The Role of Non-Coding RNAs Involved in Nickel-Induced Lung Carcinogenic Mechanisms


Department of Environmental Medicine, New York University School of Medicine, New York, NY 10010, USA
*
Author to whom correspondence should be addressed.
Inorganics 2019, 7(7), 81; https://0-doi-org.brum.beds.ac.uk/10.3390/inorganics7070081
Submission received: 29 May 2019
/
Revised: 19 June 2019
/
Accepted: 25 June 2019
/
Published: 28 June 2019
(This article belongs to the Special Issue Bioinorganic Chemistry of Nickel)
{kind=link}
Abstract
:Nickel is a naturally occurring element found in the Earth’s crust and an International Agency for Research on Cancer (IARC)-classified human carcinogen. While low levels found in the natural environment pose a minor concern, the extensive use of nickel in industrial settings such as in the production of stainless steel and various alloys complicate human exposure and health effects. Notably, interactions with nickel macromolecules, primarily through inhalation, have been demonstrated to promote lung cancer. Mechanisms of nickel-carcinogenesis range from oxidative stress, DNA damage, and hypoxia-inducible pathways to epigenetic mechanisms. Recently, non-coding RNAs have drawn increased attention in cancer mechanistic studies. Specifically, nickel has been found to disrupt expression and functions of micro-RNAs and long-non-coding RNAs, resulting in subsequent changes in target gene expression levels, some of which include key cancer genes such as p53, MDM2, c-myc, and AP-1. Non-coding RNAs are also involved in well-studied mechanisms of nickel-induced lung carcinogenesis, such as the hypoxia-inducible factor (HIF) pathway, oxidative stress, DNA damage and repair, DNA hypermethylation, and alterations in tumor suppressors and oncogenes. This review provides a summary of the currently known epigenetic mechanisms involved in nickel-induced lung carcinogenesis, with a particular focus on non-coding RNAs.
1. Introduction
1.1. Nickel Overview
Nickel is a commonly occurring metal on Earth, existing as both soluble and insoluble compounds in soil, fumes, and water. Used as a component in many products such as watches, coins, belt buckles, earrings, mobile phones, and various medical devices, it is as ubiquitous in industrial usage as it is in nature [1]. Nickel is a transition metal that normally exists in nature in a +2 oxidative state [2]. While nickel plays fundamental roles for plants, bacteria, archaea, and unicellular eukaryotes, there are no enzymes in the human body that require nickel to function. As a result, it is only biologically significant as a toxicant for humans [3]. Inhalation is the most prominent route of exposure, with oral and dermal routes considered much less important [4]. On a cellular level, insoluble particulate nickel enters cells by phagocytosis, while nickel carbonyl is a lipophilic compound that can pass through the plasma membrane. Ni(II) can be transported into cells through calcium channels and/or divalent cation transporters such as DMT-1 [5]. Nickel is primarily excreted through urine with elimination following first order kinetics without any evidence of dose-dependent excretion. Excretion may also occur in saliva and sweat, which may be more significant in hot environments [6]. Workplace-related nickel exposure tends to be via inhalation of airborne fumes and dusts containing nickel and its associated compounds, while non-workplace-related nickel exposures are often from diet or dermal contact [7]. Dietary exposure is more significant in vegetables (particularly spinach), cocoa and nuts because of their higher nickel content. Chronic nickel exposure can lead to dermatitis, pulmonary fibrosis, and asthma [8,9]. Nickel sensitivity is a well-documented issue that affects millions of people. It is caused by triggering T-lymphocyte-driven delayed-type hypersensitivity reaction by Ni2+, followed by leukocyte infiltration at the site of exposure [1].
1.2. Nickel-Induced Carcinogenesis
The most scrutinized effect of nickel exposure is carcinogenesis. In 1990, the International Agency for Research on Cancer (IARC) classified soluble nickel as a “group 1” human carcinogen [10]. In 1990, a report from the IARC that aimed to identify which chemical forms of nickel caused elevated risks of cancer mortality in occupationally exposed workers found that exposure to high levels of oxidic nickel compounds, exposure to sulfidic nickel in combination with oxidic nickel, and exposure to water-soluble nickel, alone or together with less soluble compounds, led to higher mortality from cancers of the lung and nasal sinuses [11]. While some studies have since suggested that nickel workers processing and refining sulfidic nickel ores have demonstrated an increased pulmonary and sinonasal cancer risk, some studies involving low level nickel exposure did not observe an increased incidence of cancer, suggesting that there may be threshold-like responses in tumor incidence [12,13,14,15,16,17].
