
Dinuclear Lanthanide (III) Coordination Polymers in a Domino Reaction

 , ,
, , 
Abstract
:
1. Introduction


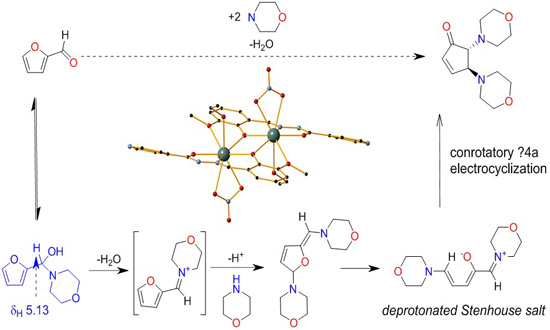
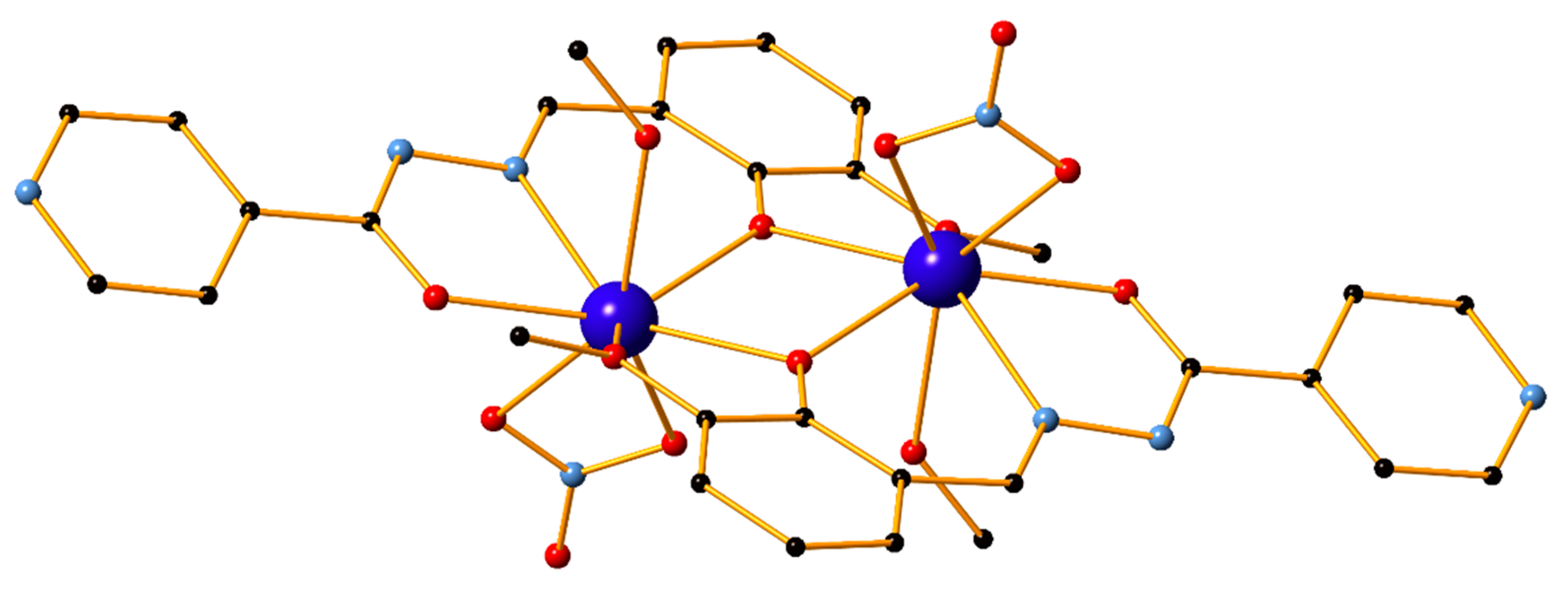
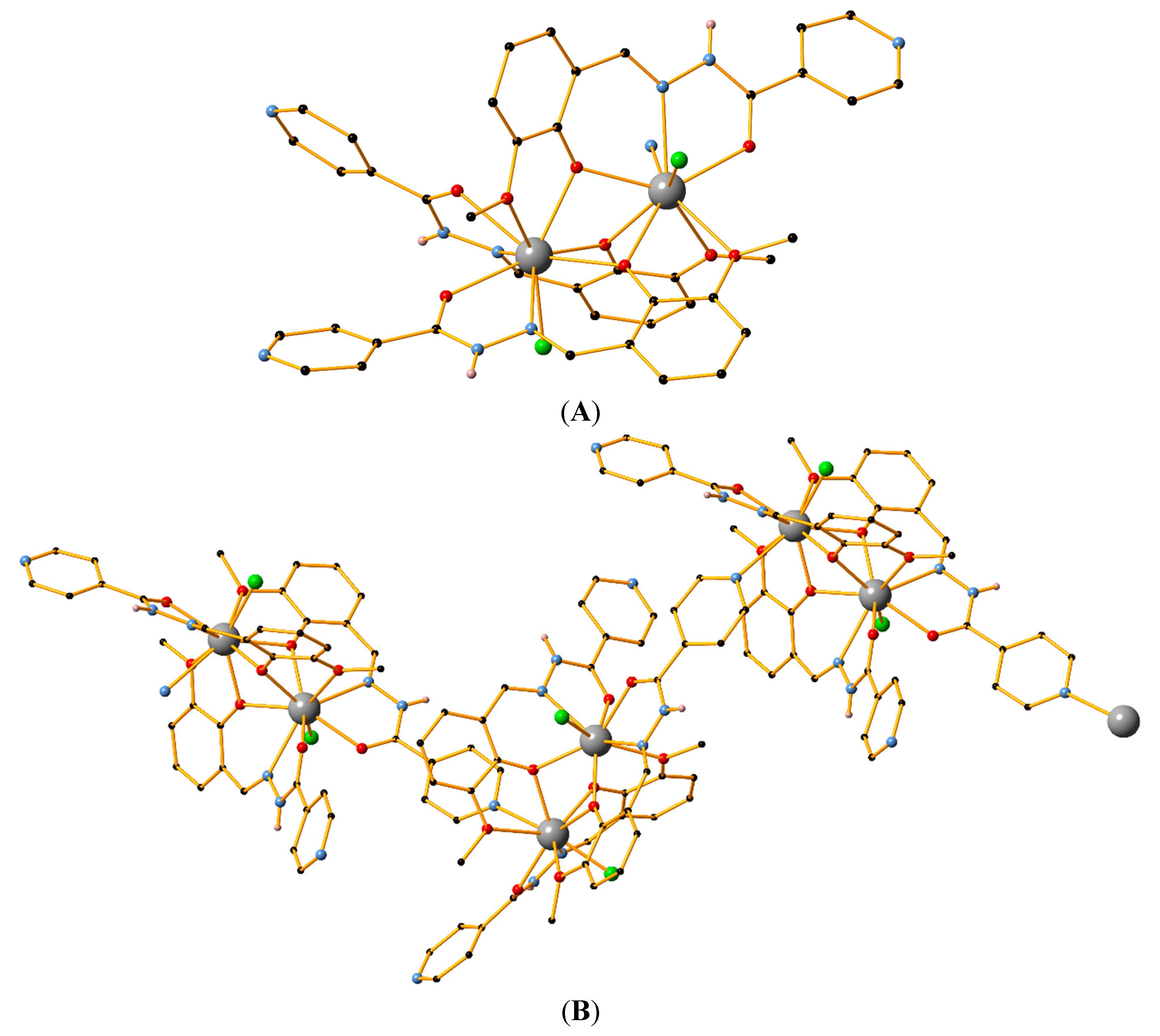
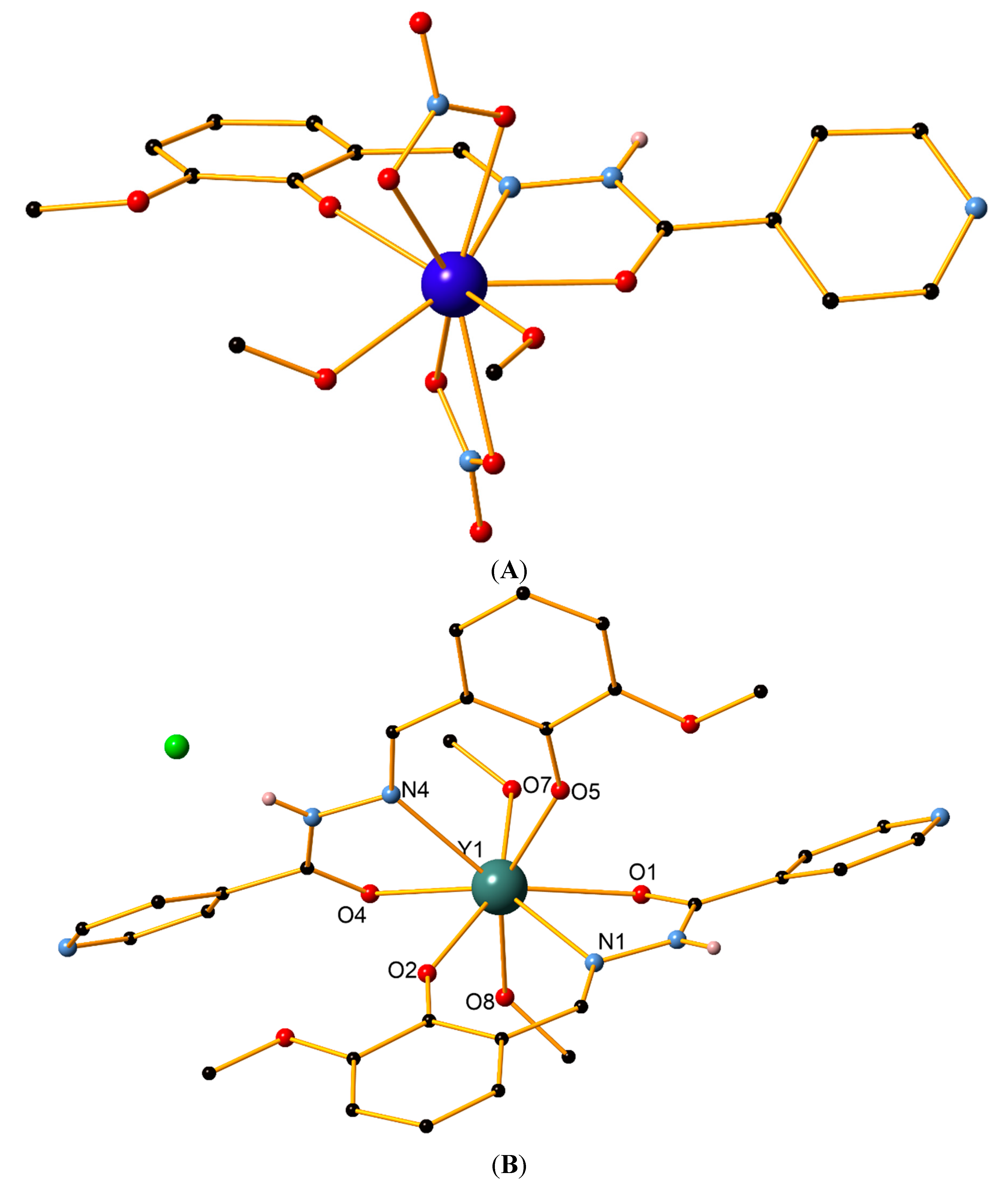
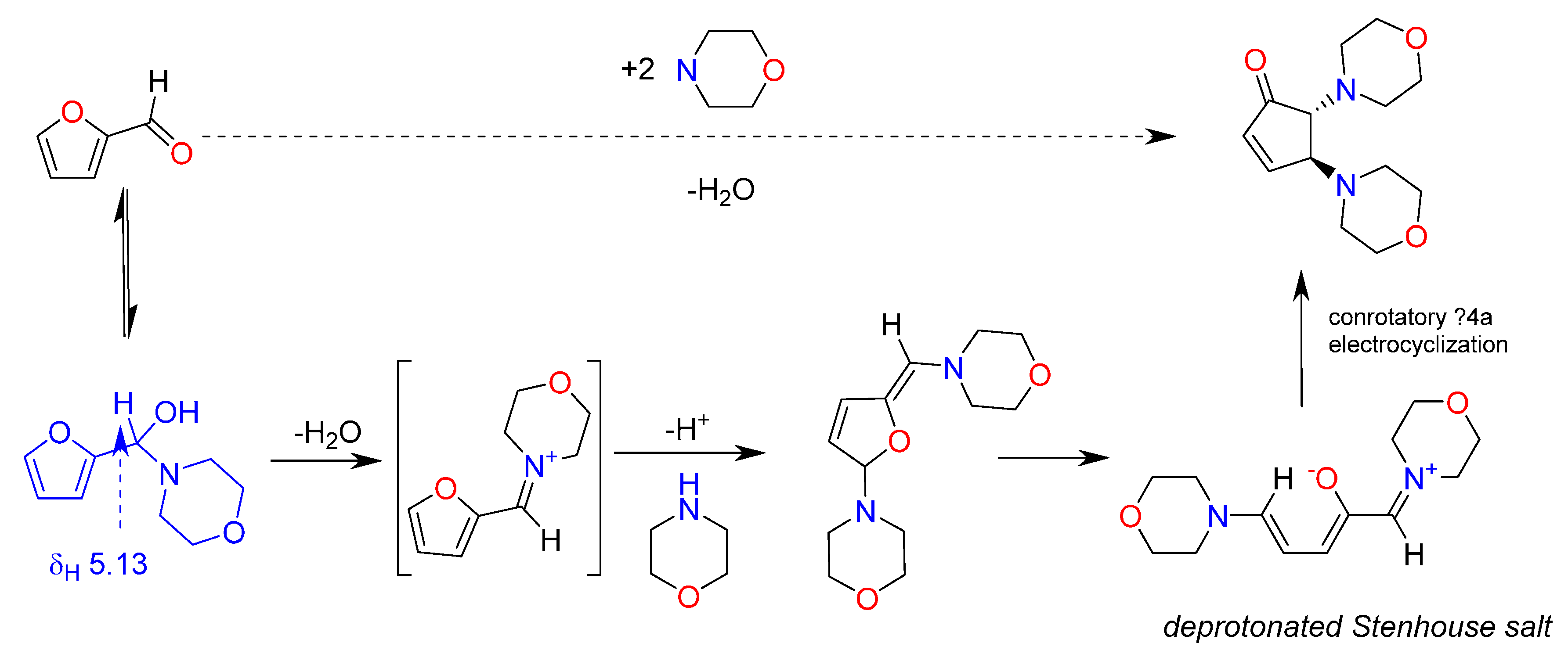
2. Results and Discussion



| Entry | Catalyst | Loading/% b | Time/h | Yield/% |
|---|---|---|---|---|
| 1 | 1-Dy | 10 | 2 | 100 |
| 2 | 1-Dy | 5 | 2 | 96 |
| 3 | 1-Dy | 2.5 | 2 | 95 |
| 4 | 1-Dy | 1 | 2 | 80 |
| 5 | 2-Dy | 10 | 2 | 100 |
| 6 | 2-Dy | 5 | 2 | 90 |
| 7 | 2-Dy | 2.5 | 2 | 88 |
| 8 | 2-Dy | 1 | 2 | 79 |
| 9 | 1-Gd | 2.5 | 2 | 80 |
| 10 | 2-Gd | 2.5 | 2 | 54 |
| 11 | 1-Y | 2.5 | 2 | 100 |
| 12 | 1-Y | 1 | 2 | 89 |
| 13 | 1-Y | 0.5 | 2 | 68 |
| 14 | 2-Y | 2.5 | 2 | 100 |
| 15 | 2-Y | 1 | 2 | 85 |
| 16 | 2-Y | 0.5 | 2 | 38 |


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}

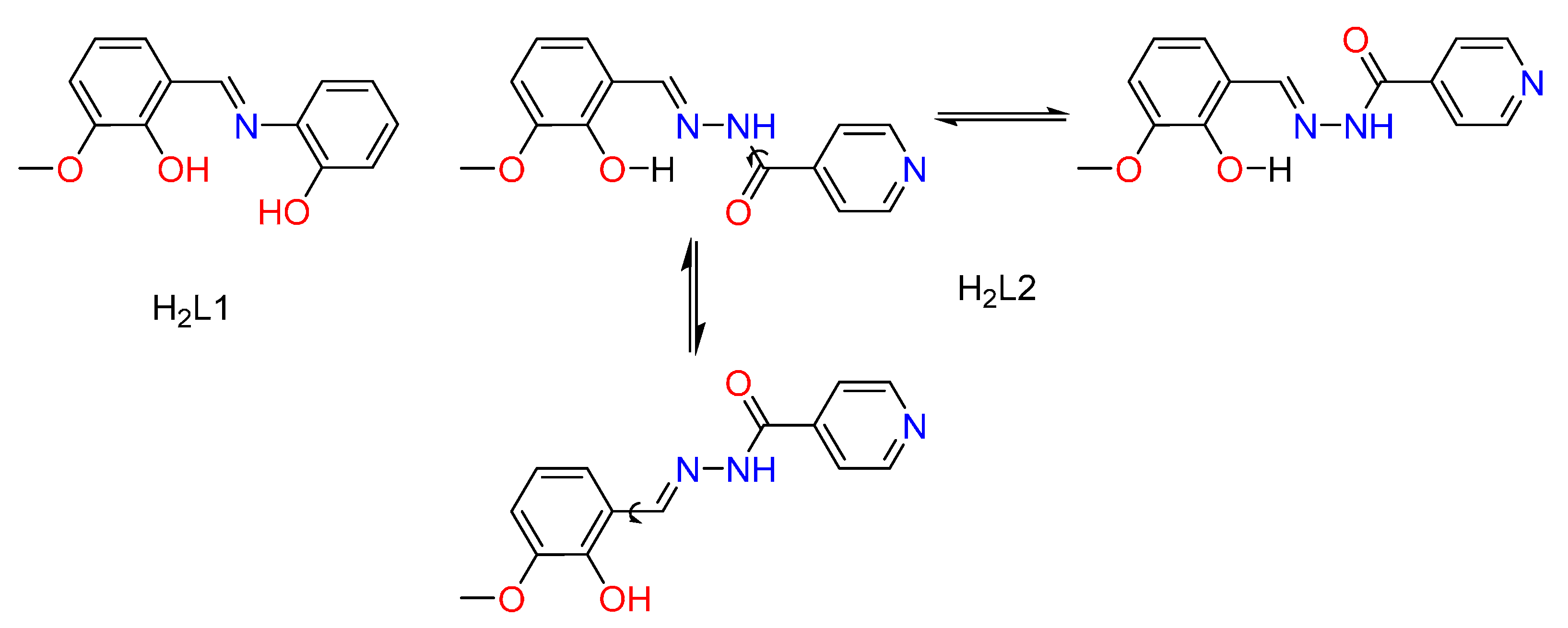
2.1. Solution Studies
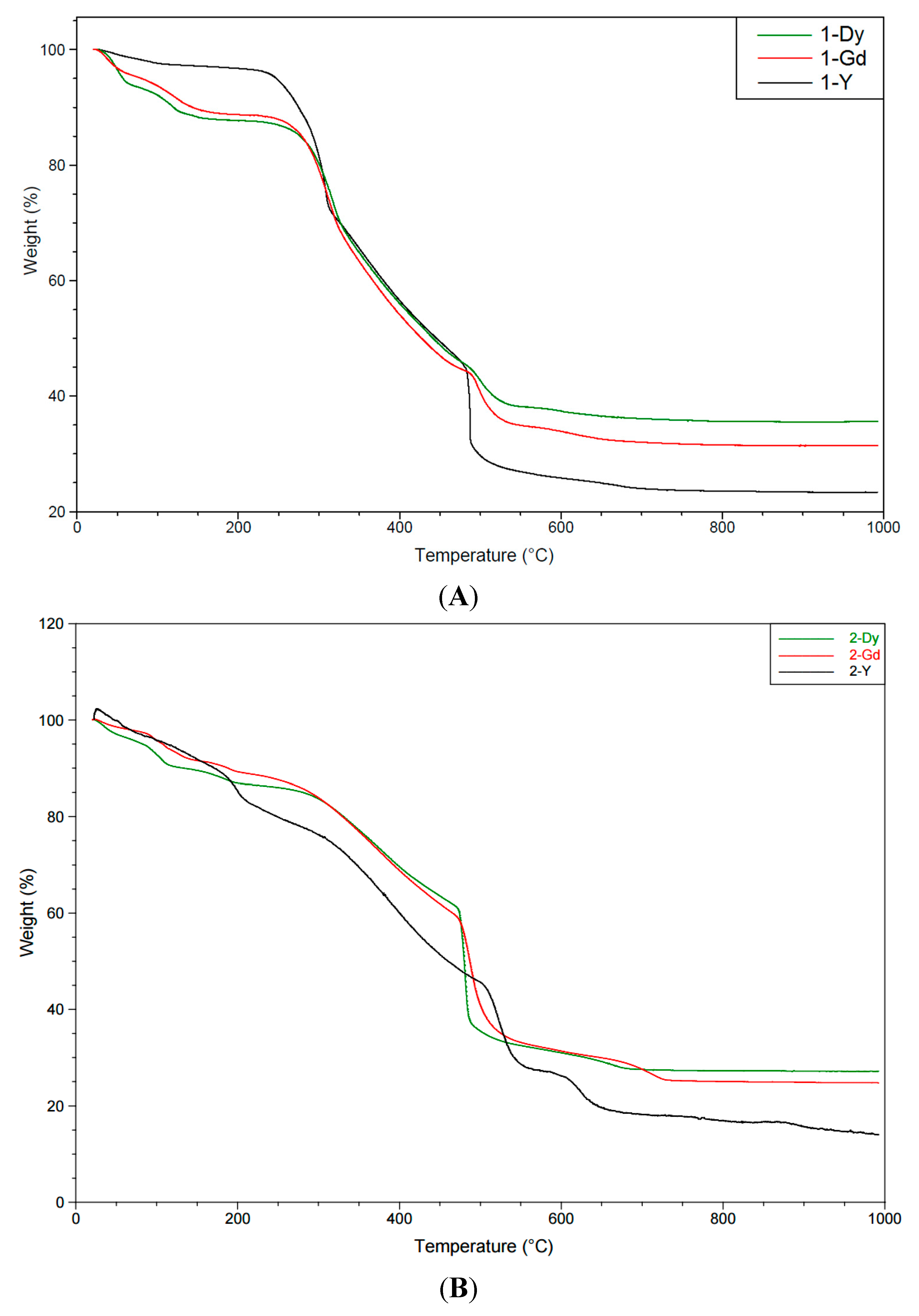
2.2. Thermal and Powder XRD Studies

3. Experimental Section
3.1. Materials
3.2. Instrumentation
3.3. Crystallography
3.4. Ligand Synthesis
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Zheng, Z. Ligand-controlled self-assembly of polynuclear lanthanide-oxo/hydroxo complexes: From synthetic serendipity to rational supramolecular design. Chem. Commun. 2001, 2521–2529. [Google Scholar] [CrossRef]
- D’Alessio, D.; Sobolev, A.N.; Skelton, B.W.; Fuller, R.O.; Woodward, R.C.; Lengkeek, N.A.; Fraser, B.H.; Massi, M.; Ogden, M.I. Lanthanoid “bottlebrush” clusters: Remarkably elongated metal-oxo core structures with controllable lengths. J. Am. Chem. Soc. 2014, 136, 15122–15125. [Google Scholar] [CrossRef] [PubMed]
