccia 3, 50019 Sesto Fiorentino, Italy
*
Authors to whom correspondence should be addressed.
Inorganics 2022, 10(1), 12; https://0-doi-org.brum.beds.ac.uk/10.3390/inorganics10010012
Submission received: 14 December 2021
/
Revised: 11 January 2022
/
Accepted: 11 January 2022
/
Published: 13 January 2022
(This article belongs to the Section Coordination Chemistry)
Abstract
:Iodine-dense polyiodide phases are interesting materials for a number of potential uses, including batteries and solid-state conductors. The incorporation of transition metal cations is considered a promising way to enhance the stability, tune the properties, and influence the architecture of polyiodides. However, several interesting metals, including Cu(II), may suffer redox processes, which generally make them not compatible with the I2/I− redox couple. Herein L, a simple derivative of cyclen, is proposed as a Cu(II) ligand capable of protecting the +2 oxidation state of the metal even in the presence of polyiodides. With a step by step approach, we report the crystal structure of free L; then we present spectrophotometric verification of Cu(II) complex stability, stoichiometry, and formation kinetic in DMF solution, together with Cu(II) binding mode elucidation via XRD analysis of [Cu(L)Cl]ClO4∙CH3CN crystals; afterwards, the stability of the CuL complex in the presence of I− is demonstrated in DMF solution, where the formation of a Cu:L:I− ternary complex, rather than reduction to Cu(I), is observed; lastly, polyiodide crystals are prepared, affording the [Cu(L)I]2I3I5 crystal structure. This layered structure is highly peculiar due to its chiral arrangement, opening further perspective for the crystal engineering of polyiodide phases.
1. Introduction
Polyiodide chemistry is a long-lasting research area. In terms of scientific interest, it has been tied throughout history with inorganic, theoretical, and supramolecular research, while it is well known that iodine and polyiodides were used in applications soon after iodine discovery (iodimetry, iodometry, Lugol solution, etc.) [1,2].
Contemporary application-oriented research on polyiodides focuses on batteries, [3,4] solar cells [5,6], solid state conductors [7,8], and even high-energy, iodine-dispersing agents [9].
Our contribution to the field has been mostly directed towards the supramolecular chemistry of polyiodides, aimed at obtaining ordered solids with high iodide density organized in extended networks. We demonstrated that, with suitable organic ligands, it is possible to obtain crystal phase architectures featuring alternating planes of ligands and polyiodides [10], high density polyiodide-based clathrates self-assembled around suitable ligands [11,12], and even complex architectures such as solid-state pseudopolyrotaxanes with a [3]-catenane axle [13]. The involvement of metal centers in polyiodide stabilization could also be of high interest, both to exploit metal cations as structural elements and to, potentially, improve their thermal stability and tune the electronic properties of the material. Accordingly, we recently proposed Cu(II) complexes of tetraazacyclophanes [14,15], showing how the choice of substituents on the macrocycle could shift packing forces from an H-bond-based arrangement, featuring charge-dense, noninteracting polyiodides, to an I⋯I dominated crystal featuring polyiodide chains periodically decorated with Cu(II) complexes.
It is well known that Cu(II) is prone to reduction to Cu(I) in the presence of I− and that metal ion redox potentials can be tuned by complexation. In the case of the abovementioned pyridinophanes, no formation of CuI has been detected. In view of further developments, we decided to check whether simpler, i.e., alkyl, tetraazamacrocycles could also be used to stabilize Cu(II) in the presence of polyiodides. In this study a simple derivative of cyclen has been employed (Scheme 1). The ligand bears two tosylated ethylenamino arms, which were added in view of the ability of tosyl to stabilize polyiodides [16] and other polyhalides [17]. The possibility to employ simpler, readily available (potentially commercial) ligands to prepare metal-coordinated polyiodides could hasten the development of this chemistry.
2. Results and Discussion
2.1. Crystal Structure of L
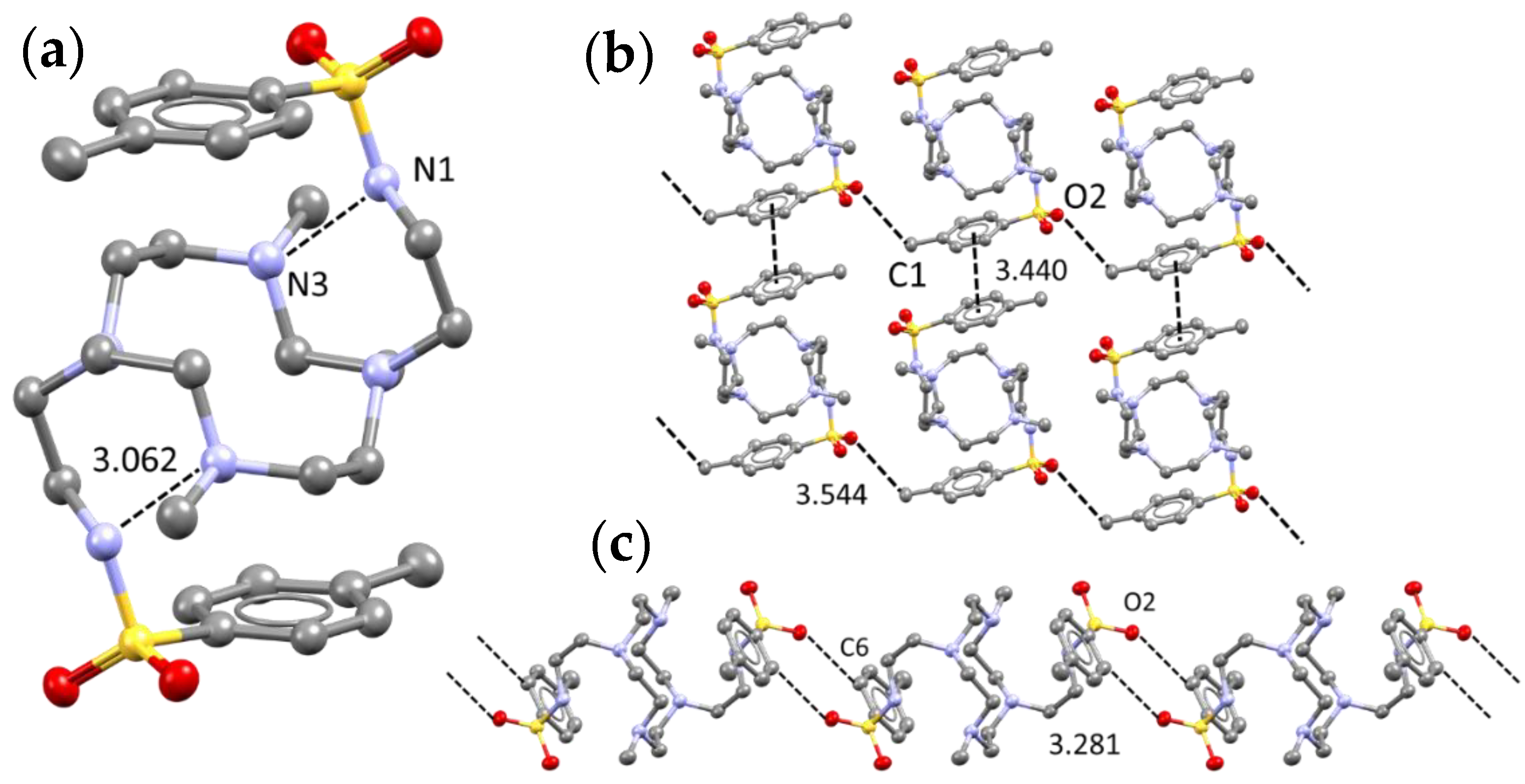
The free ligand adopts a centrosymmetric conformation featuring the two tosyl rings on opposite sides with respect to the macrocycle plane (Figure 1a). Common amide NH–sulfone O=S intermolecular H bonding, a usual feature for sulfonamides (moreover observed in other studies dealing with polyiodide stabilization using similar moieties) [16] are entirely absent here. The reason, and the main driving force for the observed ligand folding, lies in strong N1-N3 intramolecular hydrogen bonding (N1⋯N3 3.062(3) Å). The main intermolecular interaction is a mutual CHortho⋯O=S-CHortho⋯O=S involving O2 and C6 with their symmetry-related neighbors (O2⋯C6 3.281(3) Å) (Figure 1c). Further CH⋯O contacts, namely C1⋯O2 (3.544(5) Å) are also present. Tosyl groups also engage in π-π stacking interactions, being perfectly parallel with their neighbors (as required by crystal symmetry) with a C6 (tosyl)-C6 (tosyl) interplanar distance of 3.440 Å (Figure 1b).
