Fluorescent Bis(guanidine) Copper Complexes as Precursors for Hydroxylation Catalysis †

 , ,
, ,
Abstract
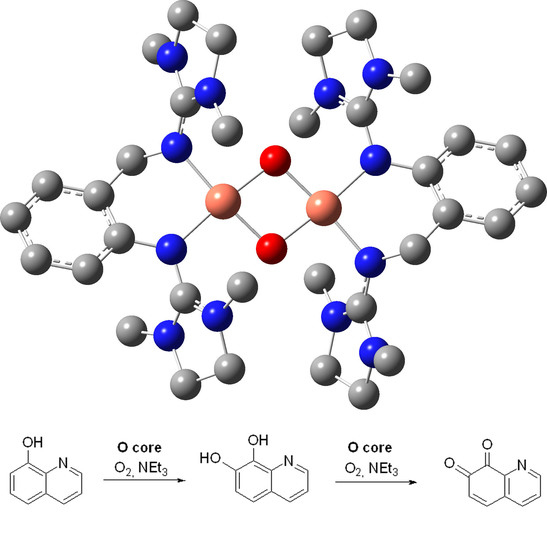
:
1. Introduction
2. Results
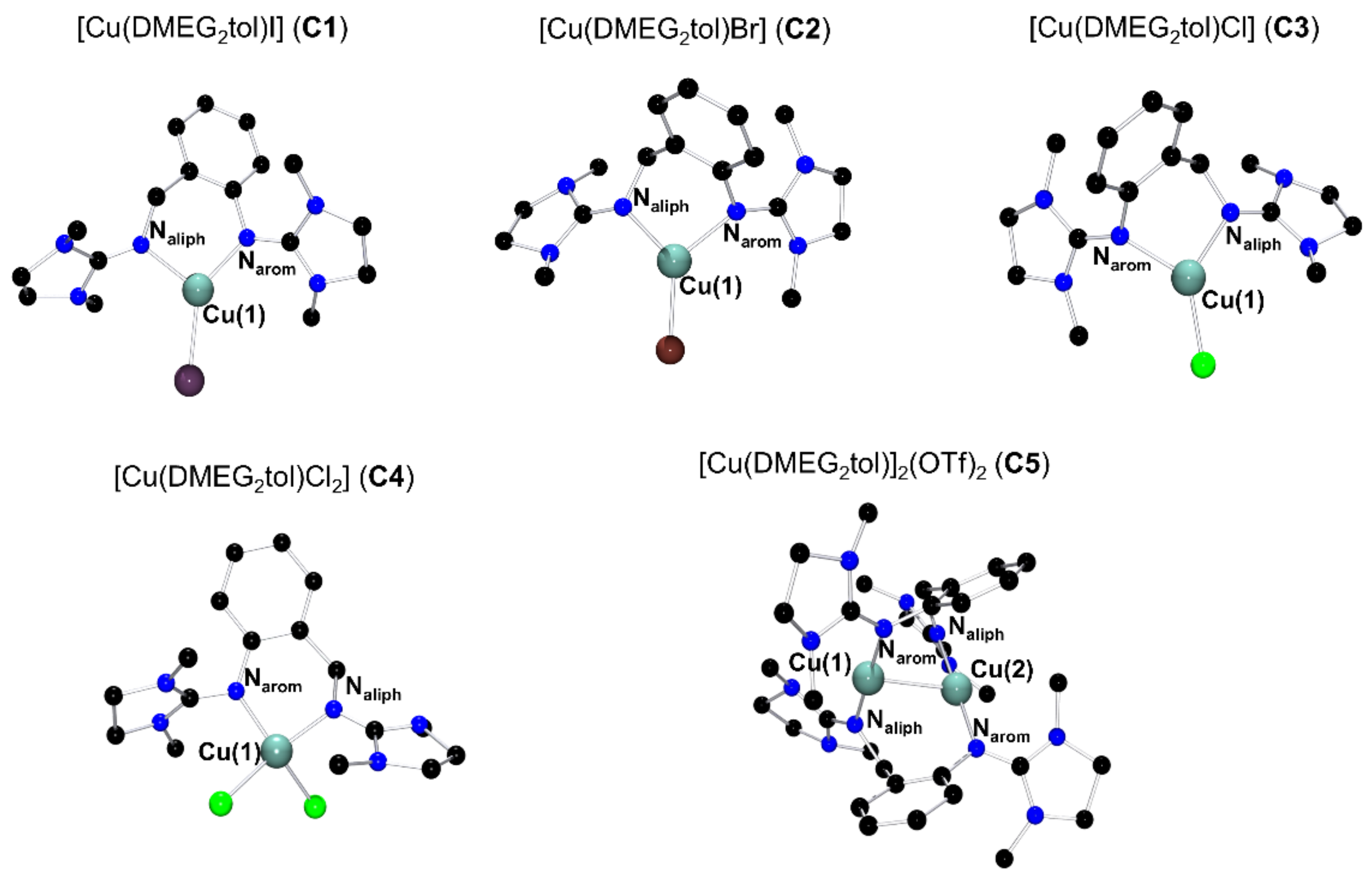
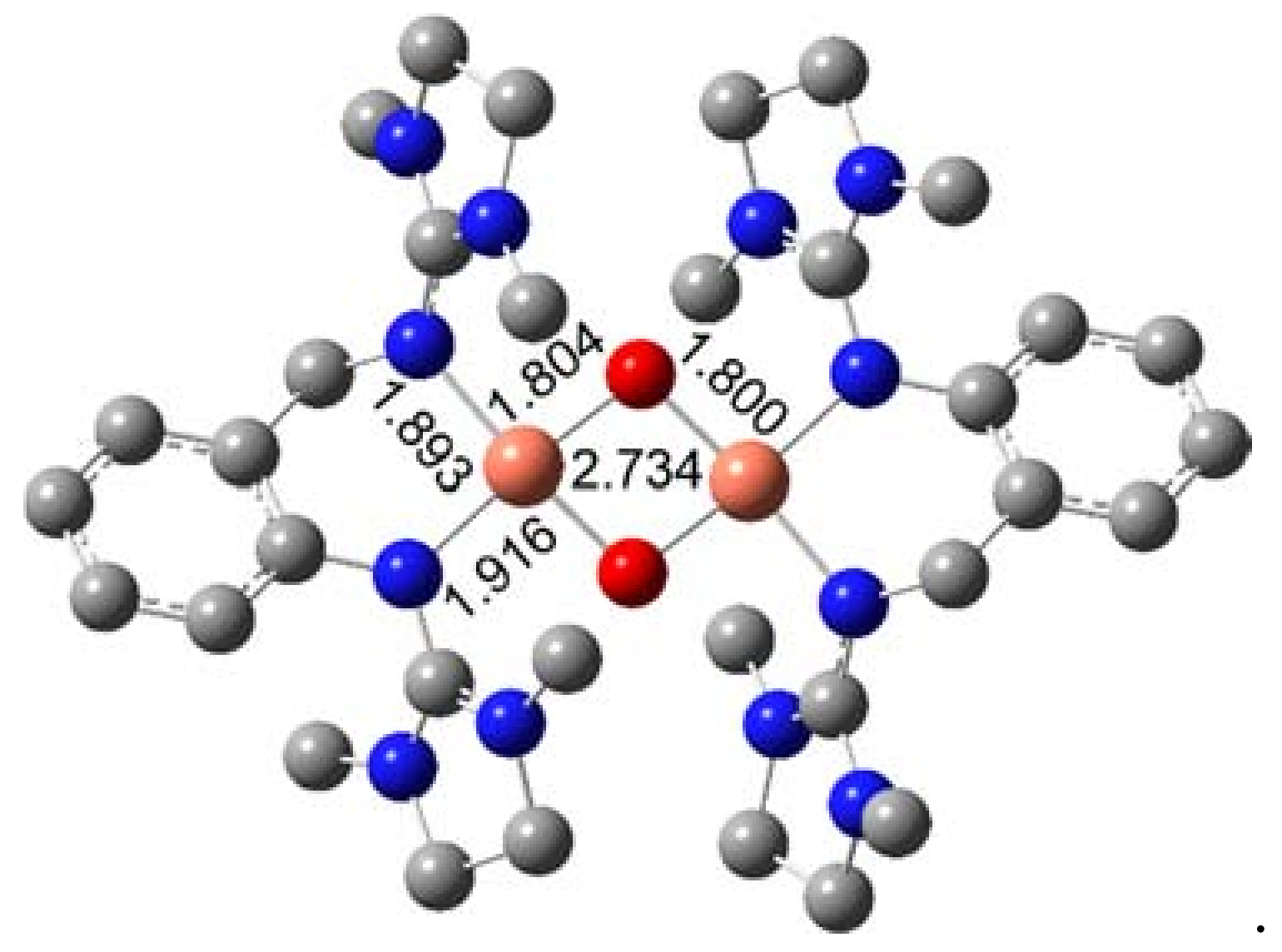
2.1. Molecular Structures in the Solid State
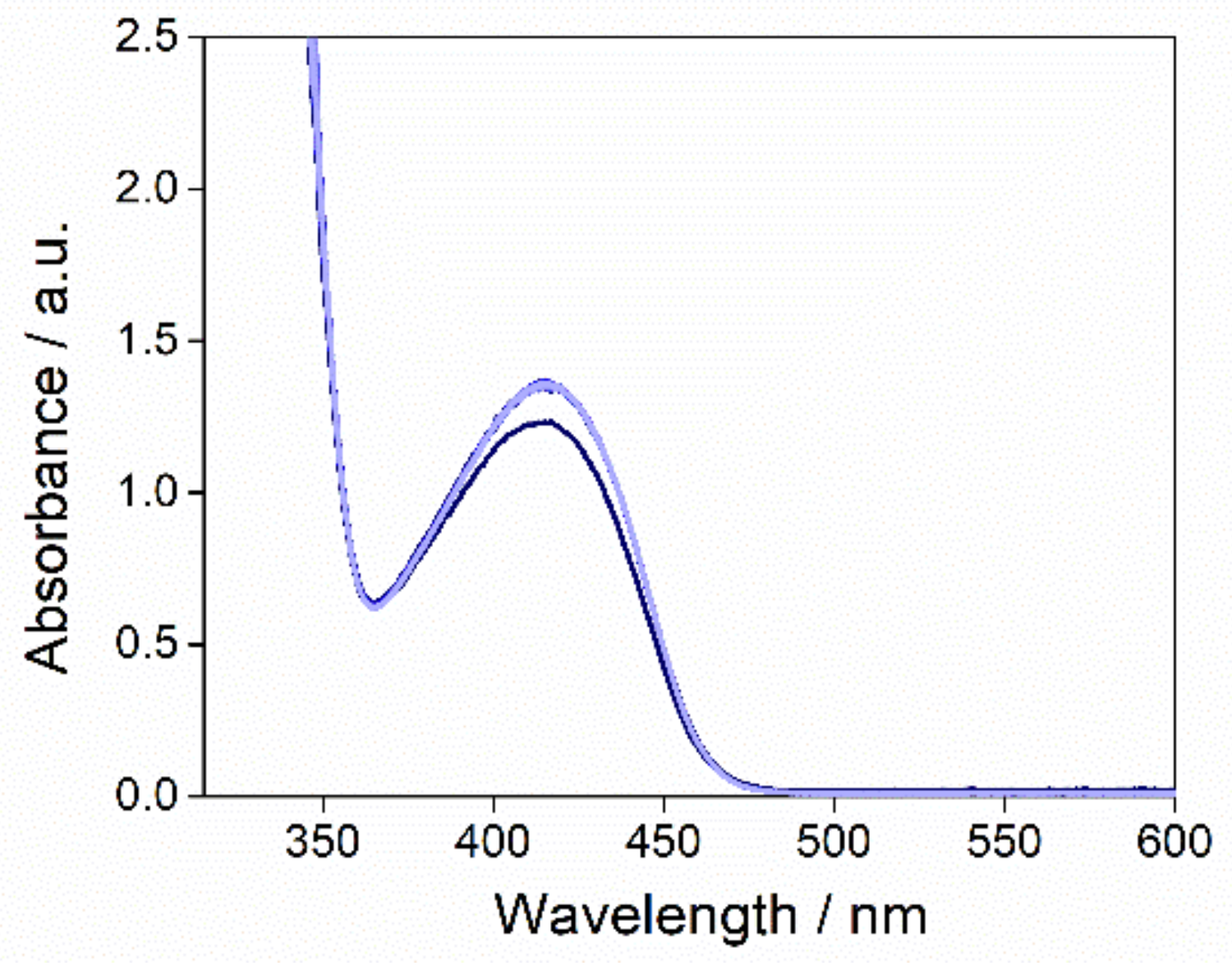
2.2. Oxygen Activation
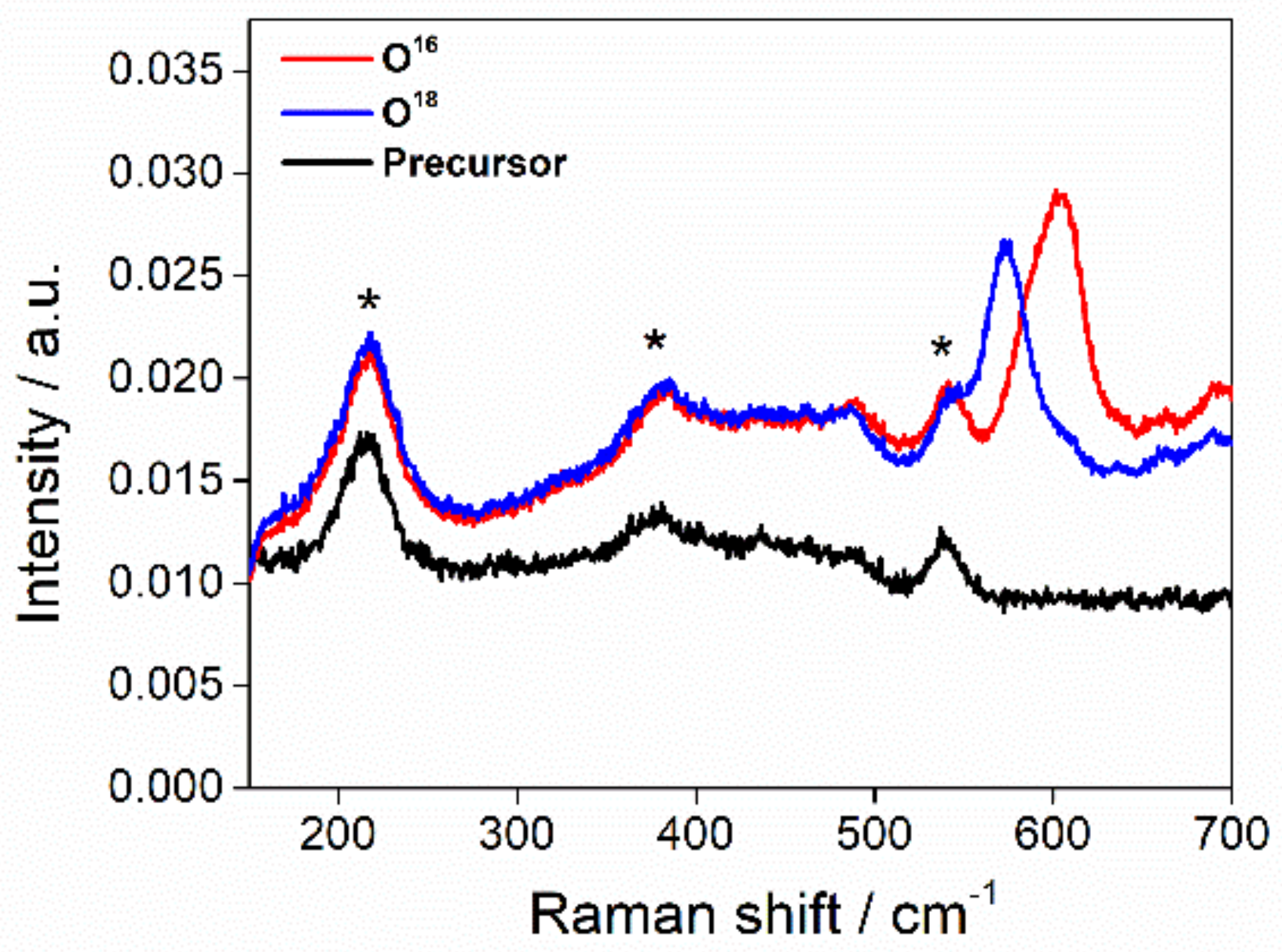
2.3 Raman Spectroscopy
2.4. Density Functional Theory
2.5. Catalysis
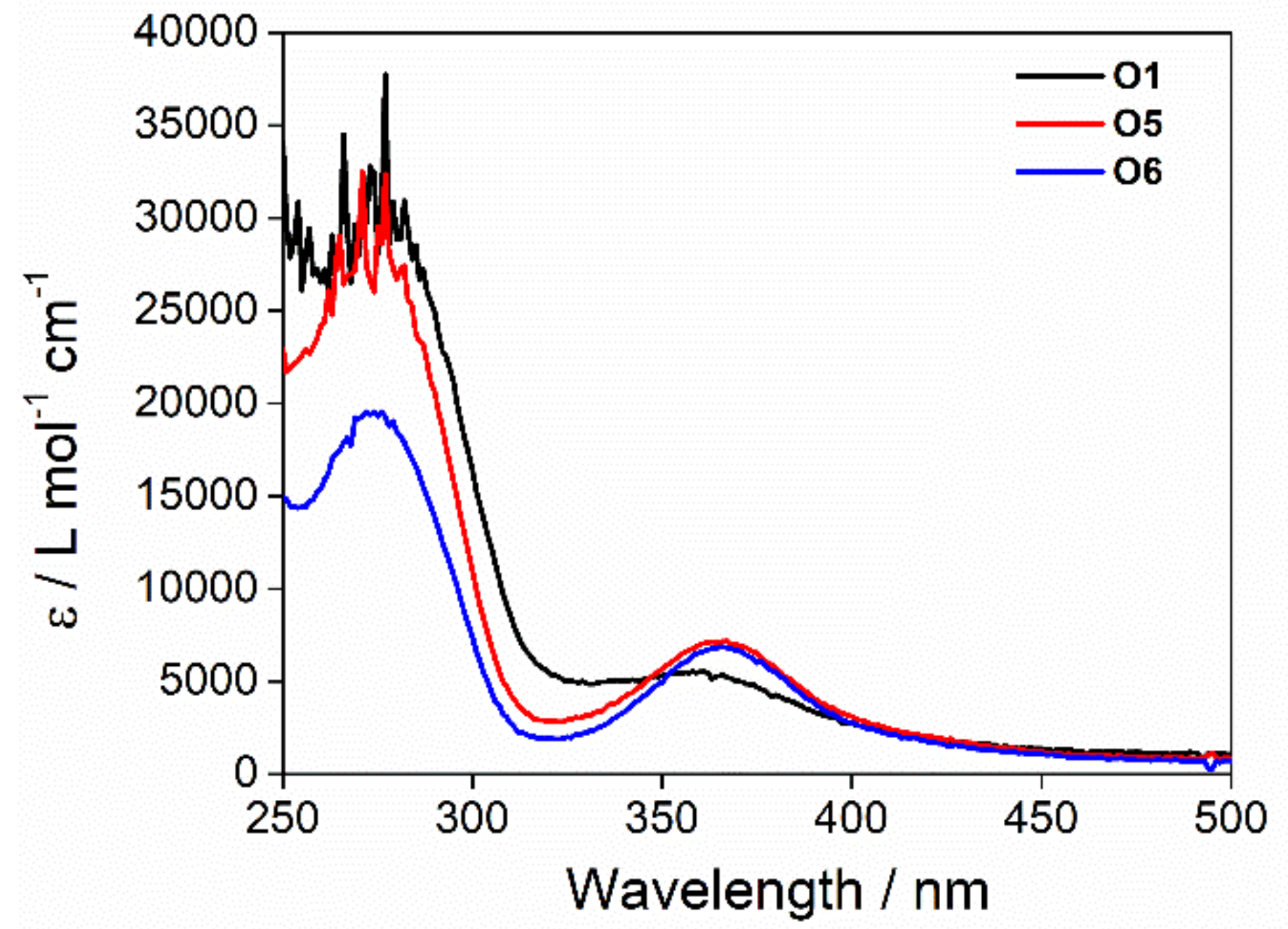
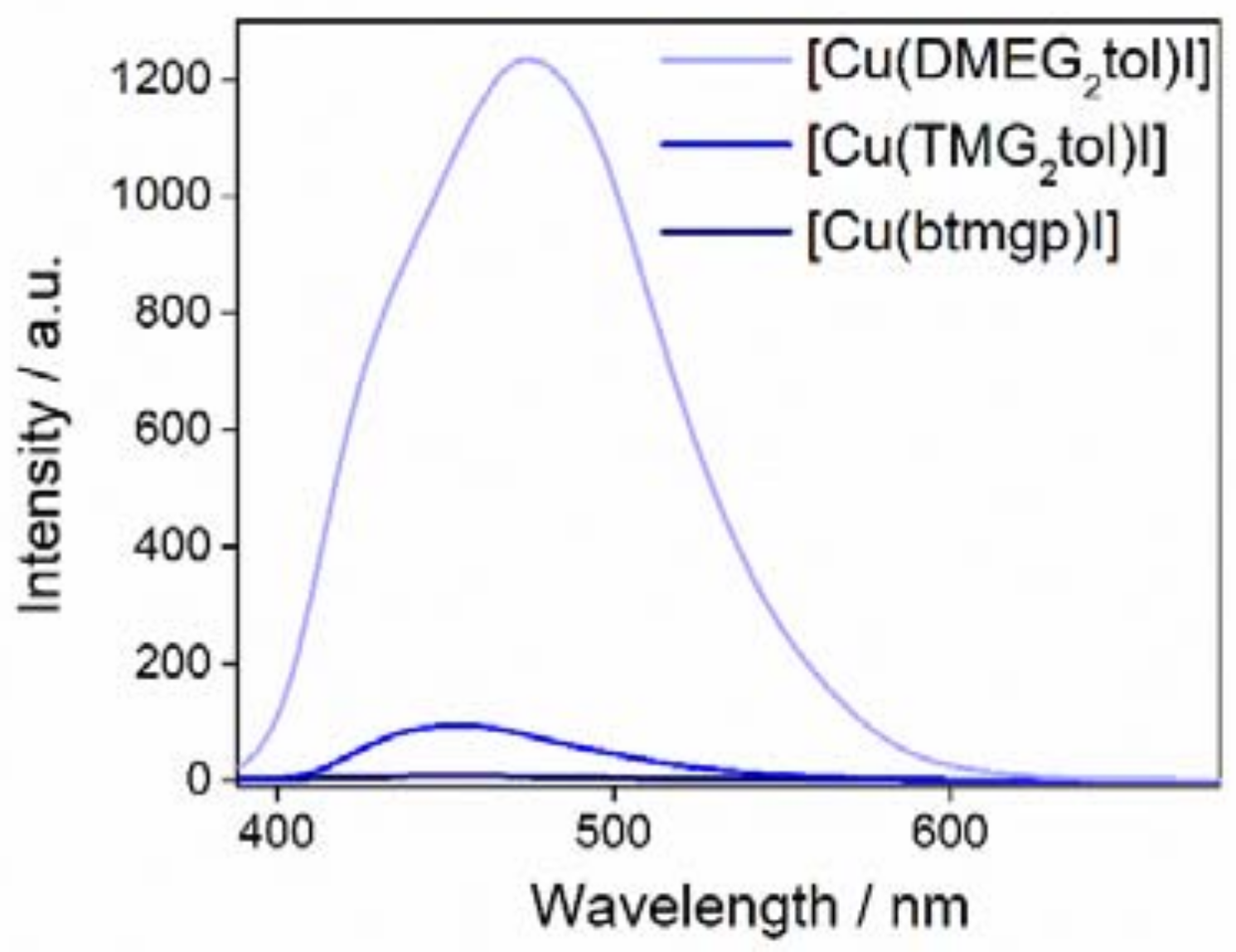
2.6. Fluorescence Measurements
3. Discussion
4. Materials and Methods
4.1. General Analytic Methods
4.1.1. NMR Spectroscopy
4.1.2. IR-Spectroscopy
4.1.3. Mass-Spectrometry
4.1.4. UV/Vis Spectroscopy
4.1.5. Fluorescence Spectroscopy
4.1.6. Elemental Analysis
4.1.7. X-ray Diffraction Analysis
4.1.8 Raman Spectroscopy
4.1.9. Computational Details
4.2. Synthesis and Characterization
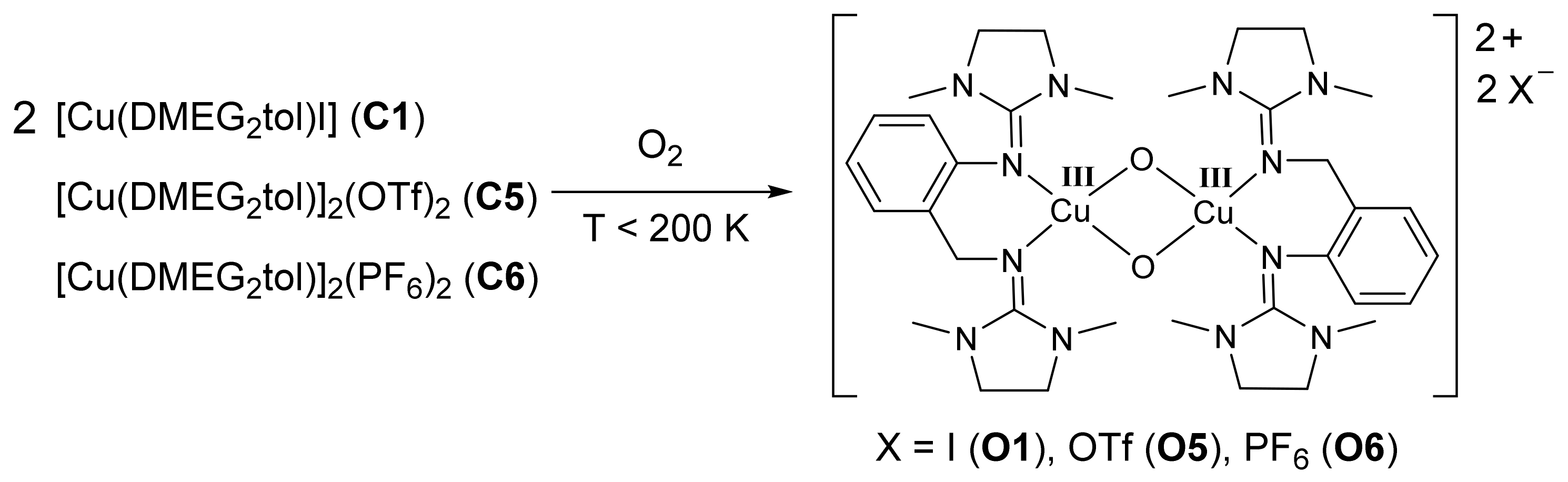
4.2.1. Synthesis of the Oxido Complexes O1, O5, and O6
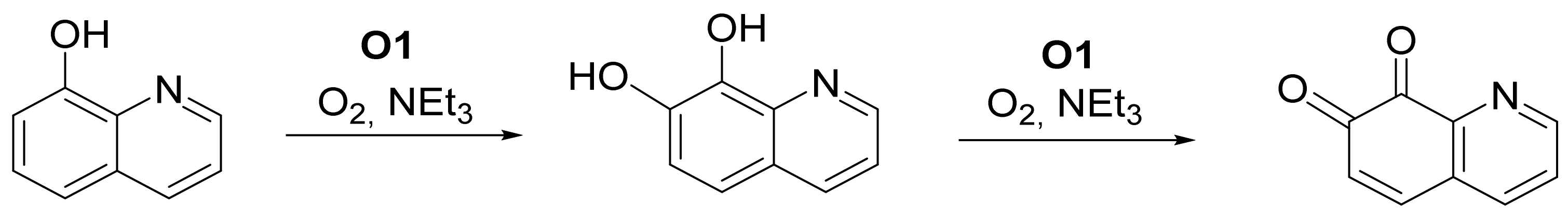
4.2.2. Catalytic Conversion of 8-Hydroxyquinoline with the Oxido Species
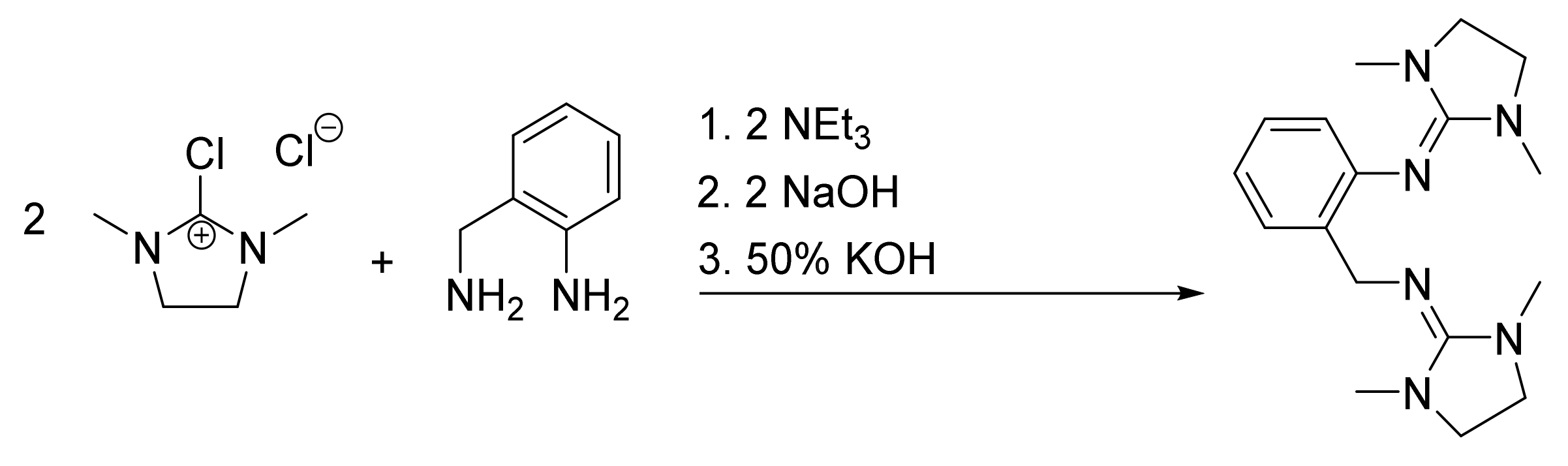
Synthesis of 2,2’-(((Dimethylamino)methylene)amino)benzyl)-1,1,3,3-dimethylethyleneguanidine (DMEG2tol) (L1)

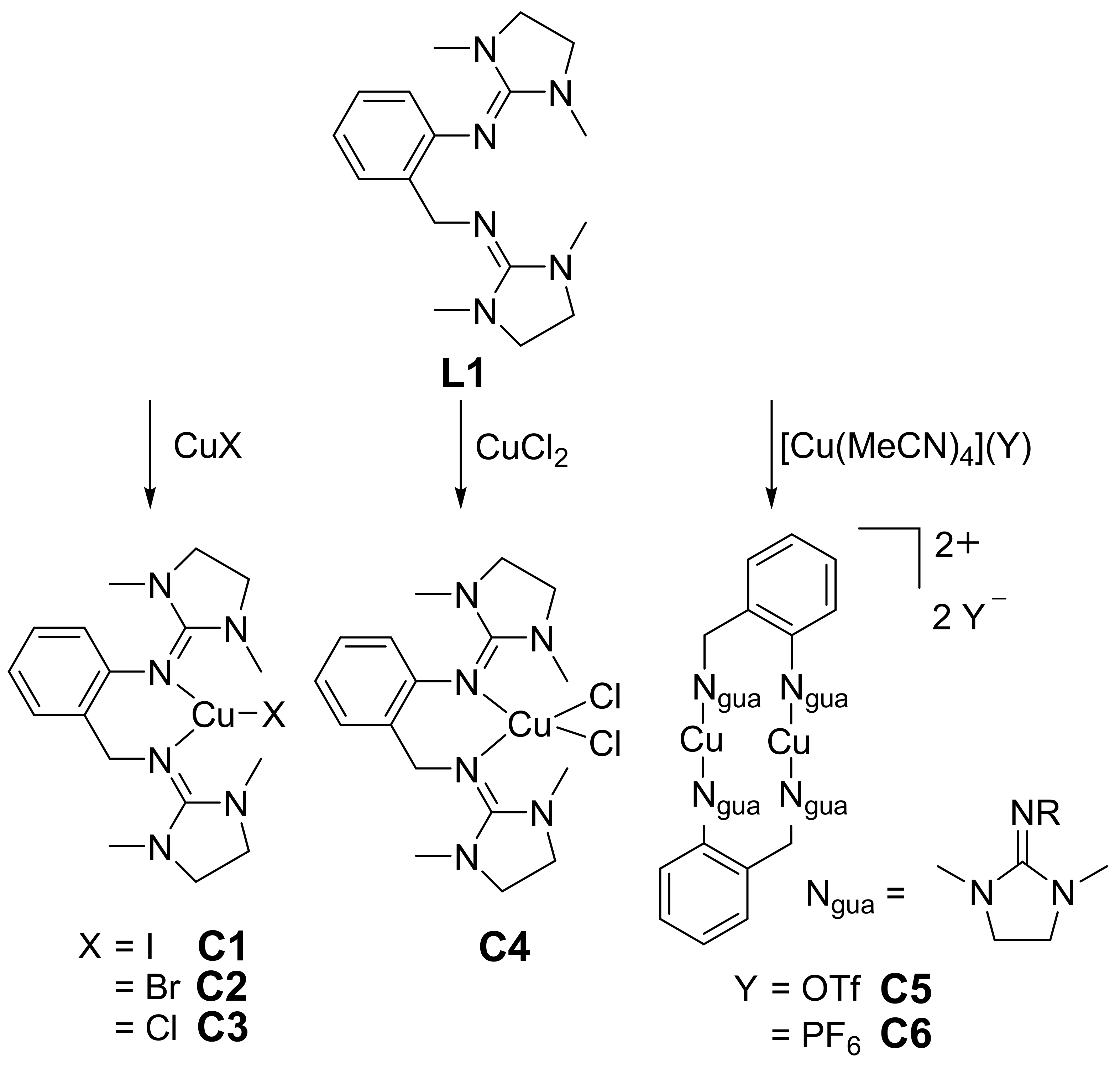
[Cu(DMEG2tol)I] (C1)

[Cu(DMEG2tol)Br] (C2)

[Cu(DMEG2tol)Cl] (C3)

[Cu(DMEG2tol)Cl2] (C4)

[Cu(DMEG2tol)]2(OTf)2 (C5)

[Cu(DMEG2tol)]2(PF6)2 (C6)

5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Punniyamurthy, T.; Velusamy, S.; Iqbal, J. Recent advances in transition metal catalyzed oxidation of organic substrates with molecular oxygen. Chem. Rev. 2005, 105, 2329–2363. [Google Scholar] [CrossRef] [PubMed]
- Itoh, S.; Fukuzumi, S. Monooxygenase activity of type 3 copper proteins. Acc. Chem. Res. 2007, 40, 592–600. [Google Scholar] [CrossRef] [PubMed]
- Yamazaki, S.-I.; Itoh, S. Kinetic evaluation of phenolase activity of tyrosinase using simplified catalytic reaction system. J. Am. Chem. Soc. 2003, 125, 13034–13035. [Google Scholar] [CrossRef] [PubMed]
- Hamann, J.N.; Tuczek, F. New catalytic model systems of tyrosinase: Fine tuning of the reactivity with pyrazole-based N-donor ligands. Chem. Commun. 2014, 50, 2298–2300. [Google Scholar] [CrossRef] [PubMed]
- Rolff, M.; Schottenheim, J.; Decker, H.; Tuczek, F. Copper-O2 reactivity of tyrosinase models towards external monophenolic substrates: Molecular mechanism and comparison with the enzyme. Chem. Soc. Rev. 2011, 40, 4077–4098. [Google Scholar] [CrossRef] [PubMed]
- Bijelic, A.; Pretzler, M.; Molitor, C.; Zekiri, F.; Rompel, A. The Structure of a Plant Tyrosinase from Walnut Leaves Reveals the Importance of “Substrate-Guiding Residues” for Enzymatic Specificity. Angew. Chem. Int. Ed. 2015, 54, 14677–14680. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matoba, Y.; Kumagai, T.; Yamamoto, A.; Yoshitsu, H.; Sugiyama, M. Crystallographic Evidence That the Dinuclear Copper Center of Tyrosinase Is Flexible during Catalysis. J. Biol. Chem. 2006, 281, 8981–8990. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mauracher, S.G.; Molitor, C.; Al-Oweini, R.; Kortz, U.; Rompel, A. Latent and active abPPO4 mushroom tyrosinase cocrystallized with hexatungstotellurate(VI) in a single crystal. Acta Crystallogr. D 2014, 70, 2301–2315. [Google Scholar] [CrossRef] [PubMed]