- Thielemann, D.T.; Wagner, A.T.; Lan, Y.; Oña-Burgos, P.; Fernández, I.; Rösch, E.S.; Kölmel, D.K.; Powell, A.K.; Bräse, S.; Roesky, P.W. Peptoid-ligated pentadecanuclear yttrium and dysprosium hydroxy clusters. Chem. Eur. J. 2015, 21, 2813–2820. [Google Scholar] [CrossRef] [PubMed]
- Wu, M.; Jiang, F.; Kong, X.; Yuan, D.; Long, L.; Al-Thabaiti, S.A.; Hong, M. Two polymeric 36-metal pure lanthanide nanosize clusters. Chem. Sci. 2013, 4, 3104–3109. [Google Scholar] [CrossRef]
- Peng, J.-B.; Kong, X.-J.; Zhang, Q.-C.; Orendáč, M.; Prokleška, J.; Ren, Y.-P.; Long, L.-S.; Zheng, Z.; Zheng, L.-S. Beauty, symmetry, and magnetocaloric effect—Four-shell keplerates with 104 lanthanide atoms. J. Am. Chem. Soc. 2014, 136, 17938–17941. [Google Scholar] [CrossRef] [PubMed]
- Woodruff, D.N.; Winpenny, R.E.P.; Layfield, R.A. Lanthanide single-molecule magnets. Chem. Rev. 2013, 113, 5110–5148. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.; Guo, Y.-N.; Tang, J. Recent advances in dysprosium-based single molecule magnets: Structural overview and synthetic strategies. Coord. Chem. Rev. 2013, 257, 1728–1763. [Google Scholar] [CrossRef]
- Amoroso, A.J.; Pope, S.J.A. Using lanthanide ions in molecular bioimaging. Chem. Soc. Rev. 2015, 44, 4723–4442. [Google Scholar] [CrossRef] [PubMed]
- Bünzli, J.-C.G. On the design of highly luminescent lanthanide complexes. Coord. Chem. Rev. 2015, 293–294, 19–47. [Google Scholar] [CrossRef]
- Eliseeva, S.V.; Bünzli, J.-C.G. Lanthanide luminescence for functional materials and bio-sciences. Chem. Soc. Rev. 2010, 39, 189–227. [Google Scholar] [CrossRef] [PubMed]
- Anastasiadis, N.C.; Granadeiro, C.M.; Klouras, N.; Cunha-Silva, L.; Raptopoulou, C.P.; Psycharis, V.; Bekiari, V.; Balula, S.S.; Escuer, A.; Perlepes, S.P. Dinuclear lanthanide(III) complexes by metal-ion-assisted hydration of di-2-pyridyl ketone azine. Inorg. Chem. 2013, 52, 4145–4147. [Google Scholar] [CrossRef] [PubMed]
