NHC-Based Iron Sensitizers for DSSCs


 ,
,  , ,
, ,
Abstract
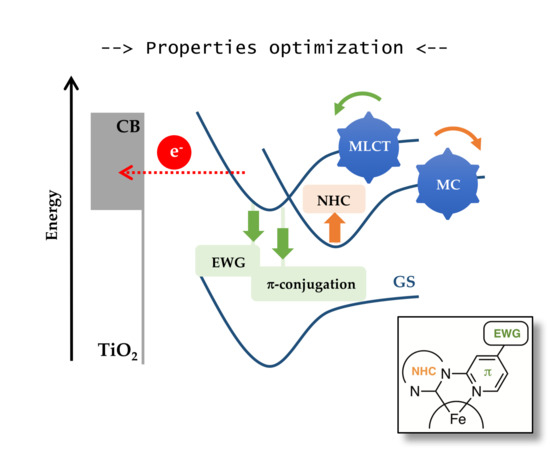
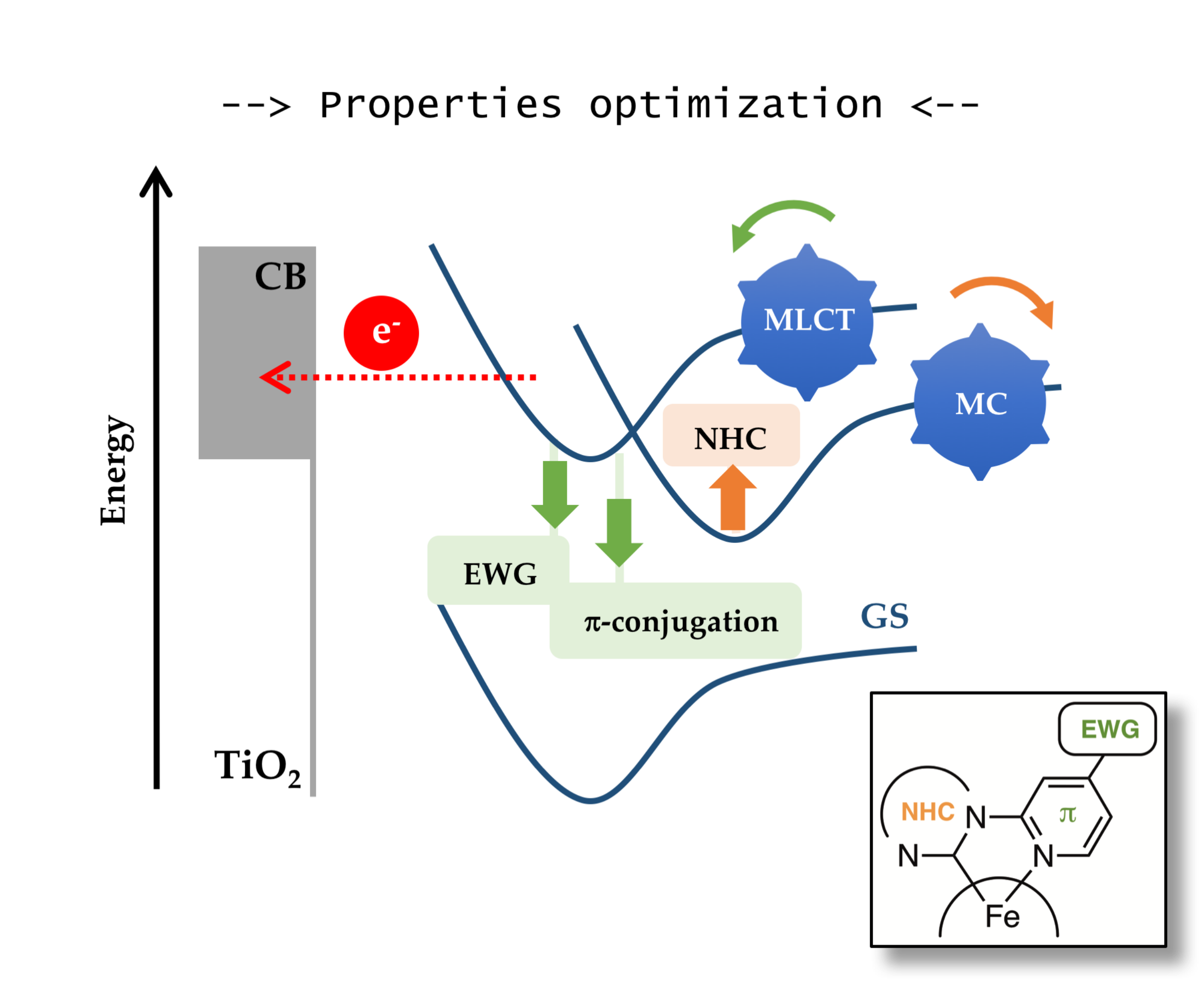
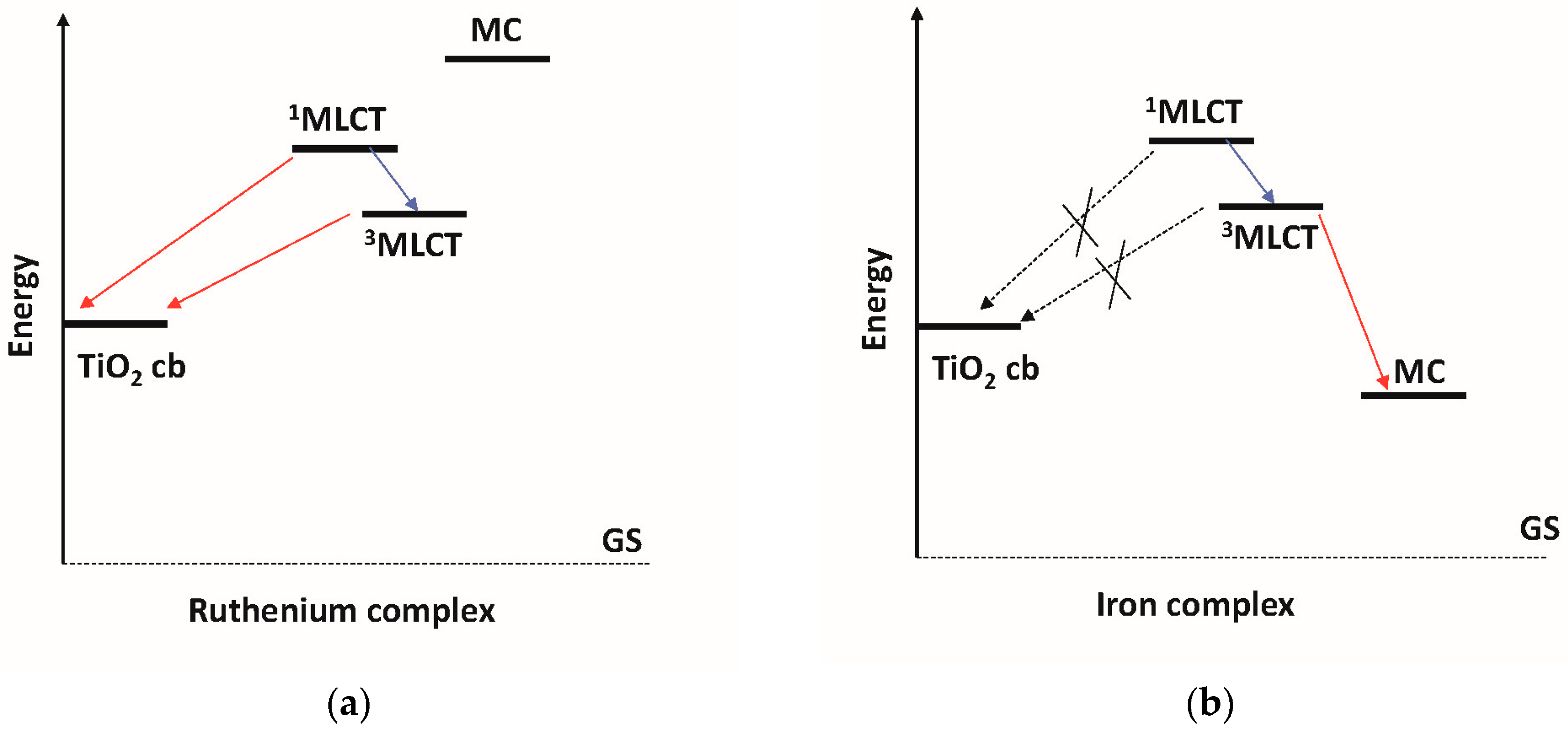
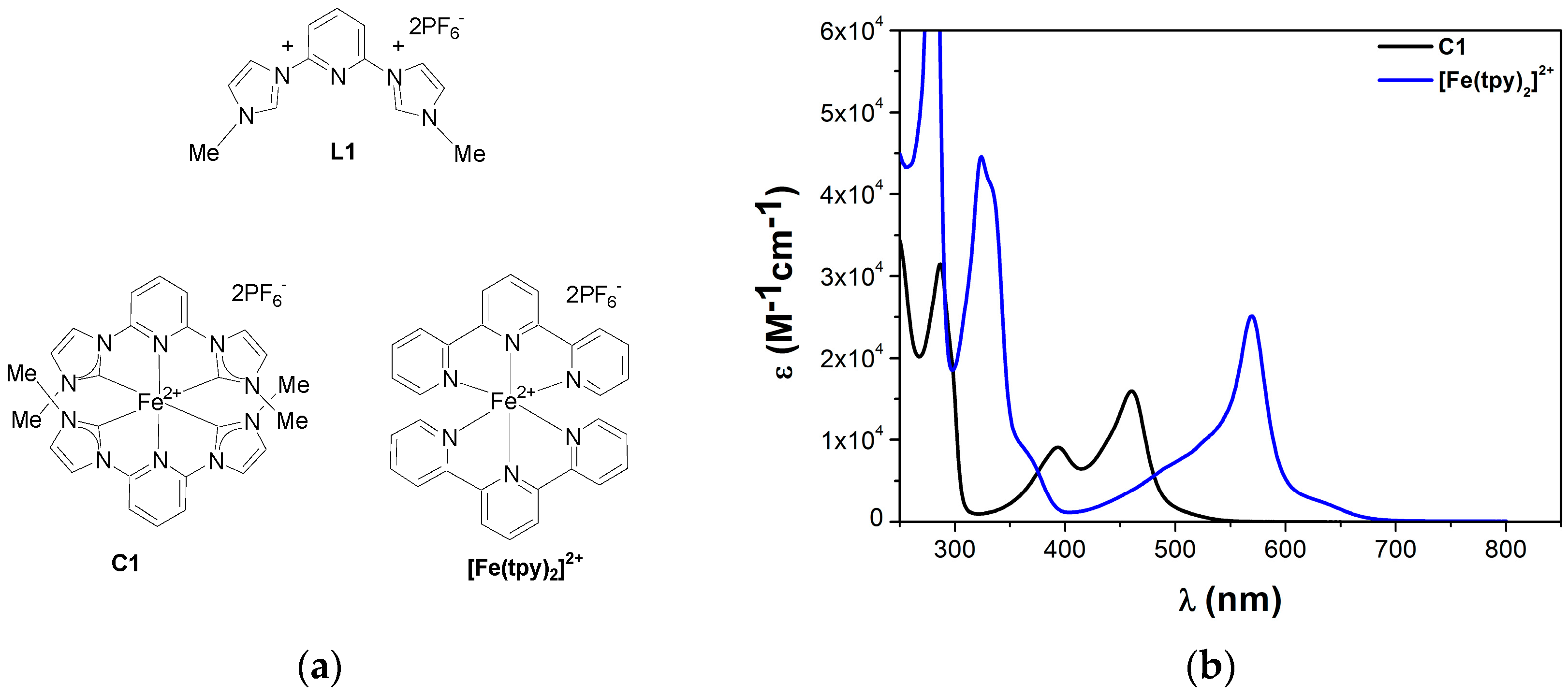
:
1. Introduction
2. Design of Sensitizers
2.1. NHC-Based Homoleptic Complexes
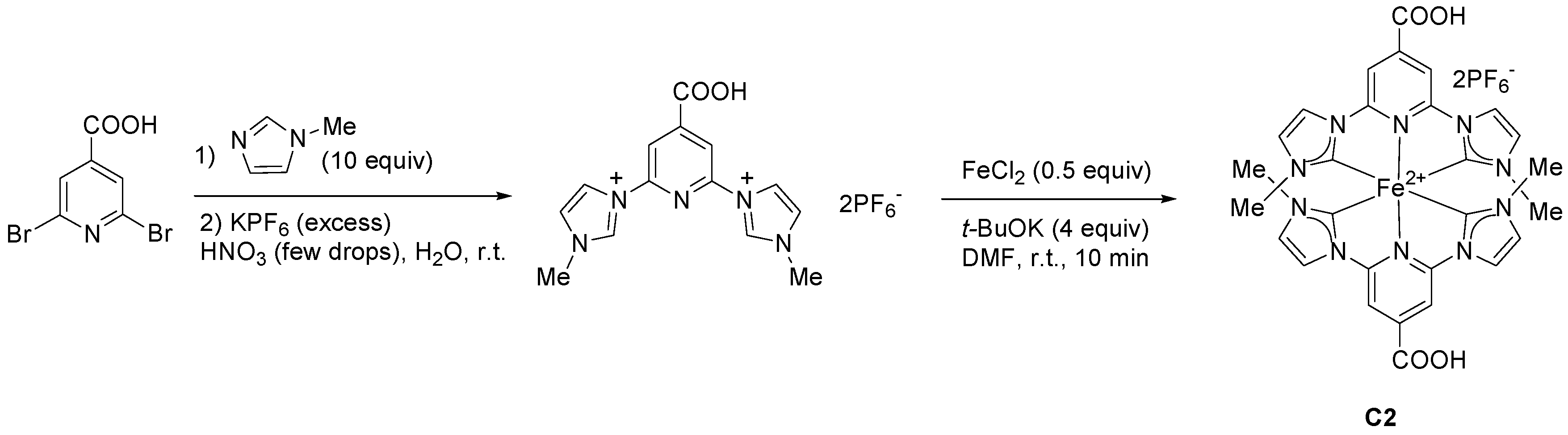
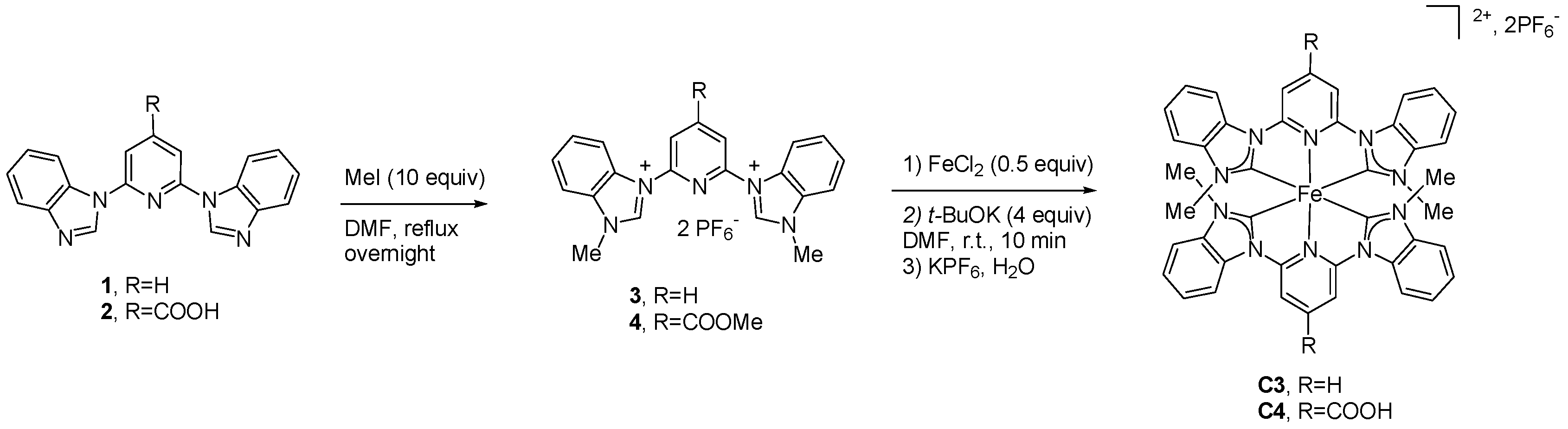
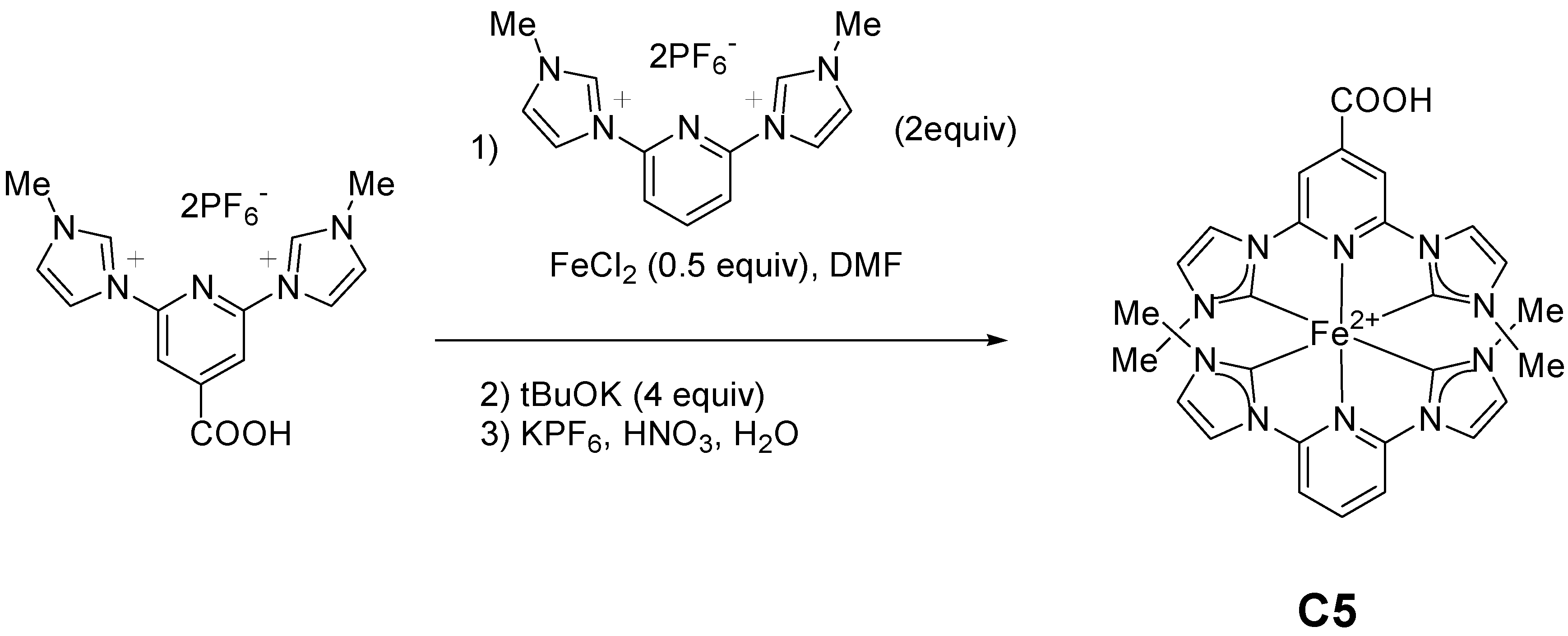
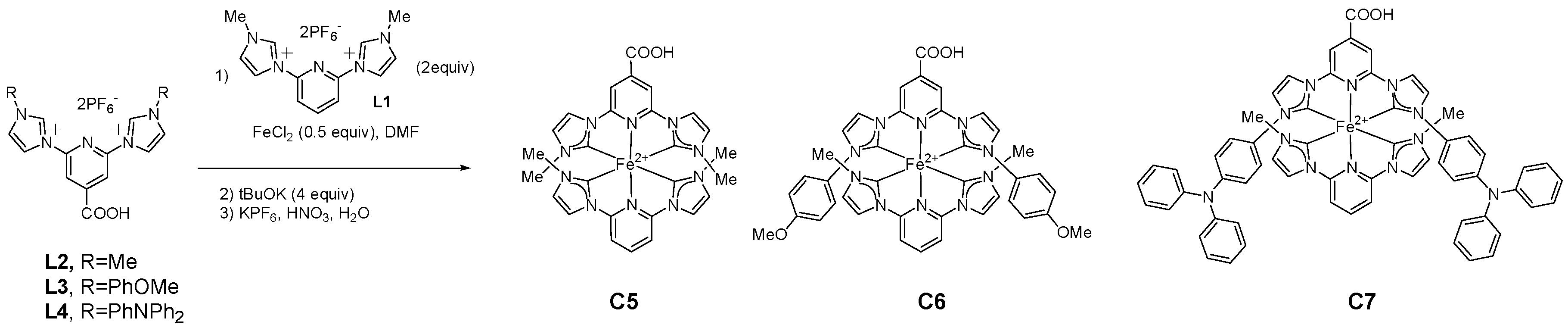
2.1.1. Effects of Carboxylic Groups
2.1.2. Effects of Alkyl Substituents
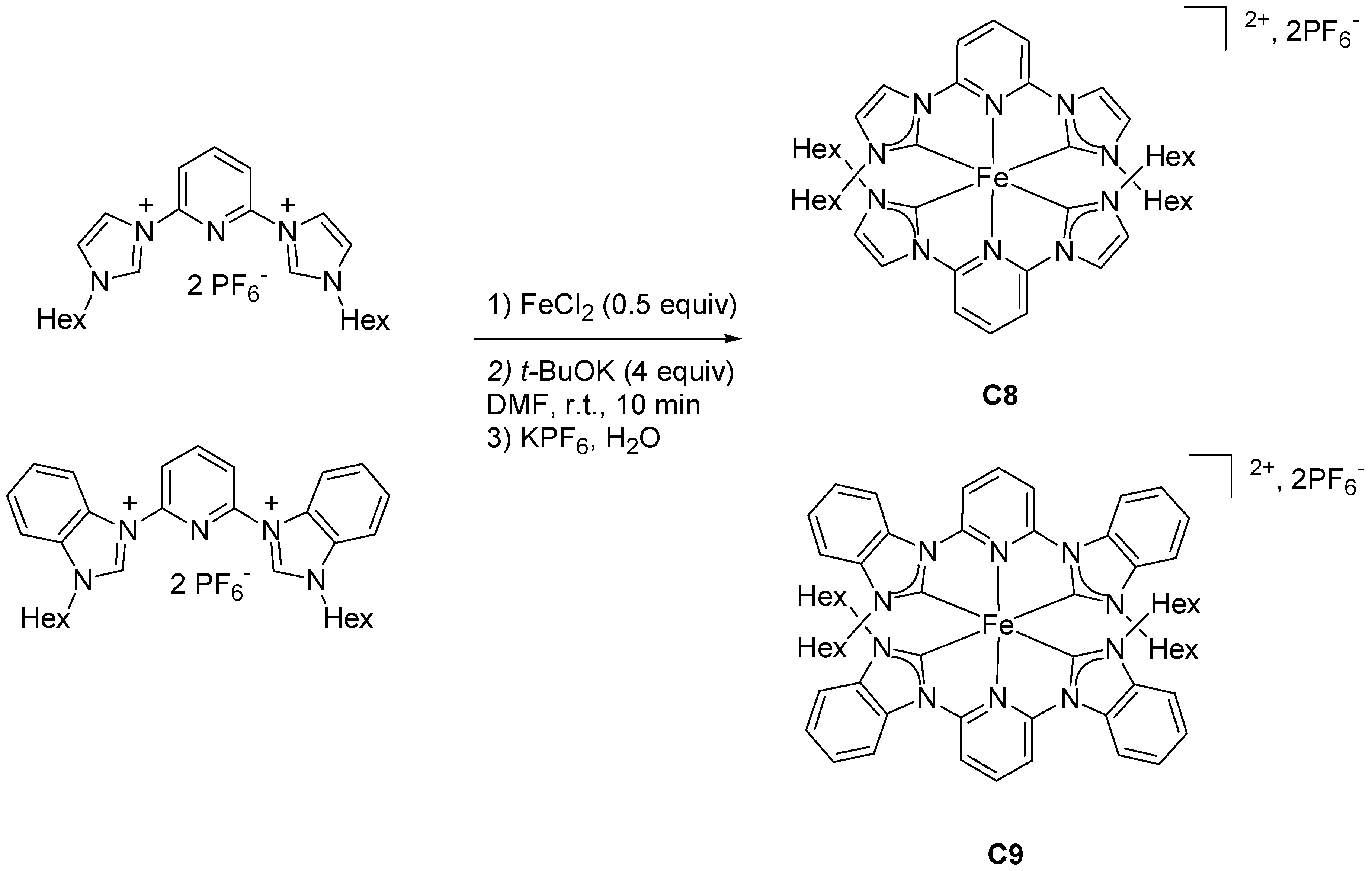
2.1.3. Effects of NHC Counts
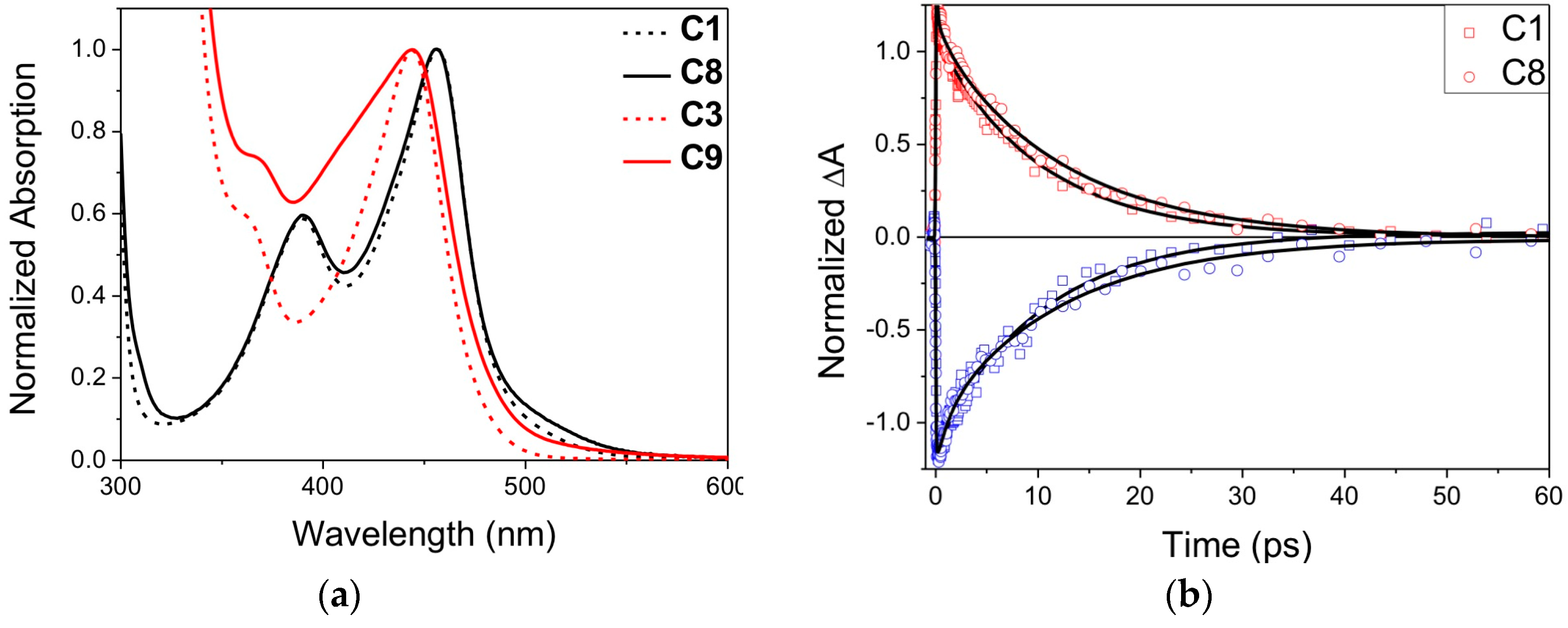
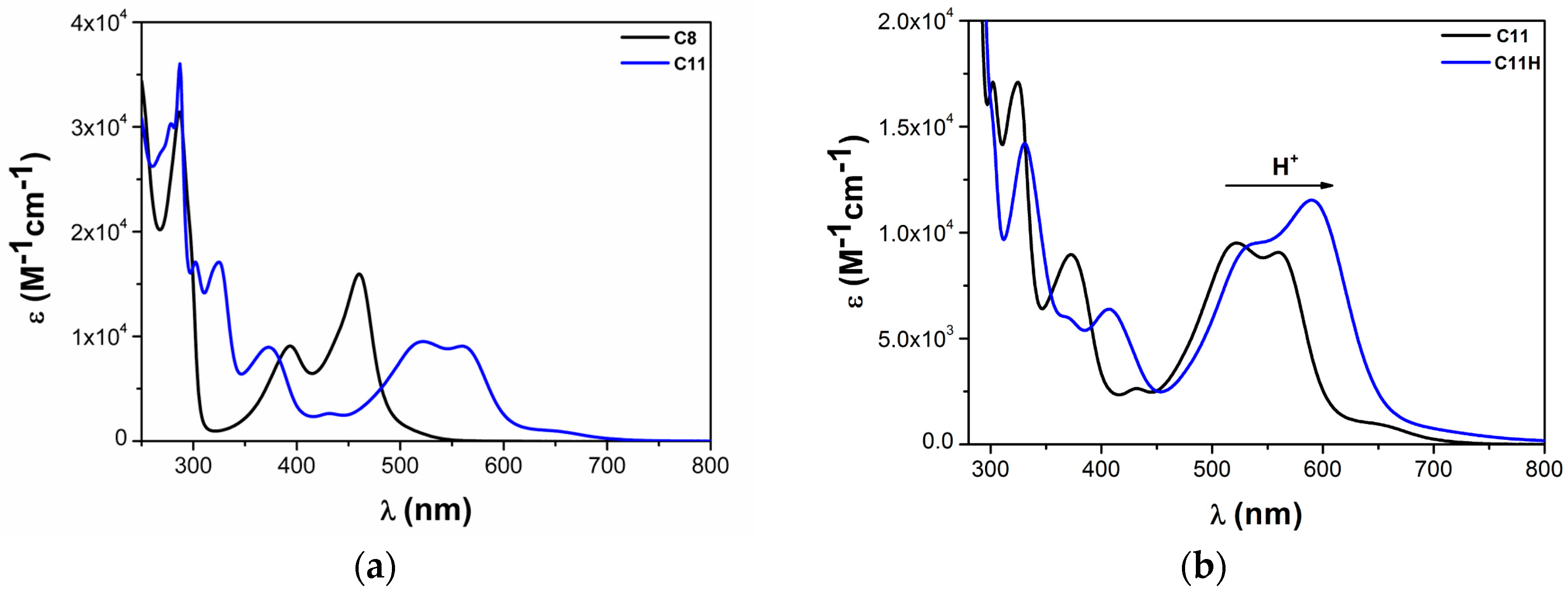
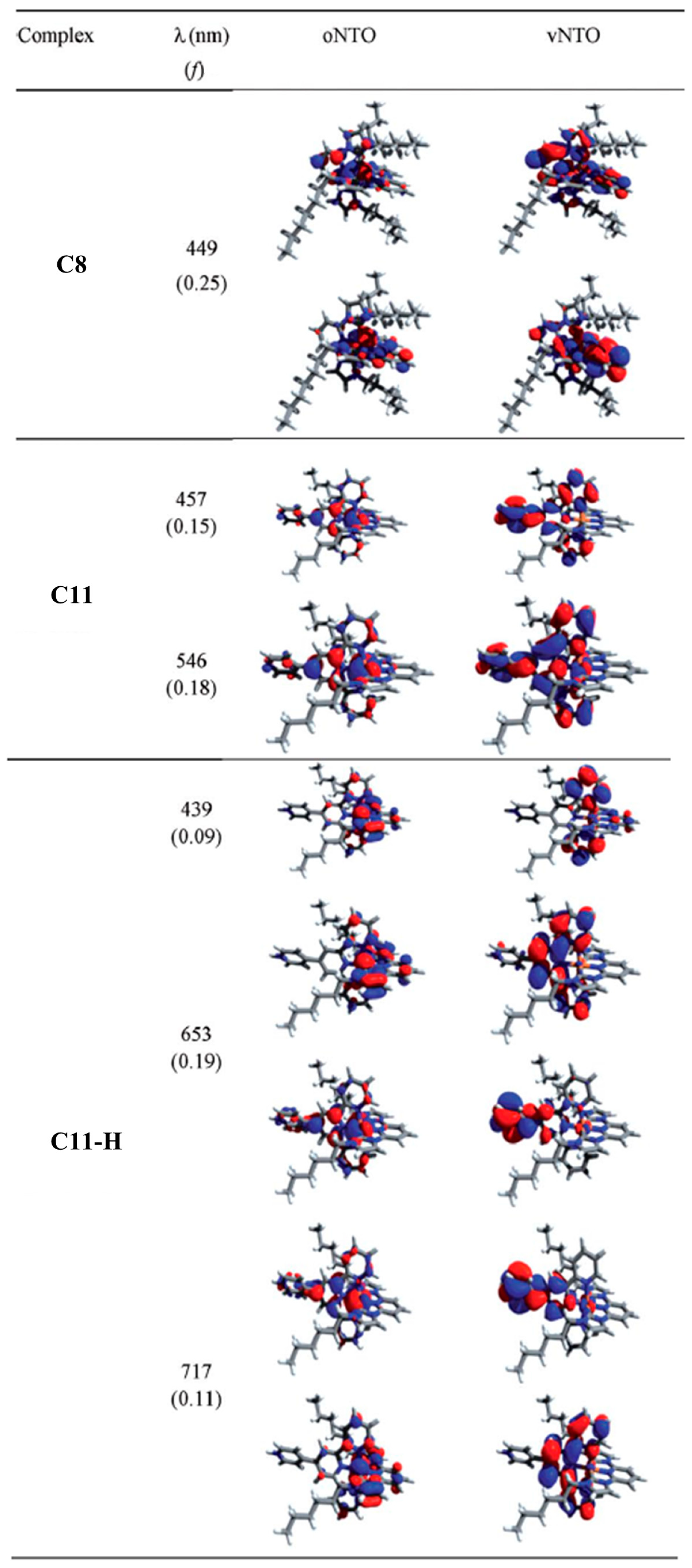
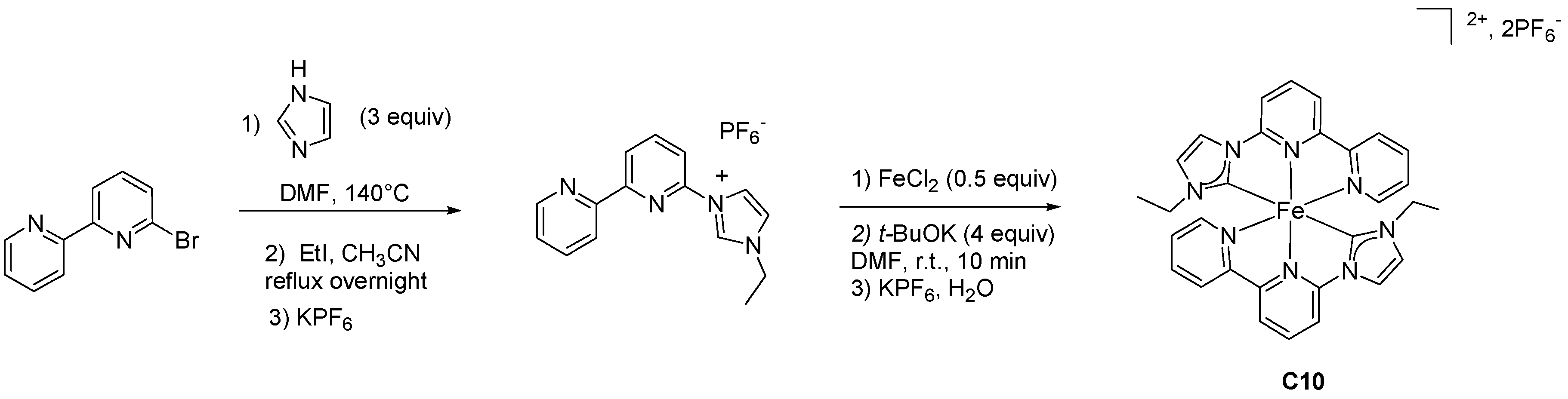
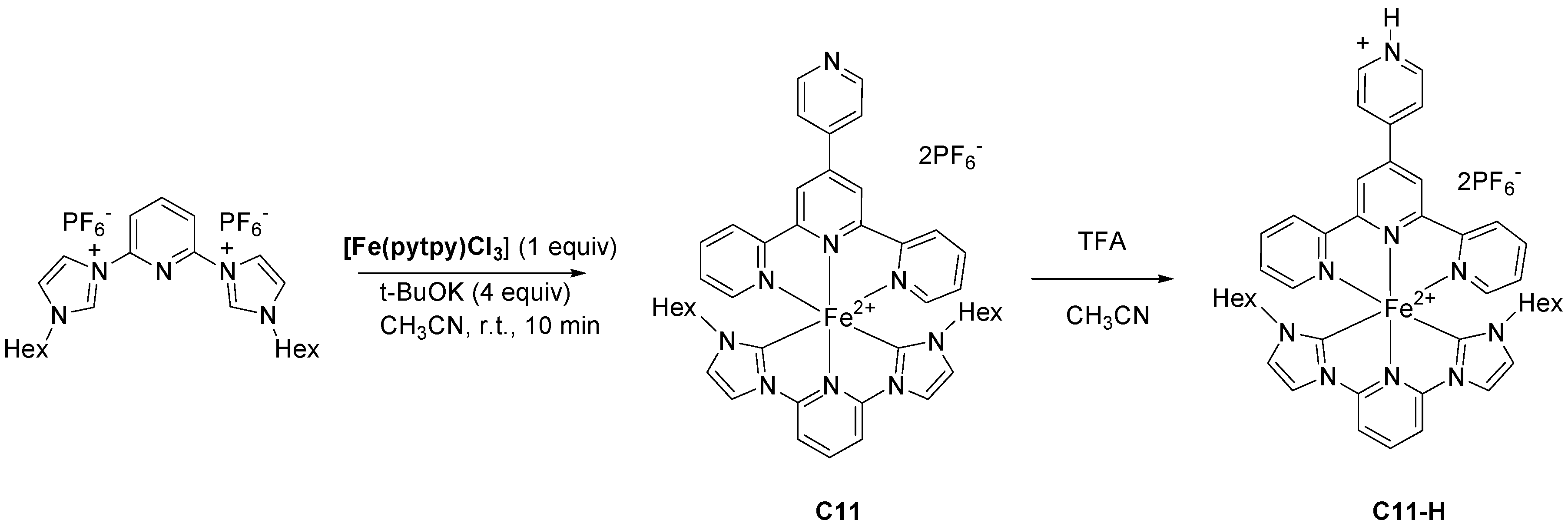
2.2. Heteroleptic Complexes
3. Photovoltaic Properties of Iron–NHC Complexes
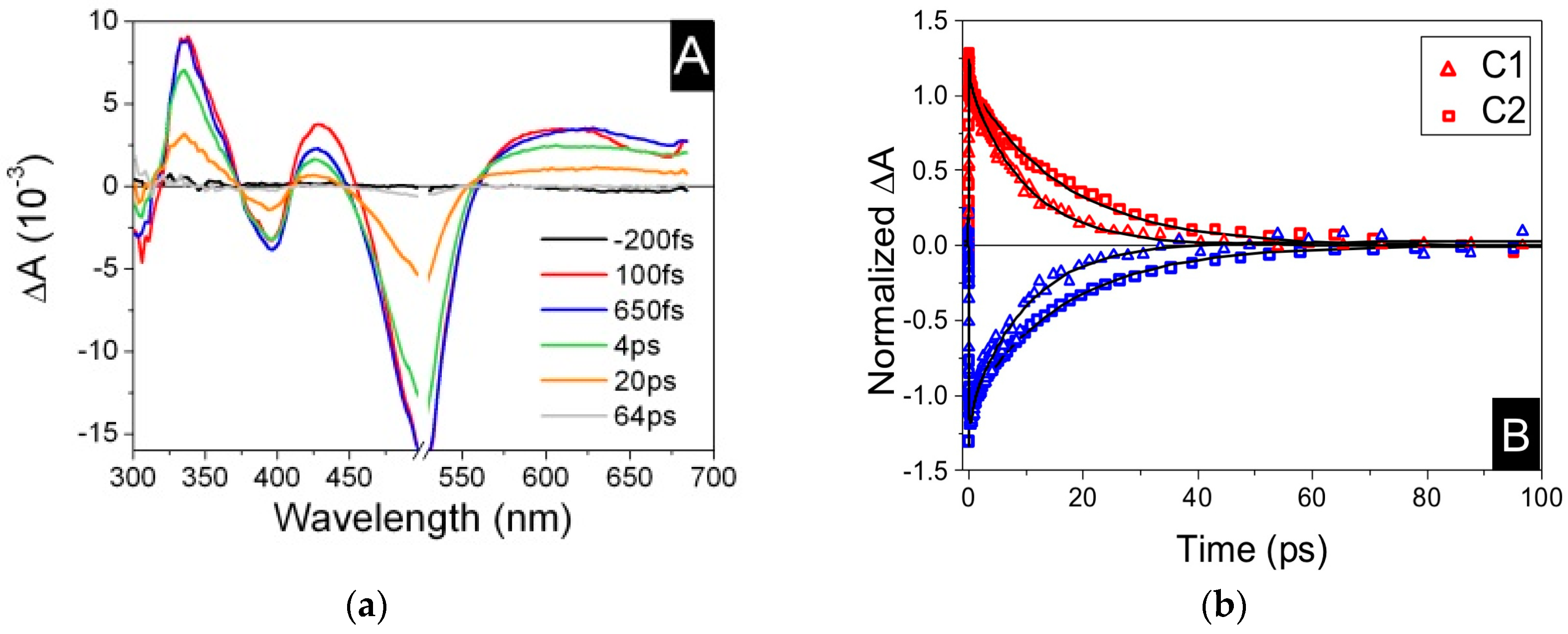
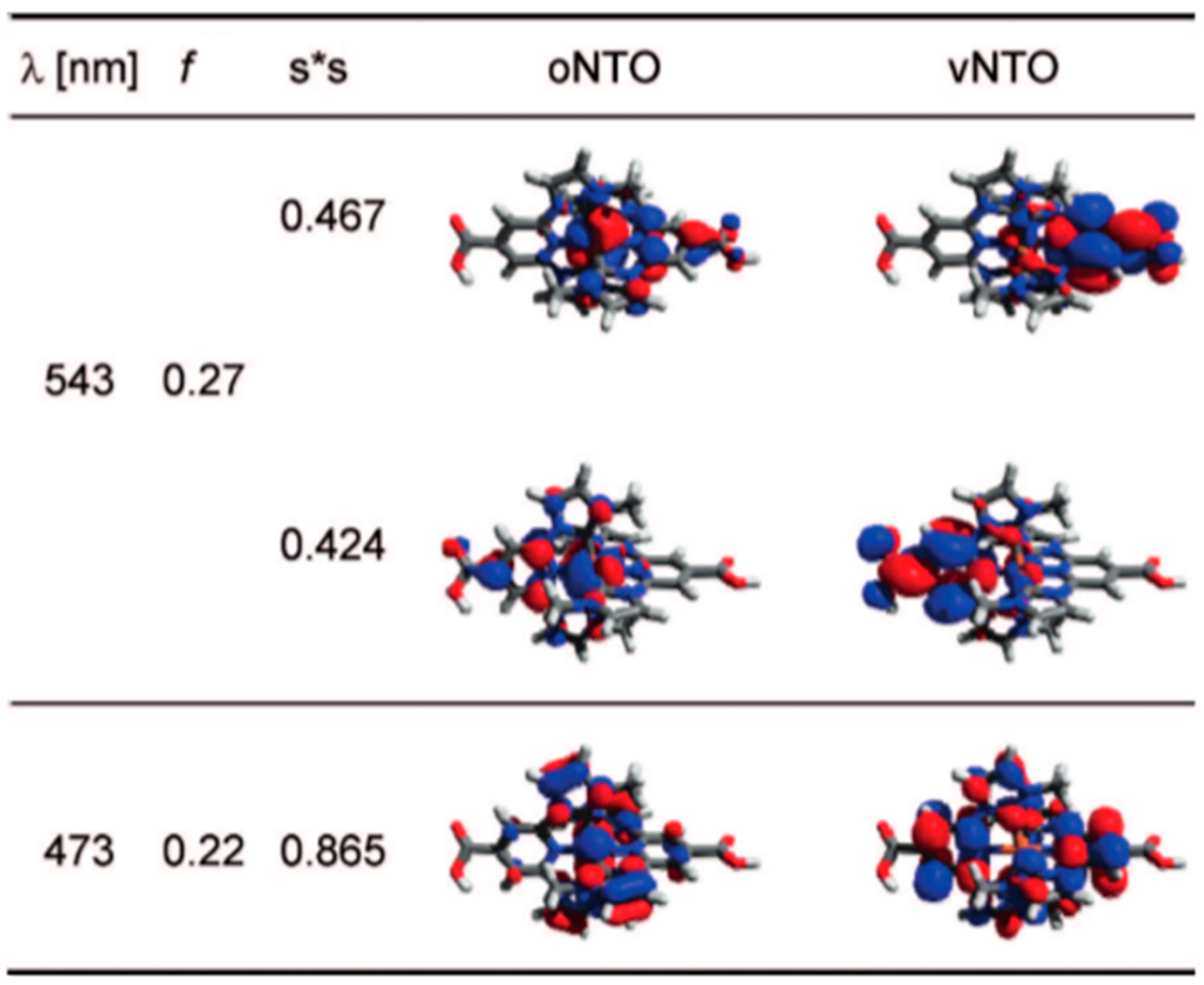
3.1. Homoleptic Complexes
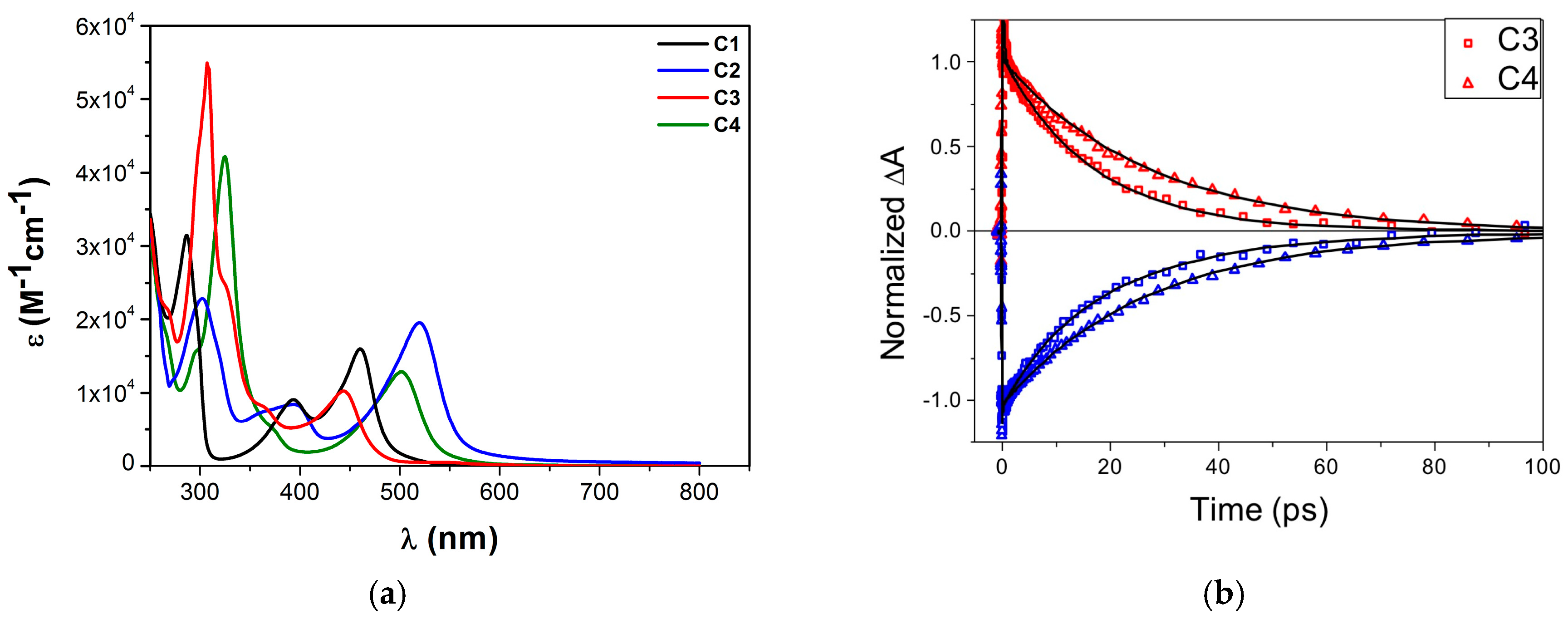
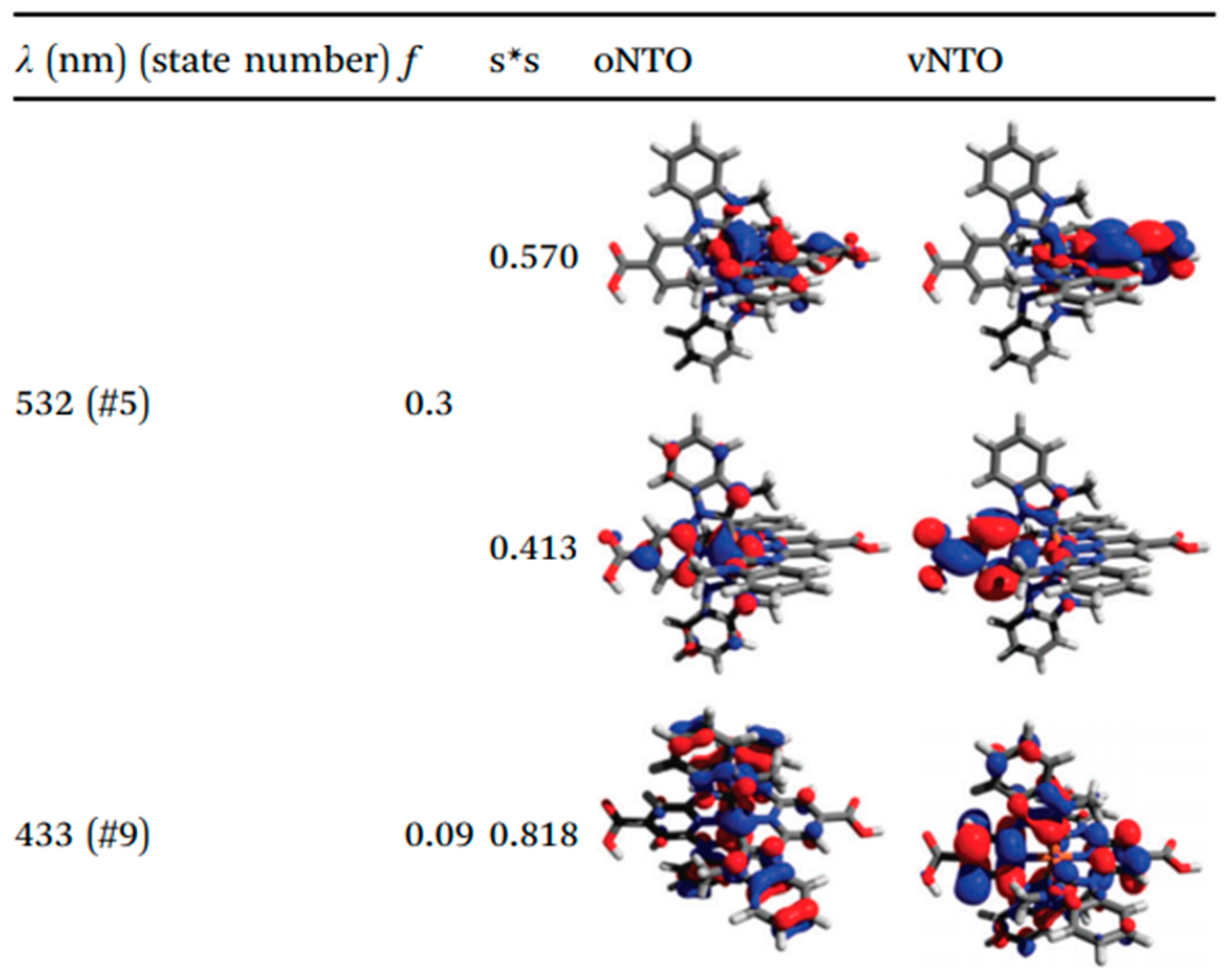
3.2. Heteroleptic Complexes
4. Conclusions and Outlook
Author Contributions
Funding
Conflicts of Interest
References
- Jung, H.S.; Lee, J.-K. Dye Sensitized Solar Cells for Economically Viable Photovoltaic Systems. J. Phys. Chem. Lett. 2013, 4, 1682–1693. [Google Scholar] [CrossRef] [PubMed]
- Hagfeldt, A.; Boschloo, G.; Sun, L.; Kloo, L.; Pettersson, H. Dye-Sensitized Solar Cells. Chem. Rev. 2010, 110, 6595–6663. [Google Scholar] [CrossRef] [PubMed]
- Hardin, B.E.; Hoke, E.T.; Armstrong, P.B.; Yum, J.-H.; Comte, P.; Torres, T.; Fréchet, J.M.J.; Nazeeruddin, M.K.; Grätzel, M.; McGehee, M.D. Increased Light Harvesting in Dye-Sensitized Solar Cells with Energy Relay Dyes. Nat. Photonics 2009, 3, 406–411. [Google Scholar] [CrossRef]
