
An Experimental and Theoretical Study of the Optical Properties of (C2H7N4O)2BiCl5 for an Optoelectronic Application

 ,
,  ,
,
Abstract
:1. Introduction
2. Results and Discussion
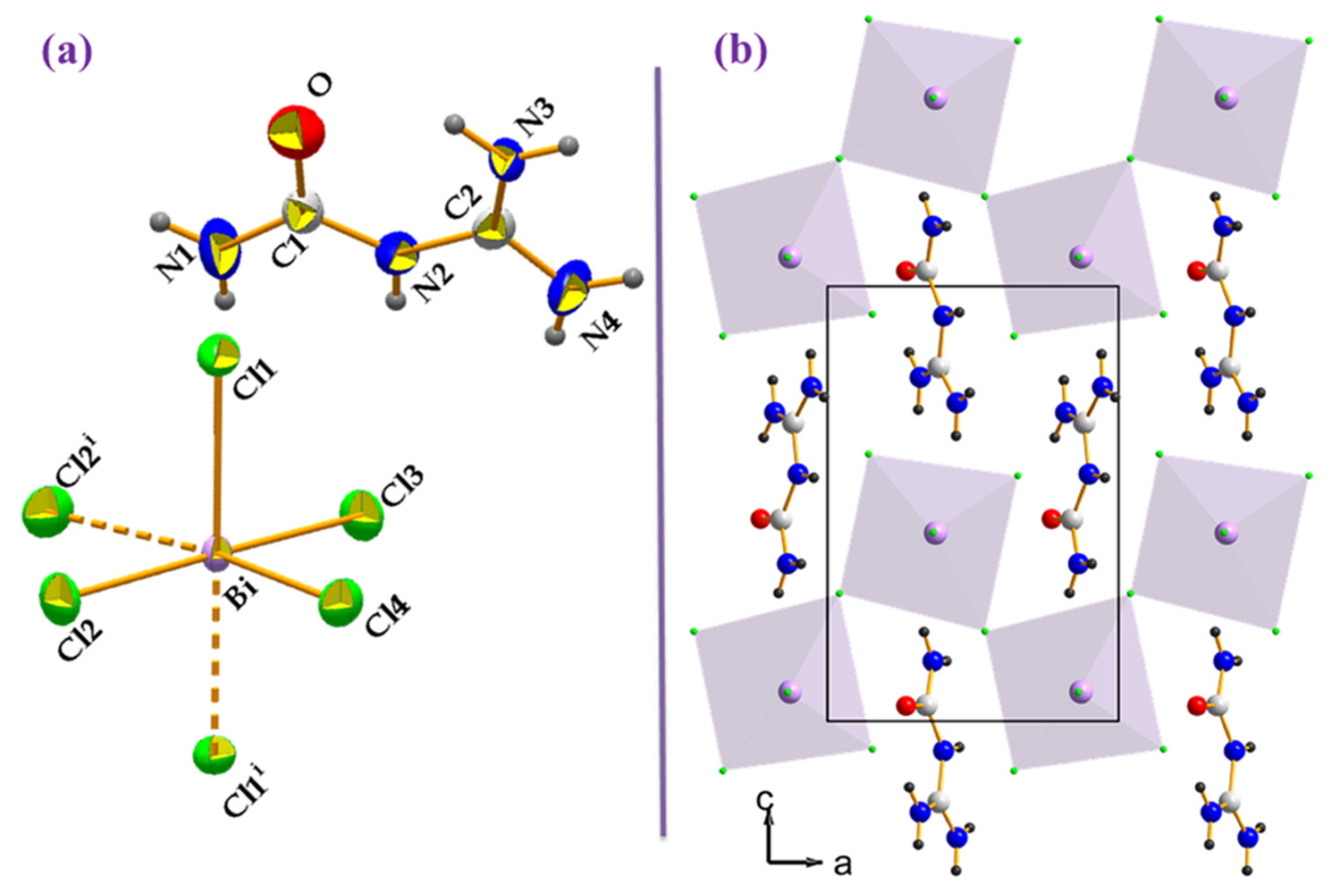
2.1. Structural Summary
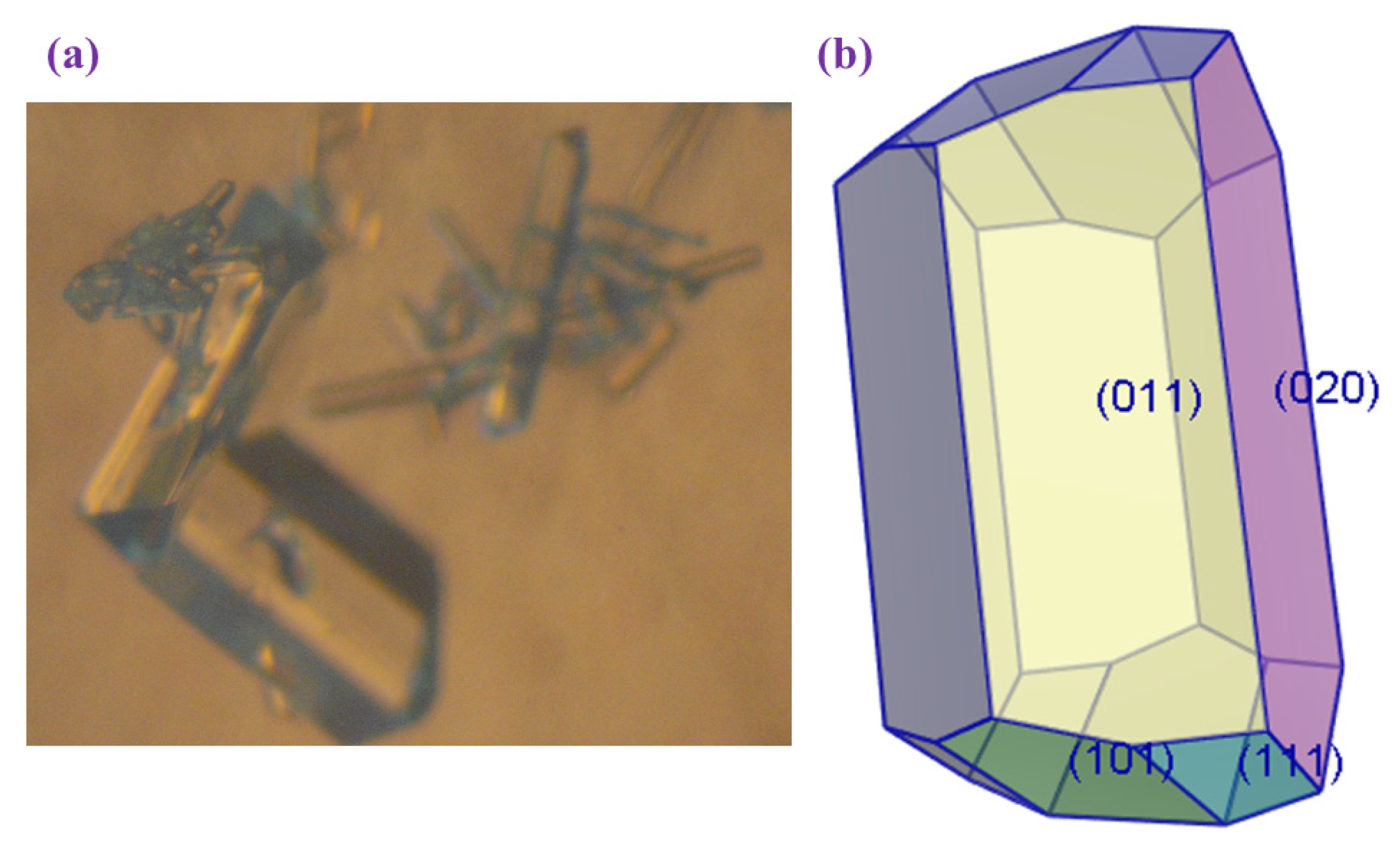
2.2. Crystal Morphology
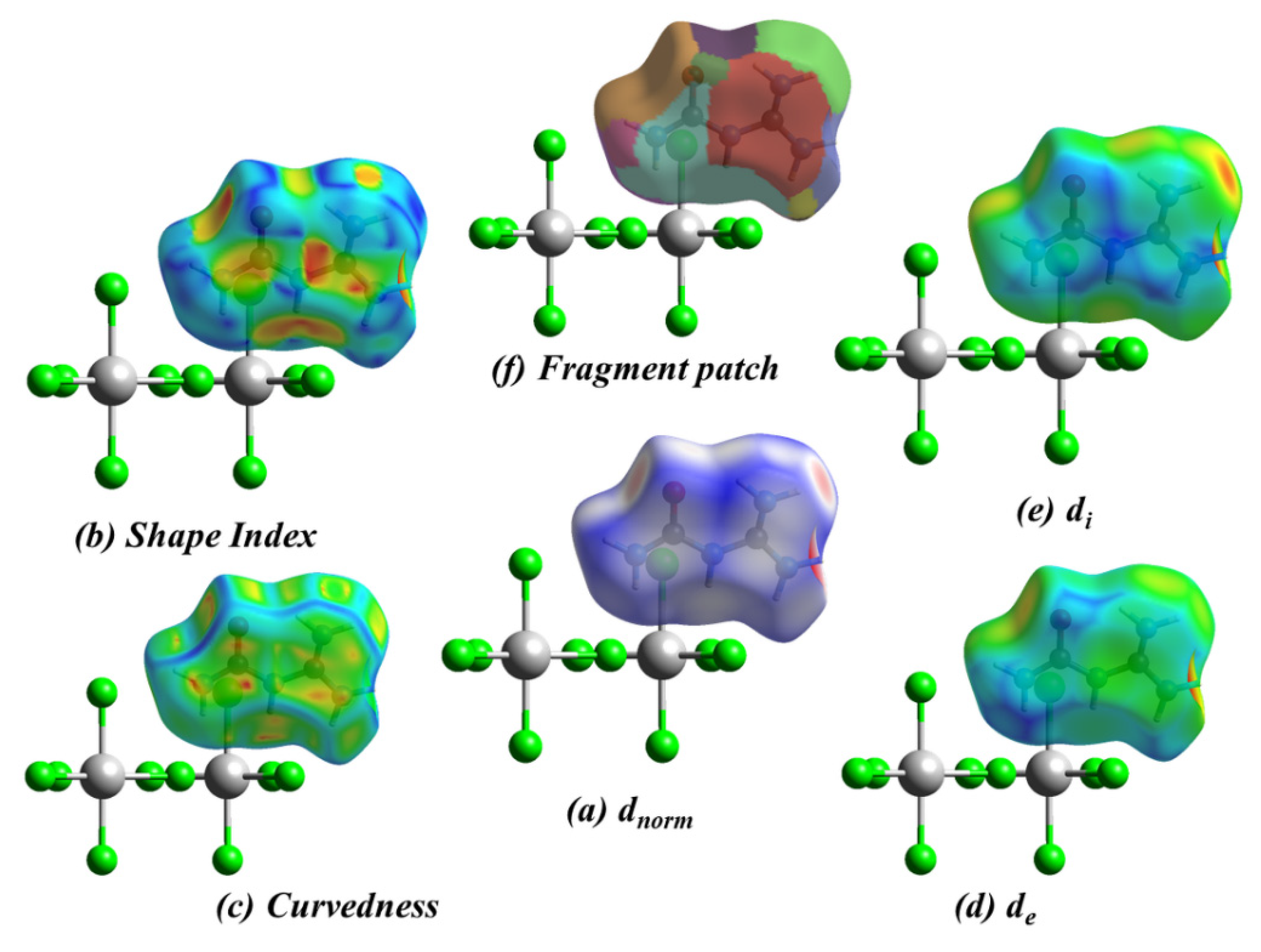
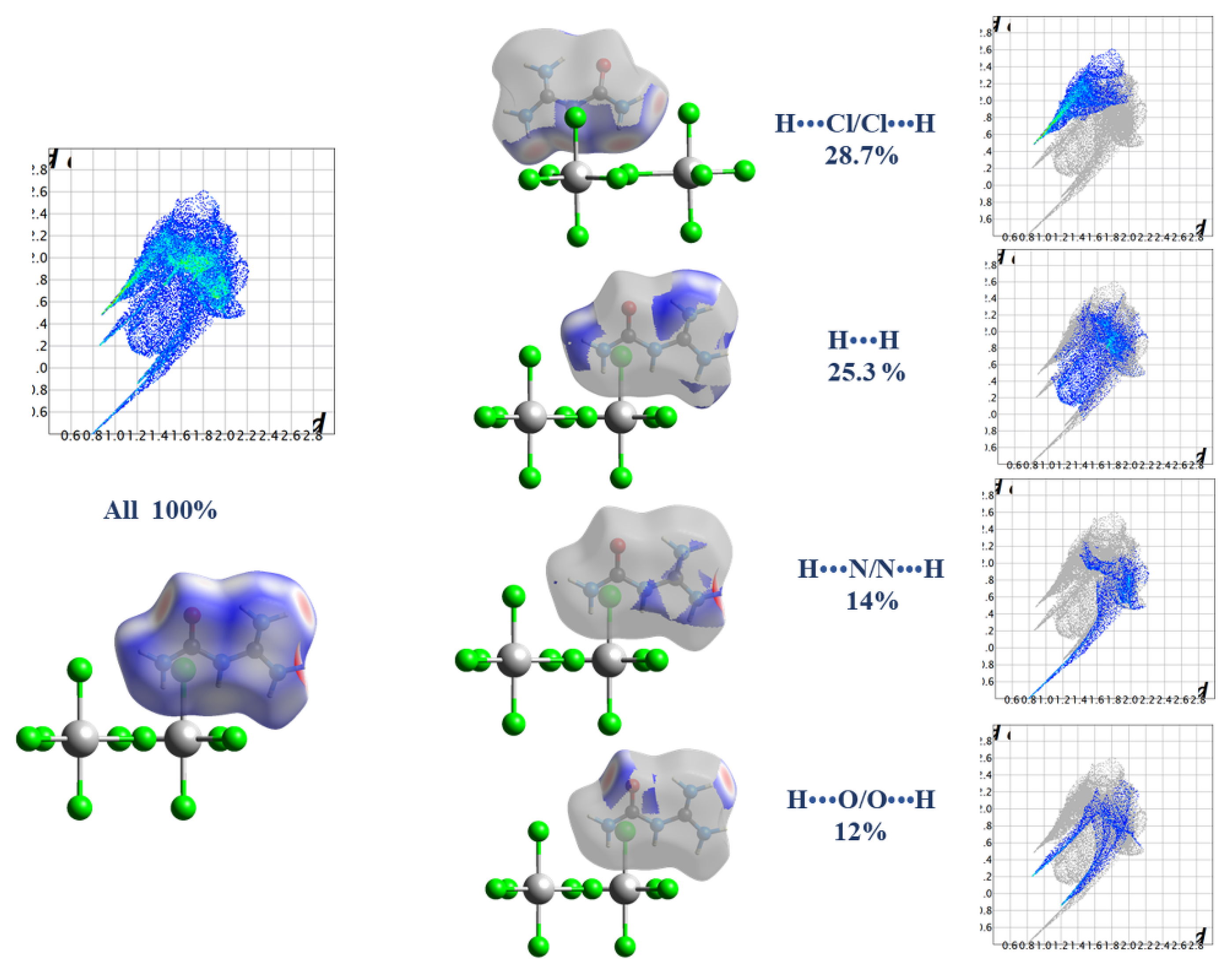
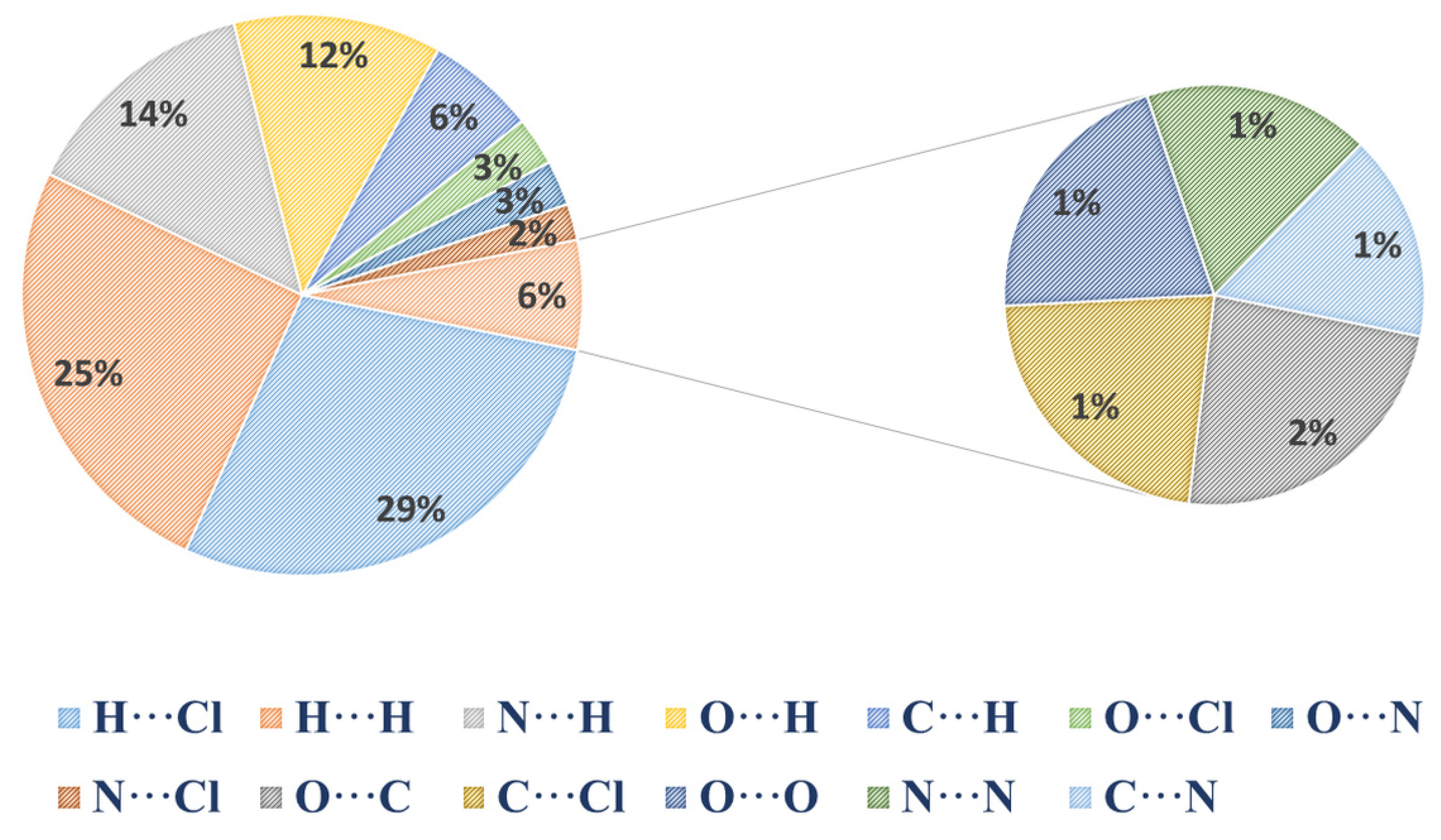
2.3. Hirshfeld Surface Analysis, Two-Dimensional Fingerprint Plots, and Enrichment Ratios (E)
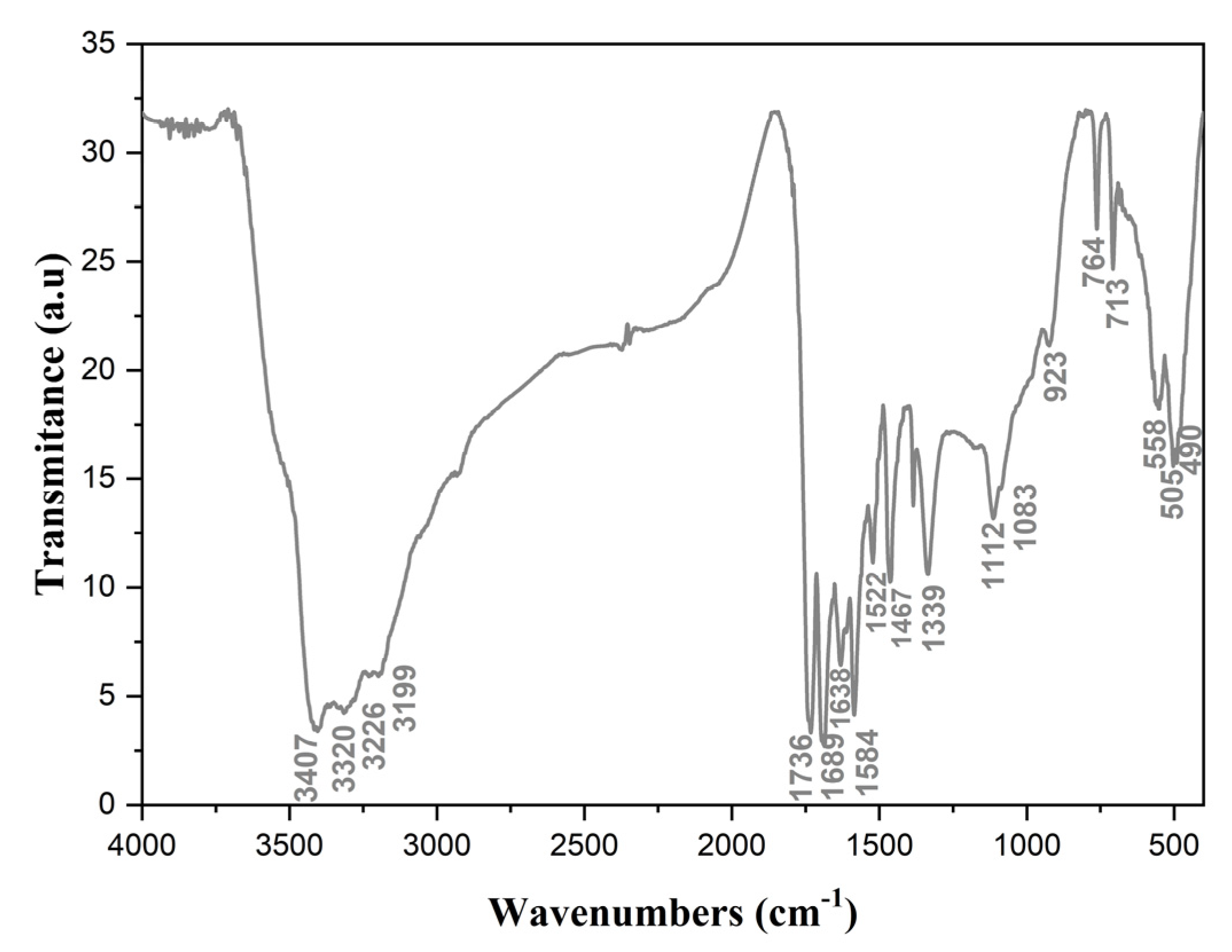
2.4. Vibrational Properties
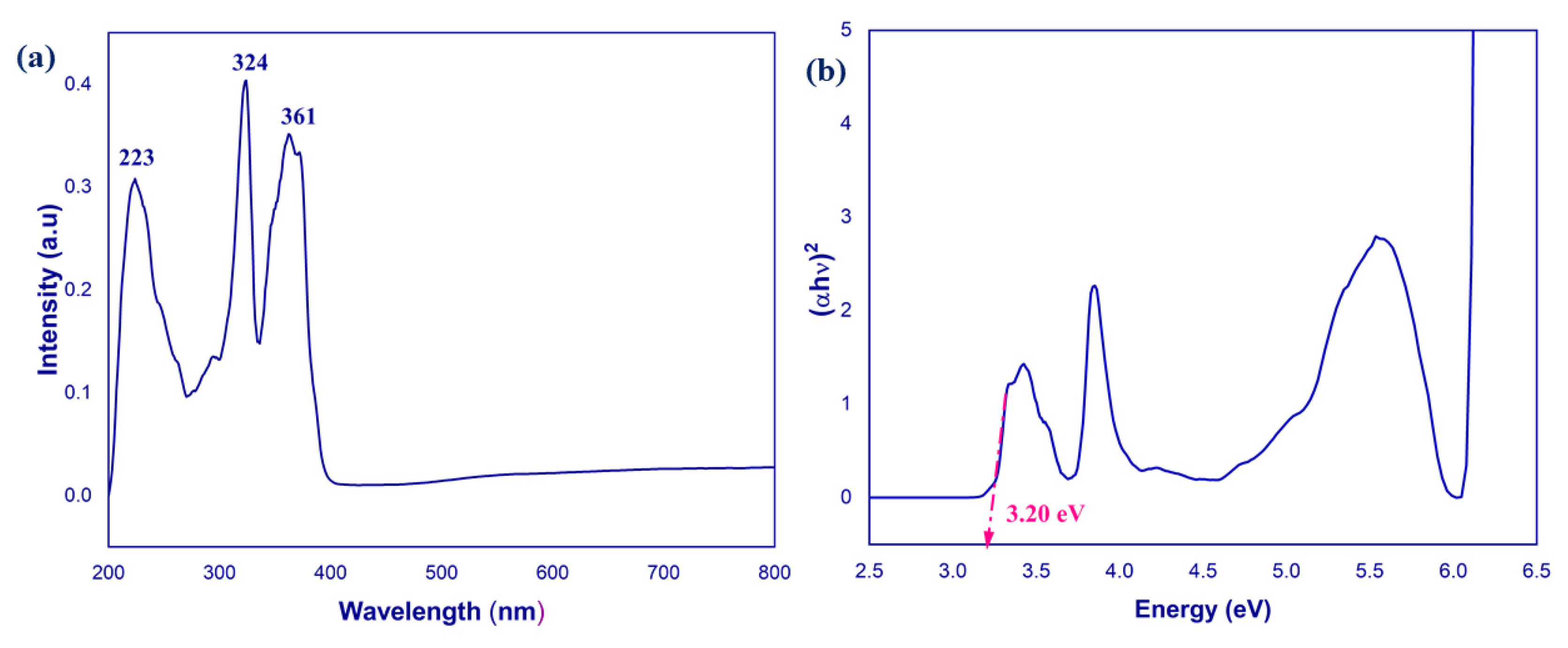
2.5. UV–Visible Spectum
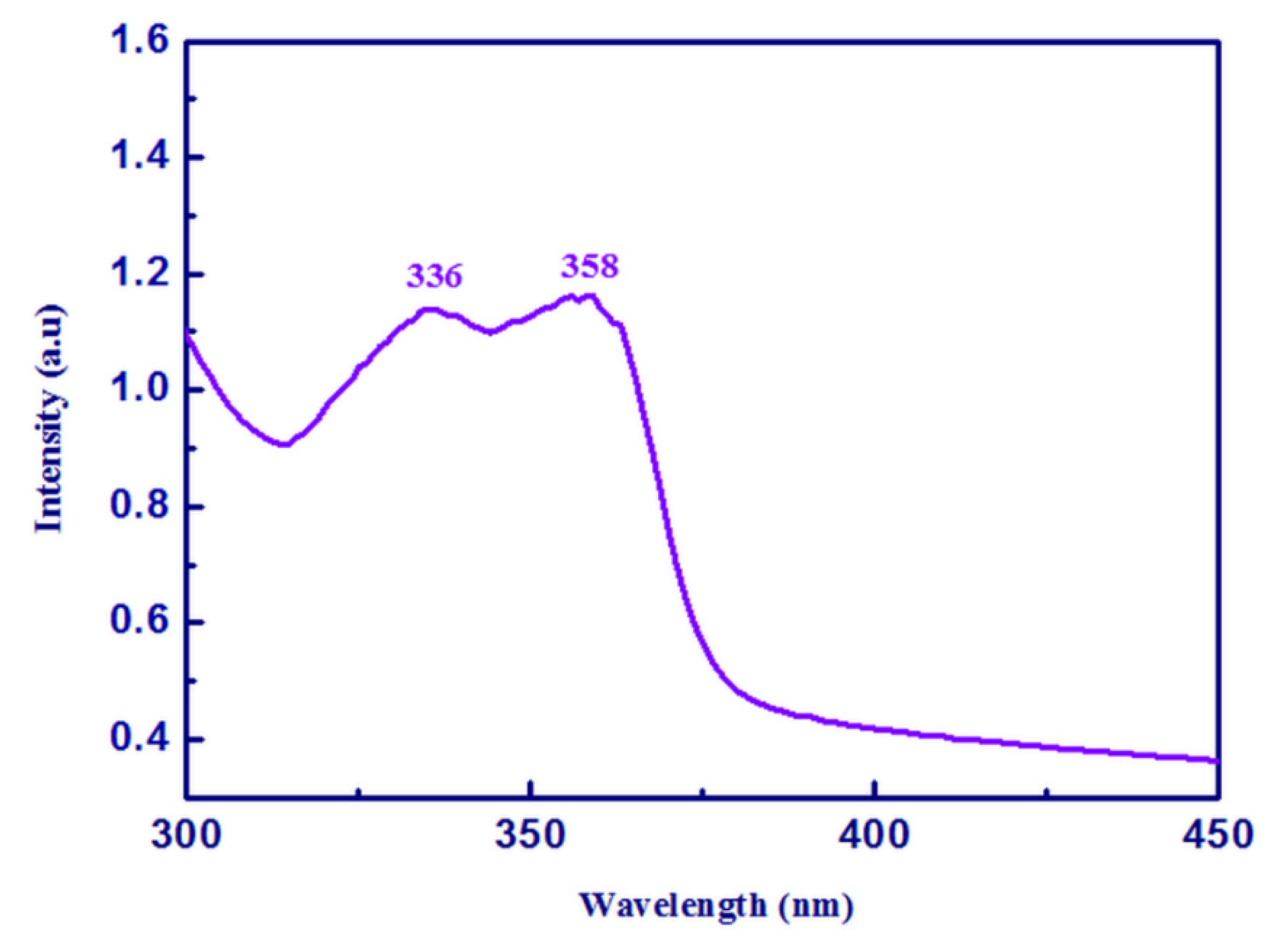
2.6. Photoluminscence Spectrum
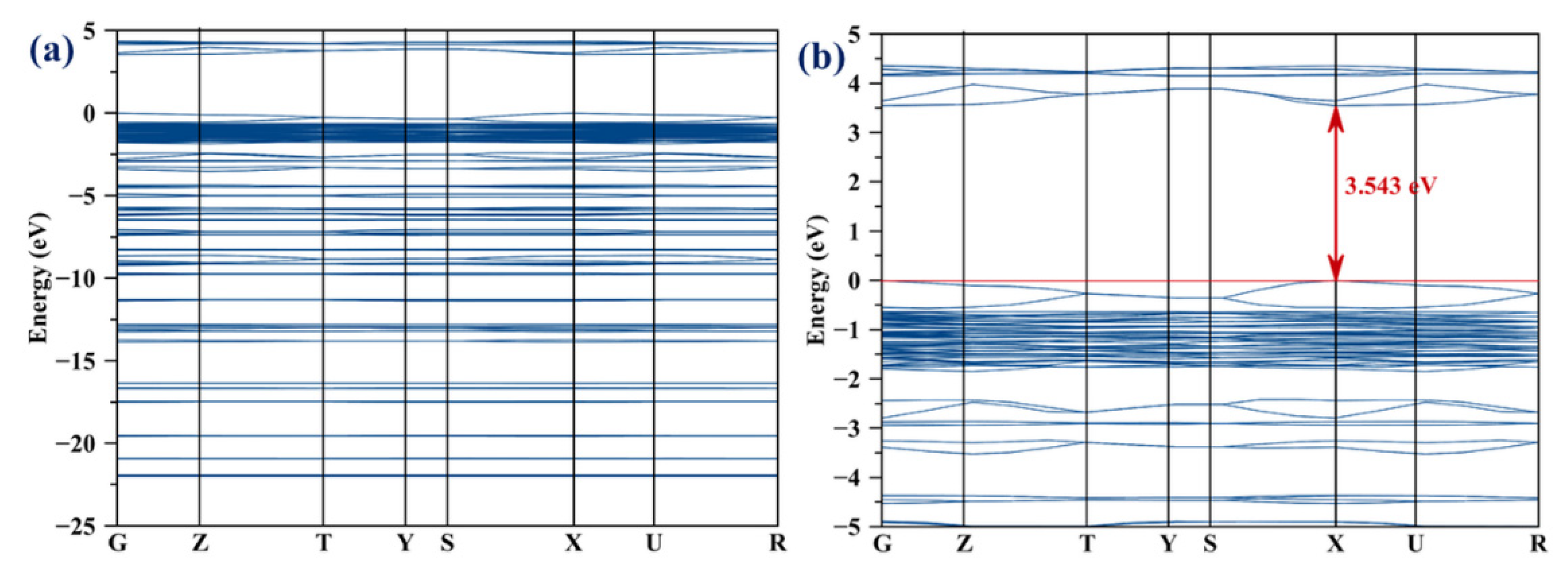
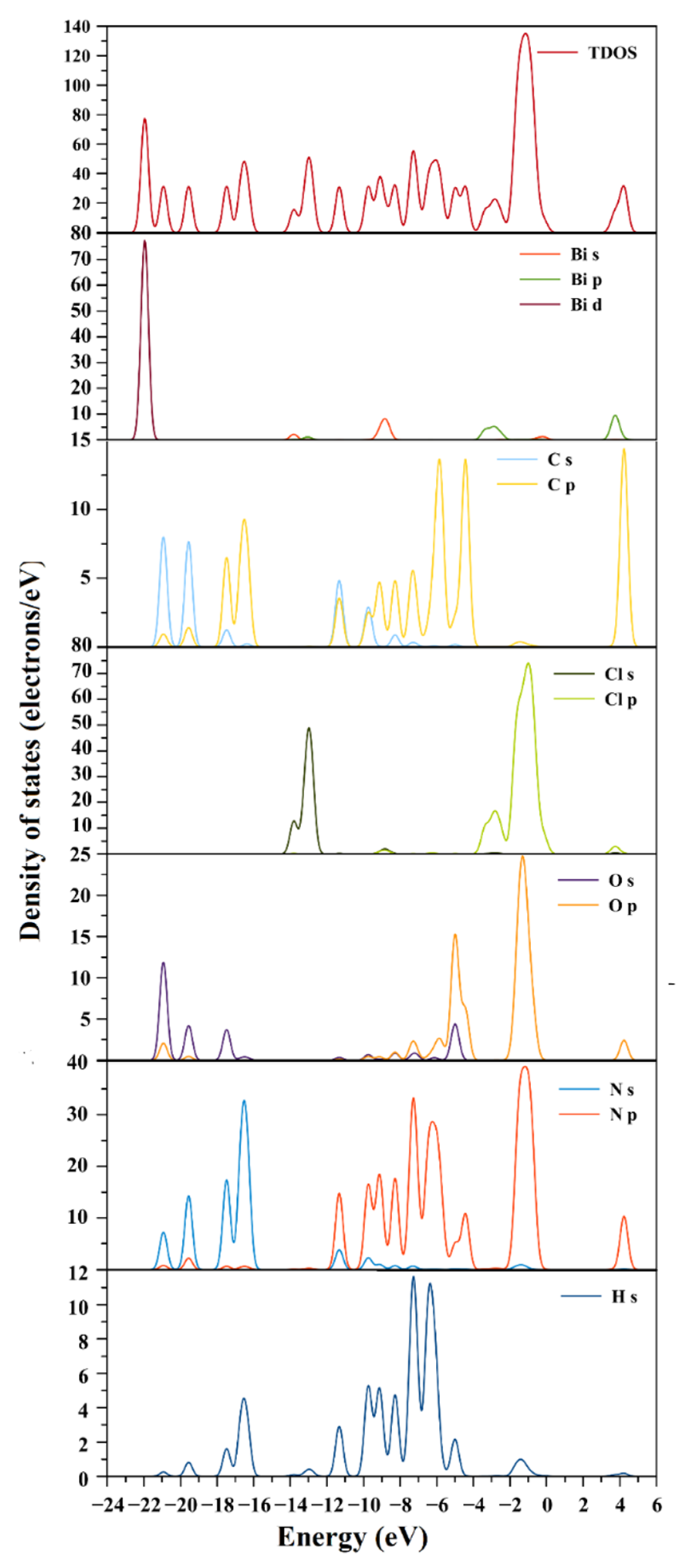
2.7. Electronic Structure
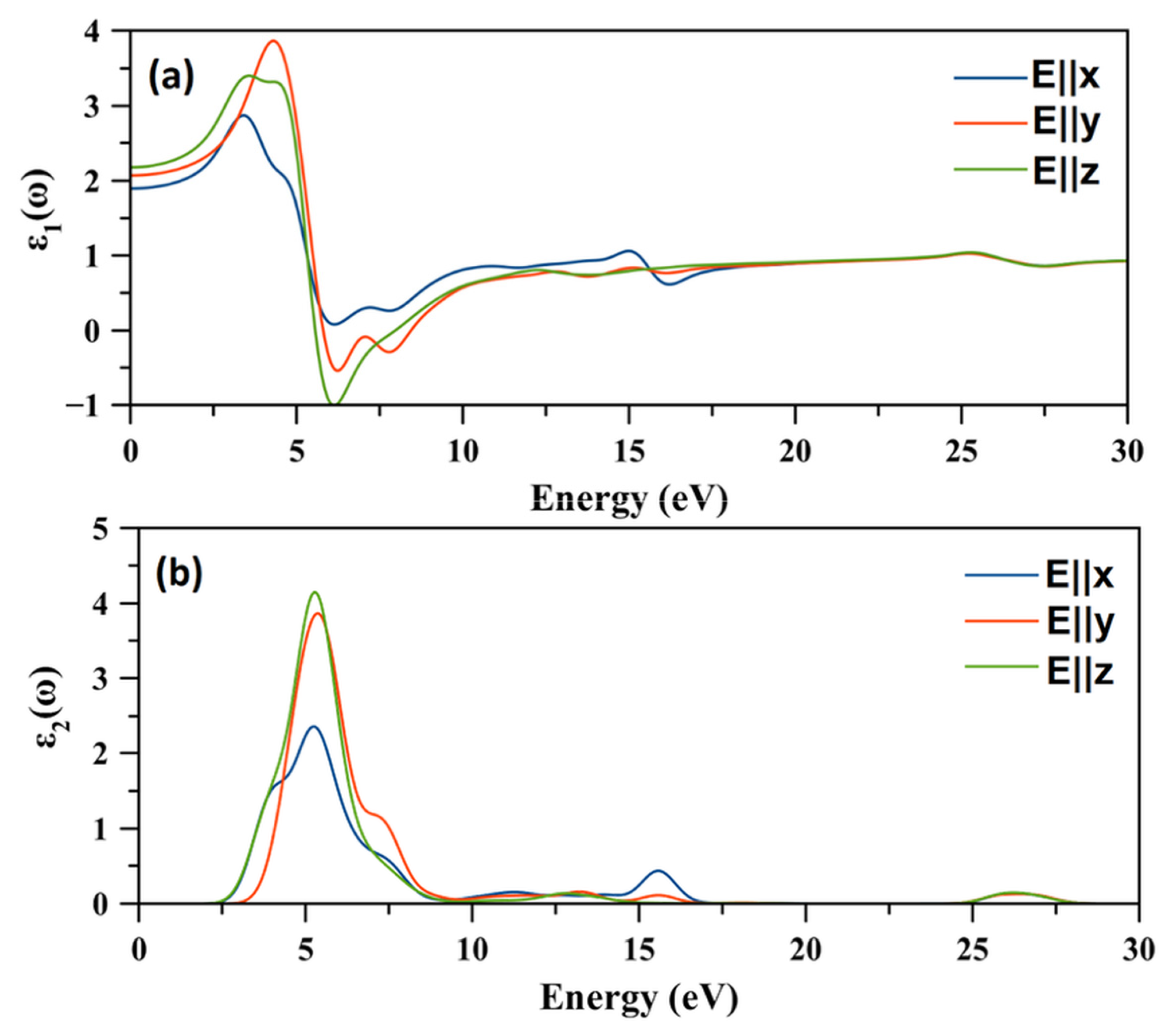
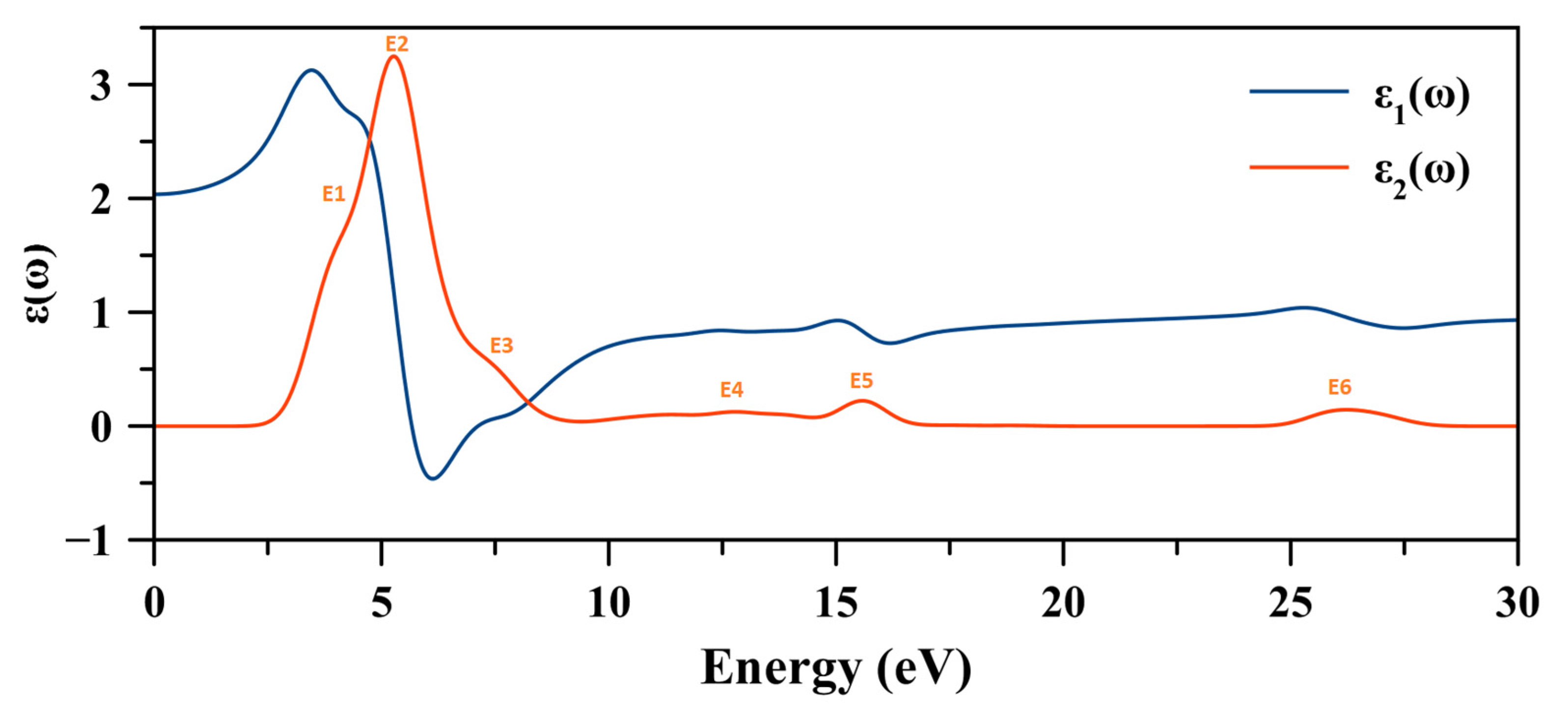
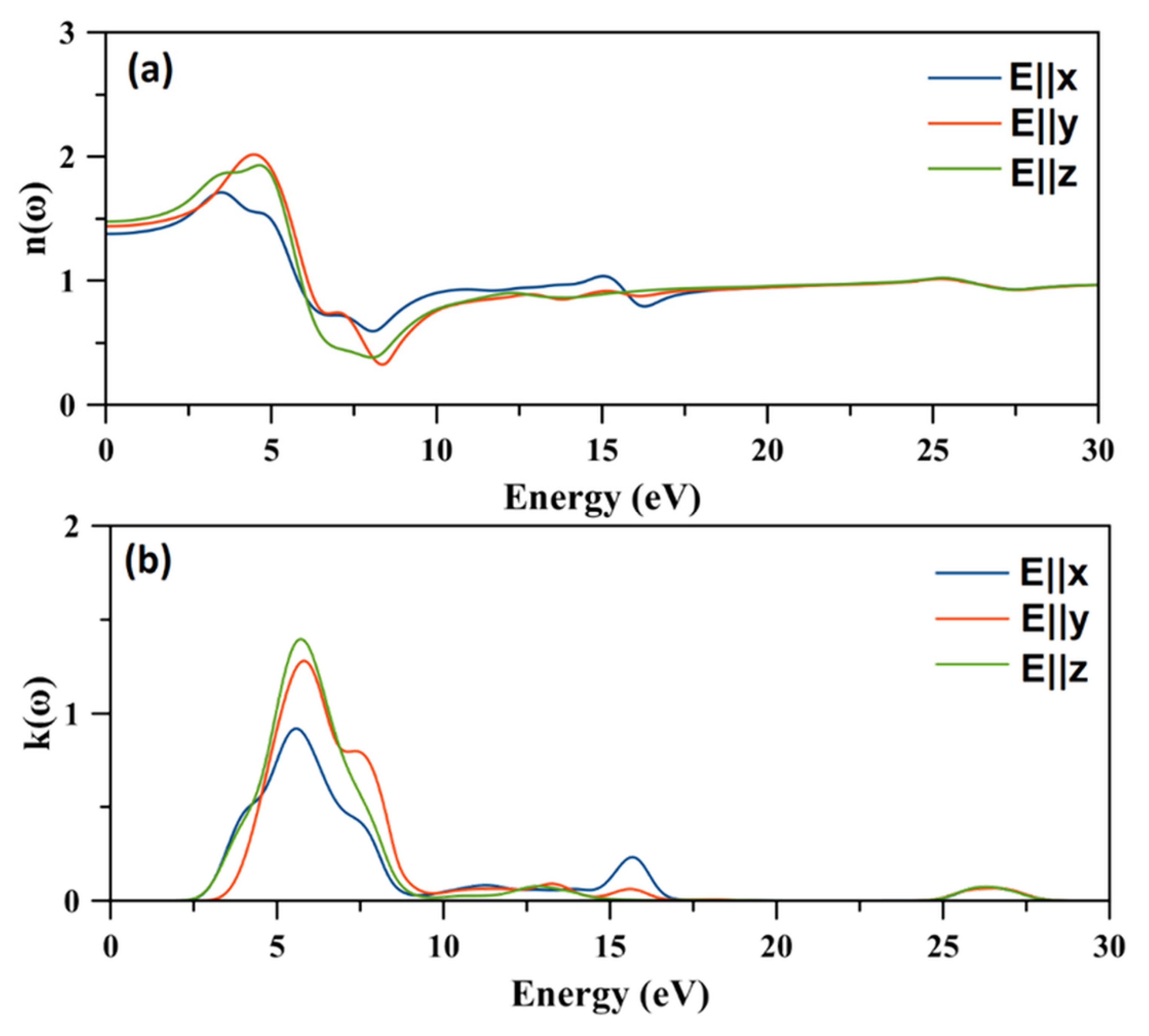
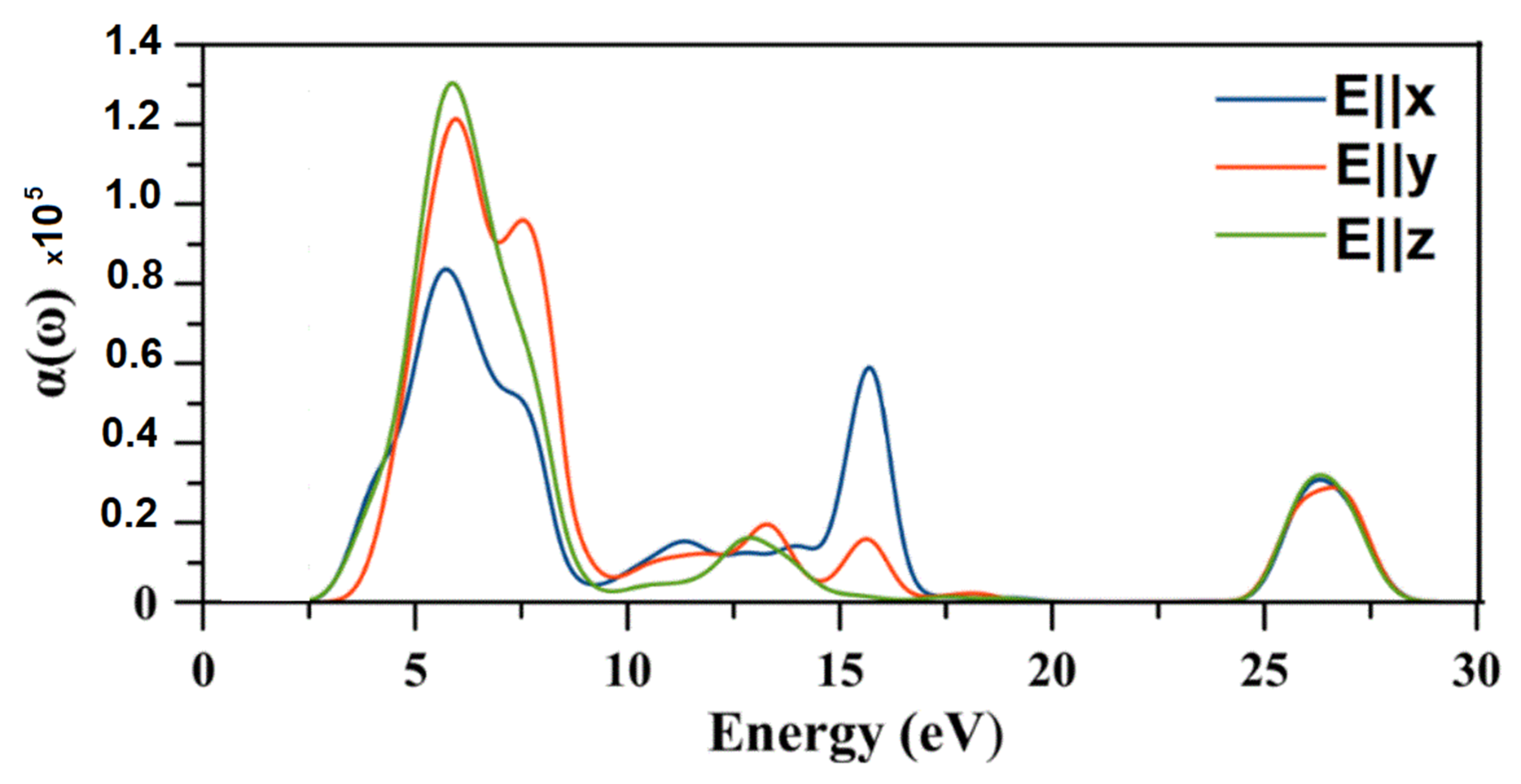
2.8. Optical Properties
3. Materials and Methods
3.1. Materials
3.2. Physical Measurements
3.3. Computational Details
3.4. Hirshfeld Surfaces Analysis, Enrichment Ratio (E), and Morphology Simulation
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Trabelsie, S.; Samet, A.; Dammak, H.; Michaud, F.; Santos, L.; Abid, Y.; Chaabouni, S. Optical properties of a new luminescent hybrid material [C6N2H5]3BiCl6 involving a resonance energy transfer (RET). Opt. Mater. 2019, 89, 355–360. [Google Scholar] [CrossRef]
- Barkaoui, H.; Abid, H.; Yangui, A.; Triki, S.; Boukheddaden, K.; Abid, Y. Yellowish white-light emission involving resonant energy transfer in a new one-dimensional hybrid material:(C9H10N2) PbCl4. J. Phys. Chem. C 2018, 122, 24253–24261. [Google Scholar] [CrossRef]
- Kotov, V.Y.; Ilyukhin, A.B.; Baranchikov, A.E.; Ishmetova, R.I.; Rusinov, G.L.; Kozyukhin, S.A. Synthesis, crystal structure and optical properties of 1,1′-(1,n-alkanediyl)bis(3-methylimidazolium) halobismuthates. J. Mol. Struct. 2018, 1151, 186–190. [Google Scholar] [CrossRef]