- Herres-Pawlis, S.; Verma, P.; Haase, R.; Kang, P.; Lyons, C.T.; Wasinger, E.C.; Flörke, U.; Henkel, G.; Stack, T.D.P. Phenolate hydroxylation in a bis(μ-oxo)dicopper(III) complex: Lessons from the guanidine/amine series. J. Am. Chem. Soc. 2009, 131, 1154–1169. [Google Scholar] [CrossRef] [PubMed]
- Spada, A.; Palavicini, S.; Monzani, E.; Bubacco, L.; Casella, L. Trapping tyrosinase key active intermediate under turnover. Dalton Trans. 2009, 6468–6471. [Google Scholar] [CrossRef] [PubMed]
- Hamann, J.N.; Herzigkeit, B.; Jurgeleit, R.; Tuczek, F. Small-molecule models of tyrosinase: From ligand hydroxylation to catalytic monooxygenation of external substrates. Coord. Chem. Rev. 2017, 334, 54–66. [Google Scholar] [CrossRef]
- Elwell, C.E.; Gagnon, N.L.; Neisen, B.D.; Dhar, D.; Spaeth, A.D.; Yee, G.M.; Tolman, W.B. Copper-Oxygen Complexes Revisited: Structures, Spectroscopy, and Reactivity. Chem. Rev. 2017, 117, 2059–2107. [Google Scholar] [CrossRef] [PubMed]
- Liebhäuser, P.; Hoffmann, A.; Herres-Pawlis, S. Tyrosinase Models: Synthesis, Spectroscopy, Theory, and Catalysis. In Reference Module in Chemistry, Molecular Sciences and Chemical Engineering; Elsevier: Waltham, MA, USA, 2016; p. 1. [Google Scholar] [CrossRef]
- Mirica, L.M.; Ottenwaelder, X.; Stack, T.D.P. Structure and spectroscopy of copper-dioxygen complexes. Chem. Rev. 2004, 104, 1013–1045. [Google Scholar] [CrossRef] [PubMed]
- Lewis, E.A.; Tolman, W.B. Reactivity of Dioxygen-Copper Systems. Chem. Rev. 2004, 104, 1047–1076. [Google Scholar] [CrossRef] [PubMed]
- Cowley, R.E.; Tian, L.; Solomon, E.I. Mechanism of O2 activation and substrate hydroxylation in noncoupled binuclear copper monooxygenases. Proc. Natl. Acad. Sci. USA. 2016, 113, 12035–12040. [Google Scholar] [CrossRef] [PubMed]
- Citek, C.; Lyons, C.T.; Wasinger, E.C.; Stack, T.D.P. Self-assembly of the oxy-tyrosinase core and the fundamental components of phenolic hydroxylation. Nat. Chem. 2012, 4, 317–322. [Google Scholar] [CrossRef] [PubMed]
- Wendt, F.; Näther, C.; Tuczek, F. Tyrosinase and catechol oxidase activity of copper(I) complexes supported by imidazole-based ligands: Structure-reactivity correlations. J. Biol. Inorg. Chem. 2016, 21, 777–792. [Google Scholar] [CrossRef] [PubMed]
- Wilfer, C.; Liebhäuser, P.; Hoffmann, A.; Erdmann, H.; Grossmann, O.; Runtsch, L.; Paffenholz, E.; Schepper, R.; Dick, R.; Bauer, M.; et al. Efficient Biomimetic Hydroxylation Catalysis with a Bis(pyrazolyl)imidazolylmethane Copper Peroxide Complex. Chem. Eur. J. 2015, 21, 17639–17649. [Google Scholar] [CrossRef] [PubMed]
- Chiang, L.; Keown, W.; Citek, C.; Wasinger, E.C.; Stack, T.D.P. Simplest Monodentate Imidazole Stabilization of the oxy-Tyrosinase Cu2O2 Core: Phenolate Hydroxylation through a Cu(III) Intermediate. Angew. Chem. 2016, 128, 10609–10613. [Google Scholar] [CrossRef]