- Sauvage, J.P.; Collin, J.P.; Chambron, J.C.; Guillerez, S.; Coudret, C.; Balzani, V.; Barigelletti, F.; De Cola, L.; Flamigni, L. Ruthenium(II) and Osmium(II) Bis(Terpyridine) Complexes in Covalently-Linked Multicomponent Systems: Synthesis, Electrochemical Behavior, Absorption Spectra, and Photochemical and Photophysical Properties. Chem. Rev. 1994, 94, 993–1019. [Google Scholar] [CrossRef]
- Dixon, I.M.; Lebon, E.; Sutra, P.; Igau, A. Luminescent Ruthenium–Polypyridine Complexes & Phosphorus Ligands: Anything but a Simple Story. Chem. Soc. Rev. 2009, 38, 1621. [Google Scholar] [PubMed]
- Beley, M.; Gros, P.C. Ruthenium Polypyridine Complexes Bearing Pyrroles and π-Extended Analogues. Synthesis, Spectroelectronic, Electrochemical, and Photovoltaic Properties. Organometallics 2014, 33, 4590–4606. [Google Scholar] [CrossRef]
- Nazeeruddin, M.K.; De Angelis, F.; Fantacci, S.; Selloni, A.; Viscardi, G.; Liska, P.; Ito, S.; Takeru, B.; Grätzel, M. Combined Experimental and DFT-TDDFT Computational Study of Photoelectrochemical Cell Ruthenium Sensitizers. J. Am. Chem. Soc. 2005, 127, 16835–16847. [Google Scholar] [CrossRef] [PubMed]
- Fantacci, S.; De Angelis, F.; Selloni, A. Absorption Spectrum and Solvatochromism of the [Ru(4,4’-COOH-2,2’-Bpy)2(NCS)2] Molecular Dye by Time Dependent Density Functional Theory. J. Am. Chem. Soc. 2003, 125, 4381–4387. [Google Scholar] [CrossRef] [PubMed]
- Hagfeldt, A.; Peter, L. Dye-Sensitized Solar Cells; Kalyanasundaram, K., Ed.; EPFL Press: Lausanne, Switzerland, 2010. [Google Scholar]
- Listorti, A.; Creager, C.; Sommeling, P.; Kroon, J.; Palomares, E.; Fornelli, A.; Breen, B.; Barnes, P.R.F.; Durrant, J.R.; Law, C.; et al. The Mechanism behind the Beneficial Effect of Light Soaking on Injection Efficiency and Photocurrent in Dye Sensitized Solar Cells. Energy Environ. Sci. 2011, 4, 3494–3501. [Google Scholar] [CrossRef]