2.2. Cu(II) Complexes: Solution and Solid State
The parent ligand 1 is water soluble and forms strong complexes of 1:1 stoichiometry with Cu(II). The addition of Cu(II) to 1 to afford [Cu(1)]2+ happens in water with log β = 17.89(3) (T = 298 K, I = 0.5 M KNO3) [18]. Accordingly, strong complexes of 1:1 stoichiometry are also expected for L.
However, due to the presence of the tosyl groups, L is only sparingly soluble in water, with the exception of very acidic pH regions (<2.5) and cannot be studied potentiometrically. Although it is expected that Cu(II) binding can be followed by its visible d-d transition, the addition of Cu(II) to acidic ligand solution produces little to no color change, implying excessive proton competition at this pH and/or kinetic sluggishness.
Spectrophotometric studies were therefore performed in DMF, an aprotic solvent which allows L to easily dissolve in adequate quantities (mM concentration range).
By adding Cu(II) to a DMF solution of L, the appearance of a blue/cyan complex is observed immediately. However, as kinetic issues are commonplace with cyclen and its derivatives, a kinetic experiment aimed at establishing a reliable equilibrium time for complex formations under experimental conditions, was performed.
Indeed, despite the complexation kinetic being much faster in DMF (Figure 2), spectral changes over time are observed. Namely, for a 1:1 L:Cu(II) 1.8 mM DMF solution, 1 h is found as an acceptable timeframe to properly achieve equilibrium.
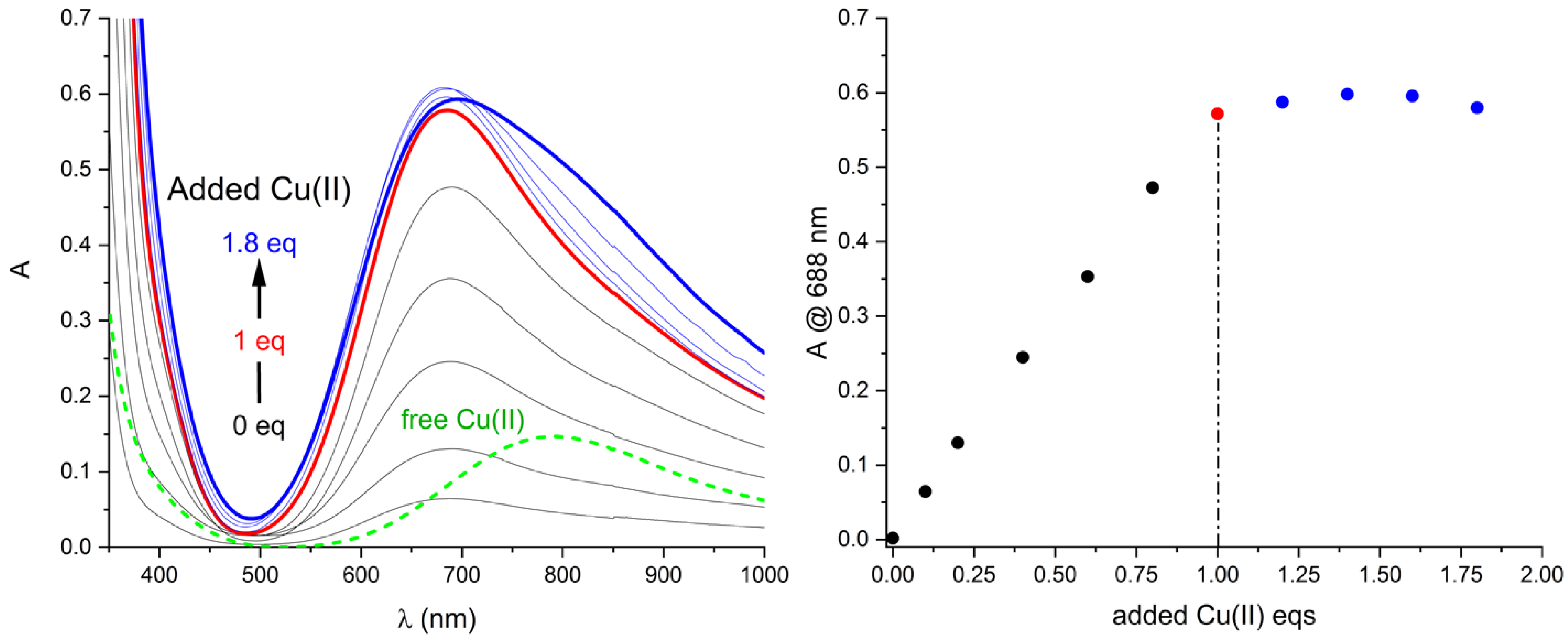
According to these findings, in order to check the complex stoichiometry, 11 solutions spanning 0–1.8 eqs of added Cu(II) to a DMF solution of the ligand ([L] = 1.8 mM) were prepared and left to equilibrate overnight.
Results are shown in Figure 3.
While the ligand does not exhibit any band in the VIS region, the appearance of a VIS band is observed upon the addition of Cu(II) with a maximum at 688 nm. The band is relatively broad, although cyclen and its derivatives are known for cis/trans bis(solvent) equilibria among possible octahedral complexes. The band raises steadily until exactly one equivalent of Cu(II) has been added. Afterwards, further addition of Cu(II) affects the spectrum only slightly and the band profile is observed to contain the free Cu(II) component. This establishes that a strong (i.e., quantitatively formed under experimental conditions) complexe of 1:1 stoichiometry is unambiguously formed.
The Cu:L complex has been isolated in the solid state from acetonitrile solvent (which better allows for a slow solvent evaporation technique in comparison to DMF), crystallizing as [Cu(L)Cl]ClO4∙CH3CN.