- Schottenheim, J.; Fateeva, N.; Thimm, W.; Krahmer, J.; Tuczek, F. Catalytic Conversion of Monophenols to Ortho-Quinones in a Tyrosinase-Like Fashion: Towards More Biomimetic and More Efficient Model Systems. Z. Anorg. Allg. Chem. 2013, 639, 1491–1497. [Google Scholar] [CrossRef]
- Réglier, M.; Jorand, C.; Waegell, B. Binuclear copper complex model of tyrosinase. J. Chem. Soc. Chem. Commun. 1990, 107, 1752–1755. [Google Scholar] [CrossRef]
- Rolff, M.; Schottenheim, J.; Peters, G.; Tuczek, F. The first catalytic tyrosinase model system based on a mononuclear copper(I) complex: Kinetics and mechanism. Angew. Chem. Int. Ed. 2010, 49, 6438–6442. [Google Scholar] [CrossRef] [PubMed]
- Becker, J.; Gupta, P.; Angersbach, F.; Tuczek, F.; Näther, C.; Holthausen, M.C.; Schindler, S. Selective Aromatic Hydroxylation with Dioxygen and Simple Copper Imine Complexes. Chem. Eur. J. 2015, 21, 11735–11744. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilfer, C.; Liebhäuser, P.; Erdmann, H.; Hoffmann, A.; Herres-Pawlis, S. Biomimetic Hydroxylation Catalysis Through Self-Assembly of a Bis(pyrazolyl)methane Copper-Peroxo Complex. Eur. J. Inorg. Chem. 2015, 2015, 494–502. [Google Scholar] [CrossRef]
- Hamann, J.N.; Schneider, R.; Tuczek, F. Catalytic oxygenation of various monophenols by copper(I) complexes with bis(pyrazolyl)methane ligands: Differences in reactivity. J. Coord. Chem. 2015, 68, 3259–3271. [Google Scholar] [CrossRef]
- Halfen, J.A.; Mahapatra, S.; Wilkinson, E.C.; Kaderli, S.; Young, V.G.; Que, L.; Zuberbühler, A.D.; Tolman, W.B. Reversible Cleavage and Formation of the Dioxygen O–O Bond Within a Dicopper Complex. Science 1996, 271, 1397–1400. [Google Scholar] [CrossRef] [PubMed]
- Que, J.L.; Tolman, W.B. Bis(μ-oxo)dimetal “Diamond” Cores in Copper and Iron Complexes Relevant to Biocatalysis. Angew. Chem. Int. Ed. 2002, 41, 1114–1137. [Google Scholar] [CrossRef]
- Henson, M.J.; Mukherjee, P.; Root, D.E.; Stack, T.D.P.; Solomon, E.I. Spectroscopic and Electronic Structural Studies of the Cu(III) 2 Bis-μ-oxo Core and Its Relation to the Side-On Peroxo-Bridged Dimer. J. Am. Chem. Soc. 1999, 121, 10332–10345. [Google Scholar] [CrossRef]
- Schottenheim, J.; Gernert, C.; Herzigkeit, B.; Krahmer, J.; Tuczek, F. Catalytic Models of Tyrosinase: Reactivity Differences between Systems Based on Mono- and Binucleating Ligands. Eur. J. Inorg. Chem. 2015, 2015, 3501–3511. [Google Scholar] [CrossRef]
- Herzigkeit, B.; Flöser, B.M.; Engesser, T.A.; Näther, C.; Tuczek, F. Tyrosinase Model Systems Supported by Pyrazolylmethylpyridine Ligands: Electronic and Steric Factors Influencing the Catalytic Activity and Impact of Complex Equilibria in Solution. Eur. J. Inorg. Chem. 2018, 2018, 3058–3069. [Google Scholar] [CrossRef]
- Askari, M.S.; Esguerra, K.V.N.; Lumb, J.-P.; Ottenwaelder, X. A Biomimetic Mechanism for the Copper-Catalyzed Aerobic Oxygenation of 4-tert-Butylphenol. Inorg. Chem. 2015, 54, 8665–8672. [Google Scholar] [CrossRef] [PubMed]
- Huang, Z.; Askari, M.S.; Esguerra, K.V.N.; Dai, T.-Y.; Kwon, O.; Ottenwaelder, X.; Lumb, J.-P. A bio-inspired synthesis of oxindoles by catalytic aerobic dual C–H functionalization of phenols. Chem. Sci. 2016, 7, 358–369. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Esguerra, K.V.N.; Fall, Y.; Petitjean, L.; Lumb, J.-P. Controlling the catalytic aerobic oxidation of phenols. J. Am. Chem. Soc. 2014, 136, 7662–7668. [Google Scholar] [CrossRef] [PubMed]