- Listorti, A.; O’Regan, B.; Durrant, J.R. Electron Transfer Dynamics in Dye-Sensitized Solar Cells. Chem. Mater. 2011, 23, 3381–3399. [Google Scholar] [CrossRef]
- Lobello, M.G.; Fantacci, S.; De Angelis, F. Computational Spectroscopy Characterization of the Species Involved in Dye Oxidation and Regeneration Processes in Dye-Sensitized Solar Cells. J. Phys. Chem. C 2011, 115, 18863–18872. [Google Scholar] [CrossRef]
- McCusker, J.K.; Walda, K.N.; Dunn, R.C.; Simon, J.D.; Magde, D.; Hendrickson, D.N. Subpicosecond 1MLCT. Fwdarw. 5T2 Intersystem Crossing of Low-Spin Polypyridyl Ferrous Complexes. J. Am. Chem. Soc. 1993, 115, 298–307. [Google Scholar] [CrossRef]
- Ferrere, S.; Gregg, B.A. Photosensitization of TiO2 by [FeII(2,2′-Bipyridine-4,4′-Dicarboxylic Acid)2(CN)2]: Band Selective Electron Injection from Ultra-Short-Lived Excited States. J. Am. Chem. Soc. 1998, 120, 843–844. [Google Scholar] [CrossRef]
- Ferrere, S. New Photosensitizers Based upon [Fe(L)2(CN)2] and [Fe(L)3] (L = Substituted 2,2′-Bipyridine): Yields for the Photosensitization of TiO2 and Effects on the Band Selectivity. Chem. Mater. 2000, 12, 1083–1089. [Google Scholar] [CrossRef]
- Dixon, I.M.; Khan, S.; Alary, F.; Boggio-Pasqua, M.; Heully, J.-L. Probing the Photophysical Capability of Mono and Bis(Cyclometallated) Fe(ii) Polypyridine Complexes Using Inexpensive Ground State DFT. Dalton Trans. 2014, 43, 15898–15905. [Google Scholar] [CrossRef] [PubMed]
- Housecroft, C.E.; Constable, E.C. The Emergence of Copper(i)-Based Dye Sensitized Solar Cells. Chem. Soc. Rev. 2015, 44, 8386–8398. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sandroni, M.; Kayanuma, M.; Planchat, A.; Szuwarski, N.; Blart, E.; Pellegrin, Y.; Daniel, C.; Boujtita, M.; Odobel, F. First Application of the HETPHEN Concept to New Heteroleptic Bis(Diimine) Copper(i) Complexes as Sensitizers in Dye Sensitized Solar Cells. Dalton Trans. 2013, 42, 10818. [Google Scholar] [CrossRef] [PubMed]
