Exploring Mechanisms in Ni Terpyridine Catalyzed C–C Cross-Coupling Reactions—A Review


1
A. E. Arbuzov Institute of Organic and Physical Chemistry, Kazan Scientific Center of Russian Academy of Sciences, 8, Arbuzov Str., 420088 Kazan, Russia
2
A. M. Butlerov Chemistry Institute, Kazan Federal University, Kremlevskaya str. 18, 420008 Kazan, Russia
3
Department of Chemistry, Lehigh University, 6 E. Packer Ave., Bethlehem, PA 18015, USA
4
Department für Chemie, Institut für Anorganische Chemie, Universität zu Köln, Greinstraße 6, D-50939 Köln, Germany
*
Authors to whom correspondence should be addressed.
Inorganics 2018, 6(1), 18; https://0-doi-org.brum.beds.ac.uk/10.3390/inorganics6010018
Submission received: 9 November 2017
/
Revised: 30 December 2017
/
Accepted: 18 January 2018
/
Published: 23 January 2018
(This article belongs to the Special Issue Revealing Reaction Mechanisms in Homogeneous Transition Metal Catalysis)
Abstract
:In recent years, nickel has entered the stage for catalyzed C–C cross-coupling reactions, replacing expensive palladium, and in some cases enabling the use of new substrate classes. Polypyridine ligands have played an important role in this development, and the prototypical tridentate 2,2′:6′,2′′-terpyridine (tpy) stands as an excellent example of these ligands. This review summarizes research that has been devoted to exploring the mechanistic details in catalyzed C–C cross-coupling reactions using tpy-based nickel systems.
1. Introduction
Polypyridine ligands such as 2,2′-bipyridine (bpy) and 2,2′:6′,2′′-terpyridine (tpy) are common ligands in coordination chemistry and molecular catalysis, and they generally generate stable well-defined complexes [1,2,3].
Tpy and the first nickel(II) complexes [Ni(tpy)2]X2·H2O (X = Br or I) were prepared by Morgan and Burstall in the 1930s [4,5]. The terpyridine molecule, because of its three pyridine nitrogen donor atoms, can form stable complexes of the [M(tpy)]n+ type with transition metals. Nickel(II) complexes of the composition [Ni(tpy)X2] (X = halides and pseudohalides) [6,7,8,9,10] have been structurally characterized by X-ray crystallographic analysis as monomeric trigonal–bipyramidal complexes [8,9,10], or as dimeric X-bridged species containing octahedrally configured nickel(II) [10,11], depending on the nature of the X coligand. In dilute solutions, monomeric species can be expected [6], and they can be pentacoordinate or hexacoordinate if further ligands are available [8,10,12]. For example, the recrystallization of [Ni(tpy)Cl2]·3H2O from a water/ethanol mixture gave the isomeric [Ni(tpy)Cl(H2O)2]Cl·H2O in single crystalline form [12] nickel(II) complexes of the composition [Ni(tpy)2]2+, where all the X type coligands are outer sphere, and show octahedral coordination [3,13].
Nickel(II) complexes have received high attention as (pre)catalysts in the field of transition metal catalyzed C–C cross-coupling reactions [14,15,16,17,18,19,20,21,22,23,24,25,26,27]. This area has been dominated by palladium catalysts in the last 20 years, which became manifest in the 2010 Nobel prize for R. Heck, E. Negishi, and A. Suzuki [28,29]. Importantly, the introduction of nickel-based catalysts to C–C cross-coupling catalysis has broadened the substrate scope due to its different reactivity, and lowered the cost relative to palladium-catalyzed reactions [21,22,23,24,25,30,31]. Although many C–C cross-coupling reactions can be catalyzed by palladium and nickel compounds, there are often large mechanistic differences between the two metals under similar reaction conditions. Palladium catalysts normally shuttle between two of the three even electron oxidation states (0, +II, and +IV) during C–C coupling reactions, and two-electron processes such as oxidative addition and reductive elimination are common mechanistic steps [14,15,24,25]. For nickel, single electron pathways between the oxidation states (0, +I, +II, +III, and +IV) often occur in addition to oxidative addition and reductive elimination [24,25,26,31,32].
Out of the many applications of nickel(II) catalysts in C–C or C–E cross-coupling reactions, we will focus in this contribution on the Negishi-type reactions [15,18,19,25,26,33] using organometallic zinc reagents, and on electrochemically driven C–C cross-coupling reactions. Also, instead of providing a literature overview of the many transformation that have been achieved using these methods, we will focus on selected studies that have provided mechanistic insight into these reactions.
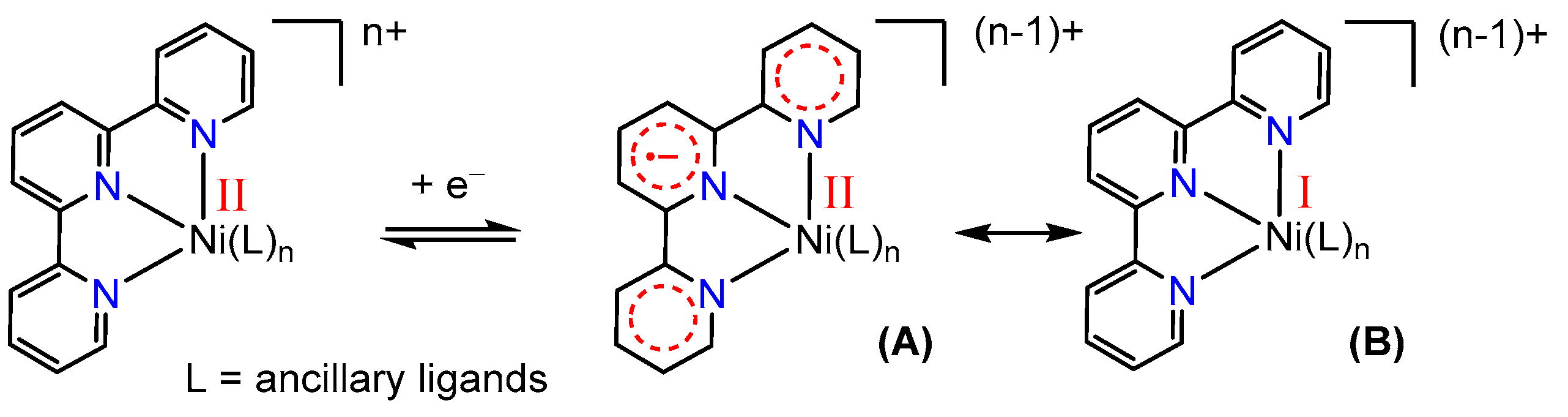
Non-innocent ligands [34,35,36,37,38,39] such as tpy contribute strongly to the electron inventory of such nickel complexes, and redox chemistry may often occur at the ligand in addition to the abovementioned oxidation states (0, +I, +II, +III, and +IV). For instance, since tpy is an excellent electron acceptor through its extended π system, a species resulting from the reduction of a nickel(II)-tpy complex can be described in two different resonance forms: as a divalent nickel bound to a radical anionic (tpy)•− ligand [Ni(II)(tpy•−)](n−1)+ (A), or as a monovalent nickel [Ni(I)(tpy)](n−1)+ complex (B) (Scheme 1) [40,41].
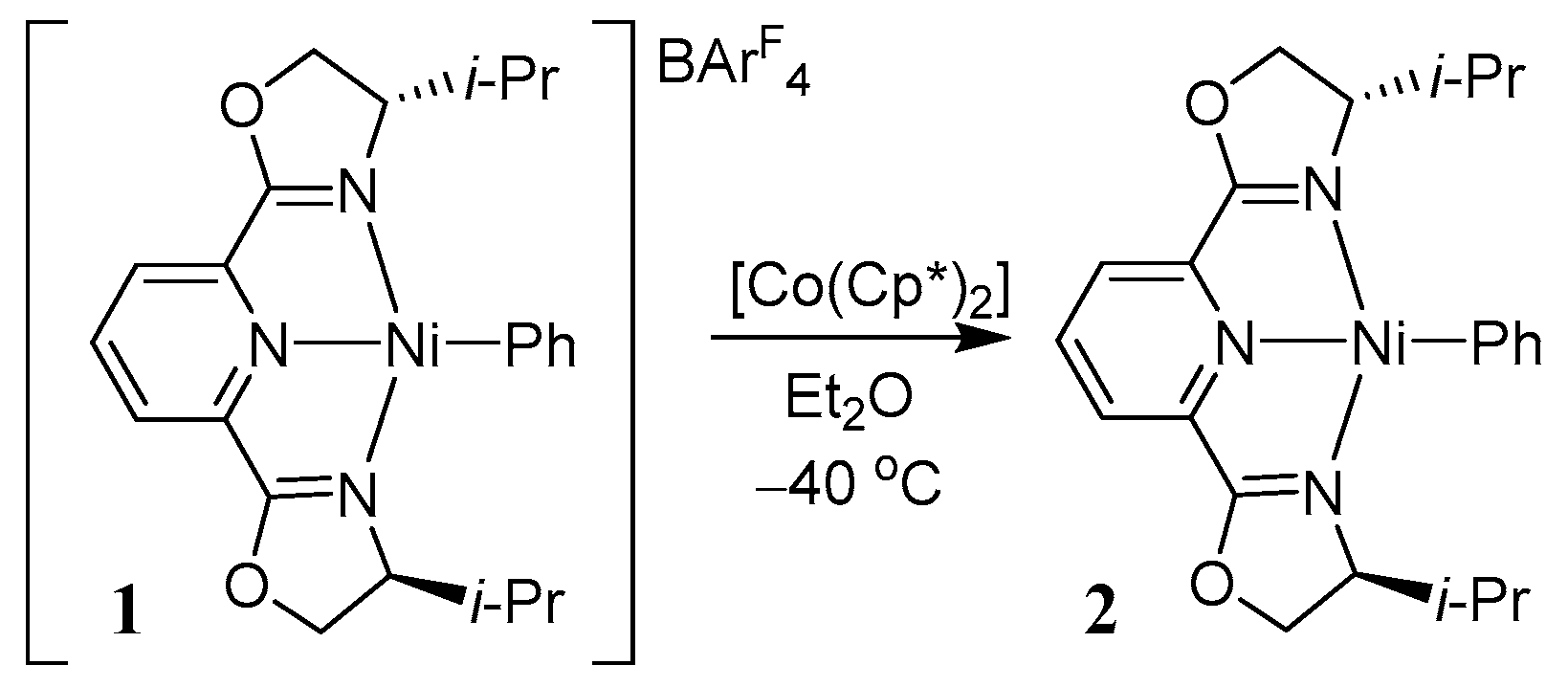
The fundamental studies on the electronic structures of terpyridine nickel complexes have also prompted others to explore details of related pybox complexes (pybox = pyridine-2,6-bisoxazolidine). Nickel pybox complexes are known to display high activity for alkyl–alkyl cross-coupling reactions, and can even promote stereoconvergent cross-couplings of racemic alkyl halides [26,42,43]. Recently, Fu et al. found that upon reduction of the nickel(II) complex [Ni(i-Pr-pybox)(Ph)](BArF4), the reduced species [Ni(i-Pr-pybox)(Ph)]• 2 could be isolated and characterized (Scheme 2) [26]. Spectroscopic studies of this complex in analogy to what was previously observed for related terpyridine complexes [44] indicated that the proper electronic structure consisted of a nickel(II) center bearing a reduced pybox ligand [Ni(II)(i-Pr-pybox•−)(Ph)] [26].
2. Nickel–tpy Catalyzed C–C Cross-Coupling
The simple organometallic low-valent species [Ni(tpy)(Me)] was reported to catalyze the cross-coupling of unactivated alkyl halides with primary alkylzinc reagents about 10 years ago (Scheme 3) [44,45,46,47].
When propargylic secondary bromides and chlorides were used as the electrophiles, secondary alkylzinc reagents could be coupled, producing two adjacent secondary carbon centers using the nickel–terpyridine catalyst system (Scheme 4) [48].
2.1. First Mechanistic Investigations on the [Ni(tpy)(Me)] Catalyst
The reaction of a nickel(0) precursor with MeI in the presence of a tpy derivative provided the cationic methylnickel(II) complex 3 (Scheme 5). In a related reaction, the treatment of [Ni(tmeda)(Me)2] with the same terpyridine ligand gave the neutral complex 4 [45,46]. Reaction of the cationic nickel(II) complex 3 with an alkylzinc reagent gave only a 8% yield of the cross-coupled product (Scheme 5). On the other hand, the neutral methylnickel(I) complex 4 reacted with n-heptyliodide to give the corresponding coupling product in 90% yield (Scheme 6). These results suggest that the Ni-tpy catalytic system does not include a simple Ni(0)/Ni(II) mechanism comprising of an oxidative addition of alkyl halides at nickel(0), transmetalation of a nickel alkyl halide complex, and lastly, reductive elimination of carbon–carbon bonds (Scheme 7).
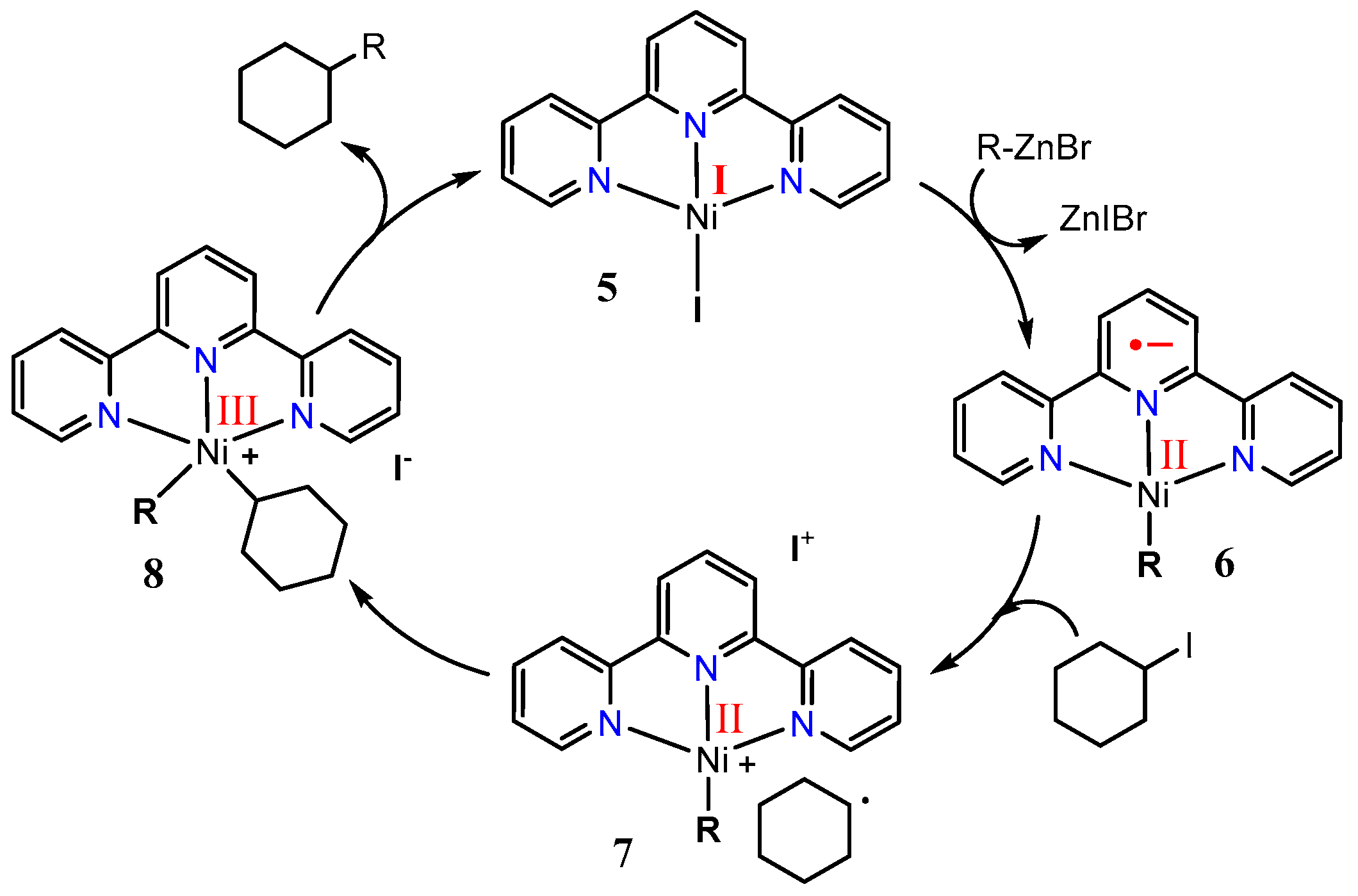
A plausible catalytic cycle for the nickel–terpyridine-catalyzed cross-coupling reaction of alkyl iodides with alkylzinc reagents is shown in Scheme 7. The in situ-formed low-valent nickel complex 5 undergoes transmetalation with organozinc reagents to give the corresponding organonickel species 6. Single electron transfer (SET) from the nickel complex 6 to alkyl iodides forms alkyl radicals and the nickel(II) complex 7. These radicals add oxidatively [46,49] to the Ni(II) complex, giving a nickel(III) species 8 as an intermediate addition, which then undergoes reductive elimination to yield coupling products with generation of a nickel(I) species 5 to complete the catalytic cycle.
Studies were performed on [Ni(tpy)(Me)] in order to determine its electronic structure. Electron paramagnetic resonance (EPR) spectroscopy, in combination with density functional theory (DFT) calculations on the electronic nature of the neutral complex [Ni(tpy)(Me)], revealed that the species should be described as divalent nickel bound to a radical anionic (tpy)•− ligand [Ni(II)(tpy•−)(Me)], rather than the monovalent resonance structure [Ni(I)(tpy)(Me)] (Scheme 1) [44,47].
2.2. Investigations of the Low-Valent Systems [Ni(tpy)(aryl)], [Ni(tpy)I], [Ni(tpy)Br], and [Ni(tpy)Cl]
After initial characterization of the reduced/low valent [Ni(tpy)(Me)] complex, further mechanistic work was devoted to understanding the nature of the so far proposed reduced species [Ni(tpy)I] [45,46,47], [Ni(tpy)Br] [50,51,52,53], and aryl derivatives [Ni(tpy)(aryl)]. Cationic complexes [Ni(II)(R-tpy)(aryl)]+ (R-tpy = substituted terpyridines) have been prepared from suitable precursors and studied structurally and spectroscopically in detail [51]. Their completely reversible first electrochemical reductions are centered at potentials near −1.5 V, and by using spectroelectrochemical (UV–Vis and EPR) methods, the reduced species [Ni(tpy)(aryl)] could be generated and studied. As found before for the Me derivate [Ni(tpy)(Me)], the aryl complexes [Ni(tpy)(aryl)] were best described as divalent nickel bound to a radical anionic (tpy)•− ligand [Ni(II)(tpy•−)(aryl)], and not as [Ni(I)(tpy)(aryl)] [50,51].
[Ni(tpy)Br] was prepared in a comproportionation reaction from [Ni(II)(dme)Br2] (dme = 1,2-dimethoxy-ethane) and [Ni(0)(COD)2] (COD = 1,5-cyclooctadiene) in the presence of the tpy ligand, and its structure was studied in detail. EPR and quantum chemical calculations clearly point to a complex containing monovalent nickel [Ni(I)(tpy)Br] and a neutral tpy ligand, in marked contrast to the methyl and aryl derivatives, as described above [52].
The low-temperature solid-state powder EPR spectrum of [Ni(tpy)Br]• exhibits an axial signal with g║= 2.256 and g⊥ = 2.091 consistent with a metal-centered ground state [52]. In DMF solution, an isotropic signal can be observed with giso = 2.139. Thus, both in the crystaline form and in solution, signals for a radical with substantial metal character are observed for [Ni(tpy)Br]• [52]. In contrast to this, [Ni(tpy)(Me)]•, exhibits an isotropic signal at 298 K with giso = 2.021, and a rhombic spectrum with g1 = 2.056, g2 = 2.021, and g3 = 1.999 at 77 K in a glassy frozen solution [44]. The spectra of [Ni(tpy)(Mes)]• (Mes = 2,4,6-trimethylphenyl) are very similar with giso = 2.0006, g1 = 2.009, g2 = 2.002, and g3 = 1.991 at 298 K or 110 K, respectively (Table 1).
Two parameters in Table 1 are most revealing for the character of the complex radicals. Both the high giso value and the high g anisotropy ∆g observed for [Ni(tpy)Br]• characterize this complex as having the unpaired electron largely centered at the metal in contrast to the Me and aryl derivatives [50,51].