1.3. Non-Coding RNA
While 80% of the human genome can be transcribed into RNA, only 2% of the genes code for proteins [18]. Non-coding RNAs are classified as either housekeeping non-coding RNAs, such as small nuclear RNAs, transfer RNAs, ribosomal RNAs, and small nucleolar RNAs, or regulatory non-coding RNAs, such as microRNAs (miRNAs), piwiRNAs, and long non-coding RNAs (lncRNAs) based on their size.
MicroRNAs are small non-coding RNAs that are 18–25 nucleotides in length and function by targeting complementary mRNAs at the 3′-untranslated region (3′-UTR) to cause degradation, translation inhibition or regulating gene expression at the post-transcriptional level [19]. They are specifically expressed in different human cells. Dysregulation of miRNA is related to various types of cancer and is involved in multiple processes, such as cell proliferation and differentiation, apoptosis, cancer initiation and progression, and cell metastasis [20]. MicroRNAs are now providing important information for cancer diagnosis, treatment, and prognosis. A lot of miRNAs have been identified with different roles in lung carcinogenesis as summarized by Uddin et al. [21], such as miR-21 in apoptosis, miR-31 in cell division, and miR-101, miR-15 and miR-16 in cell proliferation [22,23,24,25].
Long non-coding RNAs (lncRNAs) are RNAs longer than 200 nucleotides. Unlike miRNAs, lncRNAs are poorly conserved and regulate gene expression at different levels including histone modification, transcription regulation, post-transcription regulation, translation regulation and pseudogene regulation [26]. They also serve as scaffolds for cellular components and decoys for miRNAs and other coding RNAs [27,28]. lncRNAs have been given more attention recently since it was discovered that they play an essential role in pathological progressions including cancer invasion and metastasis [29,30,31]. Many lncRNAs have been reported to be associated with non-small cell lung cancer (NSCLC), such as CAR10, MALAT1, HOTAIR and so on [32,33,34]. Studies have shown that lncRNA was also involved in nickel-induced tumorigenesis [35,36]. A new lncRNA, termed nickel-related gene (NRG1), was the first to be identified as a lncRNA induced in Ni-induced lung cancer but the mechanisms that regulate nickel-induced NRG1 level is not known [37].
The molecular mechanisms by which nickel causes tumorigenesis can be summarized as DNA hypermethylation, oxidative stress and DNA damage, hypoxia-inducible signaling pathways, tumor suppressor genes, oncogene alterations, etc. [37]. In this review, we will discuss the molecular mechanisms involving non-coding RNA, especially miRNA and lncRNA, in nickel-induced lung cancer. Although the number of ncRNAs studied in this area so far is limited, this review reveals a current state of knowledge and provides directions for future research in ncRNAs involved in nickel-induced carcinogenesis.
2. Mechanisms
2.1. DNA Hypermethylation
While only considered weakly mutagenic, nickel is known to promote tumorigenesis through epigenetic mechanisms including DNA methylation, histone acetylation, and miRNAs. To date, DNA methylation is the primary epigenetic event studied in nickel-induced lung cancer, and has been shown to induce various human diseases and cancers through gene inactivation, including fragile X mental retardation [38,39,40,41,42,43]. Specifically, DNA methyltransferases (DNMTs) act to transfer a methyl group from S-adenosyl methionine (SAM) to the fifth carbon on cytosine. Notably, as is the case in most cancers, nickel is capable of eliciting global hypermethylation, thereby suppressing key tumor suppressor genes and inducing senescence as a part of its carcinogenic mechanism [44]. As a case in point, while 5-azacytidine and nickel have both been shown to inhibit DNA methyltransferase activity, the effects of nickel are only transient, in that after a recovery period following nickel exposure, levels of methyltransferases will recuperate and surge, leading to genome-wide increases in DNA methylation levels [38,45,46]. In fact, nickel-induced DNA hypermethylation has been confirmed both in vitro and in vivo. For example, in human bronchial epithelial (Beas-2B) cells, a commonly-used cell line for studies involving lung cancer, nickel has been found to induce the suppression of E-cadherin [47]. E-cadherin is a prominent tumor suppressor gene and cell-adhesion molecule. Reductions in E-cadherin levels have been evidenced to trigger dysfunctions of the cell–cell adhesion system, leading to cancer cell metastasis and invasion [48]. In addition, nickel has also been shown to induce the silencing of p16 through CpG site hypermethylation and thereby escape cell senescence [44]. Specifically, using p53 heterozygous mice and Wistar rats, treatment with nickel sulfide promoted promoter hypermethylation of p16, a cyclin-dependent kinase inhibitor and tumor suppressor gene [49]. Previous studies have demonstrated that nickel induces site-specific DNA hypermethylation. Specifically, instead of active euchromatin, nickel selectively targets inactive heterochromatin regions such as the long arm of chromosome X in Chinese hamster ovary cells [50]. In detail, the gpt gene was inserted near an active euchromatin region and the telomere in G10 and G12 cells, respectively [51]. The successful silencing of the gpt gene is validated by exhibition of high resistance to 6TG. Interestingly, while nickel was shown to induce silencing of the gpt gene in both G10 and G12 cells, the silencing effect was much more significant in G12 cells, indicating that nickel specifically targets genes in heterochromatic regions and spread heterochromatinization through DNA condensation [39,52]. The heterochromatin’s condensed structure dictates that genes in this region have minimal activity, but its overall structure is intrinsic for the maintenance of overall genomic stability and that the organization of heterochromatin is often compromised in cancer [53,54].