- Mitzi, D.B.; Brock, P. Structure and optical properties of several organic−inorganic hybrids containing corner-sharing chains of bismuth iodide Octahedra. Inorg. Chem. 2001, 40, 2096–2104. [Google Scholar] [CrossRef]
- Hassen, S.; Chebbi, H.; Arfaoui, Y.; Robeyns, K.; Steenhaut, T.; Hermans, S.; Filinchuk, Y. Spectroscopic and structural studies, thermal characterization, optical proprieties and theoretical investigation of 2-aminobenzimidazolium tetrachlorocobaltate (II). Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2020, 240, 118612. [Google Scholar] [CrossRef] [PubMed]
- Adonin, S.A.; Rakhmanova, M.E.; Samsonenko, D.G.; Sokolov, M.N.; Fedin, V.P. Bi (III) halide complexes containing 4,4′-vinylenedipyridinium cation: Synthesis, structure and luminescence in solid state. Polyhedron 2015, 98, 1–4. [Google Scholar] [CrossRef]
- Lambarki, F.; Ouasri, A.; Zouihri, H.; Rhandour, A. Crystal structure, Hirshfeld and vibrational study at ambient temperature of propylammonium pentachlorobismuthate [n-C3H7NH3] 2BiCl5 (III). J. Mol. Struct. 2017, 1142, 275–284. [Google Scholar] [CrossRef]
- Chański, M.; Białońska, A.; Jakubas, R.; Piecha-Bisiorek, A. Structural characterization and properties of bis (1, 4-H2-1, 2, 4-triazolium) pentachlorobismuthate (III) and cocrystal of ammonium chloride with tris (1, 4-H2-1, 2, 4-triazolium) hexachlorobismuthate (III). Polyhedron 2014, 71, 69–74. [Google Scholar] [CrossRef]
- Ganguly, P.; Desiraju, G.R. Van der Waals and polar intermolecular contact distances: Quantifying supramolecular synthons. Chem.–Asian J. 2008, 3, 868–880. [Google Scholar] [CrossRef]
- Ouerghi, Z.; Roisnel, T.; Fezai, R.; Kefi, R. Physico-chemical characterization, Hirshfeld surface analysis and opto-electric properties of a new hybrid material: Tris (2-amino-5-chloropyridinium) hexachlorobismuthate (III). J. Mol. Struct. 2018, 1173, 439–447. [Google Scholar] [CrossRef]