Recently, the Cl derivative [Ni(tpy)Cl] was added to this list. The molecular structure from XRD clearly points to a Ni(I) description [Ni(I)(tpy)Cl] with neutral tpy, as for the Br derivative [41]. Unfortunately, EPR spectra were not reported. For [Ni(tpy)I], quantum chemical calculations show similar electron distribution as that found for the Br derivative [52]. In summary, it can be assumed that the largely superior σ-donor character of the Me or aryl coligands raise the orbital at the nickel above the lowest π* orbitals of the tpy ligand, and reduction leads to a (d8)(π*1) configured species, while for the weaker ligands Cl, Br, and I, a (d8)(1) configuration is found [41,52].
2.3. Studies on the Trivalent Species [Ni(tpy)(CnFm)2]+(CnFm = CF3 or C2F5)
Vicic showed that trifluoromethyl ligands can support the five-coordinate nickel(II) species 9, which can be chemically oxidized with [ferrocenium][PF6] to generate the transient nickel(III) species 10 (Scheme 8) [54]. Once formed, however, 10 undergoes a reductive homolysis of a trifluoromethyl ligand to afford the cationic nickel(II) species 11. Spectroelectrochemical EPR studies supported the intermediacy of 10, but its short-lived nature precluded any fundamental studies of its reactivity. Quite clearly, a •CF3 radical is cleaved from 10, which was shown through spin-trapping studies using PBN (N-t-Bu-α-phenylnitrone) [54].
2.4. Electrochemistry and Character of the Trivalent Species [Ni(tpy)(C4F8)]+
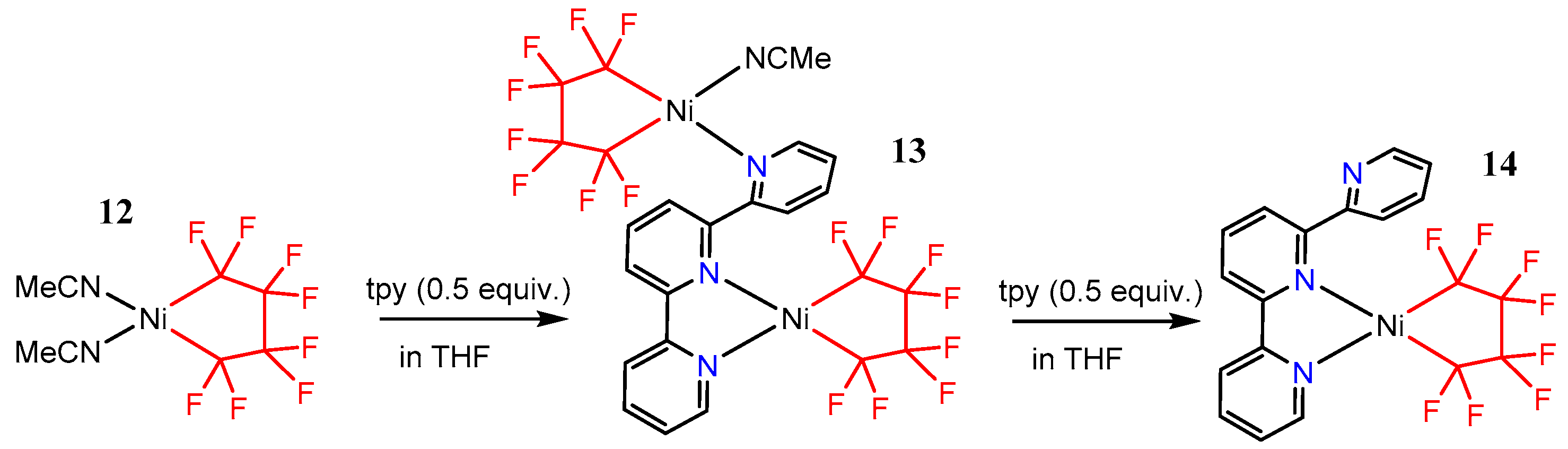
It was found that the addition of terpyridine to [Ni(MeCN)2(C4F8)] 12 led cleanly to the formation of [Ni(tpy)(C4F8)] 14 (Scheme 9) [55]. Surprisingly, in the solid state, the terpyridine ligand in 14 coordinates to nickel in a η2-fashion. However, solutions of 14 are paramagnetic, supporting an equilibrium with the η3-binding mode. Interestingly, when 0.5 equiv. of terpyridine is added to 12, the bimetallic species 13 can be isolated reproducibly, which is a testament to the lability of the acetonitrile ligands. Terpyridine is known to bind to more than one metal center [56,57,58,59,60,61], but 13 represented the first such adduct with nickel.
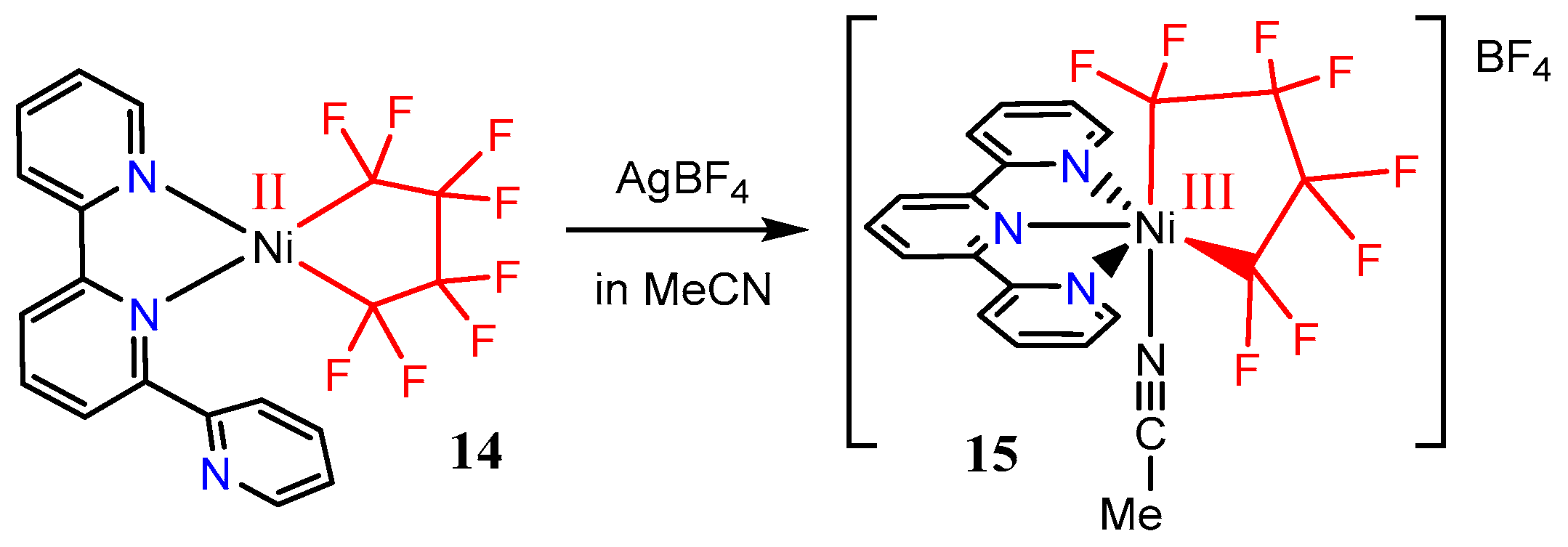
Oxidation of the [Ni(tpy)(C4F8)] complex 14 was explored. The η3-binding of the terpyridine ligand generates a high-spin nickel complex which, upon addition of Ag[BF4], affords the bluish/purple nickel(III) species 15 (Scheme 10). Unlike complex 10, which readily loses a trifluoromethyl radical, there is no evidence that 15 loses a perfluoroalkyl radical, even upon standing for hours in MeCN solution. The stability of 15 facilitated its characterization by X-ray crystallography, which identified the addition of an acetonitrile to the coordination sphere.
Variable temperature EPR spectra and magnetic moment data were obtained for 15, confirming its unpaired electron. Cyclic voltammetry experiments on 6 identifies a redox potential of +1.34 V for the Ni(III)/Ni(IV) couple. So, the [C4F82−] ligand is exceptionally suitable for stabilizing high valent nickel. Unlike [Ni(tpy)(CF3)2]+ complexes, which readily lose [•CF3] radicals [54], the [Ni(tpy)(C4F8)]+ analogue is solution stable, and can also be isolated in the solid state. The unique stability afforded by the [C4F82−] ligand now provides a family of related fluoroalkyl nickel complexes spanning the +II to +IV oxidation states for future fundamental studies [55].
2.5. Further Ni–tpy Catalyzed C–C Cross-Coupling Reactions
In the last 10 years, nickel–tpy systems have been used successfully for a number of C–C cross-couplings and related reactions.
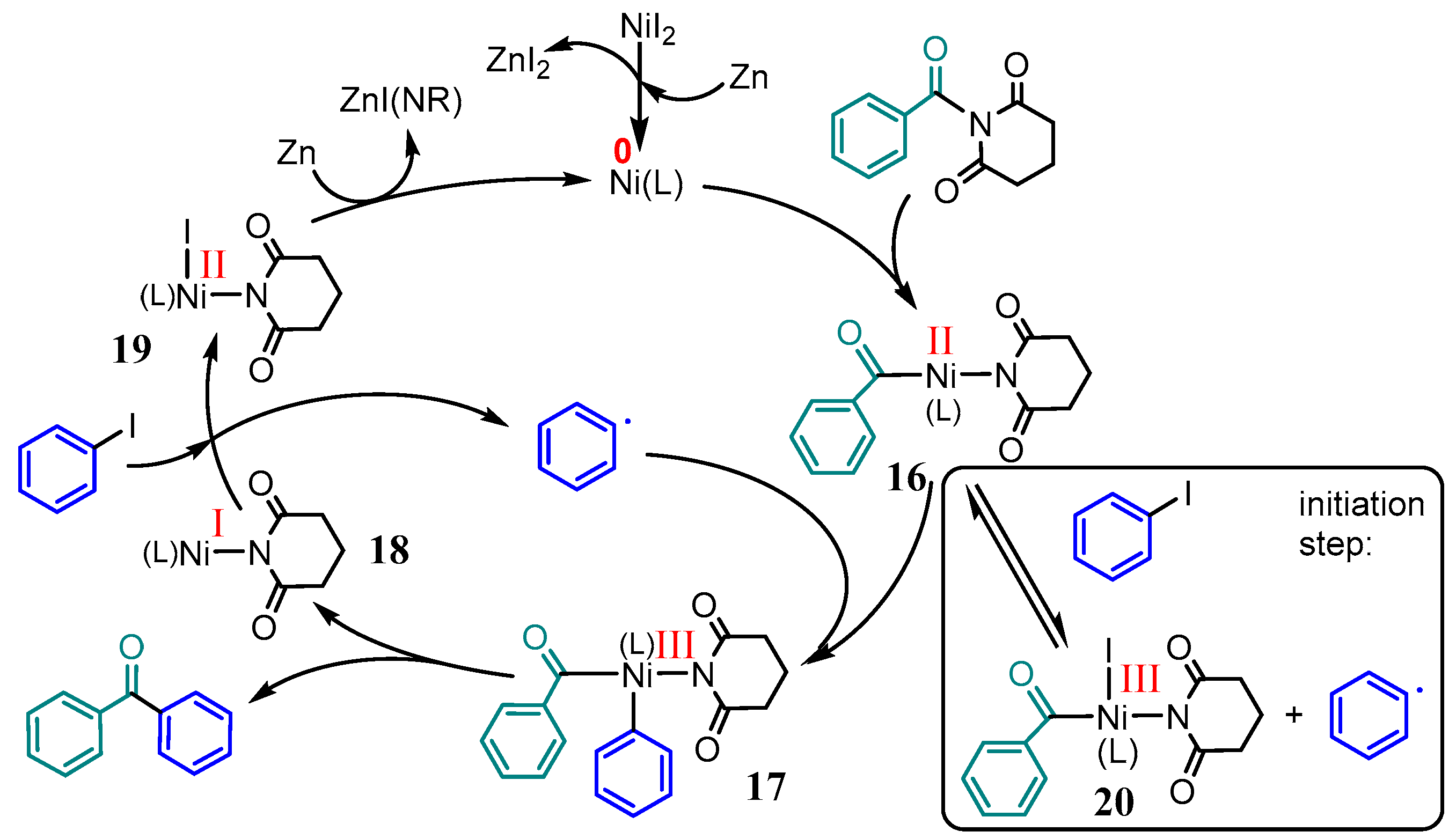
Recently, Han et al. reported on Ni-catalyzed reductive cross-coupling reactions between two electrophiles, amides and aryl iodides [62]. The nickel–tpy catalysts system turned out to be superior to catalysts containing the bidentate ligands 2,2′-bipyridine, 2,2′-biquinoline, 1,10-phenanthroline, and its 5,6-dione. In a mechanistic proposal (Scheme 11), the starting NiI2 is reduced in the presence of tpy (L) to [Ni(0)(L)] by the added metallic Zn, and the encompassing C–N bond activation of the amide is described as an oxidative addition to yield an [Ni(II)(L)(acyl) (amide)] species 16. This complex reacts with an aryl radical stemming from the aryl iodide substrate, and forms the [Ni(III)(L)(acyl)(amide) (aryl)] species 17 through a radical oxidative addition. This species cleaves the C–C coupling product, leaving an [Ni(I)(L)(amide)] complex 18. The Ni(I) species reacts with aryl iodide to produce the aforementioned aryl radical, and the resulting [Ni(II)(L)(amide)I] 19 is subsequently reduced to [Ni(0)(L)] by metallic Zn to restart the cycle. In addition to the production of the aryl radical through complex 18, 16 could also react with Ph-I to yield this radical in an initiation step forming another Ni(III) species 20. As a proof for the radical oxidative addition reaction, the addition of TEMPO (TEMPO = 2,2,6,6-Tetramethylpiperidin-1-yl)oxyl) inhibited the reaction and yielded the acyl–TEMPO adduct. Furthermore, [Ni(COD)2] was also successfully used as the catalyst precursor, underpinning the role of the metallic zinc [62].
An earlier study using [Ni(glyme)Cl2] (glyme = ethyleneglycoldimethylether) and various bi- and tridentate aromatic imine ligands as catalysts systems for the Negishi-type cross-coupling of secondary alkylzinc halides and aryl/heteroaryl iodides also found terpyridines superior to bidentate bpy or phen ligands, but also slightly better than the tridentate 2,6-di(1H-pyrazol-1-yl)pyridine, with the exception of the 4′-NO2– and 4′-CF3–tpy derivatives [63]. This indicates that the redox potentials of the various Ni–tpy species are important, which is in line with our observations.
Elemental manganese has been used in effective Ni-catalyzed reductive dimerization of alkyl halides, tosylates, and trifluoroacetates applying a [Ni(glyme)Cl2]/4,4′,4′′-(t-Bu)3tpy catalyst system [64]. Interestingly, the addition of NaI promotes the reaction, but the reason for this is not yet clear.
An interesting Ni-catalyzed reductive Csp3–Csp3 homocoupling using elemental zinc was reported to yield an alkyl chain axle [2]rotaxane [65]. The authors firstly optimized the homocoupling reaction of 1-bromo-3-phenoxypropane, and found the Ni-(R,R)-Ph-pybox system slightly superior to Ni–tpy (pybox = pyridine-2,6-bisoxazolidine, see also introduction). Then, they coordinated Ni(II) to a pybox-containing macrocycle (*pybox), and added 1-bromo-3-O–C6H4–C(t-Bu–Ph)3-propyl. With the addition of Zn, the thread (t-Bu–Ph)3C–C6H4–O–(CH2)6–O–C6H4–C(t-Bu–Ph3) was formed at the catalyst Ni center, and interlocked with the macrocycle. Mechanistically, it was proposed that Zn reduces the initial Ni(II) complex to Ni(0), which undergoes oxidative addition to a [Ni(II)(*pybox)Br(propyl-OR)] species. This is reduced to [Ni(I)(*pybox)(propyl-OR)], cleaving bromide. This complex undergoes oxidative addition, yielding a [Ni(III)(*pybox)Br((propyl-OR)2], which subsequently cleaves the C–C coupling product, the [2]rotaxane and a Ni(I) species in a reductive elimination. This not further described Ni(I) species is reduced by Zn to Ni(0) and coordinates to *pybox to restart the cycle. [65]
When using [Ni(tpy)(Py)(MeCN)2](PF6) as the catalyst precursor, Vannucci et al. were able to perform efficient photoredox-assisted reductive coupling of two carbon electrophiles [66]. Their mechanistic proposal starts with two subsequent reductions of the catalyst precursor to Ni(I)– and Ni(0)–tpy species by the iridium/TEOA photosystem (TEOA = triethanolamine). The Ni(0) complex undergoes oxidative addition of aryl halides to yield a Ni(II)Br(aryl) complex. At the same time, this species is supposed to react with alkyl halides through single electron transfer (SET) under the formation of an alkyl radical and a halide, thus recovering the Ni(I) species. Very similar to the report by Han [62], and our findings [44], the next step comprises a radical oxidative addition of an alkyl radical [49] stemming from the second substrate to the Ni(II) complex. The resulting Ni(III) species undergoes reductive elimination to yield the C–C coupling product and a Ni(I) species, which is then reduced by the photosystem to recover the active Ni(0) species [66].
In 2010, C–H alkylations of 1,3-azoles with alkyl halides were reported by Hirano and Miura (Scheme 12) [67]. C–H functionalizations using alkyl halides as a coupling partner are relatively rare compared with using unsaturated reactants, because of the difficulty in suppressing the β-hydrogen elimination of the alkylmetal intermediates. The nickel–terpyridine combination, in the presence of LiOt-Bu, was effective at suppressing such β-hydrogen eliminations.
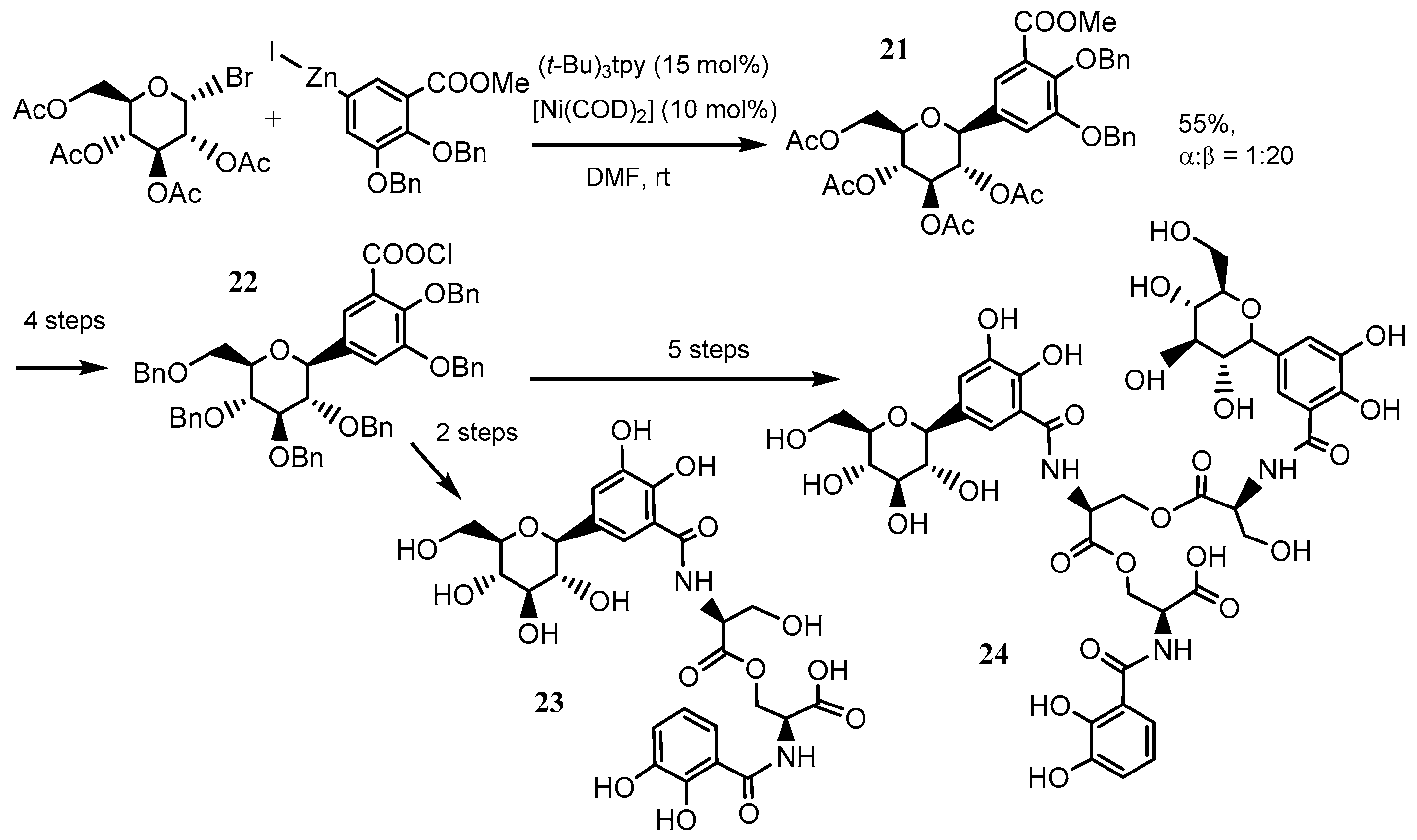
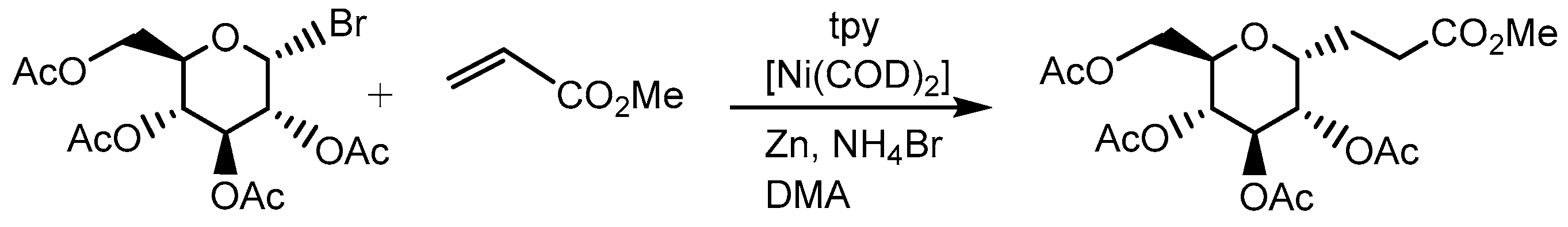
Gagné et al. achieved a total synthesis of a family of salmochelins, which are metabolites of the ferric-binding siderophores produced by E. coli and S. enterica. A key step in the synthesis was the nickel-catalyzed cross-coupling reaction of a bromoglucose derivative with an arylzinc reagent employing the terpyridine ligand on nickel [68]. The key coupling reaction proceeded with excellent diastereoselectivity in a good yield to give 21 (Scheme 13). The C-aryl glycoside 21 was successfully converted into salmochelin S1 23 and S2 24 by subsequent condensation and deprotection processes (Scheme 13) [69]. Moreover, Gagné also found that the combination of terpyridine and [Ni(0)(COD)2] (COD = 1,5 cyclooctadiene) can catalyze the reductive coupling of glycosyl bromides with activated alkenes (Scheme 14) [70].
3. Electrochemical Cross-Coupling Using Ni–tpy Systems
In recent years, the benefits of electrochemistry in nickel-catalysed reductive C–C cross-coupling reactions has been demonstrated [71,72,73,74,75,76,77]. In the same way, electrochemically induced P–C cross-coupling under the action of Ni complexes with α-diimines has been successful, and was described in detail [50,78,79,80,81,82,83]. Nickel complexes of α-diimines in high oxidation states catalyze ligand-directed C–H functionalization under electrochemical conditions [84].
The most important features of such electrocatalytic reactions include the possibility of using stable, simple, commercially available precatalysts, their compatibility with organic halides, and high functional group tolerances. In addition, compared with the more conventional chemical cross-coupling methods, electrochemical reactions have the advantage of providing an additional driving force (cathodic potential) for the reaction processes, which thus limits the need for additional means of activation, such as heating of the reaction medium [85,86,87,88,89].
3.1. Electrochemical Properties of Ni–tpy Complexes
Knowledge of mechanism and key intermediates is important for the rationalization and control of catalytic processes. In Ni–tpy catalyzed reactions, the nickel oxidation states +IV, +III, +II, +I and 0 have to be considered in combination with neutral (tpy) or reduced forms of the ligand (tpy•− or tpy2−).