Hypermethylation at the promoter regions of non-coding RNAs upon nickel treatment has been reported in several recent studies. One study observed that nickel led to a dose (0–0.5 mM) and time-dependent (0–12 h) reduction of lncRNA maternally expressed gene 3 (MEG3), in both Beas-2B and BEP2D cells (both human bronchial epithelial cells). Ectopic expression of MEG3 in Beas-2B cells inhibited cell transformation by nickel and knockdown of MEG3 resulted in spontaneous cell transformation of Beas-2B cells [35]. MEG3 is a 1.6 kb lncRNA and is expressed in many normal tissues, while loss of MEG3 expression has been seen in tumors and was reported to be related to tumor development and progression [55,56,57]. Zhou et al. found that the MEG3 promoter region was hypermethylated in normal human bronchial epithelial cells upon nickel exposure (0.5 µM, from 0–12 h), and treatment of cells with the DNA methylation inhibitor 5-aza-2-deoxycytidine (5-Aza) (5 µM, 72 h) removed the methylation status and increased the expression level of MEG3. They also revealed that the hypermethylation effect was mainly mediated by DNMT3b since nickel exposure upregulated DNMT3b specifically, and knockout of DNMT3b restored MEG3 expression in NiCl2-treated Beas-2B cells [35].
Another study found that DNMT1 was upregulated in NiS-transformed 16HBE cells (NSTCs), a human bronchial epithelial cell line [58], along with a significant inhibition of miR-152 expression [59]. MicroRNA-152 is a member of the miR-148/152 family that has sequence complementarity to the 3′-UTR of DNMT1. Other members in the family include miR-148a and miR-148b. Ectopic expression of miR-152 in NSTCs suppressed cell growth, and knockdown of miR-152 increased cell growth of non-treated 16HBE cells. Interestingly, they first found that miR-152 repressed DNMT1 expression by targeting its 3′-UTR, and then discovered that downregulation of miR-152 in NSTCs was mediated through hypermethylation at the promoter region by DNMT1, indicating a double-negative loop between miR-152 and DNMT1. They speculated that NiS exposure firstly induced upregulation of DNMT1 via other mechanisms such as oxidative stress, and then led to hypermethylation of miR-152 at the CpG island promoter region, resulting in its downregulation, which further promoted DNMT1 expression by reduced methylation at the 3′-UTR by miR-152. It was also shown that miR-152 expression suppressed cell growth and a reduction of miR-152 expression lead to a 30% increase in cell proliferation [59].
MicroRNA-203 is another miRNA that was found to be downregulated in NSTCs [60]. Ectopic expression of miR-203 reduced NSTC cell migration and tumor growth in nude mice. It was found that the CpG island and first exon area of miR-203 was hypermethylated, however the mechanisms that mediate the hypermethylation effect was not elucidated in the study. Intriguingly, besides 5-Aza, TSA (Trichostatin A, a histone deacetylase inhibitor)-treated NSTCs also partially restored the expression level of miR-203, suggesting that downregulation of miR-203 is possibly mediated by DNA hypermethylation and/or other epigenetic silencing mechanisms.