- Zhu, S.; Jiang, M.; Ye, J.; Xie, H.; Qiu, Y. Optical properties of photovoltaic materials: Organic-inorganic mixed halide perovskites CH3NH3Pb (I1-yXy) 3 (X=Cl, Br). Comput. Theor. Chem. 2018, 1144, 1–8. [Google Scholar] [CrossRef]
- Lyu, M.; Yun, J.-H.; Cai, M.; Jiao, Y.; Bernhardt, P.V.; Zhang, M.; Wang, Q.; Du, A.; Wang, H.; Liu, G. Organic–inorganic bismuth (III)-based material: A lead-free, air-stable and solution-processable light-absorber beyond organolead perovskites. Nano Res. 2016, 9, 692–702. [Google Scholar] [CrossRef] [Green Version]
- Oswald, I.W.H.; Mozur, E.M.; Moseley, I.P.; Ahn, H.; Neilson, J.R. Hybrid Charge-Transfer Semiconductors: (C7H7)SbI4, (C7H7)BiI4, and Their Halide Congeners. Inorg. Chem. 2019, 58, 5818–5826. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qiu, Y.; Liu, W.; Chen, W.; Chen, W.; Zhou, G.; Hsu, P.-C.; Zhang, R.; Liang, Z.; Fan, S.; Zhang, Y. Efficient solar-driven water splitting by nanocone BiVO4-perovskite tandem cells. Sci. Adv. 2016, 2, e1501764. [Google Scholar] [CrossRef] [Green Version]
- Ferjani, H. Structural, Hirshfeld surface analysis, morphological approach, and spectroscopic study of new hybrid iodobismuthate containing tetranuclear 0D cluster Bi4I16 4 (C6H9N2) 2 (H2O). Crystals 2020, 10, 397. [Google Scholar] [CrossRef]
- Rhaiem, T.B.; Elleuch, S.; Boughzala, H.; Abid, Y. A new luminescent organic-inorganic hybrid material based on cadmium iodide. Inorg. Chem. Commun. 2019, 109, 107572. [Google Scholar] [CrossRef]
- Ferjani, H.; Bechaieb, R.; Abd El-Fattah, W.; Fettouhi, M. Broad-band luminescence involving fluconazole antifungal drug in a lead-free bismuth iodide perovskite: Combined experimental and computational insights. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2020, 237, 118354. [Google Scholar] [CrossRef]
- Intissar, H.; Khan, Y.; Aouaini, F.; Seo, J.H.; Koo, H.-J.; Turnbull, M.M.; Walker, B.; Naïli, H. A Copper-Based 2D Hybrid Perovskite Solar Absorber as a Potential Eco-Friendly Alternative to Lead Halide Perovskites. J. Mater. Chem. C 2022, 10, 3738–3745. [Google Scholar]