- Casella, L.; Gullotti, M.; Radaelli, R.; Di Gennaro, P. A tyrosinase model system. Phenol ortho-hydroxylation by a binuclear three-coordinate copper(I) complex and dioxygen. J. Chem. Soc. Chem. Commun. 1991, 1611. [Google Scholar] [CrossRef]
- Garcia-Bosch, I.; Company, A.; Frisch, J.R.; Torrent-Sucarrat, M.; Cardellach, M.; Gamba, I.; Güell, M.; Casella, L.; Que, L.; Ribas, X.; et al. O2 activation and selective phenolate ortho hydroxylation by an unsymmetric dicopper µ-η1:η1-peroxido complex. Angew. Chem. 2010, 122, 2456–2459. [Google Scholar] [CrossRef]
- Hoffmann, A.; Citek, C.; Binder, S.; Goos, A.; Rübhausen, M.; Troeppner, O.; Ivanović-Burmazović, I.; Wasinger, E.C.; Stack, T.D.P.; Herres-Pawlis, S. Catalytic phenol hydroxylation with dioxygen: Extension of the tyrosinase mechanism beyond the protein matrix. Angew. Chem. Int. Ed. 2013, 52, 5398–5401. [Google Scholar] [CrossRef] [PubMed]
- Liebhäuser, P.; Keisers, K.; Hoffmann, A.; Schnappinger, T.; Sommer, I.; Thoma, A.; Wilfer, C.; Schoch, R.; Stührenberg, K.; Bauer, M.; et al. Record Broken: A Copper Peroxide Complex with Enhanced Stability and Faster Hydroxylation Catalysis. Chem. Eur. J. 2017, 23, 12171–12183. [Google Scholar] [CrossRef] [PubMed]
- Solem, E.; Tuczek, F.; Decker, H. Tyrosinase versus Catechol Oxidase: One Asparagine Makes the Difference. Angew. Chem. Int. Ed. 2016, 55, 2884–2888. [Google Scholar] [CrossRef] [PubMed]
- Battaini, G.; Carolis, M.D.; Monzani, E.; Tuczek, F.; Casella, L. The phenol ortho-oxygenation by mononuclear copper(i) complexes requires a dinuclear µ-η2∶η2-peroxodicopper(II) complex rather than mononuclear CuO2 species. Chem. Commun. 2003, 726–727. [Google Scholar] [CrossRef]
- Palavicini, S.; Granata, A.; Monzani, E.; Casella, L. Hydroxylation of phenolic compounds by a peroxodicopper(II) complex: Further insight into the mechanism of tyrosinase. J. Am. Chem. Soc. 2005, 127, 18031–18036. [Google Scholar] [CrossRef] [PubMed]
- Bienemann, O.; Hoffmann, A.; Herres-Pawlis, S. (Guanidine)copper complexes: Structural variety and application in bioinorganic chemistry and catalysis. Rev. Inorg. Chem. 2011, 31, 83–108. [Google Scholar] [CrossRef]
- Schatz, M.; Raab, V.; Foxon, S.P.; Brehm, G.; Schneider, S.; Reiher, M.; Holthausen, M.C.; Sundermeyer, J.; Schindler, S. Combined spectroscopic and theoretical evidence for a persistent end-on copper superoxo complex. Angew. Chem. Int. Ed. 2004, 43, 4360–4363. [Google Scholar] [CrossRef] [PubMed]
- Würtele, C.; Gaoutchenova, E.; Harms, K.; Holthausen, M.C.; Sundermeyer, J.; Schindler, S. Crystallographic characterization of a synthetic 1:1 end-on copper dioxygen adduct complex. Angew. Chem. Int. Ed. 2006, 45, 3867–3869. [Google Scholar] [CrossRef] [PubMed]
- Maiti, D.; Lee, D.-H.; Gaoutchenova, K.; Würtele, C.; Holthausen, M.C.; Narducci Sarjeant, A.A.; Sundermeyer, J.; Schindler, S.; Karlin, K.D. Reactions of a copper(II) superoxo complex lead to C–H and O–H substrate oxygenation: Modeling copper-monooxygenase C–H hydroxylation. Angew. Chem. 2008, 120, 88–91. [Google Scholar] [CrossRef]