2.2. Hypoxia-Inducible Pathway
The hypoxia-inducible signaling pathway is another important route for nickel-induced carcinogenesis, in which the transcription factor hypoxia-inducible-factor-1 (HIF-1) is activated [61]. HIF-1 is a critical regulator of genes that facilitate cell survival and adaptation in hypoxic conditions [62,63,64]. It is a heterodimeric transcriptional factor that consists of two subunits, HIF-1α and HIF-1β, both of which are required for HIF-1 to function; HIF-1β is integral in HIF-1 heterodimer formation, while HIF-1α is the key regulatory subunit and is responsible for HIF-1 transcriptional activity [65]. HIF-1 overexpression has been observed in various cancers [66]. Studies suggested that nickel can replace iron in HIF prolyl hydroxylases and thus inhibits the association of HIF-1α with Von-Hippel-Lindau (VHL) E3 ubiquitin ligase, which in turn stabilizes the HIF protein [67,68,69]. It was reported that elevated HIF-1 aided in tumor progression by inducing glycolytic activity and increasing production of lactic acid in tumor cells (Warburg effect) [70]. A collection of hypoxia-inducible genes were also induced by nickel including glycolytic enzymes and glucose transporters, and tumor oncogenes such as NDRG1 (N-myc Downstream-Regulated Gene 1/Cap43), and some other HIF-dependent genes involved in carcinogenesis such as BCL-2 (B-Cell Lymphoma 2) binding protein Nip3, EGLN1 (Egl-9 Family Hypoxia Inducible Factor 1), HIG1 (Hypoxia-Induced Gene 1) and P4H (Prolyl 4-Hydroxylase) [61]. High fidelity DNA repair mechanisms such as base excision repair and nucleotide repair were found to be compromised by the hypoxic conditions induced by nickel [71,72].
MicroRNA-210 is known as a hypoxia-sensitive miRNA and is one of the target miRNAs for HIF-1α. These are termed as hypoxia-regulated microRNAs (HRMs), and include miR-21, 23, 24, 26a, 213, etc. [73]. One study reported that nickel exposure (NiCl2, 1 mM, 4 h) induced overexpression of miR-210 with stabilization of the HIF-1α protein, resulting in a metabolism shift [74]. In fact, HIF-1α has been demonstrated to be directly recruited to the promoter region of miR-210 during hypoxic conditions to induce miRNA expression [75]. Similar with the DNMT1/miR-152 double-negative loop, another study revealed a non-canonical miR-210 targeting site in the HIF-1α 3′-UTR region, suggesting a potential negative feedback loop in HIF-1α/miR-210 signaling [76].
MEG3, as mentioned, is a long non-coding RNA that was found to be downregulated by DNMT3b under nickel exposure. It has been suggested that MEG3 is a regulator of HIF-1α and nickel-induced HIF-1α accumulation independent of iron metabolism through inhibiting MEG3 [35]. Instead of directly targeting HIF-1α at the promoter region, MEG3 regulates HIF-1α at the protein translational level, since alterations in transcription, mRNA stability and protein degradation were not observed in MEG3 ectopically expressed Beas-2B cells compared with vector control cells in the study. It was further investigated that MEG3 induced protein translation by S6 phosphorylation via the Akt/p70S6K/S6 pathway, and they identified the upstream regulator of the pathway, PHLPP1 (PH Domain and Leucine Rich Repeat Protein Phosphatase 1), and c-Jun (a component of the transcription factor activator protein 1), a direct target of MEG3 and a negative regulator for PHLPP1. Together, nickel induces DNMT3b expression and hypermethylation of the MEG3 promoter, reduces MEG3 binding with the c-Jun transcription factor and increases its activity, which negatively regulates PHLPP1 by binding to its promoter, and in turn activates the Akt/p70S6K/S6 pathway, resulting in increased protein translation of HIF-1α.
2.3. Oxidative Stress
Oxidative stress was suggested to be another essential component in nickel-induced carcinogenesis [77]. Oxidative stress is known as the imbalance of oxidants, such as overproduction of reactive oxygen species (ROS) and nitrogen species (NOS), and antioxidants in favor of the oxidants [78,79]. Sustained high levels of ROS can cause oxidative damage to nucleic acids, lipids, and proteins, alter oxidative equilibrium, and regulate cell viability [80]. Oxidative damage to DNA is known to be one of the most important mechanisms in cancers [81].