- Ferjani, H.; Boughzala, H. New Quasi-One-Dimensional Organic-Inorganic Hybrid Material: 1, 3-Bis (4-piperidinium) propane Pentachlorobismuthate (III) Synthesis, Crystal Structure, and Spectroscopic Studies. J. Mater. 2014, 2014, 253602. [Google Scholar] [CrossRef]
- Ferjani, H.; Boughzala, H.; Driss, A. Poly [bis (1-carbamoylguanidinium)[tri-μ-chlorido-dichloridobismuthate (III)]]. Acta Crystallogr. Sect. E Struct. Rep. Online 2012, 68, m615. [Google Scholar] [CrossRef] [Green Version]
- Bravais, A. Etudes Cristallographiques; Gauthier-Villars: France, Paris, 1866. [Google Scholar]
- Donnay, J.; Harker, D. Springer Handbook of Crystal Growth. Am. Mineral. 1937, 22, 446–467. [Google Scholar]
- Renjith, R.; Mary, Y.S.; Panicker, C.Y.; Varghese, H.T.; Pakosińska-Parys, M.; Van Alsenoy, C.; Manojkumar, T. Spectroscopic (FT-IR, FT-Raman), first order hyperpolarizability, NBO analysis, HOMO and LUMO analysis of 1,7,8,9-tetrachloro-10,10-dimethoxy-4-[3-(4-phenylpiperazin-1-yl)propyl]-4-azatricyclo[5.2.1.02,6]dec-8-ene-3,5-dione by density functional methods. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2014, 124, 500–513. [Google Scholar] [CrossRef] [PubMed]
- Jones, W.J. The infra-red spectrum and structure of guanidine. Trans. Faraday Soc. 1959, 55, 524–531. [Google Scholar] [CrossRef]
- Jones, W.J.; Orville-Thomas, W. The infra-red spectrum and structure of dicyandiamide. Trans. Faraday Soc. 1959, 55, 193–202. [Google Scholar] [CrossRef]
- Scoponi, M.; Polo, E.; Bertolasi, V.; Carassiti, V.; Bertelli, G. Crystal structure and spectroscopic analyses of guanylurea hydrochloride. Evidence of the influence of hydrogen bonding on the π-electron delocalization. J. Chem. Soc. Perkin Trans. 1991, 2, 1619–1624. [Google Scholar] [CrossRef]
- Matulková, I.; Fridrichová, M.; Císařová, I.; Vaněk, P.; Uhlík, F.; Němec, I. Vibrational spectroscopic and crystallographic study of the novel guanylurea salts with sulphuric and selenic acids. J. Mol. Struct. 2017, 1131, 294–305. [Google Scholar] [CrossRef]