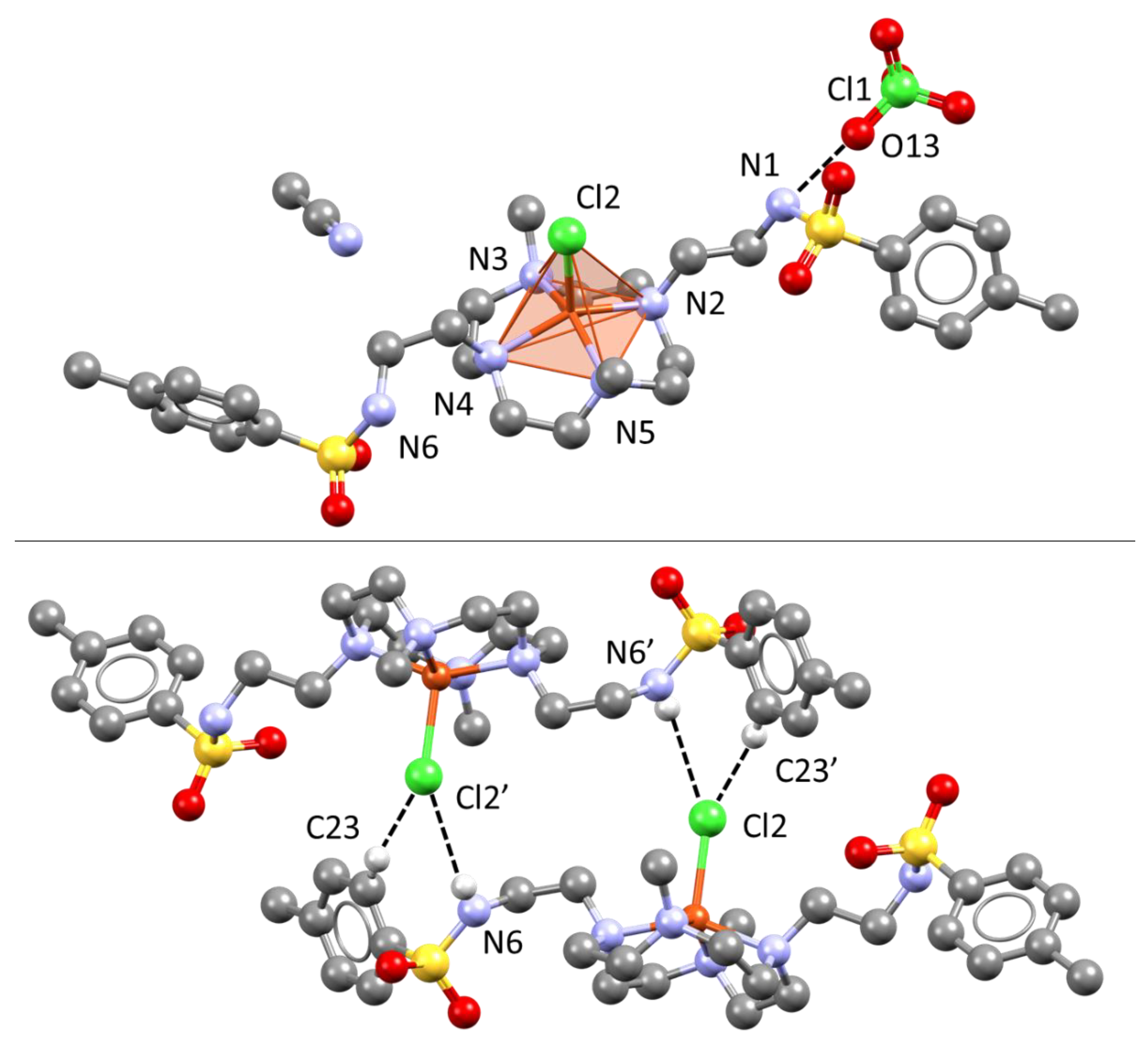
Cu(II) is found in a square pyramidal environment constituted by the 4 N donors of the macrocycle, defining the basal plane, and the chloride anion in the apical position (Figure 4 top). Despite the coordination environment not at all being distorted towards the trigonal bipyramid geometry (τ = 1), all the Cu⋯N distances are found to be different (in the 2.041(4)–2.081(4) Å range, Table 1). Asymmetry of all chelate rings was previously reported also for the parent ligand 1 in its Cu(II) and Ni(II) complexes [18].
Stacking interactions among tosyl moieties are absent in this crystal structure, which seems to be governed by H-bonding/CH⋯anion contacts. For instance, sulfonamide N6H and the C23Hortho form short contacts with coordinated Cl2 (3.319(4) and 3.604(5) Å, respectively), Figure 5 bottom. Cl2 is involved in a third stabilizing contact with HC15 (3.486(6) Å, Figure S1). The second sulfonamide group is engaged in H-bonding to the ClO4− counterion (N1⋯O13 2.976(6) Å), which is found in a pocket, defined by the backs of the macrocycles, where it establishes multiple CH⋯O contacts (Figure S1).
2.3. Solution Stability of [Cu(L)]2+ Complex in the Presence of Iodide
Iodide is known to react with Cu(II) according to the redox equilibrium:
which generally happens with the involvement of an I-CuII-I intermediate.
2Cu2+ + 4I− → 2CuI + I2,
When active, this process is easily detected as discoloration of Cu(II) solutions is observed, often accompanied (in most solvents) by CuI precipitation.
Stability of the [Cu(L)]2+ complex in the presence of I− was checked in the DMF solution by adding equivalents of I− to the preformed 1:1 Cu:L complex. L and Cu concentrations were: [L] = 1.8 mM, [Cu] = 1.44 mM. The complex stability was assessed in a 0.8:1 M:L ratio, and not 1:1, to ensure no potentially interfering free Cu(II) exists in the medium (complexation appears quantitative, as detailed above).