As outlined in Section 2.2, reduced [Ni(tpy)(R)] complexes with R = alkyl or aryl clearly show structural and spectroscopic properties in line with a ligand-reduced character [Ni(II)(tpy•−)(R)], rather than with a monovalent nickel atom [Ni(I)(tpy)(Me)] [44,45,48,49]. In contrast to this, monovalent halide complexes [Ni(tpy)(X)] with X = Cl, Br, or I are best described as Ni(I) (d9) systems [41,42,43,45,49,50].
The complex [Ni(II)(tpy)2]2+ is reversibly reduced in three one-electron steps, and can also be reversibly oxidized (Scheme 15) [40,41,52,90,91,92,93]. For the oxidized species a [Ni(III)(tpy)2]3+ character has recently been assumed from calculations [41]; however, further spectroscopic evidence is not available from this report. The controlled potential oxidation of [Ni(tpy)2](ClO4)2 to the [Ni(III)(tpy)2]3+ complex was tried in 1977 [93], and the visible absorption spectrum and magnetic moment of the obtained material assumed to be [Ni(tpy)2](ClO4)3 were in line with a Ni(III) species. However, these spectroscopic results were devaluated by the elemental analysis, which was far off the calculated values. The authors ascribed this to the high reactivity of [Ni(III)(tpy)3]3+ in air, decomposing the isolated compound upon workup and isolation.
Most of the studies agree that the reduction sequence is largely ligand centered: [Ni(II)(tpy)2]2+/[Ni(II)(tpy•−)(tpy)]+/[Ni(II)(tpy•−)2]0/[Ni(II)(tpy2−)(tpy•−)]− [40,41,52,90,91,92,93]. A recent study revealed that this sequence is true for six coordinated [Ni(tpy)2]n complexes [41]. Presumably, the octahedral coordination through two tpy ligands (2 × η3 coordinate tpy) favors a Ni(II) d8 configuration over a (probably Jahn-Teller distorted) Ni(I) d9 system, and details of quantum chemical calculations reveal a significant interaction of the ligand (tpy) π radical anions and the unpaired electrons at the Ni(II) [41]. On the other hand, a four-coordinate species [Ni(tpy)2]0 with two η2 coordinating tpy ligands has been proposed for the doubly-reduced state [41,94]. Quantum chemical calculations reveal two isomers with a pseudo-tetrahedral structure and an [Ni(I)(tpy•−)(tpy0)] electron configuration with an S = 0 ground state. This is due to an antiferromagnetic coupling of the Ni(I), and the tpy•− radical and the calculations are in agreement with magnetic data [41].
Thus, the number of tpy ligands and the way they coordinate to Ni determines the redox potentials, the character of the reduced species, and therefore the reactivity of catalytically relevant complex species [52].
3.2. Aromatic Perfluoroalkylation Using Ni–tpy Under Electrocatalytic Conditions
In recent years, extensive efforts have led to new methods for the introduction of the trifluoromethyl and perfluoroalkyl groups into aromatic and heteroaromatic ring systems. [Ni(II)(tpy)] complexes were found to be effective catalyst precursors in olefin fluoroalkylationns and cross-couplings of fluoroalkylhalides and arylhalides.
A one-step catalytic method for aromatic perfluoroalkylation catalyzed by electrochemically reduced metal complexes, including Ni(tpy), has recently been developed [72]. Cross-coupling proceeds under mild conditions with the decisive role of the sacrificial anode metal. The reaction is successful with bromo- and iodobenzene and perfluoroalkyl iodides as substrates (Scheme 16).
The most effective catalysts systems in this study turned out to be Ni(II), Co(II), and Cu(II) complexes of bpy and tpy (and related diimines), in conjunction with a copper anode, and Cu(II) complex species were therefore considered in the proposed mechanistic cycle (Scheme 17). Furthermore, the success of the catalytic process depends on the reduction potential of the catalyst. The more negative reduction potential, the more effective the catalyst. Since [Ni(tpy)Br2] is reduced at the lowest potential (−1.30 V), compared with the other metal complexes studied in this work (e.g., (−1.12 V for [Ni(bpy)Br2]), the yields of the cross-coupling product with [Ni(tpy)Br2] participation are relatively higher (up to 73%) [72].
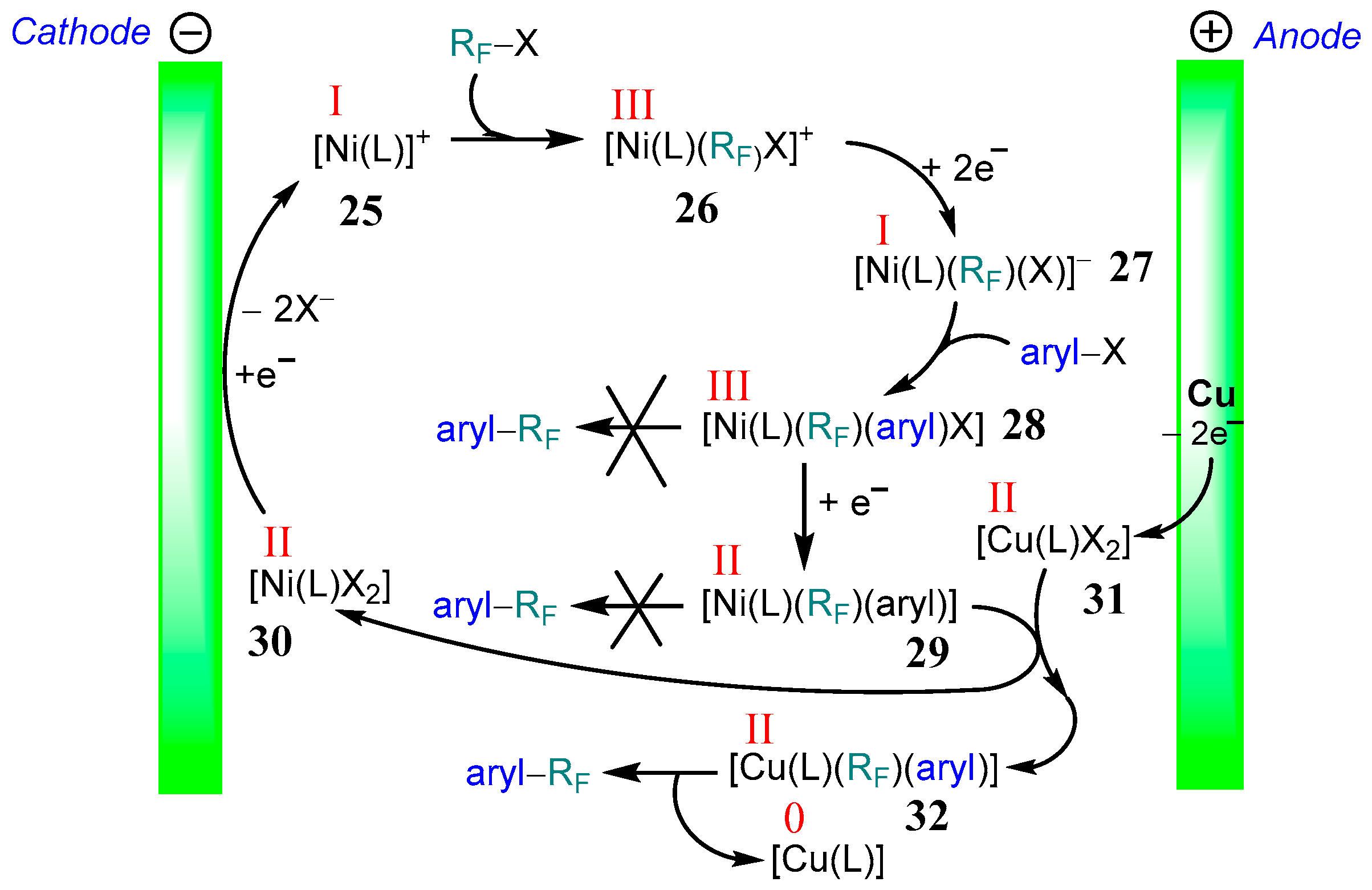
The proposed catalytic cycle (Scheme 17) involves a monovalent species [Ni(I)(L)] 25 (L = tpy or bpy), which is generated at the cathode. Oxidative addition leads to complexes [Ni(III)(L)(RF)X]+ 26; two electron reductions yield [Ni(I)(L)(RF)X]− 27. The oxidative addition of aryl–X substrates leads then to formal Ni(III) species 28, which are reduced again to yield the stable so-called σ-complexes [Ni(II)(L)(aryl)(RF)] 29. Importantly, neither the Ni(III) species [Ni(III)(L)(RF)X(aryl)] 28 nor the Ni(II) complexes [Ni(II)(L)(aryl)(RF)] 29 undergo reductive elimination to yield cross-coupling products aryl–RF [95,96]. Instead, the Ni(II) σ-complexes 29 transmetalate copper halide species to yield [Cu(II)(L)(RF)(aryl)] 32. They finally cleave the cross-coupling products through reductive elimination [72]. The halide complexes [Ni(L)X2] 30 were reductively reconverted to the initial Ni(I) species 25. To support the described mechanism, detailed cyclic voltammetry (CV) experiments and stoichiometric reactions were performed. It was shown that Cu(II) complexes such as [Cu(bpy)Cl2], [Cu(dmphen)Cl2] (dmphen = 2,9-dimethyl-1,10-phenanthroline), or [Cu(dppe)Cl2] (dppe = 1,2-bis(diphenylphosphano)ethane) under reductive conditions do not react with the RF–I with a noticeable rate, and their reduction behavior (potentials, reversibility) is not changed by the addition of RF–I. In contrast to this, the CVs of the corresponding nickel and cobalt complexes are sensitive to the presence of RF–I. The reduction wave of M(II) to M(I) for both Ni and Co complexes experienced a twofold increase in current with a loss of reversibility [52,72], which indicates the occurrence of oxidative addition RF–X to [M(I)(L)] without the cyclic regeneration of the catalyst. Regeneration of the catalyst would have led to a strong catalytic current in the presence of substrate [97,98], but this was not observed. Thus, the observed changes in CVs can be described by an ECE (electrochemical-chemical-electrochemical) scheme [50,52].
The advantages of this Cu-assisted method are as follows: it is a one-step catalytic reaction carrying out under mild conditions, at room temperature, and the cross-couplings are successful not only with iodobenzene, but also with bromobenzene and perfluoroalkyl iodides.
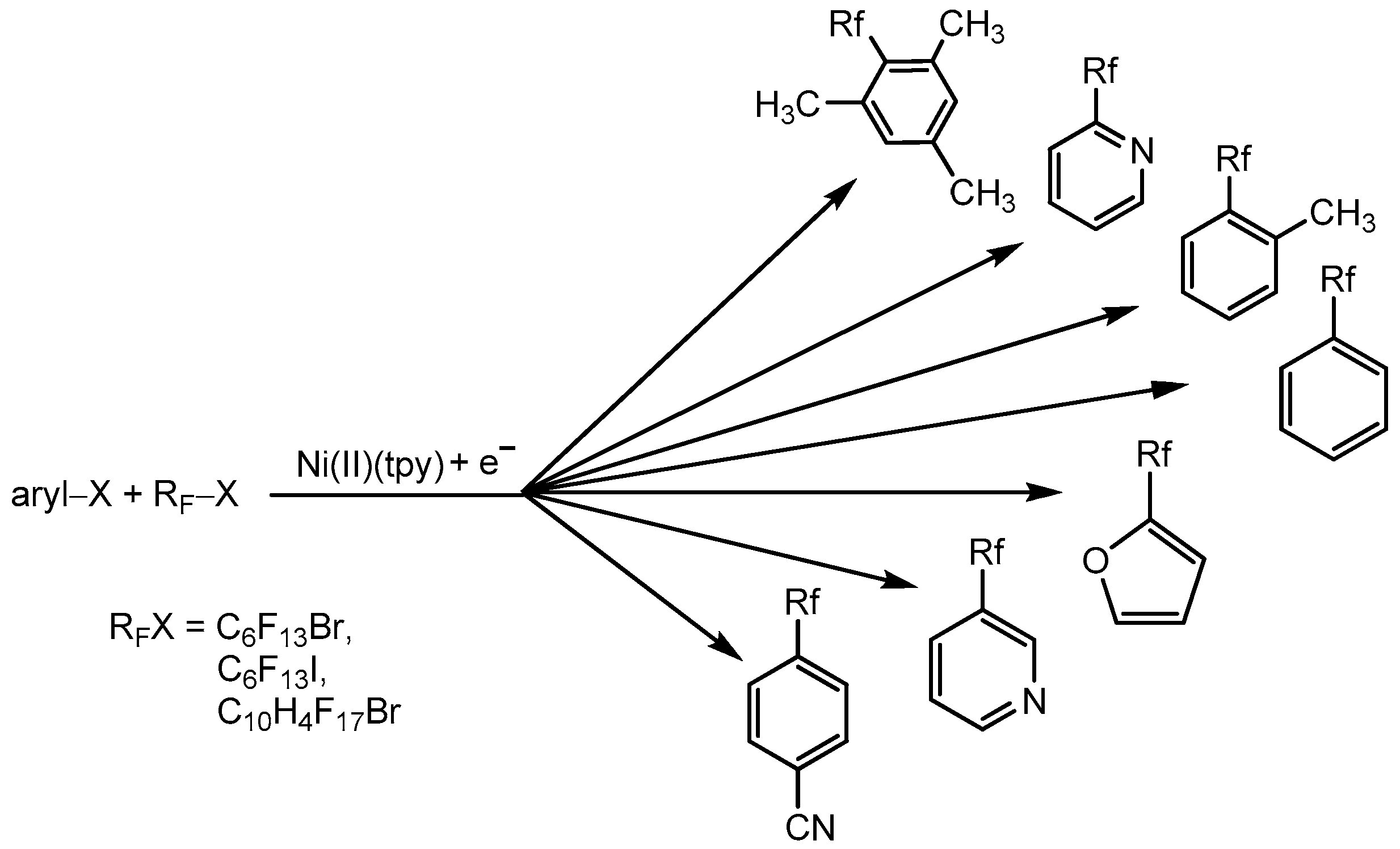
Later, the method was extended to various aromatic halides (Scheme 18) [71]. So, the single-stage synthesis of perfluoroalkylated arenes via the cross-coupling of bromo (in some cases, chloro) arenes or heteroarenes (derivatives of benzene, pyridine, and furan) and organic perfluoroalkyl halides involving [Ni(tpy)] complexes (and other metal and α-diimines ligands) in a low oxidation state under mild conditions was achieved. Perfluoroalkylated products are obtained in good yields in the presence of the [Ni(I)(L)] catalyst (1–10%) that was electrochemically generated from [Ni(II)(L)] at room temperature without chemical reductant. In the general case, it has been found that the success of the catalytic process depends on the reduction potential of the catalyst. The more negative reduction potential, the more effective the catalyst.
Very effective cross-coupling process was found for 4-bromobenzonitrile, 2-bromofuran, 2-bromopyridine, 2-chloropyridine, or 3-bromopyridine substrates, and perfluoro-n-hexyl iodide, perfluoro-n-hexyl bromide, or 1-bromo-1H,1H,2H,2H-perfluorodecan partners (Scheme 18). Thus, the method is suitable for different arenes, except thiophene and derivatives with NH2– and OCH3– groups. The most effective catalysts are complexes characterized by negative reduction potentials, and [Ni(tpy)Br2] is one of the best catalysts. There is no simple explanation for this observation, but perhaps a more negative reduction potential determines the greater force of the reducing agent and a faster oxidative addition step rate, which is a determining factor in the catalytic cycle. In any case, these catalysts were effective for mediating the assumed steps of the catalytic cycle: oxidative addition and reductive elimination [71].
3.3. Electrochemical Nickel-Induced Fluoroalkylation of Olefins
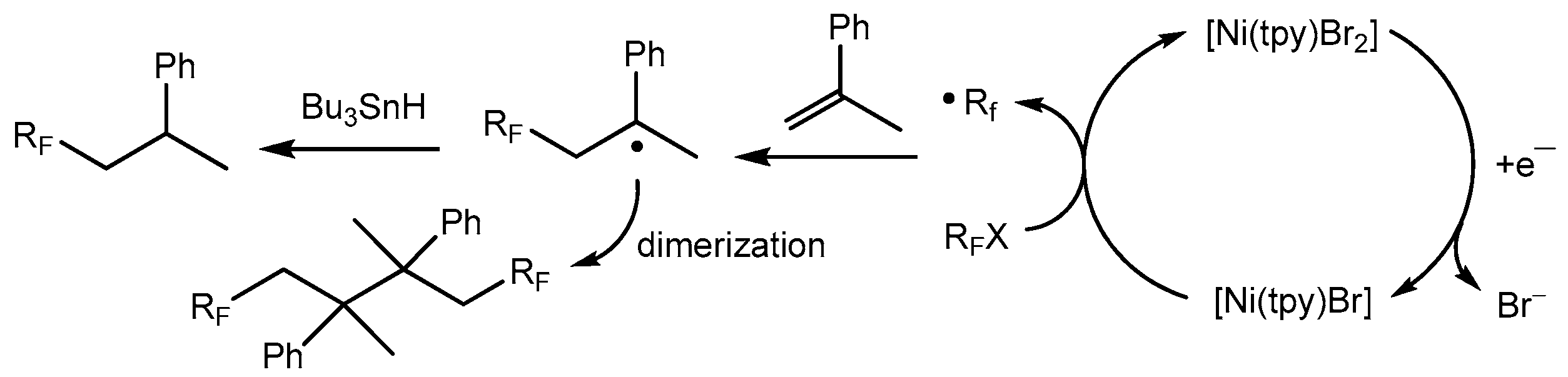
Electrochemical one electron reduction of [Ni(tpy)Br2] leads to an active species that can catalyze the addition of perfluoroalkyl radicals to olefins [53,99,100]. The electrocatalytic generation of a Ni(I) catalyst by reduction of a Ni(II)–tpy precursor complex in the presence of olefinic substrates and fluoroalkyl halides leads to new organic products derived from addition-dimerization processes (Scheme 19), showing a direct evidence of the importance of the four-coordinate low valent [Ni(tpy)Br] complex.
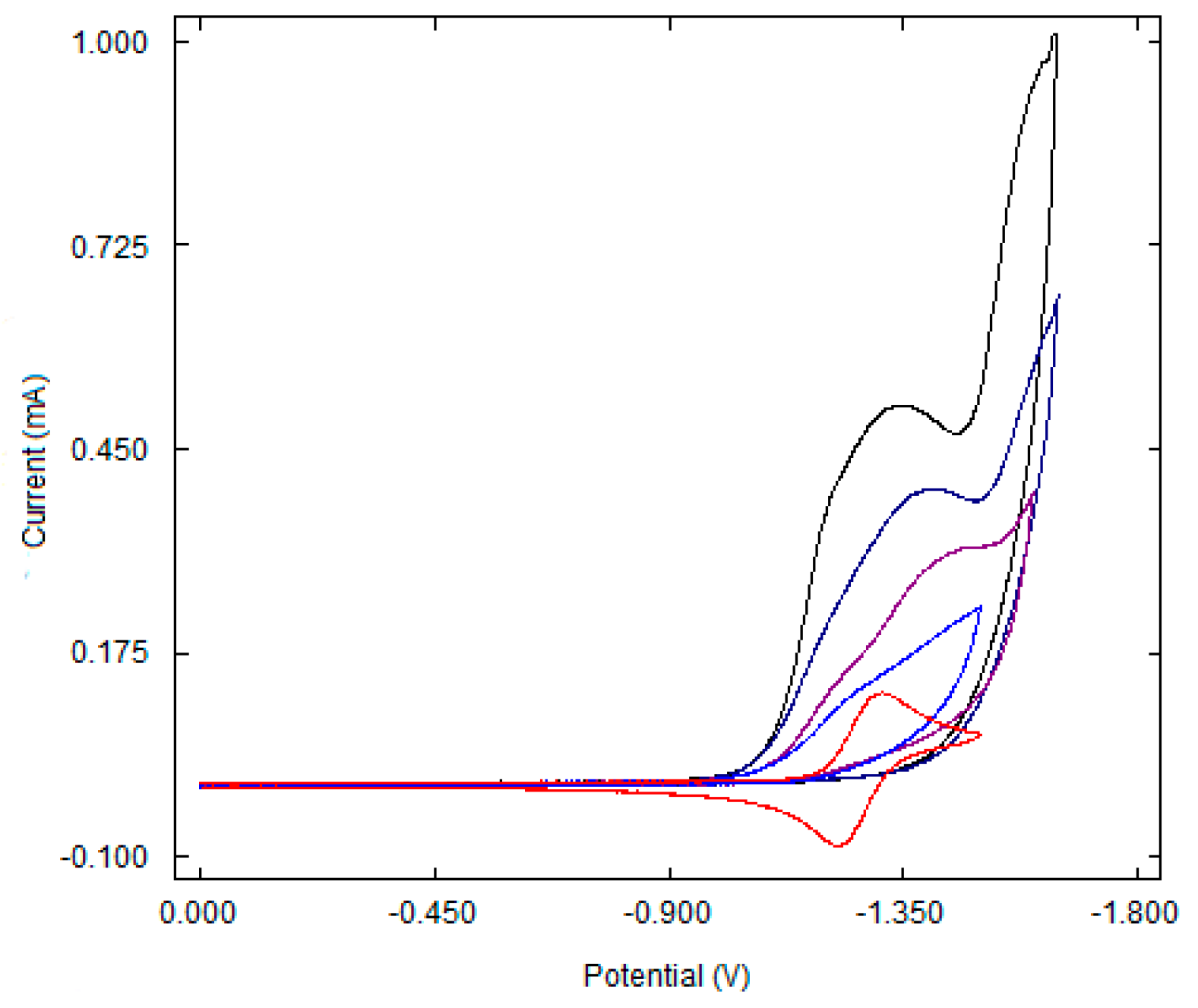
Due to the presence of two chiral carbon atoms in the dimerization products, three isomers were characterized by a variety of analytical techniques, including multi-dimensional NMR methods and X-ray single crystal diffraction [71,101]. The cyclic voltammetry study of the [Ni(tpy)Br2] complex, along with fluoroalkyl halides, demonstrated that [Ni(I)(L)Br] is the active form of the catalyst. The cyclic voltammetry and preparative electrolysis data give experimental support to the reactions highlighted in Scheme 20 [53]. The cyclic voltammograms of [Ni(tpy)Br2] under the reaction conditions are shown in Figure 1. The addition of a C6F13I substrate increases the current of the first reduction waves in the complex, suggesting that the Ni(I) species are the active forms of the catalyst.
The proposed mechanism of the process involves electrochemical reduction of the [Ni(II)(L)Br2] complex to [Ni(I)(L)Br], followed by an oxidation by the fluoroorganic halide to regenerate [Ni(II)(L)BrX] (Scheme 20). The perfluoroalkyl radicals produced by these redox events react with α-methyl styrene to form an addition product, which undergoes either a dimerization process or forms the monomer product under the treatment of tributyltin hydride. An alternative and more complex mechanism would involve coordination of the olefinic substrate [53].
Acknowledgments
This work was partially supported by the grant of the Russian Science Foundation No. 14-23-00016 (Y.H.B., electrosynthesis), the German Science Foundation KL1194/5-1, and KL1194/6-1 (A.K.), and the Office of Basic Energy Sciences of the U.S. Department of Energy (DE-SC0009363) (D.A.V.).
Author Contributions
All three authors have contributed equally to the writing of this manuscript.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Winter, A.; Newkome, G.R.; Schubert, U.S. Catalytic Applications of Terpyridines and their Transition Metals. ChemCatChem 2011, 3, 1384–1406. [Google Scholar] [CrossRef]
- Kaes, C.; Katz, A.; Hosseini, M.W. Bipyridine: The Most Widely Used Ligand. A Review of Molecules Comprising at Least Two 2,2′-Bipyridine Units. Chem. Rev. 2000, 100, 3553–3590. [Google Scholar] [CrossRef] [PubMed]
- Constable, E.C. 2,2′:6′,2″-Terpyridines: From chemical obscurity to common supramolecular motifs. Chem. Soc. Rev. 2007, 36, 246–253. [Google Scholar] [CrossRef] [PubMed]
- Morgan, G.; Burstall, F.H. Dehydrogenation of Pyridine by Anhydrous Ferric Chloride. J. Chem. Soc. 1932, 20–30. [Google Scholar] [CrossRef]