- Monat, J.E.; McCusker, J.K. Femtosecond Excited-State Dynamics of an Iron(II) Polypyridyl Solar Cell Sensitizer Model. J. Am. Chem. Soc. 2000, 122, 4092–4097. [Google Scholar] [CrossRef]
- Zhang, W.; Alonso-Mori, R.; Bergmann, U.; Bressler, C.; Chollet, M.; Galler, A.; Gawelda, W.; Hadt, R.G.; Hartsock, R.W.; Kroll, T.; et al. Tracking Excited-State Charge and Spin Dynamics in Iron Coordination Complexes. Nature 2014, 509, 345–348. [Google Scholar] [CrossRef] [PubMed]
- Jakubikova, E.; Bowman, D.N. Fe(II)-Polypyridines as Chromophores in Dye-Sensitized Solar Cells: A Computational Perspective. Acc. Chem. Res. 2015, 48, 1441–1449. [Google Scholar] [CrossRef] [PubMed]
- Auböck, G.; Chergui, M. Sub-50-Fs Photoinduced Spin Crossover in [Fe(Bpy)3]2+. Nat. Chem. 2015, 7, 629–633. [Google Scholar] [CrossRef] [PubMed]
- Meyer, T.J. Photochemistry of Metal Coordination Complexes: Metal to Ligand Charge Transfer Excited States. Pure Appl. Chem. 1986, 58, 1193–1206. [Google Scholar] [CrossRef]
- Consani, C.; Prémont-Schwarz, M.; ElNahhas, A.; Bressler, C.; van Mourik, F.; Cannizzo, A.; Chergui, M. Vibrational Coherences and Relaxation in the High-Spin State of Aqueous [FeII(Bpy)3]2+. Angew. Chem. Int. Ed. 2009, 48, 7184–7187. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Harlang, T.; Canton, S.E.; Chábera, P.; Suárez-Alcántara, K.; Fleckhaus, A.; Vithanage, D.A.; Göransson, E.; Corani, A.; Lomoth, R.; et al. Towards Longer-Lived Metal-to-Ligand Charge Transfer States of Iron(II) Complexes: An N-Heterocyclic Carbene Approach. Chem. Commun. 2013, 49, 6412. [Google Scholar] [CrossRef] [PubMed]