- Wiesner, S.; Wagner, A.; Hübner, O.; Kaifer, E.; Himmel, H.-J. Thermochromism of Cu(I) Tetrakisguanidine Complexes: Reversible Activation of Metal-to-Ligand Charge-Transfer Bands. Chem. Eur. J. 2015, 21, 16494–16503. [Google Scholar] [CrossRef] [PubMed]
- Wiesner, S.; Wagner, A.; Kaifer, E.; Himmel, H.-J. A Valence Tautomeric Dinuclear Copper Tetrakisguanidine Complex. Chem. Eur. J. 2016, 22, 10438–10445. [Google Scholar] [CrossRef] [PubMed]
- Wiesner, S.; Wagner, A.; Kaifer, E.; Himmel, H.-J. The control of the electronic structure of dinuclear copper complexes of redox-active tetrakisguanidine ligands by the environment. Dalton Trans. 2016, 45, 15828–15839. [Google Scholar] [CrossRef] [PubMed]
- Rösener, T.; Bienemann, O.; Sigl, K.; Schopp, N.; Schnitter, F.; Flörke, U.; Hoffmann, A.; Döring, A.; Kuckling, D.; Herres-Pawlis, S. A Comprehensive Study of Copper Guanidine Quinoline Complexes: Predicting the Activity of Catalysts in ATRP with DFT. Chem. Eur. J. 2016, 22, 13550–13562. [Google Scholar] [CrossRef] [PubMed]
- Stanek, J.; Sackers, N.; Fink, F.; Paul, M.; Peters, L.; Grunzke, R.; Hoffmann, A.; Herres-Pawlis, S. Copper Guanidinoquinoline Complexes as Entatic State Models of Electron-Transfer Proteins. Chem. Eur. J. 2017, 23, 15738–15745. [Google Scholar] [CrossRef] [PubMed]
- Dicke, B.; Hoffmann, A.; Stanek, J.; Rampp, M.S.; Grimm-Lebsanft, B.; Biebl, F.; Rukser, D.; Maerz, B.; Göries, D.; Naumova, M.; et al. Transferring the entatic-state principle to copper photochemistry. Nat. Chem. 2018, 10, 355–362. [Google Scholar] [CrossRef] [PubMed]
- Herres-Pawlis, S.; Binder, S.; Eich, A.; Haase, R.; Schulz, B.; Wellenreuther, G.; Henkel, G.; Rübhausen, M.; Meyer-Klaucke, W. Stabilisation of a highly reactive bis(μ-oxo)dicopper(III) species at room temperature by electronic and steric constraint of an unconventional nitrogen donor ligand. Chem. Eur. J. 2009, 15, 8678–8682. [Google Scholar] [CrossRef] [PubMed]
- Herres-Pawlis, S.; Neuba, A.; Seewald, O.; Seshadri, T.; Egold, H.; Flörke, U.; Henkel, G. A Library of Peralkylated Bis-guanidine Ligands for Use in Biomimetic Coordination Chemistry. Eur. J. Org. Chem. 2005, 2005, 4879–4890. [Google Scholar] [CrossRef]
- Herres, S.; Heuwing, A.J.; Flörke, U.; Schneider, J.; Henkel, G. Hydroxylation of a methyl group: Synthesis of [Cu2(btmmO)2I]+ and of [Cu2(btmmO)2]2+ containing the novel ligand {bis(trimethylmethoxy)guanidino}propane (btmmO) by copper-assisted oxygen activation. Inorg. Chim. Acta 2005, 358, 1089–1095. [Google Scholar] [CrossRef]
- Hoffmann, A.; Wern, M.; Hoppe, T.; Witte, M.; Haase, R.; Liebhäuser, P.; Glatthaar, J.; Herres-Pawlis, S.; Schindler, S. Hand in Hand: Experimental and Theoretical Investigations into the Reactions of Copper(I) Mono- and Bis(guanidine) Complexes with Dioxygen. Eur. J. Inorg. Chem. 2016, 2016, 4744–4751. [Google Scholar] [CrossRef] [Green Version]