- Robinson, J.R.; Gu, J.; Carroll, P.J.; Schelter, E.J.; Walsh, P.J. Exchange processes in Shibasaki’s rare earth alkali metal BINOLate frameworks and their relevance in multifunctional asymmetric catalysis. J. Am. Chem. Soc. 2015, 137, 7135–7144. [Google Scholar] [CrossRef] [PubMed]
- Roesky, P.W.; Canseco-Melchor, G.; Zulys, A. A pentanuclear yttrium hydroxo cluster as an oxidation catalyst. Catalytic oxidation of aldehydes in the presence of air. Chem. Commun. 2004, 738–739. [Google Scholar] [CrossRef] [PubMed]
- Bi, Y.; Wang, X.-T.; Liao, W.; Wang, X.; Deng, R.; Zhang, H.; Gao, S. Thiacalix[4]arene-supported planar Ln(4) (Ln = TbIII, DyIII) clusters: Toward luminescent and magnetic bifunctional materials. Inorg. Chem. 2009, 48, 11743–11747. [Google Scholar] [CrossRef] [PubMed]
- Alexandropoulos, D.I.; Fournet, A.; Cunha-Silva, L.; Mowson, A.M.; Bekiari, V.; Christou, G.; Stamatatos, T.C. Fluorescent naphthalene diols as bridging ligands in LnIII cluster chemistry: Synthetic, structural, magnetic, and photophysical characterization of LnIII8 “Christmas Stars”. Inorg. Chem. 2014, 53, 5420–5422. [Google Scholar] [CrossRef] [PubMed]
- Pointillart, F.; le Guennic, B.; Golhen, S.; Cador, O.; Maury, O.; Ouahab, L. A redox-active luminescent ytterbium based single molecule magnet. Chem. Commun. 2013, 49, 615–617. [Google Scholar] [CrossRef] [PubMed]
- Mazarakioti, E.C.; Poole, K.M.; Cunha-Silva, L.; Christou, G.; Stamatatos, T.C. A new family of Ln7 clusters with an ideal D3h metal-centered trigonal prismatic geometry, and SMM and photoluminescence behaviors. Dalton Trans. 2014, 43, 11456–11460. [Google Scholar] [CrossRef] [PubMed]
- Bag, P.; Rastogi, C.K.; Biswas, S.; Sivakumar, S.; Mereacre, V.; Chandrasekhar, V. Homodinuclear lanthanide {Ln2} (Ln = Gd, Tb, Dy, Eu) complexes prepared from an o-vanillin based ligand: Luminescence and single-molecule magnetism behavior. Dalton Trans. 2015, 44, 4328–4340. [Google Scholar] [CrossRef] [PubMed]