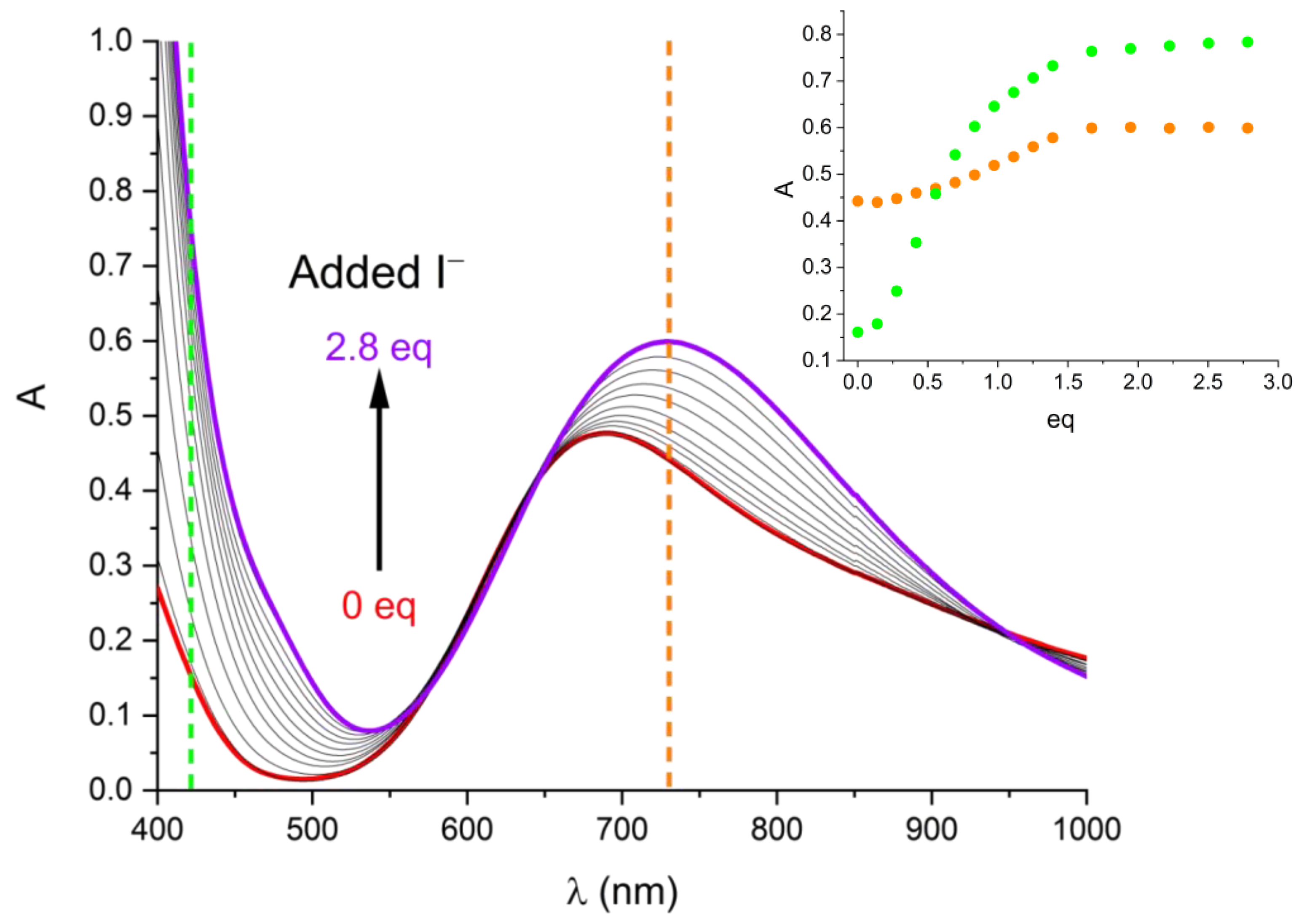
Instead of discoloration, a diagnostic of Cu(I) formation, the intensity of the absorption band was observed to increase and its maximum to shift towards higher wavelengths (from 688 to 730 nm), with the tail of the UV band (λ < 550 nm) also deeply affected, as shown in Figure 5. The emergence of an isobestic point and characteristic sigmoidal profiles of spectral changes upon addition of I− equivalents (Figure 5 inset) demonstrate that an equilibrium is at work. The observed reaction is [Cu(L)]2+ + I− = [Cu(L)I]+, where the entering iodide is supposed to occupy the axial position on Cu(II) as observed for Cl− in the solid state. An apparent binding constant (I varies during the titration) for this reaction could be calculated by fitting experimental data with the Hypspec [19] software. A logKeff = 4.03(1) was determined.
Data indicates not only that [Cu(L)]2+ is redox stable in the presence of I−, but also that it readily binds to I−, giving a [Cu(L)I]+ tecton of appreciable stability. This is in line with what was observed before with pyridine-bearing azacyclophanes [14,15] and, hence, allows for the usage of [Cu(L)I]+ as a reliable building block for more complex polyiodides.
2.4. Crystal Structure of [Cu(L)I]2I3I5
The crystal structure is constituted by two nonequivalent [Cu(L)I]+ complexes with I3− and I5− counterions: Figure 6 displays a view of the asymmetric unit content.
Both Cu(II) centers are found in coordination environments almost completely described as square pyramids (Cu1 τ = 0.03, Cu2 τ = 0.02), each defined by the 4 N donors of a macrocycle plus an apical iodide ligand. The coordination environment closely resembles the one found in the [Cu(L)Cl]ClO4∙CH3CN crystal structure (cf. Table 2 for relevant bond lengths). This is indeed the [Cu(L)I]+ synthon anticipated by solution studies.
Interestingly, despite this compound crystallizing in a chiral space group, the two nonequivalent complexes appear to be enantiomorphic, their overlaid conformations related by a pseudo mirror plane (Figure 7a). They repeat perpendicularly to the c-axis, defining the planar array shown in Figure 7b, the strongest contacts being NH⋯O=S and CH⋯O=S hydrogen bonds (N7⋯O3 2.94(2), O7⋯N1 3.01(2) Å, C19⋯O8 3.52(2) Å, respectively), and CH⋯π contacts (C46⋯C2-C7 ring centroid 3.41 Å). Interplanar NH⋯O=S H-bonds (N6⋯O5 2.94(2) Å and N12⋯O2 2.93(2) Å, Figure 7c,d) then join couples of planar arrays which face their macrocyclic polar sides.
The H-bond network generated by sulfonamide groups interacting among themselves (Figure 6) and saturating NH and S=O groups fosters polyiodide development between layers of complexes’ planar arrays (Figure 8). A similar arrangement in the segregated layers was previously observed with flat s-tetrazine ligands [10].
Despite the interesting, layered arrangement, iodine density of the solid is not remarkably high (IN = 0.211); this is partly due to the large size of the ligand and to the extent of I⋯I interactions.
The coordinated iodide, previously found to be a key element involved in Cu(II) complexes into polyiodide superstructures (rings, chains, etc.) [14,15], was not involved in superior polyiodides or I⋯I contacts. In this sense, it seems that the incorporation of aromatic sulfonamides, previously exploited as their S=O⋯HN H-bonding, offers the opportunity to panel polyiodides [16], and in this case, reduced the possibility of the coordinated iodide to participate in supramolecular architectures. In fact, such H-bonds generate a network leaving no space around the coordinated I− for its further development in superior polyiodides (Figure S2).
While I3− units are not in contact with other polyiodides (shortest I⋯I contact 4.171(2) Å), I5− anions are all mutually interacting through the I6⋯I9′ short contact (3.691(2) Å) linking the terminal I2 units of one pentaiodide with the central one, formally charge bearing, of its neighbor. Such interactions can be considered sigma-hole/halogen bond type contacts and cause little distortion of I5− anions (Figure 9).
Accordingly, while the global polyiodide arrangement of the polyiodide network is layered, in the present case intralayer I-I interactions amount to a single I⋯I contact per pentaiodide, reducing the possibility of tighter packing and overall iodine density.
Perhaps one of the most interesting features of the [Cu(L)I]2I3I5 is the fact that it crystallizes in the P212121 chiral space group. A 2005 survey found that the P212121 space group is among the most common space groups for chiral molecules (representing ≈ 34% of all chiral organic molecules with chiral centers); conversely, crystallization of molecules not bearing a chiral center in the P212121 space group is much rarer (7.8% of achiral molecules) [20]. It has been documented, therein, that rigid molecules (defined as units with less than three consecutive rotatable acyclic single bonds) are more likely to crystallize in chiral space groups. While [Cu(L)I]+ possesses a flexible portion (the ethylenimine bridge, three consecutive rotatable bonds) it is mainly composed of three conformationally rigid domains (the macrocycle plus the two tosyl rings), rotation of sulfonamide N-S bond also being notoriously hindered [21]. The loss of the mirror plane/inversion center (as in the crystal structure of free L) due to Cu(II) coordination might also play a role in space group choice. Although it could not be anticipated and happened serendipitously, the idea of conferring chirality to the polyiodide-dense network in view of the peculiar properties exhibited by these phases could be worthy of further, and this time intended, exploration.
3. Materials and Methods
3.1. Synthesis of L
L is a common intermediate for further decoration of the parent amine 1 [22,23]. It was synthesized as summarized in Scheme 1 starting from commercial materials and following literature procedures. Prior to recrystallization, a minor impurity was found. Since it crystallized independently as long thin colorless needles, it was possible to elucidate its nature by XRD (1,4-ditosylpiperazine), although we only managed to get preliminary data due to poor crystal quality (cf. dedicated section of Supporting Materials Figure S3). Afterwards, recrystallization of the synthetic batch from acetone:ethanol 1:4 was performed, affording high purity L in 69.7% yield.