- Strassl, F.; Grimm-Lebsanft, B.; Rukser, D.; Biebl, F.; Biednov, M.; Brett, C.; Timmermann, R.; Metz, F.; Hoffmann, A.; Rübhausen, M.; et al. Oxygen Activation by Copper Complexes with an Aromatic Bis(guanidine) Ligand. Eur. J. Inorg. Chem. 2017, 2017, 3350–3359. [Google Scholar] [CrossRef]
- Bienemann, O.; Haase, R.; Jesser, A.; Beschnitt, T.; Döring, A.; Kuckling, D.; dos Santos Vieira, I.; Flörke, U.; Herres-Pawlis, S. Synthesis and Application of New Guanidine Copper Complexes in Atom Transfer Radical Polymerisation. Eur. J. Inorg. Chem. 2011, 2011, 2367–2379. [Google Scholar] [CrossRef]
- Paul, M.; Strassl, F.; Hoffmann, A.; Hoffmann, M.; Schlüter, M.; Herres-Pawlis, S. Reaction Systems for Bubbly Flows. Eur. J. Inorg. Chem. 2018, 2018, 2101–2124. [Google Scholar] [CrossRef]
- Schurr, D.; Strassl, F.; Liebhäuser, P.; Rinke, G.; Dittmeyer, R.; Herres-Pawlis, S. Decay kinetics of sensitive bioinorganic species in a SuperFocus mixer at ambient conditions. React. Chem. Eng. 2016, 1, 485–493. [Google Scholar] [CrossRef] [Green Version]
- Strassl, F.; Timmermann, J.; Schlüter, M.; Herres-Pawlis, S. Kinetics of the Activation of Oxygen. G.I.T. Lab. J. 2018, 22, 24–26. [Google Scholar]
- Rohrmüller, M.; Hoffmann, A.; Thierfelder, C.; Herres-Pawlis, S.; Schmidt, W.G. The Cu2O2 torture track for a real-life system: Cu2(btmgp)2O2(2+) oxo and peroxo species in density functional calculations. J. Comput. Chem. 2015, 36, 1672–1685. [Google Scholar] [CrossRef] [PubMed]
- Herres-Pawlis, S.; Flörke, U.; Henkel, G. Tuning of Copper(I)-Dioxygen Reactivity by Bis(guanidine) Ligands. Eur. J. Inorg. Chem. 2005, 2005, 3815–3824. [Google Scholar] [CrossRef]
- Herres-Pawlis, S.; Berth, G.; Wiedemeier, V.; Schmidt, L.; Zrenner, A.; Warnecke, H.-J. Oxygen sensing by fluorescence quenching of [Cu(btmgp)I]. J. Lumin. 2010, 130, 1958–1962. [Google Scholar] [CrossRef]
- Kantlehner, W.; Haug, E.; Mergen, W.W.; Speh, P.; Maier, T.; Kapassakalidis, J.J.; Bräuner, H.-J.; Hagen, H.; Orthoamide, X.L. Herstellung von 1,1,2,3,3-pentasubstituierten und 1,1,2,2,3,3-hexasubstituierten Guanidiniumsalzen sowie von 1,1,2,3,3-Pentaalkylguanidinen. Liebigs Ann. Chem. 1984, 1984, 108–126. [Google Scholar] [CrossRef]
- Yang, L.; Powell, D.R.; Houser, R.P. Structural variation in copper(I) complexes with pyridylmethylamide ligands: Structural analysis with a new four-coordinate geometry index, τ4. Dalton Trans. 2007, 955–964. [Google Scholar] [CrossRef] [PubMed]
- Raab, V.; Harms, K.; Sundermeyer, J.; Kovacevic, B.; Maksic, Z.B. 1,8-bis(dimethylethyleneguanidino)naphthalene: Tailoring the basicity of bisguanidine “proton sponges” by experiment and theory. J. Org. Chem. 2003, 68, 8790–8797. [Google Scholar] [CrossRef] [PubMed]
- Herres-Pawlis, S.; Haase, R.; Verma, P.; Hoffmann, A.; Kang, P.; Stack, T.D.P. Formation of Hybrid Guanidine-Stabilized Bis(μ-oxo)dicopper Cores in Solution: Electronic and Steric Perturbations. Eur. J. Inorg. Chem. 2015, 2015, 5426–5436. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dos Santos Vieira, I.; Herres-Pawlis, S. Lactide Polymerisation with Complexes of Neutral N-Donors—New Strategies for Robust Catalysts. Eur. J. Inorg. Chem. 2012, 2012, 765–774. [Google Scholar] [CrossRef]