- Harlang, T.C.B.; Liu, Y.; Gordivska, O.; Fredin, L.A.; Ponseca, C.S.; Huang, P.; Chábera, P.; Kjaer, K.S.; Mateos, H.; Uhlig, J.; et al. Iron Sensitizer Converts Light to Electrons with 92% Yield. Nat. Chem. 2015, 7, 883–889. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fredin, L.A.; Pápai, M.; Rozsályi, E.; Vankó, G.; Wärnmark, K.; Sundström, V.; Persson, P. Exceptional Excited-State Lifetime of an Iron(II)-N-Heterocyclic Carbene Complex Explained. J. Phys. Chem. Lett. 2014, 5, 2066–2071. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Persson, P.; Sundström, V.; Wärnmark, K. Fe N -Heterocyclic Carbene Complexes as Promising Photosensitizers. Acc. Chem. Res. 2016, 49, 1477–1485. [Google Scholar] [CrossRef] [PubMed]
- Shepard, S.G.; Fatur, S.M.; Rappé, A.K.; Damrauer, N.H. Highly Strained Iron(II) Polypyridines: Exploiting the Quintet Manifold to Extend the Lifetime of MLCT Excited States. J. Am. Chem. Soc. 2016, 138, 2949–2952. [Google Scholar] [CrossRef] [PubMed]
- Duchanois, T.; Etienne, T.; Cebrián, C.; Liu, L.; Monari, A.; Beley, M.; Assfeld, X.; Haacke, S.; Gros, P.C. An Iron-Based Photosensitizer with Extended Excited-State Lifetime: Photophysical and Photovoltaic Properties: An Iron-Based Photosensitizer with Extended Excited-State Lifetime. Eur. J. Inorg. Chem. 2015, 2015, 2469–2477. [Google Scholar] [CrossRef]
- Liu, Y.; Kjaer, K.S.; Fredin, L.A.; Chábera, P.; Harlang, T.; Canton, S.E.; Lidin, S.; Zhang, J.; Lomoth, R.; Bergquist, K.-E.; et al. A Heteroleptic Ferrous Complex with Mesoionic Bis(1,2,3-Triazol-5-Ylidene) Ligands: Taming the MLCT Excited State of Iron(II). Chem. Eur. J. 2015, 21, 3628–3639. [Google Scholar] [CrossRef] [PubMed]