- Rao, A.S.; Baruah, U.; Das, S.K. Stabilization of [BiCl6] 3− and [Bi2Cl10] 4− with various organic precursors as cations leading to inorganic–organic supramolecular adducts: Syntheses, crystal structures and properties of [C5H7N2] 3 [BiCl6],[C5H7N2][C5H8N2][BiCl6] and [C10H10N2] 2 [Bi2Cl10]. Inorg. Chim. Acta 2011, 372, 206–212. [Google Scholar]
- Oldenburg, K.; Vogler, A.; Mikó, I.; Horváth, O. Photoredox decomposition of tin (II), lead (II), antimony (III) and bismuth (III) iodide complexes in solution. Inorg. Chim. Acta 1996, 248, 107–110. [Google Scholar] [CrossRef]
- Clark, S.J.; Segall, M.D.; Pickard, C.J.; Hasnip, P.J.; Probert, M.I.; Refson, K.; Payne, M.C. First principles methods using CASTEP. Z. Für Krist.-Cryst. Mater. 2005, 220, 567–570. [Google Scholar] [CrossRef] [Green Version]
- Vogler, A.; Nikol, H. Photochemistry and photophysics of coordination compounds of the main group metals. Pure Appl. Chem. 1992, 64, 1311–1317. [Google Scholar] [CrossRef] [Green Version]
- Moore, C.H. Coordination Chemistry of Guanidine Derivatives; University of Nottingham: Nottingham, UK, 1989. [Google Scholar]
- Badgujar, D.M.; Wagh, R.M.; Pawar, S.J.; Sikder, A.K. Process optimization for synthesis of guanylurea dinitramide (GUDN). Propellants Explos. Pyrotech. 2014, 39, 658–661. [Google Scholar] [CrossRef]
- Wu, L.-M.; Wu, X.-T.; Chen, L. Structural overview and structure–property relationships of iodoplumbate and iodobismuthate. Coord. Chem. Rev. 2009, 253, 2787–2804. [Google Scholar] [CrossRef]
- Adonin, S.A.; Sokolov, M.N.; Fedin, V.P. Polynuclear halide complexes of Bi (III): From structural diversity to the new properties. Coord. Chem. Rev. 2016, 312, 1–21. [Google Scholar] [CrossRef]
- Ferjani, H.; Boughzala, H.; Driss, A. Synthesis, Crystal Structure, and Characterization of a New Organic-Inorganic Hybrid Material. J. Crystallogr. 2013, 2013, 658939. [Google Scholar] [CrossRef] [Green Version]
- Pelle, F.; Jacquier, B.; Denis, J.; Blanzat, B. Optical properties of Cs2NaBiCl6. J. Lumin. 1978, 17, 61–72. [Google Scholar] [CrossRef]