- Morgan, G.; Burstall, F.H. Researches on Residual Affinity and Co-ordination. Part XXX VII. Complex Metallic Salts containing 2:6-Di-2′-pyridylpyridine (2:2′:2″-Tripyridyl). J. Chem. Soc. 1937, 1649–1655. [Google Scholar] [CrossRef]
- Judge, J.S.; Reiff, W.M.; Intille, G.M.; Ballway, P.; Baker, W.A., Jr. Five-Coordinate Complexes of First Transition Series Ions with 2,2′,2″-Terpyridine. J. Inorg. Nucl. Chem. 1967, 29, 1711–1716. [Google Scholar] [CrossRef]
- Hogg, R.; Wilkins, R.G. Exchange Studies of Certain Chelate Compounds of the Transitional Metals. Part VIII. 2,2′,2″-Terpyridine Complexes. J. Chem. Soc. 1962, 341–350. [Google Scholar] [CrossRef]
- Gorczynski, A.; Walesa-Chorab, M.; Kubicki, M.; Korabik, M.; Patroniak, V. New complexes of 6,6″-dimethyl-2,2′:6′,2″-terpyridine with Ni(II) ions: Synthesis, structure and magnetic properties. Polyhedron 2014, 77, 17–23. [Google Scholar] [CrossRef]
- Constable, E.C.; Phillips, D.; Raithby, P.R. Nickel(II) chloride adducts of 4′-phenyl-2,2′:6′,2″-terpyridine. Inorg. Chem. Commun. 2002, 5, 519–521. [Google Scholar] [CrossRef]
- Baidya, N.; Olmstead, M.; Mascharak, P.K. Pentacoordinated Nickel(II) Complexes with Thiolato Ligation: Synthetic Strategy, Structures, and Properties. Inorg. Chem. 1991, 30, 929–937. [Google Scholar] [CrossRef]
- Arriortua, M.I.; Cortes, A.R.; Lezam, L.; Rojo, T.; Solans, X.; Font-Bardia, M. Crystal Structure and Magnetic Properties of [Ni(terpy)(N3)2]2·2H2O, a Nickel(II) Dinuclear Complex with Ferromagnetic Interaction. Inorg. Chim. Acta 1990, 174, 263–269. [Google Scholar] [CrossRef]
- Cortes, R.; Arriortua, M.I.; Rojo, T.; Solans, X.; Miravitelles, C.; Beltran, D. Structure of Diaquachloro(2,2′:6′,2″-terpyridyl)nickel(II) Chloride Monohydrate. Acta Crystallogr. C 1985, 41, 1733–1736. [Google Scholar] [CrossRef]
- Constable, E.C.; Lewis, J.; Liptrot, M.C.; Raithby, P.R. The coordination chemistry of 4′-phenyl-2,2′:6′,2″-terpyridine; the synthesis, crystal and molecular structures of 4′-phenyl-2,2′:6′,2″-terpyridine and bis(4′-phenyl-2,2′:6′,2″-terpyridine)nickel(II) chloride decahydrate. Inorg. Chim. Acta 1990, 178, 47–54. [Google Scholar] [CrossRef]
- Echevarren, A.M.; Homs, A. Mechanistic aspects of metal-catalyzed C,C- and C,X-bond forming reactions. In Metal-Catalyzed Cross-Coupling Reactions and More, 1st ed.; de Meijere, A., Bräse, S., Oestreich, M., Eds.; Wiley-VCH: Weinheim, Germany, 2014; pp. 1–64. ISBN 978-3-52-733154-3. [Google Scholar]
- Liu, C.; Zhang, H.; Shi, W.; Lei, A. Bond formations between two nucleophiles: Transition metal catalysed oxidative cross-coupling reactions. Chem. Rev. 2011, 111, 1780–1824. [Google Scholar] [CrossRef] [PubMed]
- Tasker, S.Z.; Standley, E.A.; Jamison, T.F. Recent advances in homogeneous nickel catalysis. Nature 2014, 509, 299–309. [Google Scholar] [CrossRef] [PubMed]
- Biswas, S.; Weix, D.J. Mechanism and selectivity in nickel-catalyzed cross-electrophile coupling of aryl halides with alkyl halides. J. Am. Chem. Soc. 2013, 135, 16192–16197. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.-X.; Liu, N. Nickel-catalyzed cross-coupling with pincer ligands. Eur. J. Inorg. Chem. 2012, 901–911. [Google Scholar] [CrossRef]
- Rosen, B.M.; Quasdorf, K.W.; Wilson, D.A.; Zhang, N.; Resmerita, A.-M.; Garg, N.K.; Percec, V. Nickel-catalyzed cross-couplings involving carbon–oxygen bonds. Chem. Rev. 2011, 111, 1346–1416. [Google Scholar] [CrossRef] [PubMed]
- Phapale, V.B.; Guisan-Ceinos, M.; Bunuel, E.; Cardenas, D.J. Nickel-catalyzed cross-coupling of alkyl zinc halides for the formation of C(sp2)–C(sp2) bonds: Scope and mechanism. Chem. Eur. J. 2009, 15, 12681–12688. [Google Scholar] [CrossRef] [PubMed]
- Zuo, Z.; Ahneman, D.T.; Chu, L.; Terrett, J.A.; Doyle, A.G.; MacMillan, S.W.C. Merging photoredox with nickel catalysis: Coupling of α-carboxyl sp3-carbons with aryl halides. Science 2014, 345, 437–440. [Google Scholar] [CrossRef] [PubMed]
- Tellis, J.C.; Primer, D.N.; Molander, G.A. Single-electron transmetalation in organoboron cross-coupling by photoredox/nickel dual catalysis. Science 2014, 345, 433–436. [Google Scholar] [CrossRef] [PubMed]
- Lloyd-Jones, G.C.; Ball, L.T. Self-control tames the coupling of reactive radicals. Science 2014, 345, 381–382. [Google Scholar] [CrossRef] [PubMed]
- Han, F.-S. Transition-metal-catalyzed Suzuki-Miyaura cross-coupling reactions: A remarkable advance from palladium to nickel catalysts. Chem. Soc. Rev. 2013, 42, 5270–5298. [Google Scholar] [CrossRef] [PubMed]
- Jana, R.; Pathak, T.P.; Sigman, M.S. Advances in transition metal (Pd,Ni,Fe)-catalyzed cross-coupling reactions using alkyl-organometallics as reaction partners. Chem. Rev. 2011, 111, 1417–1492. [Google Scholar] [CrossRef] [PubMed]
- Schley, N.D.; Fu, G.C. Nickel-catalyzed Negishi arylation of propargylic bromides: A mechanistic investigation. J. Am. Chem. Soc. 2014, 136, 16588–16593. [Google Scholar] [CrossRef] [PubMed]
- Khan, M.S.; Haque, A.; Al-Suti, M.K.; Raithby, P.R. Recent advances in the application of group-transition metal based catalysts in C–H activation and functionalization. J. Organomet. Chem. 2015, 793, 114–133. [Google Scholar] [CrossRef] [Green Version]
- Johansson Seechurn, C.C.C.; Deangelis, A.; Colacot, T.J. Introduction to new trends in cross-coupling. In New Trends in Cross-Coupling: Theory and Applications; RSC Catalysis Series; Colacot, T.J., Ed.; Royal Society of Chemistry: Cambridge, UK, 2015; pp. 1–19. ISBN 978-1-84-973896-5. [Google Scholar]
- Colacot, T.J. The Nobel Prize in chemistry: Palladium-catalysed cross-coupling. The importance of carbon–carbon coupling for real world applications. Plat. Met. Rev. 2011, 55, 84–90. [Google Scholar] [CrossRef]
- Labinger, J.A. Tutorial on oxidative addition. Organometallics 2015, 34, 4784–4795. [Google Scholar] [CrossRef]
- Su, B.; Cao, Z.-C.; Shi, Z.-J. Exploration of earth-abundant transition metals (Fe, Co, and Ni) as catalysts in unreactive chemical bond activations. Acc. Chem. Res. 2015, 48, 886–896. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Diccianni, J.B.; Katigbak, J.; Hu, C.; Zhang, Y.; Diao, T. Bimetallic C–C Bond-Forming Reductive Elimination from Nickel. J. Am. Chem. Soc. 2016, 138, 4779–4786. [Google Scholar] [CrossRef] [PubMed]
- Kaplan, P.T.; Xu, L.; Chen, B.; McGarry, K.R.; Yu, S.; Wang, H.; Vicic, D.A. Mild, safe, and versatile reagents for (CF2)n transfer and the construction of fluoroalkyl-containing rings. Organometallics 2013, 32, 7552–7558. [Google Scholar] [CrossRef]
- Lin, S.; Day, M.W.; Agapie, T. Nickel hydrides supported by a non-innocent diphosphine arene pincer: Mechanistic studies of nickel–arene H-migration and partial arene hydrogenation. J. Am. Chem. Soc. 2011, 133, 3828–3831. [Google Scholar] [CrossRef] [PubMed]
- Uyeda, C.; Steiman, T.J.; Pal, S. Catalytically active nickel–nickel bonds using redox-active ligands. Synlett 2016, 27, 814–820. [Google Scholar] [CrossRef]
- Kaim, W. The Shrinking World of Innocent Ligands: Conventional and Non-Conventional Redox-Active Ligands. Eur. J. Inorg. Chem. 2012, 343–348. [Google Scholar] [CrossRef]
- Lyaskovsky, V.; de Bruin, B. Redox Non-Innocent Ligands: Versatile New Tools to Control Catalytic Reactions. ACS Catal. 2012, 2, 270–279. [Google Scholar] [CrossRef]
- Chaudhuri, P.; Verani, C.N.; Bill, E.; Bothe, E.; Weyhermüller, T.; Wieghardt, K. Electronic structure of bis(o-iminobenzosemiquinonato)metal complexes (Cu, Ni, Pd). The art of establishing physical oxidation states in transition-metal complexes containing radical ligands. J. Am. Chem. Soc. 2001, 123, 2213–2223. [Google Scholar] [CrossRef] [PubMed]
- Jørgensen, C.K. Differences between the four halide ligands, and discussion remarks on trigonal-bipyramidal complexes, on oxidation states, and on diagonal elements of one-electron energy. Coord. Chem. Rev. 1966, 1, 164–187. [Google Scholar] [CrossRef]
- Arana, C.; Yan, S.; Keshavarz, M.; Potts, K.T.; Abruna, H.D. Electrocatalytic Reduction of Carbon Dioxide with Iron, Cobalt, and Nickel Complexes of Terdentate Ligands. Inorg. Chem. 1992, 31, 3680–3682. [Google Scholar] [CrossRef]
- Wang, M.; England, J.; Weyhermüller, T.; Wieghardt, K. Electronic Structures of “Low-Valent” Neutral Complexes [NiL2](S = 0; L = bpy, phen, tpy)—An Experimental and DFT Computational Study. Eur. J. Inorg. Chem. 2015, 1511–1523. [Google Scholar] [CrossRef]
- Choi, J.; Fu, G.C. Transition metal-catalyzed alkyl–alkyl bond formation: Another dimension in cross-coupling chemistry. Science 2017, 356, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Liang, Y.; Fu, G.C. Nickel-Catalyzed Alkyl–Alkyl Cross-Couplings of Fluorinated Secondary Electropphiles: A General Approach to the Synthesis of Compounds having a Perfluoroalkyl Substituent. Angew. Chem. Int. Ed. 2015, 54, 9047–9051. [Google Scholar] [CrossRef] [PubMed]
- Anderson, T.J.; Jones, G.D.; Vicic, D.A. Evidence for a NiI Active Species in the Catalytic Cross-Coupling of Alkyl Electrophiles. J. Am. Chem. Soc. 2004, 126, 8100–8101. [Google Scholar] [CrossRef] [PubMed]
- Jones, G.D.; McFarland, C.; Anderson, T.J.; Vicic, D.A. Analysis of key steps in the catalytic cross-coupling of alkyl electrophiles under Negishi-like conditions. Chem. Commun. 2005, 4211–4213. [Google Scholar] [CrossRef] [PubMed]
- Jones, G.D.; Martin, J.L.; McFarland, C.; Allen, O.R.; Hall, R.E.; Haley, A.D.; Brandon, R.J.; Konovalova, T.; Desrochers, P.J.; Pulay, P.; et al. Ligand Redox Effects in the Synthesis, Electronic Structure, and Reactivity of an Alkyl–Alkyl Cross-Coupling Catalyst. J. Am. Chem. Soc. 2006, 128, 13175–13183. [Google Scholar] [CrossRef] [PubMed]
- Lin, X.; Phillips, D.L. Density Functional Theory Studies of Negishi Alkyl–Alkyl Cross-Coupling Reactions Catalyzed by a Methylterpyridyl-Ni(I) Complex. J. Org. Chem. 2008, 73, 3680–3688. [Google Scholar] [CrossRef] [PubMed]
- Smith, S.W.; Fu, G.C. Nickel-Catalyzed Negishi Cross-Couplings of Secondary Nucleophiles with Secondary Propargylic Electrophiles at Room Temperature. Angew. Chem. Int. Ed. 2008, 47, 9334–9336. [Google Scholar] [CrossRef] [PubMed]
- Breitenfeld, J.; Ruiz, J.; Wodrich, M.D.; Hu, X. Bimetallic Oxidative Addition Involving Radical Intermediates in Nickel-Catalyzed Alkyl—Alkyl Kumada Coupling Reactions. J. Am. Chem. Soc. 2013, 135, 12004–12012. [Google Scholar] [CrossRef] [PubMed]
- Klein, A.; Budnikova, Y.H.; Sinyashin, O.G. Electron transfer in organonickel complexes of α-diimines: Versatile redox catalysts for C–C or C–P coupling reactions—A review. J. Organomet. Chem. 2007, 692, 3156–3166. [Google Scholar] [CrossRef]
- Hamacher, C.; Hurkes, N.; Kaiser, A.; Klein, A.; Schüren, A. Electrochemistry and Spectroscopy of Organometallic Terpyridine Nickel Complexes. Inorg. Chem. 2009, 48, 9947–9951. [Google Scholar] [CrossRef] [PubMed]
- Ciszewski, J.T.; Mikhaylov, D.Y.; Holin, K.V.; Kadirov, M.K.; Budnikova, Y.H.; Sinyashin, O.; Vicic, D.A. Redox Trends in Terpyridine Nickel Complexes. Inorg. Chem. 2011, 50, 8630–8635. [Google Scholar] [CrossRef] [PubMed]