- Kjær, K.S.; Zhang, W.; Alonso-Mori, R.; Bergmann, U.; Chollet, M.; Hadt, R.G.; Hartsock, R.W.; Harlang, T.; Kroll, T.; Kubiček, K.; et al. Ligand Manipulation of Charge Transfer Excited State Relaxation and Spin Crossover in [Fe(2,2′-Bipyridine)2(CN)2]. Struct. Dyn. 2017, 4, 044030. [Google Scholar] [CrossRef] [PubMed]
- Kjær, K.S.; Kunnus, K.; Harlang, T.C.B.; Van Driel, T.B.; Ledbetter, K.; Hartsock, R.W.; Reinhard, M.E.; Koroidov, S.; Li, L.; Laursen, M.G.; et al. Solvent Control of Charge Transfer Excited State Relaxation Pathways in [Fe(2,2′-Bipyridine)(CN)4]2−. Phys. Chem. Chem. Phys. 2018, 20, 4238–4249. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Duchanois, T.; Etienne, T.; Monari, A.; Beley, M.; Assfeld, X.; Haacke, S.; Gros, P.C. A New Record Excited State 3 MLCT Lifetime for Metalorganic Iron(ii) Complexes. Phys. Chem. Chem. Phys. 2016, 18, 12550–12556. [Google Scholar] [CrossRef] [PubMed]
- Pastore, M.; Duchanois, T.; Liu, L.; Monari, A.; Assfeld, X.; Haacke, S.; Gros, P.C. Interfacial Charge Separation and Photovoltaic Efficiency in Fe(ii)-Carbene Sensitized Solar Cells. Phys. Chem. Chem. Phys. 2016, 18, 28069–28081. [Google Scholar] [CrossRef] [PubMed]
- Hahn, A.W.; Van Kuiken, B.E.; al Samarai, M.; Atanasov, M.; Weyhermüller, T.; Cui, Y.-T.; Miyawaki, J.; Harada, Y.; Nicolaou, A.; DeBeer, S. Measurement of the Ligand Field Spectra of Ferrous and Ferric Iron Chlorides Using 2p3d RIXS. Inorg. Chem. 2017, 56, 8203–8211. [Google Scholar] [CrossRef] [PubMed]
- Zimmer, P.; Burkhardt, L.; Friedrich, A.; Steube, J.; Neuba, A.; Schepper, R.; Müller, P.; Flörke, U.; Huber, M.; Lochbrunner, S.; et al. The Connection between NHC Ligand Count and Photophysical Properties in Fe(II) Photosensitizers: An Experimental Study. Inorg. Chem. 2018, 57, 360–373. [Google Scholar] [CrossRef] [PubMed]