- Chang, L.; Besteiro, L.V.; Sun, J.; Santiago, E.Y.; Gray, S.K.; Wang, Z.; Govorov, A.O. Electronic structure of the plasmons in metal nanocrystals: Fundamental limitations for the energy efficiency of hot electron generation. ACS Energy Lett. 2019, 4, 2552–2568. [Google Scholar] [CrossRef]
- Toll, J.S. Causality and the dispersion relation: Logical foundations. Phys. Rev. 1956, 104, 1760. [Google Scholar] [CrossRef]
- Gajdoš, M.; Hummer, K.; Kresse, G.; Furthmüller, J.; Bechstedt, F. Linear optical properties in the projector-augmented wave methodology. Phys. Rev. B 2006, 73, 045112. [Google Scholar] [CrossRef] [Green Version]
- Yu, P.Y.; Cardona, M. Electronic band structures. In Fundamentals of Semiconductors; Springer: Berlin/Heidelberg, Germany, 2010; pp. 17–106. [Google Scholar]
- Cardona, M.; Peter, Y.Y. Fundamentals of Semiconductors; Springer: Berlin/Heidelberg, Germany, 2005; Volume 619. [Google Scholar]
- Crespo, C.T. The effect of the halide anion on the optical properties of lead halide perovskites. Sol. Energy Mater. Sol. Cells 2019, 195, 269–273. [Google Scholar] [CrossRef]
- Liu, S.; Wang, J.; Hu, Z.; Duan, Z.; Zhang, H.; Zhang, W.; Guo, R.; Xie, F. Role of organic cation orientation in formamidine based perovskite materials. Sci. Rep. 2021, 11, 20433. [Google Scholar] [CrossRef]
- Pazoki, M.; Johansson, M.B.; Zhu, H.; Broqvist, P.; Edvinsson, T.; Boschloo, G.; Johansson, E.M. Bismuth iodide perovskite materials for solar cell applications: Electronic structure, optical transitions, and directional charge transport. J. Phys. Chem. C 2016, 120, 29039–29046. [Google Scholar] [CrossRef]
- Alnujaim, S.; Bouhemadou, A.; Bedjaoui, A.; Bin-Omran, S.; Al-Douri, Y.; Khenata, R.; Maabed, S. Ab initio prediction of the elastic, electronic and optical properties of a new family of diamond-like semiconductors, Li2HgMS4 (M=Si, Ge and Sn). J. Alloys Compd. 2020, 843, 155991. [Google Scholar] [CrossRef]