- Chandrasekhar, V.; Bag, P.; Colacio, E. Octanuclear {LnIII8}(Ln = Gd, Tb, Dy, Ho) macrocyclic complexes in a cyclooctadiene-like conformation: Manifestation of slow relaxation of magnetization in the DyIII derivative. Inorg. Chem. 2013, 52, 4562–4570. [Google Scholar] [CrossRef] [PubMed]
- Pasatoiu, T.D.; Tiseanu, C.; Madalan, A.M.; Jurca, B.; Duhayon, C.; Sutter, J.P.; Andruh, M. Study of the luminescent and magnetic properties of a series of heterodinuclear [ZnIILnIII] complexes. Inorg. Chem. 2011, 50, 5879–5889. [Google Scholar] [CrossRef] [PubMed]
- Ehama, K.; Ohmichi, Y.; Sakamoto, S.; Fujinami, T.; Matsumoto, N.; Mochida, N.; Ishida, T.; Sunatsuki, Y.; Tsuchimoto, M.; Re, N. Synthesis, structure, luminescent, and magnetic properties of carbonato-bridged ZnII2LnIII2 complexes [(μ4-CO3)2{ZnIILnLnIII(NO3)}2] (LnIII = GdIII, TbIII, DyIII; L1 = N,N′-bis(3-methoxy-2-oxybenzylidene)-1,3-propanediaminato, L2 = N,N′-bis(3-ethoxy-2-oxybenzylidene)-1,3-propanediaminato). Inorg. Chem. 2013, 52, 12828–12841. [Google Scholar] [PubMed]
- Goura, J.; Mereacre, V.; Novitchi, G.; Powell, A.K.; Chandrasekhar, V. Homometallic FeIII4 and heterometallic {FeIII4LnIII2} (Ln = Dy, Tb) complexes—Syntheses, structures, and magnetic properties. Eur. J. Inorg. Chem. 2015, 2015, 156–165. [Google Scholar] [CrossRef]
- Ke, H.; Zhao, L.; Guo, Y.; Tang, J. A Dy6 cluster displays slow magnetic relaxation with an edge-to-edge arrangement of two Dy3 triangles. Eur. J. Inorg. Chem. 2011, 2011, 4153–4156. [Google Scholar] [CrossRef]
- Liao, S.; Yang, X.; Jones, R.A. Self-assembly of luminescent hexanuclear lanthanide salen complexes. Cryst. Growth Des. 2012, 12, 970–974. [Google Scholar] [CrossRef]