1H-NMR (CDCl3): δ = 1.99 (s, 6H), 2.37 (s, 6H), 2.44–2.92 (m, 24H), 7.26 (d, 4H), 7.77 (d, 4H). Anal. calcd. (%) for C28H46N6O4S2: C, 56.54; H, 7.79; N, 14.13; Found: C, 56.62; H 7.73; N, 14.09.
3.2. UV-Vis and NMR Spectroscopy
UV-Vis spectra were run in HPLC grade DMF dried on molecular sieves. The following experiments were conducted: (1) check of Cu(II) binding kinetic to L (1:1 M:L ratio, [L] = 1.8 mM); (2) check of Cu:L complex stoichiometry (Cu(II) equivalents, as CuSO4 DMF stock solution, added to L, [L] = 1.8 mM); (3) addition of KI equivalents (as KI DMF stock solution) to preformed CuL complex.
All spectra were recorded on a Jasco V670 spectrophotometer (Jasco Europe, Lecco, Italy) at 298 K.
Determination of (apparent) logK constants from spectroscopic data has been performed with the Hypspec software (Hyperquad suite) [19].
1H NMR spectra were recorded at 25 °C on a Bruker AV400 spectrometer (Bruker Italia Srl, Milano, Italy).
3.3. Crystals Preparation
Crystals of L are easily prepared by acetone:ethanol crystallization as specified in the synthesis of L section.
Crystals of [Cu(L)Cl]ClO4.CH3CN were prepared by adding NaClO4 to an acetonitrile solution of CuCl2 and L in 1:1 ratio. Upon solvent evaporation, deep blue crystals were obtained. Anal. calcd. (%) for C30H49N7O8S2CuCl2: C, 43.19; H, 5.92; N, 11.75; Found: C, 43.04; H, 5.88; N, 11.69.
Crystals of [Cu(L)I]2I3I5 were prepared by diffusion, inside an H-shaped tube, of a 1:1 CuCl2:L solution towards an I−/I2 1:2 mixture, using water as the solvent. Dark brown crystals were obtained upon diffusion over several weeks. Anal. calcd. (%) for C28H46N6O4S2CuI5: C, 26.01; H, 3.59; N, 6.50; Found: C, 25.95; H, 3.62; N, 6.56.
3.4. XRD Data Collection and Structure Refinement
Data collections were performed in cryogenic conditions (T = 100(2) K). Absorption correction was performed by SADABS-2016/2 [24]. The structures were solved by direct methods (SHELXLS) [25]. Anisotropic treatment for all non-H atoms. The coordinates and thermal factor for amidic hydrogens were freely refined. The refinements were performed by means of full-matrix least-squares using SHELXL Version 2014/7 [26].
3.4.1. Crystal Data for L
C28H46N6O4S2 (M = 594.83 g/mol): triclinic, space group P-1 (no. 2), a = 8.845(1)Å, b = 9.096(1) Å, c = 10.816(2) Å, α = 74.748(5)°, β = 81.762(5)°, γ = 62.824(4)°, V = 746.6(2) Å3, Z = 1, μ(CuKα) = 1.975 mm−1, Dcalc = 1.323 g/cm3, 13,945 reflections were measured (4.238° ≤ Θ ≤ 68.365°) and 2636 were unique (Rint = 0.0656), which were used in all calculations. The final R1 was 0.0712 (I > 2σ(I)), and wR2 was 0.2091 (all data).
3.4.2. Crystal Data for [Cu(L)Cl]ClO4∙CH3CN
C30H49Cl2CuN7O8S2 (M = 834.32 g/mol): triclinic, space group P-1 (no. 2), a = 7.7917(3) Å, b = 13.0527(5) Å, c = 18.4422(7) Å, α = 87.278(2)°, β = 79.279(2)°, γ = 86.248(2)°, V = 1837.7(1) Å3, Z = 2, μ(CuKα) = 3.729 mm−1, Dcalc = 1.508 g/cm3, 45,151 reflections were measured (2.440° ≤ Θ ≤ 68.903°) and 6772 were unique (Rint = 0.0735), which were used in all calculations. The final R1 was 0.0711 (I > 2σ(I)), and wR2 was 0.1753 (all data).
3.4.3. Crystal Data for [Cu(L)I]2I3I5
C56H92Cu2I10N12O8S4 (M = 2585.73 g/mol): monoclinic, space group P212121 (no. 19), a = 15.4001(3) Å, b = 17.9214(3) Å, c = 29.2725(6) Å, V = 8079.0(3) Å3, Z = 4, μ(MoKα) = 4.505 mm−1, Dcalc = 2.126 g/cm3, 85,553 reflections were measured (2.231° ≤ Θ ≤ 30.538°) and 24,493 were unique (Rint = 0.0376), which were used in all calculations. Refined as an inversion twin with equal component. The final R1 was 0.0700 (I > 2σ(I)), and wR2 was 0.1932 (all data).
3.4.4. Data Presentation
CCDC 2127462, 2127456–2127457 contains the supplementary crystallographic data for this paper. These data can be obtained free of charge via http://www.ccdc.cam.ac.uk/conts/retrieving.html (or from the CCDC, 12 Union Road, Cambridge CB2 1EZ, UK; Fax: +44 1223 336033; E-mail: [email protected]).
4. Conclusions
The new ligand L, intended for the stabilization of Cu(II)-polyiodides has been successfully prepared. The Cu(II) coordinating ability, complexation kinetics, complex stoichiometry, and ligand-binding mode have been substantiated by solution and solid-state studies. The ligand is effective in protecting the Cu(II) oxidation state even in the presence of I− in DMF solution; the formation of a 1:1:1 Cu:L:I-ternary complex has been documented with a [Cu(L)]2+ + I− = [Cu(L)I]+ apparent logK of 4.0. The polyiodide crystallization attempt afforded the interesting [Cu(L)I]2I3I5 crystalline phase, which is notably organized in alternating [Cu(L)I]+ and polyiodide layers. Direct involvement of the Cu(II)-bound iodide anion in polyiodide growth was not observed. This is in contrast with previous reports and brought about by the steric hindrance of tosyl groups and the existence of prevailing H-bonding directionality brought about by sulfonamide groups. Notably, the achiral constituents of the [Cu(L)I]2I3I5 crystal structures organized themselves in a chiral manner. A 2005 survey found that the P212121 space group is among the most common space groups for chiral molecules (representing ≈ 34% of all chiral organic molecules with chiral centers); conversely, the crystallization of molecules not bearing a chiral center in the P212121 space group is much rarer (7.8% of achiral molecules) [20]. It has been documented, therein, that rigid molecules (defined as units with less than three consecutive rotatable acyclic single bonds) are more likely to crystallize in chiral space groups. While [Cu(L)I]+ possesses a flexible portion (the ethylenimine bridge, three consecutive rotatable bonds) it is mainly composed of three conformationally rigid domains (the macrocycle plus the two tosyl rings), and the rotation of sulfonamide N-S bond also being notoriously hindered [21]. The loss of the mirror plane/inversion center (as in the crystal structure of free L) due to Cu(II) coordination might also play a role in space group choice. Although it happened serendipitously, the idea of conferring chirality to the polyiodide-dense network could be worthy of further, and this time intended, exploration. Polyiodides’ crystals explicitly engineered towards chirality will be subject to further studies.