- Perdew, J.P.; Ruzsinszky, A.; Csonka, G.I.; Vydrov, O.A.; Scuseria, G.E.; Constantin, L.A.; Zhou, X.; Burke, K. Restoring the density-gradient expansion for exchange in solids and surfaces. Phys. Rev. Lett. 2008, 100, 136406. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vanderbilt, D. Soft self-consistent pseudopotentials in a generalized eigenvalue formalism. Phys. Rev. B 1990, 41, 7892. [Google Scholar] [CrossRef]
- Monkhorst, H.J.; Pack, J.D. Special points for Brillouin-zone integrations. Phys. Rev. B 1976, 13, 5188. [Google Scholar] [CrossRef]
- Spackman, M.; Jayatilaka, D. Hirshfeld Surface Analysis. CrystEngComm 2009, 11, 19–32. [Google Scholar] [CrossRef]
- Spackman, M.A.; Spackman, P.R.; Thomas, S.P. Beyond Hirshfeld Surface Analysis: Interaction Energies, Energy Frameworks and Lattice Energies with CrystalExplorer. Complementary Bond. Anal. Gruyter 2021, 329–352. [Google Scholar] [CrossRef]
- Jelsch, C.; Ejsmont, K.; Huder, L. The enrichment ratio of atomic contacts in crystals, an indicator derived from the Hirshfeld surface analysis. IUCrJ 2014, 1, 119–128. [Google Scholar] [CrossRef]
- Materials Studio, Version 7.0; Accelrys Software Inc.: San Diego, CA, USA, 2013.

















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| hkl | Multiplicity | dhkl | Distance | Total Facet Area | % Total Facet Area |
|---|---|---|---|---|---|
| (020) | 2 | 10.35 | 9.66 | 817.08 | 28.93 |
| (011) | 4 | 9.73 | 10.27 | 1.148620 × 103 | 40.67 |
| (101) | 4 | 6.13 | 16.30 | 481.35 | 17.05 |
| (111) | 8 | 5.88 | 17.00 | 377.30 | 13.36 |
| D-H···A | D-H (Å) | H···A (Å) | D···A (Å) | D-H···A (°) |
|---|---|---|---|---|
| N1-H1A···Cl1 ii | 0.86 | 2.607 | 3.271 (8) | 135 |
| N1-H1B···Cl2 iv | 0.86 | 2.499 | 3.329 (7) | 162 |
| N2-H2···Cl4i v | 0.86 | 2.702 | 3.524 (7) | 160 |
| N3-H3A···O v | 0.86 | 2.21 | 3.053 (8) | 167 |
| N3-H3B··· O | 0.86 | 2.08 | 2.734 (8) | 132 |
| N4-H4A···Cl1 vi | 0.86 | 2.525 | 3.347 (7) | 160 |
| N4-H4B···Cl4 iv | 0.86 | 2.594 | 3.421 (7) | 162 |
| Atoms | H | C | N | O | Cl |
|---|---|---|---|---|---|
| H | 25.3 | - | - | - | - |
| C | 6.3 | / | - | - | - |