- Sarwar, M.; Madalan, A.M.; Tiseanu, C.; Novitchi, G.; Maxim, C.; Marinescu, G.; Luneau, D.; Andruh, M. A new synthetic route towards binuclear 3d-4f complexes, using non-compartmental ligands derived from o-vanillin. Syntheses, crystal structures, magnetic and luminescent properties. New J. Chem. 2013, 37, 2280–2292. [Google Scholar] [CrossRef]
- Mondal, K.C.; Kostakis, G.E.; Lan, Y.; Powell, A.K. Magnetic properties of five planar defect dicubanes of [LnIII4(μ3-OH)2(L)4(HL)2]·2THF (Ln = Gd, Tb, Dy, Ho and Er). Polyhedron 2013, 66, 268–273. [Google Scholar] [CrossRef]
- Gómez, V.; Vendier, L.; Corbella, M.; Costes, J.-P. Tetranuclear [Co–Gd]2 complexes: Aiming at a better understanding of the 3d-Gd magnetic interaction. Inorg. Chem. 2012, 51, 6396–6404. [Google Scholar] [CrossRef] [PubMed]
- Athanasopoulou, A.A.; Pilkington, M.; Raptopoulou, C.P.; Escuer, A.; Stamatatos, T.C. Structural aesthetics in molecular nanoscience: A unique Ni26 cluster with a “rabbit-face” topology and a discrete Ni18 “molecular chain”. Chem. Commun. 2014, 50, 14942–14945. [Google Scholar] [CrossRef] [PubMed]
- Tziotzi, T.G.; Tzimopoulos, D.I.; Lis, T.; Inglis, R.; Milios, C.J. Dodecanuclear [MnLn] species: Synthesis, structures and characterization of magnetic relaxation phenomena. Dalton Trans. 2015, 44, 11696–11699. [Google Scholar] [CrossRef] [PubMed]
- Meng, Z.-S.; Guo, F.-S.; Liu, J.-L.; Leng, J.-D.; Tong, M.-L. Heterometallic cubane-like {M2Ln2} (M = Ni, Zn; Ln =, Gd, Dy) and {Ni2Y2} aggregates. Synthesis, structures and magnetic properties. Dalton Trans. 2012, 41, 2320–2329. [Google Scholar] [CrossRef] [PubMed]
- Fan, L.-L.; Guo, F.-S.; Yun, L.; Lin, Z.-J.; Herchel, R.; Leng, J.-D.; Ou, Y.-C.; Tong, M.-L. Chiral transition metal clusters from two enantiomeric schiff base ligands. Synthesis, structures, CD spectra and magnetic properties. Dalton Trans. 2010, 39, 1771–1780. [Google Scholar] [CrossRef] [PubMed]