The carcinogenic effect of nickel is theorized to be caused in part by its role in the generation of reactive oxidative species. Nickel is known to play a role in the generation of R· and RO· radicals in conjunction with glutathione [82]. Oxidative stress-related biomarkers can be observed in nickel electroplating workers exposed to nickel, such as increased lipid peroxidation, and reduced anti-oxidative enzymes as GSH (Glutathione), SOD (Superoxide Dismutase), and oxidative DNA damage marker 8-OH-dG [83]. In vitro, Beas-2B lung epithelial cells exposed to nickel nanoparticles, NiO nanoparticles, and NiCl2 were shown to exhibit chromosomal damage, with NiO being the most potent in causing DNA strand breaks and increasing intracellular ROS [84]. Both water soluble NiCl2 and insoluble Ni3S2 have been shown to enhance the formation of intracellular oxidants after 6 h of exposure, and free radicals have been observed in the nucleus when cells have been exposed to Ni3S2 for more than 18 h [85]. In addition, nickel and ROS exhibit synergistic inhibition towards both DNA polymerization and ligation and cause protein fragmentation, resulting in impaired DNA repair [86]. Nickel is also shown to induce transcriptional downregulation of homology-dependent DNA double-strand break repair (HDR) and mismatch repair (MMR) pathways [87]. MicroRNA regulation of oxidative stresses has been observed in several diseases including cancers, such as miRNA-34a-5p, miRNA-1915-3p, miRNA-638, and miRNA-150-3p in hepatocellular carcinoma (HCC) [88].
Hypoxic conditions favor the increase of reactive oxygen species and oxidative stress, and the hypoxia sensitive miRNA, miR-210, was found to respond to oxidative stress induced by H2O2 in H9C2 cardiomyocytes. It indirectly regulates the iron–sulfur cluster assembly protein (ISCU) and proved its activity in mitochondrial electron transport chains and energy metabolism, which are important mechanisms in the progression of ischemic heart disease [89]. Another hypoxia-related non-coding RNA, MEG3, was found to be downregulated in H2O2-treated RF/6A cells and played an important role in diabetes-induced microvascular abnormalities. Increased expression of miR-152 was observed in hypertrophic conditions and in H2O2-treated HL-1 cells, indicating its role in oxidative stress-related cardiac hypertrophy [90]. However, the exact role of these miRNAs in the nickel-induced oxidative stress pathways in lung carcinogenesis was not clarified and needs further investigation.
2.4. Tumor Suppressor Gene and Proto-Oncogene Alterations
Cell proliferation, differentiation, immortalization and transformation are key steps in cell carcinogenesis. Carcinogenic mechanisms induced by nickel will ultimately lead to activation of proto-oncogenes or inactivation of tumor suppressor genes. p53 is one of the most studied tumor suppressors in various cancers. Although nickel-induced mutations in the p53 gene were observed in several studies, indicating that nickel is a possible mutagen in mammalian cells [91,92,93], its regulation of p53 activity is not rare. This study proved that both water soluble and insoluble nickel were able to alter the expression of p53 and lead to cell transformation [61]. MEG3 has been described as a tumor suppressor [56], and found to exert its antitumor effect by modulating the activity of p53, Mdm2 (Mouse double minute 2 homolog), Rb (Retinoblastoma) and other cell cycle regulators [94]. MEG3 inhibited NSCLC cell proliferation and apoptosis by MEG3–p53 pathways [95], and it can be increased by chemotherapeutic agents such as paclitaxel (PTX), a first-line chemotherapy drug for NSCLC [96]. It was suggested that MEG3 can regulate p53 activity independent of p21 signaling [97,98], or via regulating the activity of MDM2, the main inhibitor of p53 [99,100,101].
In fact, lncRNA MEG3 also interferes with other miRNAs and functions as competing endogenous RNAs (ceRNAs) to bind and compete for miRNA target genes [102]. c-Myc is a known oncogene related to cell transformation, immortalization, differentiation, and cell apoptosis. MEG3 was found to compete with miR-27a for PHLPP2 to downregulate c-Myc and thereby reduced bladder cancer invasion [103]. MicroRNA-27a is an inhibitor for PHLPP2 [104], and it was reported to be upregulated in NSCLC and was possibly associated with lung carcinogenesis by targeting the TGF-β (Transforming Growth Factor beta) signaling pathway [105]. Reduction of MEG3 via nickel exposure is likely to attenuate its competing effect with miR-27a and thus suppress PHLPP2 and increase c-Myc and TGF-β signaling, inducing epithelial–mesenchymal transition (EMT) and transformation in lung carcinogenesis.