- Duchanois, T.; Etienne, T.; Beley, M.; Assfeld, X.; Perpète, E.A.; Monari, A.; Gros, P.C. Heteroleptic Pyridyl-Carbene Iron Complexes with Tuneable Electronic Properties: Tuneable Pyridyl-Carbene Iron Complexes. Eur. J. Inorg. Chem. 2014, 2014, 3747–3753. [Google Scholar] [CrossRef]
- Benkö, G.; Kallioinen, J.; Korppi-Tommola, J.E.I.; Yartsev, A.P.; Sundström, V. Photoinduced Ultrafast Dye-to-Semiconductor Electron Injection from Nonthermalized and Thermalized Donor States. J. Am. Chem. Soc. 2002, 124, 489–493. [Google Scholar] [CrossRef] [PubMed]
- Pastore, M.; De Angelis, F. First-Principles Modeling of a Dye-Sensitized TiO2/IrO2 Photoanode for Water Oxidation. J. Am. Chem. Soc. 2015, 137, 5798–5809. [Google Scholar] [CrossRef] [PubMed]
- Piccinin, S.; Rocca, D.; Pastore, M. Role of Solvent in the Energy Level Alignment of Dye-Sensitized NiO Interfaces. J. Phys. Chem. C 2017, 121, 22286–22294. [Google Scholar] [CrossRef]
- Fredin, L.A.; Wärnmark, K.; Sundström, V.; Persson, P. Molecular and Interfacial Calculations of Iron(II) Light Harvesters. ChemSusChem 2016, 9, 667–675. [Google Scholar] [CrossRef]
- Liu, L. Propriétés Photo-Physiques de Nouveaux Matériaux Moléculaires Pour La Conversion de Photons En Énergie. Ph.D. Thesis, Université de Strasbourg, Strasbourg, France, 2017. [Google Scholar]


























{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
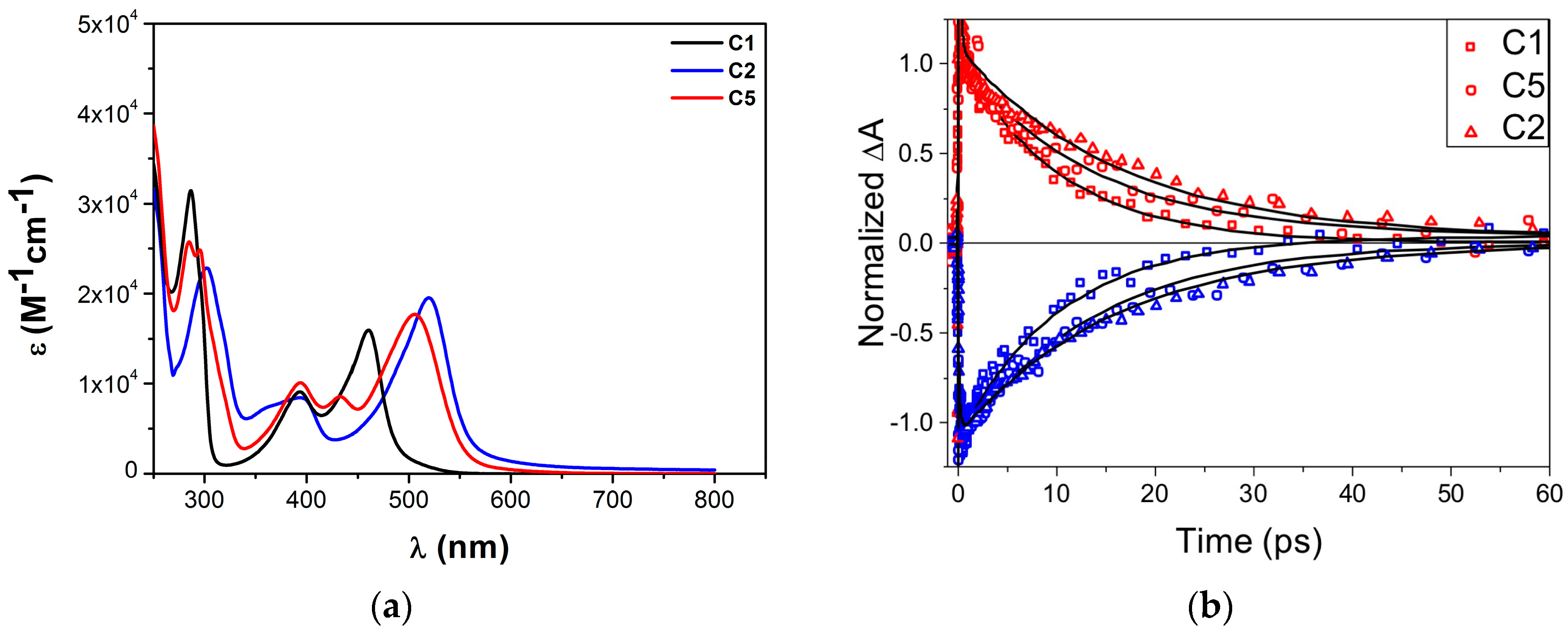
| Complex | ΔE (eV) | 3MLCT Lifetime (ps) |
|---|---|---|
| C1 | 0.78 1 | 9.5 ± 1.0 1 |
| C2 | 0.08 1 | 16.5 ± 1.0 1 |
| C3 | - | 16.4 ± 0.4 2 |
| C4 | −0.12 2 | 26 ± 1 2 |
| Dye | JSc (mA/cm2) | Voc (mV) | FF | PCE% | Dye Load (μmol/cm2) b |
|---|---|---|---|---|---|
| C2 | 0.41 | 457 | 0.68 | 0.13 | 0.09 |
| C4 | 0.12 | 368 | 0.71 | 0.03 | 0.06 |
| N719 | 13.25 | 687 | 0.67 | 6.1 | 0.08 |