- Thio, Y.; Toh, S.W.; Xue, F.; Vittal, J.J. Self-assembly of a 15-nickel metallamacrocyclic complex derived from the l-glutamic acid Schiff base ligand. Dalton Trans. 2014, 43, 5998–6001. [Google Scholar] [CrossRef] [PubMed]
- Berkoff, B.; Griffiths, K.; Abdul-Sada, A.; Tizzard, G.J.; Coles, S.; Escuer, A.; Kostakis, G.E. A new family of high nuclearity CoII/DyIII coordination clusters possessing robust and unseen topologies. Dalton Trans. 2015, 44, 12788–12795. [Google Scholar] [CrossRef] [PubMed]
- Nemec, I.; Machata, M.; Herchel, R.; Boča, R.; Trávníček, Z. A new family of Fe2Ln complexes built from mononuclear anionic Schiff base subunits. Dalton Trans. 2012, 41, 14603–14610. [Google Scholar] [CrossRef] [PubMed]
- Costes, J.-P.; Duhayon, C. An ionic dysprosium complex made of a hexanuclear Dy6 cationic cluster and a mononuclear Dy anionic unit. Eur. J. Inorg. Chem. 2014, 2014, 4745–4749. [Google Scholar] [CrossRef]
- Loukopoulos, E.; Berkoff, B.; Abdul-Sada, A.; Tizzard, G.J.; Coles, S.J.; Escuer, A.; Kostakis, G.E. A disk-like CoII3DyIII4 coordination cluster exhibiting single molecule magnet behavior. Eur. J. Inorg. Chem. 2015, 2015, 2646–2649. [Google Scholar] [CrossRef]
- Andruh, M. The exceptionally rich coordination chemistry generated by Schiff-base ligands derived from o-vanillin. Dalton Trans. 2015, 44, 16633–16653. [Google Scholar] [CrossRef] [PubMed]
- Griffiths, K.; Gallop, C.W.D.; Abdul-Sada, A.; Vargas, A.; Navarro, O.; Kostakis, G.E. Heteronuclear 3 d/DyIII coordination clusters as catalysts in a domino reaction. Chem. Eur. J. 2015, 21, 6358–6361. [Google Scholar] [CrossRef] [PubMed]