- Mikhaylov, D.; Gryaznova, T.; Dudkina, Y.; Khrizanphorov, M.; Latypov, S.; Kataeva, O.; Vicic, D.A.; Sinyashin, O.G.; Budnikova, Y. Electrochemical nickel-induced fluoroalkylation: Synthetic, structural and mechanistic study. Dalton Trans. 2012, 41, 165–172. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.-P.; Wang, H.; Klein, A.; Biewer, C.; Stirnat, K.; Yamaguchi, Y.; Xu, L.; Gomez-Benitez, V.; Vicic, D.A. A Five-Coordinate Nickel(II) Fluoroalkyl Complex as a Precursor to a Spectroscopically Detectable Ni(III) Species. J. Am. Chem. Soc. 2013, 135, 8141–8144. [Google Scholar] [CrossRef] [PubMed]
- Yu, S.; Dudkina, Y.; Wang, H.; Kholin, K.V.; Kadirov, M.K.; Budnikova, Y.H.; Vicic, D.A. Accessing perfluoroalkyl nickel(II), (III), and (IV) complexes bearing a readily attached [C4F8] ligand. Dalton Trans. 2015, 44, 19443–19446. [Google Scholar] [CrossRef] [PubMed]
- Coogan, M.P.; Fernandez-Moreira, V.; Kariuki, B.M.; Pope, S.J.A.; Thorp-Greenwood, F.L. A Rhenium Tricarbonyl 4′-Oxo-terpy Trimer as a Luminescent Molecular Vessel with a Removable Silver Stopper. Angew. Chem. Int. Ed. 2009, 48, 4965–4968. [Google Scholar] [CrossRef] [PubMed]
- Effendy; Marchetti, F.; Pettinari, C.; Pettinari, R.; Skelton, B.W.; White, A.H. Synthesis and structural characterization of adducts of silver(I) oxyanion salts, AgX (X = ClO4, NO3), with Ph2E(CH2)xEPh(‘dpex’; E = P, As; x = 1–3) and oligodentate aromatic N-bases derivative of 2,2′-bipyridyl, ‘L’, AgX:dpex:L (2:1:1) or (1:1:1). Inorg. Chim. Acta 2007, 360, 1414–1423. [Google Scholar] [CrossRef]
- Feng, H.; Zhou, X.-P.; Wu, T.; Li, D.; Yin, Y.-G.; Ng, S.W. Hydrothermal synthesis of copper complexes of 4′-pyridyl terpyridine: From discrete monomer to zigzag chain polymer. Inorg. Chim. Acta 2006, 359, 4027–4035. [Google Scholar] [CrossRef]
- Hannon, M.J.; Painting, C.L.; Plummer, E.A.; Childs, L.J.; Alcock, N.W. Competing Supramolecular Interactions Give a New Twist to Terpyridyl Chemistry: Anion- and Solvent-Induced Formation of Spiral Arrays in Silver(I) Complexes of a Simple Terpyridine. Chem. Eur. J. 2002, 8, 2225–2238. [Google Scholar] [CrossRef]
- Hou, L.; Li, D. A novel photoluminescent Ag–terpyridyl complex: One-dimensional linear metal string with double-helical structure. Inorg. Chem. Commun. 2005, 8, 128–130. [Google Scholar] [CrossRef]
- Ma, Z.; Xing, Y.; Yang, M.; Hu, M.; Liu, B.; da Silva, M.F.C.G.; Pombeiro, A.J.L. The double-helicate terpyridine silver(I) compound [Ag2L2](SO3CF3)(L = 4′-phenyl-terpyridine) as a building block for di- and mononuclear complexes. Inorg. Chim. Acta 2009, 362, 2921–2926. [Google Scholar] [CrossRef]
- Ni, S.; Zhang, W.; Mei, H.; Han, J.; Pan, Y. Ni-Catalyzed Reductive Cross-Coupling of Amides with Aryl Iodide Electrophiles via C–N Bond Activation. Org. Lett. 2017, 19, 2536–2539. [Google Scholar] [CrossRef] [PubMed]
- Joshi-Pangu, A.; Ganesh, M.; Biscoe, M.R. Nickel-Catalyzed Negishi Cross-Coupling Reactions of Secondary Alkylzinc Halides and Aryl Iodides. Org. Lett. 2011, 13, 1218–1221. [Google Scholar] [CrossRef] [PubMed]
- Prinsell, M.R.; Everson, D.A.; Weix, D.J. Nickel-catalyzed, sodium iodide-promoted reductive dimerization of alkyl halides, alkylpseudohalides, and allylic acetates. Chem. Commun. 2010, 46, 5743–5745. [Google Scholar] [CrossRef] [PubMed]
- Goldup, S.M.; Leigh, D.A.; McBurney, R.T.; McGonigal, P.R.; Plant, A. Ligand-assisted nickel-catalysed sp3–sphomocoupling of unactivated alkyl bromides and its application to the active template synthesis of rotaxanes. Chem. Sci. 2010, 1, 383–386. [Google Scholar] [CrossRef]
- Paul, A.; Smith, M.D.; Vannucci, A.K. Photoredox-Assisted Reductive Cross-Coupling: Mechanistic Insight into Catalytic Aryl–Alkyl Cross-Couplings. J. Org. Chem. 2017, 82, 1996–2003. [Google Scholar] [CrossRef] [PubMed]
- Yao, T.; Hirano, K.; Satoh, T.; Miura, M. Palladium- and Nickel-Catalyzed Direct Alkylation of Azoles with Unactivated Alkyl Bromides and Chlorides. Chem. Eur. J. 2010, 16, 12307–12311. [Google Scholar] [CrossRef] [PubMed]
- Gong, H.; Gagné, M.R. Diastereoselective Ni-Catalyzed Negishi Cross-Coupling Approach to Saturated, Fully Oxygenated C–Alkyl and C–Aryl Glycosides. J. Am. Chem. Soc. 2008, 130, 12177–12183. [Google Scholar] [CrossRef]
- Yu, X.; Dai, Y.; Yang, T.; Gagné, M.R.; Gong, H. Facile synthesis of salmochelin S1, S2, MGE, DGE, and TGE. Tetrahedron 2011, 67, 144–151. [Google Scholar] [CrossRef]
- Gong, H.; Andrews, R.S.; Zuccarello, J.L.; Lee, S.J.; Gagné, M.R. Sn-Free Ni-Catalyzed Reductive Coupling of Glycosyl Bromides with Activated Alkenes. Org. Lett. 2009, 11, 879–882. [Google Scholar] [CrossRef] [PubMed]
- Khrizanforov, M.; Khrizanforova, V.; Mamedov, V.; Zhukova, N.; Strekalova, S.; Grinenko, V.; Gryaznova, T.; Sinyashin, O.; Budnikova, Y. Single-stage synthetic route to perfluoroalkylated arenes via electrocatalytic cross-coupling of organic halides using Co and Ni complexes. J. Organomet. Chem. 2016, 820, 82–88. [Google Scholar] [CrossRef]
- Khrizanforov, M.; Gryaznova, T.; Sinyashin, O.; Budnikova, Y. Aromatic perfluoroalkylation with metal complexes in electrocatalytic conditions. J. Organomet. Chem. 2012, 718, 101–104. [Google Scholar] [CrossRef]
- Sengmany, S.; Vasseur, S.; Lajnef, A.; Le Gall, E.; Léonel, E. Beneficial Effects of Electrochemistry in Cross-Coupling Reactions: Electroreductive Synthesis of 4-Aryl- or 4-Heteroaryl-6-pyrrolylpyrimidines. Eur. J. Org. Chem. 2016, 4865–4871. [Google Scholar] [CrossRef]
- Sengmany, S.; Vitu-Thiebaud, A.; Le Gall, E.; Condon, S.; Leonel, E.; Thobie-Gautier, C.; Pipelier, M.; Lebreton, J.; Dubreuil, D. An Electrochemical Nickel-Catalyzed Arylation of 3-Amino-6-Chloropyridazines. J. Org. Chem. 2013, 78, 370–379. [Google Scholar] [CrossRef] [PubMed]
- Sengmany, S.; Le Gall, E.; Leonel, E. An Electrochemical Synthesis of Functionalized Arylpyrimidines from 4-Amino-6-Chloropyrimidines and Aryl Halides. Molecules 2011, 16, 5550–5560. [Google Scholar] [CrossRef] [PubMed]
- Sengmany, S.; Leonel, E.; Polissaint, F.; Nedelec, J.Y.; Pipelier, M.; Thobie-Gautier, C.; Dubreuil, D. Preparation of Functionalized Aryl- and Heteroarylpyridazines by Nickel-Catalyzed Electrochemical Cross-Coupling Reactions. J. Org. Chem. 2007, 72, 5631–5636. [Google Scholar] [CrossRef] [PubMed]
- Mikhaylov, D.Y.; Budnikova, Y.H. Fluoroalkylation of organic compounds. Russ. Chem. Rev. 2013, 835–864. [Google Scholar] [CrossRef]
- Budnikova, Y.H.; Gryaznova, T.V.; Grinenko, V.V.; Dudkina, Y.B.; Khrizanforov, M.N. Eco-efficient Electrocatalytic C–P bond Formation. Pure Appl. Chem. 2017, 89, 311–330. [Google Scholar] [CrossRef]
- Mikhaylov, D.Y.; Gryaznova, T.V.; Dudkina, Y.B.; Polyancev, F.M.; Latypov, S.K.; Sinyashin, O.G.; Budnikova, Y.H. Novel electrochemical pathway to fluoroalkyl phosphines and phosphine oxides. J. Fluor. Chem. 2013, 153, 178–182. [Google Scholar] [CrossRef]
- Budnikova, Y.G.; Kargin, Y.M.; Sinyashin, O.G. Electrosynthesis of mixed tertiary phosphines catalysed by nickel complexes. Mendeleev Commun. 1999, 9, 193–194. [Google Scholar] [CrossRef]
- Budnikova, Y.H.; Perichon, J.; Yakhvarov, D.G.; Kargin, Y.M.; Sinyashin, O.G. Highly reactive organonickel complexes in electrocatalytic processes. J. Organomet. Chem. 2001, 630, 185–192. [Google Scholar] [CrossRef]
- Milyukov, V.A.; Budnikova, Y.H.; Sinyashin, O.G. Organic chemistry of elemental phosphorus. Russ. Chem. Rev. 2005, 74, 781–805. [Google Scholar] [CrossRef]
- Budnikova, Y.H.; Sinyashin, O.G. Phosphorylation of aromatic C–H bonds involving metals and metal complexes. Russ. Chem. Rev. 2015, 84, 917–951. [Google Scholar] [CrossRef]
- Dudkina, Y.B.; Mikhaylov, D.Y.; Gryaznova, T.V.; Sinyashin, O.G.; Vicic, D.A.; Budnikova, Y.H. MII/MIII-Catalyzed ortho-Fluoroalkylation of 2-Phenylpyridine. Eur. J. Org. Chem. 2012, 2012, 2114–2117. [Google Scholar] [CrossRef]
- Jutand, A. Contribution of Electrochemistry to Organometallic Catalysis. Chem. Rev. 2008, 108, 2300–2347. [Google Scholar] [CrossRef] [PubMed]
- Francke, R.; Little, R.D. Redox catalysis in organic electrosynthesis: Basic principles and recent developments. Chem. Soc. Rev. 2014, 43, 2492–2521. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, J.-I.; Kataoka, K.; Horcajada, R.; Nagaki, A. Modern Strategies in Electroorganic Synthesis. Chem. Rev. 2008, 108, 2265–2299. [Google Scholar] [CrossRef] [PubMed]
- Sperry, J.B.; Wright, D.L. The application of cathodic reductions and anodic oxidations in the synthesis of complex molecules. Chem. Soc. Rev. 2006, 35, 605–621. [Google Scholar] [CrossRef] [PubMed]
- Budnikova, Y.H. Metal complex catalysis in organic electrosynthesis. Russ. Chem. Rev. 2002, 71, 111–139. [Google Scholar] [CrossRef]
- Kuehnel, M.F.; Orchard, K.L.; Dalle, K.E.; Reisner, E. Selective Photocatalytic CO Reduction in Water through Anchoring of a Molecular Ni Catalyst on CdS Nanocrystals. J. Am. Chem. Soc. 2017, 139, 7217–7223. [Google Scholar] [CrossRef] [PubMed]
- Elgrishi, N.; Chambers, M.B.; Artero, V.; Fontecave, M. Terpyridine complexes of first row transition metals and electrochemical reduction of CO2 to CO. Phys. Chem. Chem. Phys. 2014, 16, 13635–13644. [Google Scholar] [CrossRef] [PubMed]
- Arana, C.; Keshavarz, M.; Potts, K.T.; Abruna, H.D. Electrocatalytic reduction of CO and O with electropolymerized films of vinyl-terpyridine complexes of Fe, Ni and Co. Inorg. Chim. Acta 1994, 225, 285–295. [Google Scholar] [CrossRef]
- Prasad, R.; Scaife, D.B. Electro-oxidation and electro-reduction of some iron(II), cobalt(II) and nickel(II) polypyridyl complexes in acetonitrile. J. Electroanal. Chem. 1977, 84, 373–386. [Google Scholar] [CrossRef]
- Behrens, H.; Meyer, K. Über neue Darstellungsweisen von Nickel(0)-Komplexen aus Nickelocen. Z. Naturforsch. B 1966, 21, 489–490. [Google Scholar] [CrossRef]
- Churchill, M.R.; O’Brien, T.A. Crystal structure and molecular geometry of a trifluoromethyl complex of nickel: π-cyclopentadienyl-σ-trifluoromethyl(triphenylphosphine)nickel. J. Chem. Soc. A 1970, 161–167. [Google Scholar] [CrossRef]
- Dubinina, G.G.; Brennessel, W.W.; Miller, J.L.; Vicic, D.A. Exploring Trifluoromethylation Reactions at Nickel: A Structural and Reactivity Study. Organometallics 2008, 27, 3933–3938. [Google Scholar] [CrossRef]
- Savéant, J.-M. Molecular Catalysis of Electrochemical Reactions. Mechanistic Aspects. Chem. Rev. 2008, 108, 2348–2378. [Google Scholar] [CrossRef] [PubMed]
- Amatore, C.; Oturan, M.A.; Pinson, J.; Saveant, J.-M.; Thiebault, A. Electron-Transfer-Induced Reactions. A Novel Approach Based on Electrochemical Redox Catalysis. Application to Aromatic Nucleophilic Substitutions. J. Am. Chem. Soc. 1984, 106, 6318–6321. [Google Scholar] [CrossRef]
- Mikhaylov, D.Y.; Budnikova, Y.H.; Gryaznova, T.V.; Krivolapov, D.V.; Litvinov, I.A.; Vicic, D.A.; Sinyashin, O.G. Electrocatalytic fluoroalkylation of olefins. J. Organomet. Chem. 2009, 694, 3840–3843. [Google Scholar] [CrossRef]
- Mikhaylov, D.Y.; Budnikova, Y.H.; Gryaznova, T.V.; Sinyashin, O.G. Electrocatalytic fluoroalkylation of olefins. Russ. Chem. Bull. Int. Ed. 2010, 59, 1918–1920. [Google Scholar] [CrossRef]
- Dudkina, Y.B.; Gryaznova, T.V.; Osin, Y.N.; Salnikov, V.V.; Davydov, N.A.; Fedorenko, S.V.; Mustafina, A.R.; Vicic, D.A.; Sinyashin, O.G.; Budnikova, Y.H. Nanoheterogeneous catalysis in electrochemically induced olefin perfluoroalkylation. Dalton Trans. 2015, 44, 8833–8838. [Google Scholar] [CrossRef] [PubMed]
Scheme 1.
Two resonance forms for mono-reduced nickel 2,2′:6′,2′′-terpyridine (tpy) complexes.