Supplementary Materials
The following supporting information can be downloaded at: https://0-www-mdpi-com.brum.beds.ac.uk/article/10.3390/inorganics10010012/s1, Figure S1: pocket hosting the ClO4− anion in the (CuLCl)ClO4.CH3CN crystal structure; Figure S2: views representing steric hindrance generated by tosyl groups around Cu-bound iodide; Figure S3: cell content of crystallized impurity identified as 1,4-ditosylpiperazine; CIF and the checkCIF output files.
Author Contributions
Conceptualization, M.S., C.B. and A.B.; methodology, M.S. and C.B.; investigation, V.M. and M.S.; data curation, V.M., M.S. and C.B.; writing—original draft preparation, M.S. and C.B.; writing—review and editing, M.S., C.B. and A.B.; funding acquisition, C.B. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by Fondazione Cassa di Risparmio di Firenze, grant number ECR2018.0937.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Acknowledgments
Fondazione Cassa di Risparmio di Firenze is gratefully acknowledged for economic support.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Svensson, P.H.; Kloo, L. Synthesis, Structure, and Bonding in Polyiodide and Metal Iodide−Iodine Systems. Chem. Rev. 2003, 103, 1649–1684. [Google Scholar] [CrossRef] [PubMed]
- Savastano, M. Words in Supramolecular Chemistry: The Ineffable Advances of Polyiodide Chemistry. Dalton Trans. 2021, 50, 1142–1165. [Google Scholar] [CrossRef] [PubMed]
- Abbas, Q.; Fitzek, H.; Schröttner, H.; Dsoke, S.; Gollas, B. Immobilization of Polyiodide Redox Species in Porous Carbon for Battery-Like Electrodes in Eco-Friendly Hybrid Electrochemical Capacitors. Nanomaterials 2019, 9, 1413. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meng, Z.; Tian, H.; Zhang, S.; Yan, X.; Ying, H.; He, W.; Liang, C.; Zhang, W.; Hou, X.; Han, W.-Q. Polyiodide-Shuttle Restricting Polymer Cathode for Rechargeable Lithium/Iodine Battery with Ultralong Cycle Life. ACS Appl. Mater. Interfaces 2018, 10, 17933–17941. [Google Scholar] [CrossRef] [PubMed]
- Bella, F.; Galliano, S.; Falco, M.; Viscardi, G.; Barolo, C.; Grätzel, M.; Gerbaldi, C. Unveiling Iodine-Based Electrolytes Chemistry in Aqueous Dye-Sensitized Solar Cells. Chem. Sci. 2016, 7, 4880–4890. [Google Scholar] [CrossRef] [Green Version]
- Paulsson, H.; Berggrund, M.; Svantesson, E.; Hagfeldt, A.; Kloo, L. Molten and Solid Metal-Iodide-Doped Trialkylsulphonium Iodides and Polyiodides as Electrolytes in Dye-Sensitized Nanocrystalline Solar Cells. Sol. Energy Mater. Sol. Cells 2004, 82, 345–360. [Google Scholar] [CrossRef]
- Fei, Z.; Bobbink, F.D.; Păunescu, E.; Scopelliti, R.; Dyson, P.J. Influence of Elemental Iodine on Imidazolium-Based Ionic Liquids: Solution and Solid-State Effects. Inorg. Chem. 2015, 54, 10504–10512. [Google Scholar] [CrossRef]
- McDaniel, J.G.; Yethiraj, A. Grotthuss Transport of Iodide in EMIM/I3 Ionic Crystal. J. Phys. Chem. B 2018, 122, 250–257. [Google Scholar] [CrossRef] [PubMed]
- Chang, J.; Zhao, G.; Zhao, X.; He, C.; Pang, S.; Shreeve, J.M. New Promises from an Old Friend: Iodine-Rich Compounds as Prospective Energetic Biocidal Agents. Acc. Chem. Res. 2021, 54, 332–343. [Google Scholar] [CrossRef]
- Savastano, M.; Bazzicalupi, C.; García, C.; Gellini, C.; López de la Torre, M.D.; Mariani, P.; Pichierri, F.; Bianchi, A.; Melguizo, M. Iodide and Triiodide Anion Complexes Involving Anion–π Interactions with a Tetrazine-Based Receptor. Dalton Trans. 2017, 46, 4518–4529. [Google Scholar] [CrossRef] [PubMed]
- Savastano, M.; Martínez-Camarena, Á.; Bazzicalupi, C.; Delgado-Pinar, E.; Llinares, J.M.; Mariani, P.; Verdejo, B.; García-España, E.; Bianchi, A. Stabilization of Supramolecular Networks of Polyiodides with Protonated Small Tetra-Azacyclophanes. Inorganics 2019, 7, 48. [Google Scholar] [CrossRef] [Green Version]
- Savastano, M.; Bazzicalupi, C.; Gellini, C.; Bianchi, A. Genesis of Complex Polyiodide Networks: Insights on the Blue Box/I−/I2 Ternary System. Crystals 2020, 10, 387. [Google Scholar] [CrossRef]
- Savastano, M.; Bazzicalupi, C.; Gellini, C.; Bianchi, A. Infinite Supramolecular Pseudo-Polyrotaxane with Poly[3]Catenane Axle: Assembling Nanosized Rings from Mono- and Diatomic I− and I2 Tectons. Chem. Commun. 2020, 56, 551–554. [Google Scholar] [CrossRef] [PubMed]
- Martínez-Camarena, Á.; Savastano, M.; Llinares, J.M.; Verdejo, B.; Bianchi, A.; García-España, E.; Bazzicalupi, C. Stabilization of Polyiodide Networks with Cu(II) Complexes of Small Methylated Polyazacyclophanes: Shifting Directional Control from H-Bonds to I⋯I Interactions. Inorg. Chem. Front. 2020, 7, 4239–4255. [Google Scholar] [CrossRef]
- Martínez-Camarena, Á.; Savastano, M.; Blasco, S.; Delgado-Pinar, E.; Giorgi, C.; Bianchi, A.; García-España, E.; Bazzicalupi, C. Assembly of Polyiodide Networks with Cu(II) Complexes of Pyridinol-Based Tetraaza Macrocycles. Inorg. Chem. 2022, 61, 368–383. [Google Scholar] [CrossRef] [PubMed]
- Pan, F.; Englert, U. N-(6-Methyl-2-Pyridyl)Mesitylenesulfonamide: An Efficient Template for Polyiodides. Cryst. Growth Des. 2014, 14, 1057–1066. [Google Scholar] [CrossRef]