- Li, S.-W.; Batey, R.A. Mild lanthanide(III) catalyzed formation of 4,5-diaminocyclopent-2-enones from 2-furaldehyde and secondary amines: A domino condensation/ring-opening/electrocyclization process. Chem. Commun. 2007, 3759–3761. [Google Scholar] [CrossRef] [PubMed]
- Lin, P.-H.; Gorelsky, S.; Savard, D.; Burchell, T.J.; Wernsdorfer, W.; Clérac, R.; Murugesu, M. Synthesis, characterisation and computational studies on a novel one-dimensional arrangement of Schiff-base Mn3 single-molecule magnet. Dalton Trans. 2010, 39, 7650–7658. [Google Scholar] [CrossRef] [PubMed]
- Monfared, H. H.; Chamayou, A.-C.; Khajeh, S.; Janiak, C. Can a small amount of crystal solvent be overlooked or have no structural effect? Isomorphous non-stoichiometric hydrates (pseudo-polymorphs): The case of salicylaldehyde thiosemicarbazone. CrystEngComm 2010, 12, 3526–3530. [Google Scholar] [CrossRef]
- Nangia, A. Pseudopolymorph: Retain this widely accepted term. Cryst. Growth Des. 2006, 6, 2–4. [Google Scholar] [CrossRef]
- Dokorou, V.N.; Powell, A.K.; Kostakis, G.E. Two pseudopolymorphs derived from alkaline earth metals and the pseudopeptidic ligand trimesoyl-tris-glycine. Polyhedron 2013, 52, 538–544. [Google Scholar] [CrossRef]
- Lin, P.-H.; Burchell, T.J.; Clerac, R.; Murugesu, M. Dinuclear dysprosium(III) single-molecule magnets with a large anisotropic barrier. Angew. Chem. Int. Ed. 2008, 47, 8848–8851. [Google Scholar] [CrossRef] [PubMed]
- Llunell, M.D.; Casanova, J.; Cirera, P.; Alemany, S.A. SHAPE, Version 2.0; SHAPE: Barcelona, Spain, 2010.
- Coles, S.J.; Gale, P.A. Changing and challenging times for service crystallography. Chem. Sci. 2012, 3, 683–689. [Google Scholar] [CrossRef]
- Dolomanov, O.V.; Blake, A.J.; Champness, N.R.; Schröder, M. OLEX: New software for visualization and analysis of extended crystal structures. J. Appl. Crystallogr. 2003, 36, 1283–1284. [Google Scholar] [CrossRef]