- Gherman, B.F.; Cramer, C.J. Quantum chemical studies of molecules incorporating a Cu2O22+ core. Coord. Chem. Rev. 2009, 253, 723–753. [Google Scholar] [CrossRef]
- Grimm-Lebsanft, B.; Brett, C.; Strassl, F.; Rukser, D.; Biednov, M.; Biebl, F.; Naumova, M.; Hoffmann, A.; Akinsinde, L.; Brückner, D.; et al. A cryostat for low temperature resonance Raman measurements on operando oxygenated bioinorganic model complexes. Inorg. Chim. Acta 2017, 481, 176–180. [Google Scholar] [CrossRef]
- Karlin, K.D.; Itoh, S. Copper-Oxygen Chemistry; Wiley: Hoboken, NJ, USA, 2011. [Google Scholar]
- Hoffmann, A.; Herres-Pawlis, S. Theoretical Studies on Tyrosinase Models. In Encyclopedia of Inorganic and Bioinorganic Chemistry; Scott, R.A., Ed.; John Wiley & Sons, Ltd.: Chichester, UK, 2011; pp. 1–15. [Google Scholar]
- Weinhold, F.; Landis, C.R. Valency and Bonding; Cambridge University Press: Cambridge, UK, 2005. [Google Scholar]
- Glendening, E.D.; Landis, C.R.; Weinhold, F. NBO 6.0: Natural bond orbital analysis program. J. Comput. Chem. 2013, 34, 1429–1437. [Google Scholar] [CrossRef] [PubMed]
- Glendening, E.D.; Badenhoop, J.K.; Reed, A.E.; Carpenter, J.E.; Bohmann, J.A.; Morales, C.M.; Landis, C.R.; Weinhold, F. NBO 6.0.; Theoretical Chemistry Institute, University of Wisconsin: Madison, WI, USA, 2013. [Google Scholar]
- Tao, J.; Perdew, J.P.; Staroverov, V.N.; Scuseria, G.E. Climbing the Density Functional Ladder: Nonempirical Meta-Generalized Gradient Approximation Designed for Molecules and Solids. Phys. Rev. Lett. 2003, 91, 146401-1–146401-4. [Google Scholar] [CrossRef] [PubMed]
- Weigend, F.; Ahlrichs, R. Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy. Phys. Chem. Chem. Phys. 2005, 7, 3297–3305. [Google Scholar] [CrossRef] [PubMed]
- Goerigk, L.; Grimme, S. A thorough benchmark of density functional methods for general main group thermochemistry, kinetics, and noncovalent interactions. Phys. Chem. Chem. Phys. 2011, 13, 6670–6688. [Google Scholar] [CrossRef] [PubMed]
- Grimme, S.; Ehrlich, S.; Goerigk, L. Effect of the damping function in dispersion corrected density functional theory. J. Comput. Chem. 2011, 32, 1456–1465. [Google Scholar] [CrossRef] [PubMed]
- Rohrmüller, M.; Herres-Pawlis, S.; Witte, M.; Schmidt, W.G. Bis-μ-oxo and μ-η2:η2-peroxo dicopper complexes studied within (time-dependent) density-functional and many-body perturbation theory. J. Comput. Chem. 2013, 34, 1035–1045. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, A.; Herres-Pawlis, S. Donor-driven conformational flexibility in a real-life catalytic dicopper(II) peroxo complex. Phys. Chem. Chem. Phys. 2016, 18, 6430–6440. [Google Scholar] [CrossRef] [PubMed]
- Huber, S.M.; Ertem, M.Z.; Aquilante, F.; Gagliardi, L.; Tolman, W.B.; Cramer, C.J. Generating Cu(II)-oxyl/Cu(III)-oxo species from Cu(I)-α-ketocarboxylate complexes and O2: In silico studies on ligand effects and C–H-activation reactivity. Chem. Eur. J. 2009, 15, 4886–4895. [Google Scholar] [CrossRef] [PubMed]
- Teuber, H.-J.; Benz, S. Reaktionen mit Nitrosodisulfonat, XXXIV. Chinolin-chinone-(7.8). Chem. Ber. 1967, 100, 2077–2092. [Google Scholar] [CrossRef]
- SMART (Version 5.631); Bruker AXS Inc.: Madison, WI, USA.