- Martínez-Camarena, Á.; Savastano, M.; Bazzicalupi, C.; Bianchi, A.; García-España, E. Stabilisation of Exotic Tribromide (Br3−) Anions via Supramolecular Interaction with A Tosylated Macrocyclic Pyridinophane. A Serendipitous Case. Molecules 2020, 25, 3155. [Google Scholar] [CrossRef]
- Ciampolini, M.; Micheloni, M.; Nardi, N.; Paoletti, P.; Dapporto, P.; Zanobini, F. Synthesis and Characterisation of 1,7-Dimethyl-1,4,7,10-Tetra-Azacyclododecane: Crystal Structure of the Nickel(II) Bromide Monohydrate Complex of This Macrocycle; Thermodynamic Studies of Protonation and Metal Complex Formation. J. Chem. Soc. Dalton Trans. 1984, 1357–1362. [Google Scholar] [CrossRef]
- Gans, P.; Sabatini, A.; Vacca, A. Investigation of Equilibria in Solution. Determination of Equilibrium Constants with the HYPERQUAD Suite of Programs. Talanta 1996, 43, 1739–1753. [Google Scholar] [CrossRef]
- Pidcock, E. Achiral Molecules in Non-Centrosymmetric Space Groups. Chem. Commun. 2005, 3457–3459. [Google Scholar] [CrossRef]
- Baldauf, C.; Günther, R.; Hofmann, H.-J. Conformational Properties of Sulfonamido Peptides. J. Mol. Struct.-THEOCHEM 2004, 675, 19–28. [Google Scholar] [CrossRef]
- Ambrosi, G.; Formica, M.; Fusi, V.; Giorgi, L.; Guerri, A.; Micheloni, M.; Paoli, P.; Pontellini, R.; Rossi, P. A New Macrocyclic Cryptand with Squaramide Moieties: An Overstructured CuII Complex That Selectively Binds Halides: Synthesis, Acid/Base- and Ligational Behavior, and Crystal Structures. Chem. Eur. J. 2007, 13, 702–712. [Google Scholar] [CrossRef]
- Yong, L.; Guo-Ping, X.; Cheng-Tai, W. Synthesis of Novel N-Aminoethyl Cyclic Polyamines. Chin. J. Chem. 1998, 16, 448–451. [Google Scholar] [CrossRef]
- Krause, L.; Herbst-Irmer, R.; Sheldrick, G.M.; Stalke, D. Comparison of Silver and Molybdenum Microfocus X-Ray Sources for Single-Crystal Structure Determination. J Appl. Crystallogr. 2015, 48, 3–10. [Google Scholar] [CrossRef] [Green Version]
- Sheldrick, G.M. A Short History of SHELX. Acta Cryst. A 2008, 64, 112–122. [Google Scholar] [CrossRef] [Green Version]
- Sheldrick, G.M. Crystal Structure Refinement with SHELXL. Acta Crystallogr. Sect. C Struct. Chem. 2015, 71, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Macrae, C.F.; Sovago, I.; Cottrell, S.J.; Galek, P.T.A.; McCabe, P.; Pidcock, E.; Platings, M.; Shields, G.P.; Stevens, J.S.; Towler, M.; et al. Mercury 4.0: From Visualization to Analysis, Design and Prediction. J. Appl. Crystallogr. 2020, 53, 226–235. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A Visualization System for Exploratory Research and Analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [Green Version]
Scheme 1.
Synthesis of L from parent ligand 1.

Figure 1.
Overview of main interactions as found in L crystal structure; (a) intermolecular hydrogen bond; (b) π-π stacking and methyl-sulfone contacts; (c) double S=O2⋯H-C6 contact. All distances in Å.
Figure 1.
Overview of main interactions as found in L crystal structure; (a) intermolecular hydrogen bond; (b) π-π stacking and methyl-sulfone contacts; (c) double S=O2⋯H-C6 contact. All distances in Å.

Figure 2.
Top: temporal evolution of the absorption spectra of a DMF solution of L ([L] = 1.8 mM) after addition of 1 eq of Cu(II). Cu(II) only 1.8 mM in DMF. Bottom: evolution of the 688 nm spectral maximum over time showing that invariant spectrum is achieved within 1 h.
Figure 2.
Top: temporal evolution of the absorption spectra of a DMF solution of L ([L] = 1.8 mM) after addition of 1 eq of Cu(II). Cu(II) only 1.8 mM in DMF. Bottom: evolution of the 688 nm spectral maximum over time showing that invariant spectrum is achieved within 1 h.

Figure 3.
Left: titration of L ([L] = 1.8 mM) with Cu(II) equivalents. Right: behaviors of the absorption maximum. Complex band raises steadily until 1 eq of Cu(II) is added, then spectral changes are associated with free Cu(II) in solution (green-dashed spectrum: Cu(II) 1.8 mM in DMF).
Figure 3.
Left: titration of L ([L] = 1.8 mM) with Cu(II) equivalents. Right: behaviors of the absorption maximum. Complex band raises steadily until 1 eq of Cu(II) is added, then spectral changes are associated with free Cu(II) in solution (green-dashed spectrum: Cu(II) 1.8 mM in DMF).

Figure 4.
Views of the [Cu(L)Cl]ClO4∙CH3CN crystal structure. Top: asymmetric unit content and Cu(II) coordination environment. Bottom: head to tail dipolar interactions linking adjacent complexes through Cu-coordinated Cl− and sulfonamide H-bond donors.
Figure 4.
Views of the [Cu(L)Cl]ClO4∙CH3CN crystal structure. Top: asymmetric unit content and Cu(II) coordination environment. Bottom: head to tail dipolar interactions linking adjacent complexes through Cu-coordinated Cl− and sulfonamide H-bond donors.

Figure 5.
Spectral variation caused by addition of I− equivalents to a 1.44 mM solution of [Cu(L)]2+. Inset: sigmoidal profiles of spectral changes upon I− addition at selected wavelengths (730 nm, orange, and 420 nm, green).
Figure 5.
Spectral variation caused by addition of I− equivalents to a 1.44 mM solution of [Cu(L)]2+. Inset: sigmoidal profiles of spectral changes upon I− addition at selected wavelengths (730 nm, orange, and 420 nm, green).

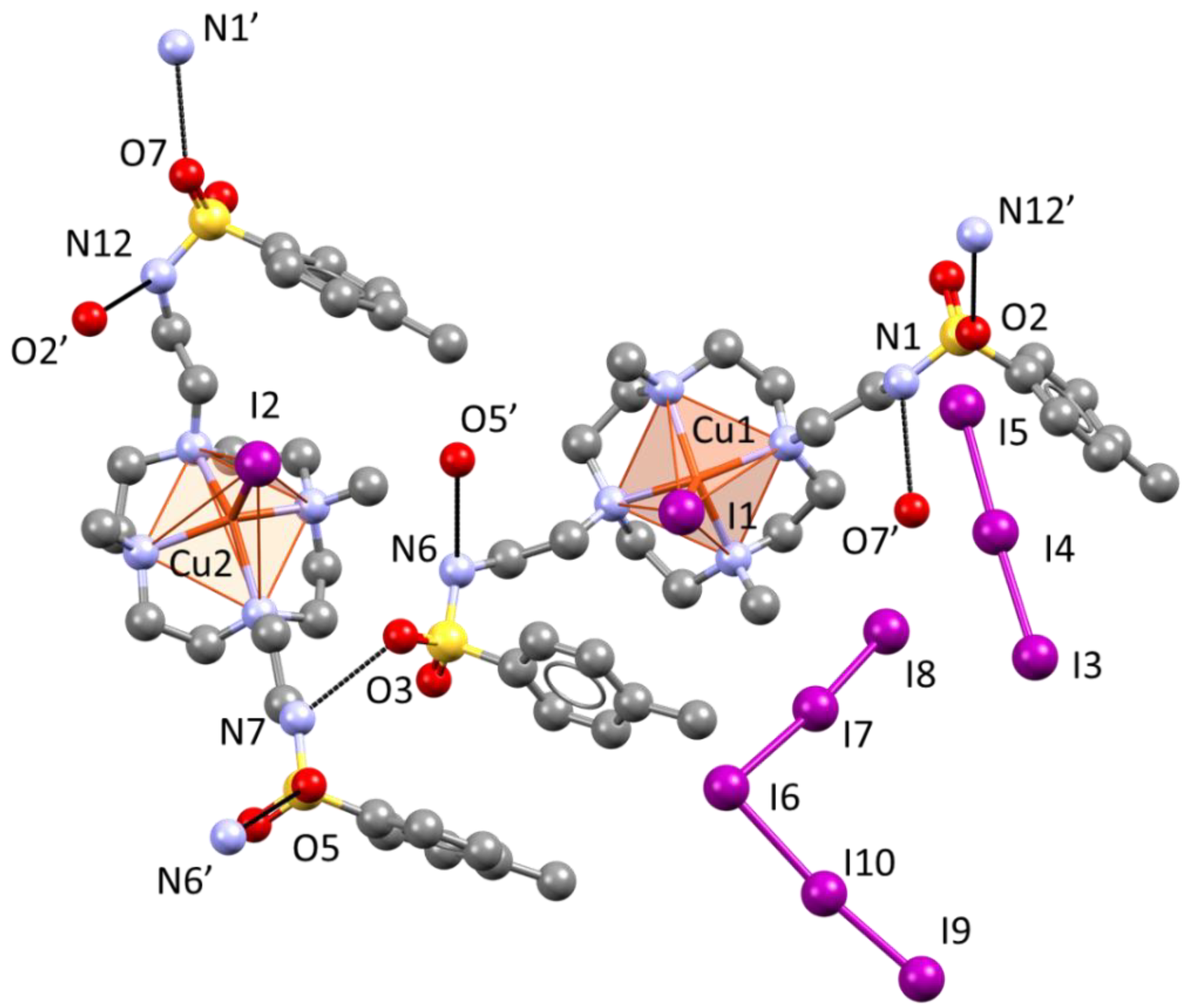
Figure 6.