Scheme 2.
Reduction of [Ni(II)(i-Pr-pybox)(Ph)])BArF4) (1) using pentamethyl cobaltocene, adapted from ref. [26]. Copyright (2014) American Chemical Society. Further permissions related to the material excerpted must be directed to the ACS.
Scheme 2.
Reduction of [Ni(II)(i-Pr-pybox)(Ph)])BArF4) (1) using pentamethyl cobaltocene, adapted from ref. [26]. Copyright (2014) American Chemical Society. Further permissions related to the material excerpted must be directed to the ACS.

Scheme 3.
Negishi-type cross-coupling of C–C bonds with [Ni(tpy)(Me)] as catalyst, adopted from ref. [45].
Scheme 3.
Negishi-type cross-coupling of C–C bonds with [Ni(tpy)(Me)] as catalyst, adopted from ref. [45].

Scheme 4.
Cross-coupling of a secondary alkyl bromide with a secondary alkylzinc reagent, adopted from ref. [48].
Scheme 4.
Cross-coupling of a secondary alkyl bromide with a secondary alkylzinc reagent, adopted from ref. [48].

Scheme 5.
Control experiment of a cationic nickel(II) complex 1 with an alkylzinc reagent [46].
Scheme 5.
Control experiment of a cationic nickel(II) complex 1 with an alkylzinc reagent [46].