- Palatinus, L.; Chapuis, G. SUPERFLIP—A computer program for the solution of crystal structures by charge flipping in arbitrary dimensions. J. Appl. Crystallogr. 2007, 40, 786–790. [Google Scholar] [CrossRef]
- Farrugia, L.J. WinGX suite for small-molecule single-crystal crystallography. J. Appl. Crystallogr. 1999, 32, 837–838. [Google Scholar] [CrossRef]
- Sheldrick, G.M. SHELXT—Integrated space-group and crystal-structure determination. Acta Crystallogr. Sect. A 2015, 71, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Sheldrick, G.M. A short history of SHELX. Acta Crystallogr. Sect. A 2008, 64, 112–122. [Google Scholar] [CrossRef] [PubMed]
- Spek, A.L. Single-crystal structure validation with the program PLATON. J. Appl. Crystallogr. 2003, 36, 7–13. [Google Scholar] [CrossRef]
- Macrae, C.F.; Edgington, P.R.; McCabe, P.; Pidcock, E.; Shields, G.P.; Taylor, R.; Towler, M.; van de Streek, J. Mercury: Visualization and analysis of crystal structures. J. Appl. Crystallogr. 2006, 39, 453–457. [Google Scholar] [CrossRef]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Loukopoulos, E.; Griffiths, K.; Akien, G.R.; Kourkoumelis, N.; Abdul-Sada, A.; Kostakis, G.E. Dinuclear Lanthanide (III) Coordination Polymers in a Domino Reaction. Inorganics 2015, 3, 448-466. https://0-doi-org.brum.beds.ac.uk/10.3390/inorganics3040448
Loukopoulos E, Griffiths K, Akien GR, Kourkoumelis N, Abdul-Sada A, Kostakis GE. Dinuclear Lanthanide (III) Coordination Polymers in a Domino Reaction. Inorganics. 2015; 3(4):448-466. https://0-doi-org.brum.beds.ac.uk/10.3390/inorganics3040448
Chicago/Turabian StyleLoukopoulos, Edward, Kieran Griffiths, Geoffrey R. Akien, Nikolaos Kourkoumelis, Alaa Abdul-Sada, and George E. Kostakis. 2015. "Dinuclear Lanthanide (III) Coordination Polymers in a Domino Reaction" Inorganics 3, no. 4: 448-466. https://0-doi-org.brum.beds.ac.uk/10.3390/inorganics3040448