| TiO2 | 0.01 | 364 | 0.51 | 0.01 | - |
| Dye | JSC (mA·cm−2) | VOC (mV) | FF | PCE% | Dye Load (μmol/cm−2) b |
|---|---|---|---|---|---|
| C2 | 0.41 | 457 | 0.68 | 0.13 | 0.09 |
| C5 | 0.33 | 400 | 0.73 | 0.10 | 0.10 |
| C6 | 0.36 | 440 | 0.73 | 0.11 | 0.12 |
| C7 | 0.36 | 390 | 0.71 | 0.10 | 0.07 |
| N719 | 13.25 | 687 | 0.67 | 6.1 | 0.08 |
| Electron Injection | ||||||
| System | Γ (εLUMO) | DOS (εLUMO) | kinj | Γ (εLUMO+1) | DOS (εLUMO+1) | kinj |
| C2@TiO2 | 2.03 × 10−4 | 129 | 1.9 × 1012 | 0.158 | 133 | 1.5 × 1015 |
| C4@TiO2 | 1.67 × 10−4 | 120 | 1.6 × 1012 | 0.120 | 136 | 1.1 × 1015 |
| C5@TiO2 | 0.010 | 130 | 9.5 ×1013 | 1.2 × 10−3 | 120 | 1.1 × 1013 |
| Recombination to the oxidized dye | ||||||
| Γ (εHOMO) | DOS (εHOMO) | kinj | ||||
| C2@TiO2 | 1.0 × 10−11 | 1 × 10−8 | 9.5 × 104 | |||
| C4@TiO2 | 5.0 × 10−10 | 3 × 10−6 | 4.8 × 107 | |||
| C5@TiO2 | 3.0 × 10−12 | 6 × 10−9 | 2.6 × 104 | |||
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Duchanois, T.; Liu, L.; Pastore, M.; Monari, A.; Cebrián, C.; Trolez, Y.; Darari, M.; Magra, K.; Francés-Monerris, A.; Domenichini, E.; et al. NHC-Based Iron Sensitizers for DSSCs. Inorganics 2018, 6, 63. https://0-doi-org.brum.beds.ac.uk/10.3390/inorganics6020063
Duchanois T, Liu L, Pastore M, Monari A, Cebrián C, Trolez Y, Darari M, Magra K, Francés-Monerris A, Domenichini E, et al. NHC-Based Iron Sensitizers for DSSCs. Inorganics. 2018; 6(2):63. https://0-doi-org.brum.beds.ac.uk/10.3390/inorganics6020063
Chicago/Turabian StyleDuchanois, Thibaut, Li Liu, Mariachiara Pastore, Antonio Monari, Cristina Cebrián, Yann Trolez, Mohamed Darari, Kevin Magra, Antonio Francés-Monerris, Edoardo Domenichini, and et al. 2018. "NHC-Based Iron Sensitizers for DSSCs" Inorganics 6, no. 2: 63. https://0-doi-org.brum.beds.ac.uk/10.3390/inorganics6020063