Scheme 6.
Intermediacy of a neutral nickel(I) complex 2 with an alkyl iodide, adopted from ref. [46].
Scheme 6.
Intermediacy of a neutral nickel(I) complex 2 with an alkyl iodide, adopted from ref. [46].

Scheme 7.
Proposed catalytic cycle of the nickel–tpy catalytic system in an alkyl–alkyl cross-coupling reaction, adopted from ref. [46], the oxidation states shown in red were the results of studies outlined in Section 2.2 and Section 2.3.
Scheme 7.
Proposed catalytic cycle of the nickel–tpy catalytic system in an alkyl–alkyl cross-coupling reaction, adopted from ref. [46], the oxidation states shown in red were the results of studies outlined in Section 2.2 and Section 2.3.

Scheme 8.
Oxidation of the isolated complex and follow-up reactions, adopted from ref. [54].
Scheme 8.
Oxidation of the isolated complex and follow-up reactions, adopted from ref. [54].

Scheme 9.
Synthesis of tpy nickel complexes bearing a readily attached [C4F82−] ligand, adopted from ref. [55].
Scheme 9.
Synthesis of tpy nickel complexes bearing a readily attached [C4F82−] ligand, adopted from ref. [55].

Scheme 10.
Stable terpyridine perfluoroalkylated Ni(III) species, adopted from ref. [55].
Scheme 10.
Stable terpyridine perfluoroalkylated Ni(III) species, adopted from ref. [55].

Scheme 11.
Proposed reaction mechanisms for the reductive cross-coupling reaction of amides and aryl iodides, adapted with permission from [62]. Copyright (2017) American Chemical Society.
Scheme 11.
Proposed reaction mechanisms for the reductive cross-coupling reaction of amides and aryl iodides, adapted with permission from [62]. Copyright (2017) American Chemical Society.

Scheme 12.
C–H alkylation of 1,3-azoles and alkyl halides (diglyme = bis(2-methoxyethyl)ether), adopted from ref. [67].
Scheme 12.
C–H alkylation of 1,3-azoles and alkyl halides (diglyme = bis(2-methoxyethyl)ether), adopted from ref. [67].

Scheme 13.
Total synthesis of salmochelins, adopted from ref. [69].
Scheme 13.
Total synthesis of salmochelins, adopted from ref. [69].

Scheme 14.
Reductive coupling of glycosyl bromides with activated alkenes, adopted from ref. [70].
Scheme 14.
Reductive coupling of glycosyl bromides with activated alkenes, adopted from ref. [70].

Scheme 15.
Proposed redox steps for [Ni(tpy)2]n+.

Scheme 16.
Electrocatalytical aromatic perfluoroalkylation catalyzed by Ni(tpy), adopted from ref. [72].
Scheme 16.
Electrocatalytical aromatic perfluoroalkylation catalyzed by Ni(tpy), adopted from ref. [72].

Scheme 17.
Catalytic cycle of Cu-assisted aromatic perfluoroalkylation with electrochemically reduced Ni–tpy complex, adopted from ref. [72].
Scheme 17.
Catalytic cycle of Cu-assisted aromatic perfluoroalkylation with electrochemically reduced Ni–tpy complex, adopted from ref. [72].

Scheme 18.
Ni-catalyzed cross-coupling of perfluoroalkyl halides and aromatic halides, adopted from ref. [71].
Scheme 18.
Ni-catalyzed cross-coupling of perfluoroalkyl halides and aromatic halides, adopted from ref. [71].