- SAINT and SADABS; Bruker AXS Inc.: Madison, WI, USA, 2008.
- XPREP; Bruker AXS Inc.: Madison, WI, USA, 2007.
- Sheldrick, G.M. Phase annealing in SHELX-90: Direct methods for larger structures. Acta Crystallogr. A 1990, 46, 467–473. [Google Scholar] [CrossRef]
- Hübschle, C.B.; Sheldrick, G.M.; Dittrich, B. ShelXle: A Qt graphical user interface for SHELXL. J. Appl. Crystallogr. 2011, 44, 1281–1284. [Google Scholar] [CrossRef] [PubMed]
- Spek, A.L. PLATON, A Multipurpose Crystallographic Tool; Utrecht University: Utrecht, The Netherlands, 2008. [Google Scholar]
- Spek, A.L. Structure validation in chemical crystallography. Acta Crystallogr. D 2009, 65, 148–155. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schulz, B.; Bäckström, J.; Budelmann, D.; Maeser, R.; Rübhausen, M.; Klein, M.V.; Schoeffel, E.; Mihill, A.; Yoon, S. Fully reflective deep ultraviolet to near infrared spectrometer and entrance optics for resonance Raman spectroscopy. Rev. Sci. Instrum. 2005, 76, 73107. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16 Rev. B.01; Gaussian. Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Hoffmann, A.; Grunzke, R.; Herres-Pawlis, S. Insights into the influence of dispersion correction in the theoretical treatment of guanidine-quinoline copper(I) complexes. J. Comput. Chem. 2014, 35, 1943–1950. [Google Scholar] [CrossRef] [PubMed]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| C1 | C2 | C3 | C4 | C5 | |
|---|---|---|---|---|---|
| Cu(1)–Ngua,aliph | 2.058(5) | 2.025(2) | 2.013(3) | 1.963(2) | 1.887(2) |
| Cu(2)–Ngua,aliph | - | - | - | - | 1.878(2) |
| Cu(1)–Ngua,arom | 1.992(4) | 2.082(2) | 2.097(3) | 1.989(2) | 1.893(2) |
| Cu(2)–Ngua,arom | - | - | - | - | 1.885(2) |
| Cu–X [a] | 2.476(1) | 2.327(1) | 2.200(1) | 2.260(1)/2.251(1) | - |
| Cu(1)…Cu(2) | - | - | - | - | 2.732(1) |
| N–Cu(1)–N | 97.5(1) | 95.4(1) | 94.8(2) | 92.6(1) | 178.1(1) |
| N–Cu(2)–N | - | - | - | - | 178.9(1) |
| τ4 [b] | - | - | - | 0.55 | - |
| ρ (guaaliph) [c] | 0.94 | 0.94 | 0.94 | 0.97 | 0.96 |
| ρ (guaarom) [c] | 0.97 | 0.96 | 0.96 | 1.00 | 0.98 |
| Guanidine twist (guaaliph) [d] | 16.2 | 17.8 | 17.6 | 16.6 | 16.8 |
| Guanidine twist (guaarom) [d] | 12.7 | 12.8 | 12.6 | 6.4 | 12.7 |
| C1 | C2 | C3 | C4 | Cation of C5 | [Cu2O2(DMEG2tol)2]2+ | |
|---|---|---|---|---|---|---|
| Bond lengths | ||||||
| Cu–Ngua,aliph | 2.038 | 2.055 | 2.043 | 1.900 | 1.901 | 1.896 |
| Cu–Ngua,arom | 2.008 | 1.996 | 2.010 | 2.011 | 1.909 | 1.916 |
| Cu–X [a] | 2.481 | 2.315 | 2.182 | 2.133/2.157 | ||
| Cu···Cu | 2.595 | 2.734 | ||||
| Cu–O | 1.804/1.800 | |||||
| NBO charges | ||||||
| Cu | 0.72 | 0.75 | 0.77 | 1.14 | 0.77 | 1.36 |
| Ngua,aliph | −0.71 | −0.69 | −0.67 | −0.61 | −0.71 | −0.64 |
| Ngua,arom | −0.71 | −0.68 | −0.67 | −0.53 | −0.74 | −0.71 |
| O | −0.96 | |||||
| CT energies | ||||||
| Ngua,aliph → Cu | 15.2 | 19.4 | 23.2 | cov. [b] | 48.7 | cov. [b] |
| Ngua,arom → Cu | 28.5 | 32.3 | 31.3 | cov. [b] | 49.7 | cov. [b] |
| C1 | C2 | C3 | C4 | C5 | |
|---|---|---|---|---|---|
| Empirical formula | C17H26ICuN6 | C17H26BrCuN6 | C17H26ClCuN6 | C17H26Cl2CuN6 | C36H52Cu2F6N12O6S2 |
| Formula mass [g mol−1] | 504.88 | 457.89 | 413.43 | 448.88 | 1054.10 |
| Crystal size [mm] | 0.23 × 0.22 × 0.19 | 0.40 × 0.32 × 0.30 | 0.38 × 0.28 × 0.14 | 0.28 × 0.24 × 0.19 | 0.34 × 0.25 × 0.24 |
| T [K] | 100(2) | 100(2) | 100(2) | 100(2) | 100(2) |
| Crystal system | orthorhombic | monoclinic | monoclinic | monoclinic | triclinic |
| Space group | Fdd2 | P21/c | P21/c | P21/n | P |
| a [Å] | 18.055(3) | 7.194(1) | 7.145(1) | 15.234(3) | 13.869(2) |
| b [Å] | 26.458(5) | 12.813(2) | 12.787(2) | 8.101(2) | 14.096(2) |
| c [Å] | 16.438(3) | 20.599(3) | 20.530(3) | 17.631(3) | 14.692(2) |
| α [°] | 90 | 90 | 90 | 90 | 112.944(2) |
| β [°] | 90 | 93.439(2) | 93.699(3) | 113.072(3) | 96.288(2) |
| γ [°] | 90 | 90 | 90 | 90 | 112.879(2) |
| V [Å3] | 7852(2) | 1895.2(4) | 1871.8(5) | 2001.8(6) | 2319.2(5) |
| Z | 16 | 4 | 4 | 4 | 2 |
| ρcalcd. [gcm−3] | 1.708 | 1.605 | 1.467 | 1.489 | 1.509 |
| μ [mm−1] | 2.698 | 3.273 | 1.322 | 1.372 | 1.087 |
| λ [Å] | 0.71073 | 0.71073 | 0.71073 | 0.71073 | 0.71073 |
| F(000) | 4032 | 936 | 864 | 932 | 1088 |
| hkl range | ±23, ±34, ±21 | ±9, ±16, ±27 | ±8, ±15, −25 ≤ l ≤ 24 | ±20, ±10, ±23 | ±18, ±19, ±19 |
| Reflections collected | 18374 | 25339 | 21427 | 20515 | 33606 |
| Independent reflections | 4665 | 4516 | 3560 | 5052 | 12115 |
| Rint. | 0.0653 | 0.0735 | 0.1084 | 0.0838 | 0.0611 |
| Number of parameters | 230 | 230 | 230 | 239 | 585 |
| R1 [I ≥ 2σ(I)] | 0.0434 | 0.0360 | 0.0414 | 0.0471 | 0.0446 |
| ωR2 (all data) | 0.1043 | 0.0900 | 0.1059 | 0.1199 | 0.1092 |
| Goodness-of-fit | 1.095 | 1.045 | 1.035 | 1.031 | 1.010 |
| Largest diff. peak, hole [e Å−3] | −1.304, 1.021 | −0.912, 0.494 | −0.524, 0.475 | −0.555, 0.934 | −0.455, 0.593 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Strassl, F.; Hoffmann, A.; Grimm-Lebsanft, B.; Rukser, D.; Biebl, F.; Tran, M.A.; Metz, F.; Rübhausen, M.; Herres-Pawlis, S. Fluorescent Bis(guanidine) Copper Complexes as Precursors for Hydroxylation Catalysis. Inorganics 2018, 6, 114. https://0-doi-org.brum.beds.ac.uk/10.3390/inorganics6040114
Strassl F, Hoffmann A, Grimm-Lebsanft B, Rukser D, Biebl F, Tran MA, Metz F, Rübhausen M, Herres-Pawlis S. Fluorescent Bis(guanidine) Copper Complexes as Precursors for Hydroxylation Catalysis. Inorganics. 2018; 6(4):114. https://0-doi-org.brum.beds.ac.uk/10.3390/inorganics6040114
Chicago/Turabian StyleStrassl, Florian, Alexander Hoffmann, Benjamin Grimm-Lebsanft, Dieter Rukser, Florian Biebl, Mai Anh Tran, Fabian Metz, Michael Rübhausen, and Sonja Herres-Pawlis. 2018. "Fluorescent Bis(guanidine) Copper Complexes as Precursors for Hydroxylation Catalysis" Inorganics 6, no. 4: 114. https://0-doi-org.brum.beds.ac.uk/10.3390/inorganics6040114