| N | 14 | 1 | 1.1 | - | - |
| O | 12 | 1.5 | 2.6 | 1.3 | - |
| Cl | 28.7 | 1.4 | 2.1 | 2.8 | 0 |
| Surface % | 55.8 | 5.1 | 10.95 | 10.75 | 17.5 |
| Random Contacts (RXX and RXY) | |||||
| H | 31.14 | - | - | - | - |
| C | 5.69 | 0.26 | - | - | - |
| N | 12.22 | 1.12 | 1.20 | - | - |
| O | 12 | 1.10 | 2.35 | 1.16 | - |
| Cl | 19.53 | 1.79 | 3.83 | 3.76 | 3.06 |
| Enrichment Ratio E | |||||
| H | 0.81 | - | - | - | - |
| C | 1.11 | / | - | - | - |
| N | 1.15 | 0.89 | 0.91 | - | - |
| O | 1 | 1.36 | 1.11 | 1.12 | - |
| Cl | 1.47 | 0.78 | 0.55 | 0.74 | / |
| Observed Wavenumbers (cm−1) | Guanylurea Hydrochloride [27] | Assignments |
|---|---|---|
| 3407 3320 | 3408 3320 | υas(NH2), υs(NH2) |
| 3220 3199 | 3230 3152 | υ(NH) |
| 1736 | 1736 | δ(NH2)Guanidine |
| 1689 | 1672 | υ(CO)Amide |
| 1638 | 1638 | υ(C=N) |
| 1584 | 1583 | δ(NH2) Amide |
| 1522 | 1523 | υa(NCN) |
| 1467 | 1460 | υ(CN)Amide |
| 1339 | 1341 | δ(NH) |
| 1112 1083 | 1117 1057 | ρ(NH2) |
| 923 | 932 | υs(NCN) |
| 764–713 | 756–716 | ω(NCN)/δ(NCN) |
| 558–505 | 535 | ω(NH2) |
| 490 | 448 428 | ρ(NCN) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ferjani, H.; Ben Smida, Y.; Onwudiwe, D.C.; Elamin, N.Y.; Ezzine, S.; Almotlaq, N.S. An Experimental and Theoretical Study of the Optical Properties of (C2H7N4O)2BiCl5 for an Optoelectronic Application. Inorganics 2022, 10, 48. https://0-doi-org.brum.beds.ac.uk/10.3390/inorganics10040048
Ferjani H, Ben Smida Y, Onwudiwe DC, Elamin NY, Ezzine S, Almotlaq NS. An Experimental and Theoretical Study of the Optical Properties of (C2H7N4O)2BiCl5 for an Optoelectronic Application. Inorganics. 2022; 10(4):48. https://0-doi-org.brum.beds.ac.uk/10.3390/inorganics10040048
Chicago/Turabian StyleFerjani, Hela, Youssef Ben Smida, Damian C. Onwudiwe, Nuha Y. Elamin, Safa Ezzine, and Norah S. Almotlaq. 2022. "An Experimental and Theoretical Study of the Optical Properties of (C2H7N4O)2BiCl5 for an Optoelectronic Application" Inorganics 10, no. 4: 48. https://0-doi-org.brum.beds.ac.uk/10.3390/inorganics10040048