Scheme 20.
Proposed reaction sequence in the electrocatalytic fluoroalkylation of α-methyl styrene using [Ni(tpy)Br2], adopted from ref. [53].
Scheme 20.
Proposed reaction sequence in the electrocatalytic fluoroalkylation of α-methyl styrene using [Ni(tpy)Br2], adopted from ref. [53].

Figure 1.
Cyclic voltammograms of [Ni(tpy)Br2] (0.01 M) in the presence of increasing quantities of С6F13I. Ratios of complexes to RFI are 1:0 (red); 1:3 (blue); 1:6 (violet); 1:12 (dark blue); 1:30 (black). Reproduced from ref. [53] with permission form The Royal Society of Chemistry.
Figure 1.
Cyclic voltammograms of [Ni(tpy)Br2] (0.01 M) in the presence of increasing quantities of С6F13I. Ratios of complexes to RFI are 1:0 (red); 1:3 (blue); 1:6 (violet); 1:12 (dark blue); 1:30 (black). Reproduced from ref. [53] with permission form The Royal Society of Chemistry.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
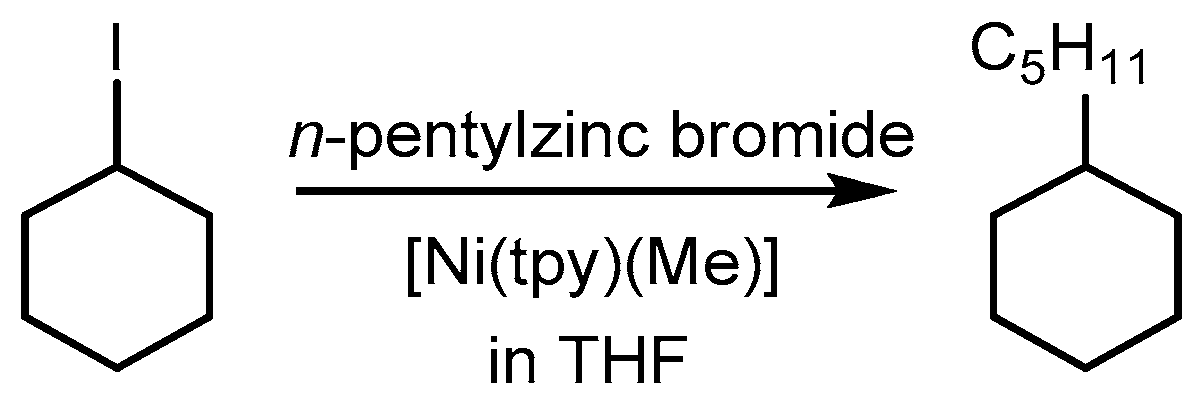{kind=link}
{kind=link}
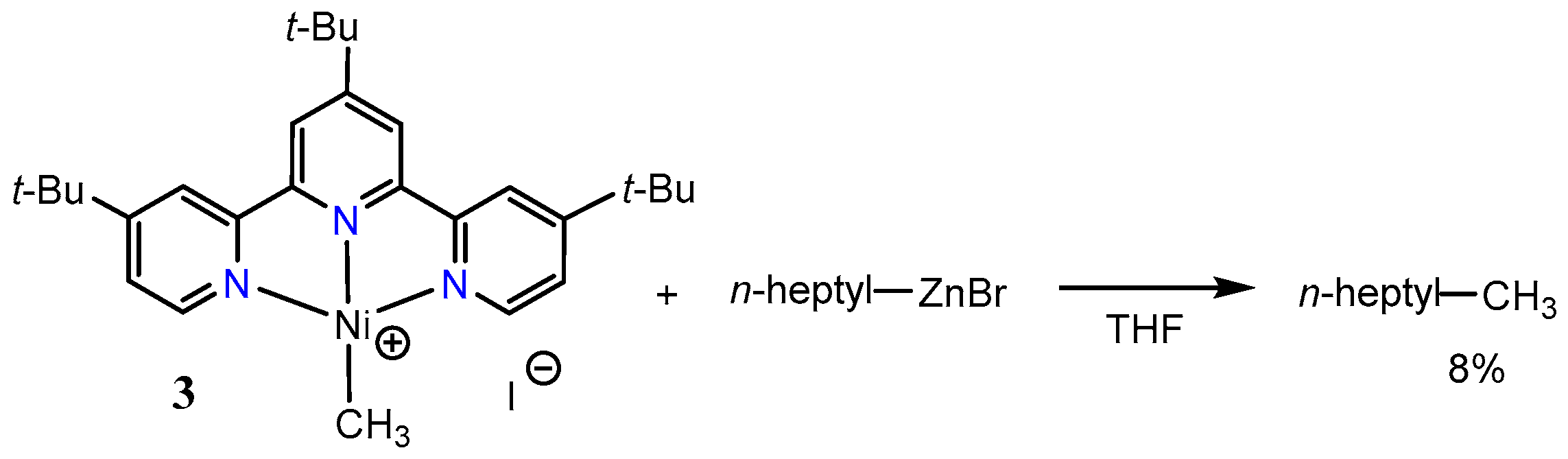{kind=link}
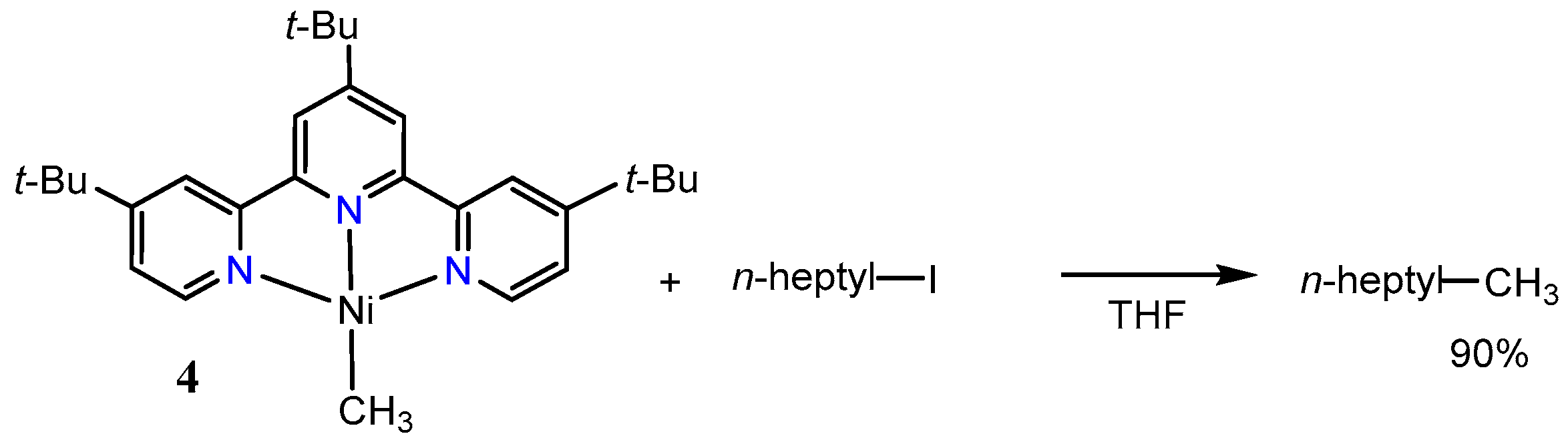{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
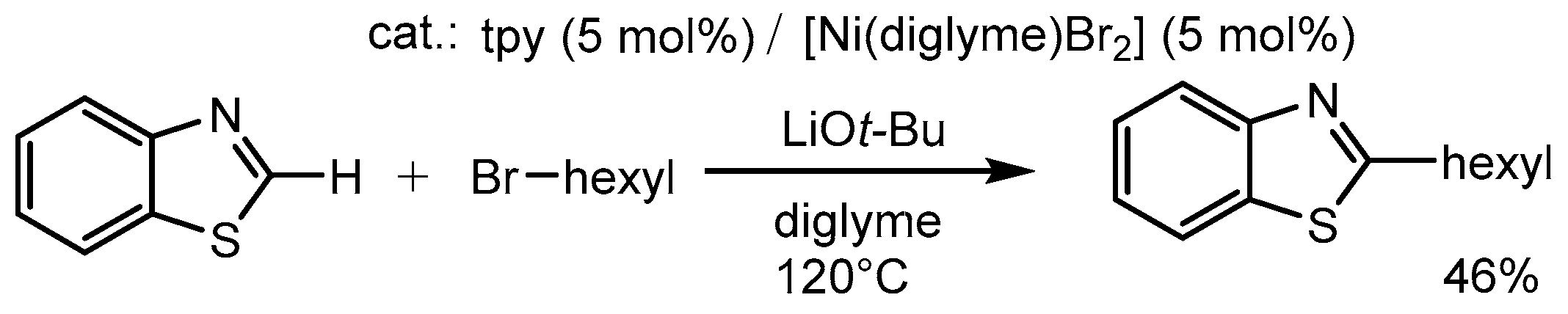{kind=link}
{kind=link}
{kind=link}
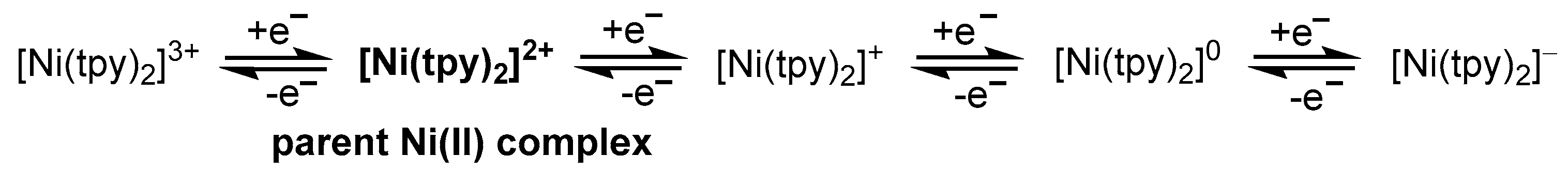{kind=link}
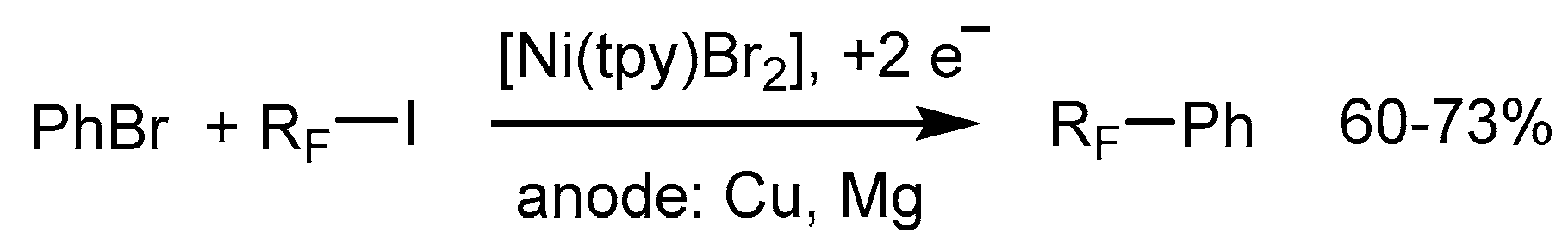{kind=link}
{kind=link}
{kind=link}
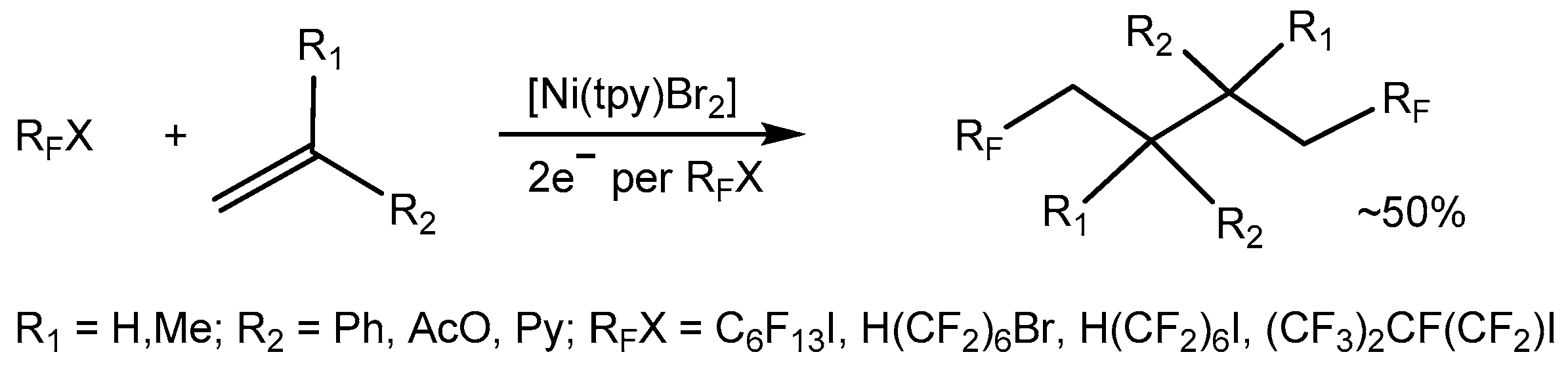{kind=link}
{kind=link}
Table 1.
Selected EPR data of reduced nickel tpy complexes a.
| Complex | giso (298 K) | gav (LT) b | g1 (LT) | g2 (LT) | g3 (LT) | ∆g (LT) c |
|---|---|---|---|---|---|---|
| [Ni(tpy)Br]• | 2.139 | 2.146 | 2.256 | 2.091 | 2.091 | 0.165 |
| [Ni(tpy)(Me)]• | 2.021 | 2.025 | 2.056 | 2.021 | 1.999 | 0.026 |
| [Ni(tpy)(Mes)]• | 2.0006 | 2.0006 | 2.009 | 2.002 | 1.991 | 0.018 |
| [((t-Bu)3tpy)Ni(Xyl)]• d | 2.0006 | 2.0007 | 2.008 | 2.003 | 1.991 | 0.017 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Budnikova, Y.H.; Vicic, D.A.; Klein, A. Exploring Mechanisms in Ni Terpyridine Catalyzed C–C Cross-Coupling Reactions—A Review. Inorganics 2018, 6, 18. https://0-doi-org.brum.beds.ac.uk/10.3390/inorganics6010018
AMA Style
Budnikova YH, Vicic DA, Klein A. Exploring Mechanisms in Ni Terpyridine Catalyzed C–C Cross-Coupling Reactions—A Review. Inorganics. 2018; 6(1):18. https://0-doi-org.brum.beds.ac.uk/10.3390/inorganics6010018
Chicago/Turabian StyleBudnikova, Yulia H., David A. Vicic, and Axel Klein. 2018. "Exploring Mechanisms in Ni Terpyridine Catalyzed C–C Cross-Coupling Reactions—A Review" Inorganics 6, no. 1: 18. https://0-doi-org.brum.beds.ac.uk/10.3390/inorganics6010018
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.