Asymmetric unit of the crystal structure of [Cu(L)I]2I3I5. Symmetry-related atoms completing dangling H-bonds (black) are also shown, hinting at the resulting network of H-bonded sulfonamides.
Figure 6.
Asymmetric unit of the crystal structure of [Cu(L)I]2I3I5. Symmetry-related atoms completing dangling H-bonds (black) are also shown, hinting at the resulting network of H-bonded sulfonamides.

Figure 7.
(a) Enantiomorphic nonequivalent complexes in the crystal structure of [Cu(L)I]2I3I5: conformations are related by a pseudo mirror plane. (b) In-plane H-bonding among [Cu(L)I]+ complexes developing normal to c-direction. (c) Top view and (d) lateral view of the couple of planes formed by [Cu(L)I]+ complexes developing parallel to the c-direction; red-circled, out-of-plane H-bonding.
Figure 7.
(a) Enantiomorphic nonequivalent complexes in the crystal structure of [Cu(L)I]2I3I5: conformations are related by a pseudo mirror plane. (b) In-plane H-bonding among [Cu(L)I]+ complexes developing normal to c-direction. (c) Top view and (d) lateral view of the couple of planes formed by [Cu(L)I]+ complexes developing parallel to the c-direction; red-circled, out-of-plane H-bonding.

Figure 8.
Left: alternated planes of polyiodides and H-bonded [Cu(L)I]+ complexes developing perpendicular to the c-axis direction. Right: breakdown of polyiodide motif viewed along the c-direction.
Figure 8.
Left: alternated planes of polyiodides and H-bonded [Cu(L)I]+ complexes developing perpendicular to the c-axis direction. Right: breakdown of polyiodide motif viewed along the c-direction.

Figure 9.
Views of the pentaiodide infinite chain linked through the I9⋯I6 halogen bond.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Bond distances and angles defining the Cu(II) coordination environment in the crystal structure of [Cu(L)Cl]ClO4∙CH3CN.
Table 1.
Bond distances and angles defining the Cu(II) coordination environment in the crystal structure of [Cu(L)Cl]ClO4∙CH3CN.
| Bond Distances (Å) | Bond Angles (°) | ||
|---|---|---|---|
| Cu1-N2 | 2.071(4) | Cl2 -Cu1 -N4 | 105.3(1) |
| Cu1-N3 | 2.047(4) | Cl2 -Cu1 -N2 | 106.7(1) |
| Cu1-N4 | 2.081(4) | Cl2 -Cu1 -N5 | 103.6(1) |
| Cu1-N5 | 2.041(4) | Cl2 -Cu1 -N3 | 106.5(1) |
| Cu1-Cl2 | 2.372(1) | N4 -Cu1 -N2 | 148.0(2) |
| N4 -Cu1 -N5 | 86.1(2) | ||
| N4 -Cu1 -N3 | 86.1(2) | ||
| N2 -Cu1 -N5 | 86.2(2) | ||
| N2 -Cu1 -N3 | 85.2(2) | ||
| N5 -Cu1 -N3 | 149.8(2) | ||
Table 2.
Bond distances and angles defining the Cu(II) coordination environment in the crystal structure of [Cu(L)I]2I3I5.
Table 2.
Bond distances and angles defining the Cu(II) coordination environment in the crystal structure of [Cu(L)I]2I3I5.
| Bond Distances (Å) | Bond Angles (°) | ||||
|---|---|---|---|---|---|
| Cu1-N2 | 2.08(1) | I1-Cu1-N2 | 107.7(3) | I2-Cu2-N8 | 107.3(3) |
| Cu1-N3 | 2.06(1) | I1-Cu1-N3 | 108.4(3) | I2-Cu2-N9 | 103.7(4) |
| Cu1-N4 | 2.13(1) | I1-Cu1-N4 | 106.4(3) | I2-Cu2-N10 | 106.9(3) |
| Cu1-N5 | 2.07(1) | I1-Cu1-N5 | 103.6(4) | I2-Cu2-N11 | 109.3(4) |
| Cu1-I1 | 2.670(2) | N2-Cu1-N3 | 86.3(5) | N8-Cu2-N9 | 85.8(5) |
| Cu2-N8 | 2.13(1) | N2-Cu1-N4 | 145.9(5) | N8-Cu2-N10 | 145.8(5) |
| Cu2-N9 | 2.05(1) | N2-Cu1-N5 | 84.6(5) | N8-Cu2-N11 | 85.5(5) |
| Cu2-N10 | 2.12(1) | N3-Cu1-N4 | 84.8(4) | N9-Cu2-N10 | 85.1(5) |
| Cu2-N11 | 2.08(1) | N3-Cu1-N5 | 147.9(5) | N9-Cu2-N11 | 147.0(5) |
| Cu2-I2 | 2.662(2) | N4-Cu1-N5 | 85.6(5) | N10-Cu2-N11 | 84.4(5) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Savastano, M.; Monini, V.; Bazzicalupi, C.; Bianchi, A. Bidimensional Polyiodide Netting Stabilized by a Cu(II) Macrocyclic Complex. Inorganics 2022, 10, 12. https://0-doi-org.brum.beds.ac.uk/10.3390/inorganics10010012
AMA Style
Savastano M, Monini V, Bazzicalupi C, Bianchi A. Bidimensional Polyiodide Netting Stabilized by a Cu(II) Macrocyclic Complex. Inorganics. 2022; 10(1):12. https://0-doi-org.brum.beds.ac.uk/10.3390/inorganics10010012
Chicago/Turabian StyleSavastano, Matteo, Valeria Monini, Carla Bazzicalupi, and Antonio Bianchi. 2022. "Bidimensional Polyiodide Netting Stabilized by a Cu(II) Macrocyclic Complex" Inorganics 10, no. 1: 12. https://0-doi-org.brum.beds.ac.uk/10.3390/inorganics10010012
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.