Other than MEG3, dysregulations of many lncRNAs in lung cancer have been identified and summarized by Khandelwal et al. [32,106], of which some have been shown to be associated with occupational PAH (Polycyclic Aromatic Hydrocarbon) exposures in coke oven workers, such as HOTAIR, TUG1, MALAT1 and GAS5 [107]. Cadmium was also found to induce the expression of lncRNA, ENST00000414355, to modulate DNA damage and repair in 16HBE cells. Other than being an upstream regulator of MEG3 and many other lncRNAs in carcinogenic signaling pathways, they can possibly be downstream of other genes; for example, TUG1 was found to be upregulated by p53 in lung cancer [108], indicating that TUG1 could be a potential lncRNA involved in the MEG3–p53 signaling pathway in nickel-induced lung carcinogenesis.
The target genes for miR-210, MNT, a MYC antagonist [109], and E2F3, a key regulator of the cell cycle [110], were of importance to study in the mechanisms of carcinogenesis. Another study revealed that miR-210 was upregulated in NSCLC human tissue samples, and six potential target genes of miR-210 were identified (IL-6, GNG11, CXCL12, ADRB2, ADCY9, and CHRM2), suggesting its role in NSCLC signaling pathways [111]. The diagnostic value of miR-210 in lung cancer was also indicated in a meta-analysis study by Yang et al. [112].
MicroRNA-203 was hypermethylated following nickel exposure both in vivo and in vitro, and inhibited miR-203 led to dysregulation of its target gene ABL1, contributing to nickel-induced cancers [60]. ABL1 is a known oncogene and plays an important role in cell differentiation, division, adhesion, and stress responses. It was proved to be the miRNA-203 direct target gene along with several other tumor proto-oncogenes, such as ABCE1, E2F3 and p63 [113,114,115,116,117]. An upregulation of miR-222 was observed in both NSTCs and Ni-induced tumor tissues of rats, and it was suggested that miR-222 could be involved in nickel-induced tumorigenesis by targeting tumor suppressor genes CDKN1C (p57) and CDKN1B (p27) [118]. Dysregulation of p27 via miR-222 was reported to be related to increased cancer cell proliferation [119]. Besides lung cancer, CDKN1C and CDKN1B were also found to be regulated by miR-222 in human hepatocellular carcinoma [120]. NiCl2 also induced expression of miR-4417 in Beas-2B cells and A549 cells, which targeted the TAB2 gene and was involved in the mechanism of nickel-induced EMT, lung fibrosis and tumor progression. Ectopic expression of miR-4417 in Beas-2B and A549 cells induced fibronectin, while nickel exposure failed to induce fibronectin in miR-4417 inhibited Beas-2B cells. Although TGF-β was involved in nickel-induced cell EMT and significantly induced miR-4417 in Beas-2B cells, its inhibitor SB525334 only partially reduced miR-4417 level and failed to restore TAB2 level in TGF-β-treated cells, indicating that nickel can induce miR-4417 through pathways independent of TGF-β [121].
3. Conclusions
Nickel is a naturally occurring element that can be found in soils, sediments, water, and air. Humans can be exposed to nickel from different sources and occupational exposure to nickel in nickel refineries and processing plants has been a big concern for workers and was reported to be related to lung cancer. The mechanisms of nickel-induced carcinogenesis that have been studied include hypoxia-inducible factor pathways and the introduction of oxidative stress which generates DNA damage and impairs DNA repair pathways. Nickel also disrupts the integrity of heterochromatin, and subsequently silences tumor suppressor genes, such as p53, and activates c-Myc, resulting in dysregulated cell differentiation, proliferation, transformation, and cancer progression.
A newly found role played by non-coding RNAs has recently been studied in nickel-induced lung carcinogenesis. With increased attention in non-coding RNAs and the development of new approaches for understanding non-coding RNAs, various new mechanisms resulting in cancer initiation and progression have been identified. Several studies have reported that nickel was able to alter the expression of miRNAs, such as miR-210, miR-203, miR-152, miR-4417, and miR-222, and lncRNAs, such as NRG1 and MEG3. These studies suggest important roles for non-coding RNAs in nickel-induced lung carcinogenesis, providing insight into the pathogenesis and mechanisms of the disease.
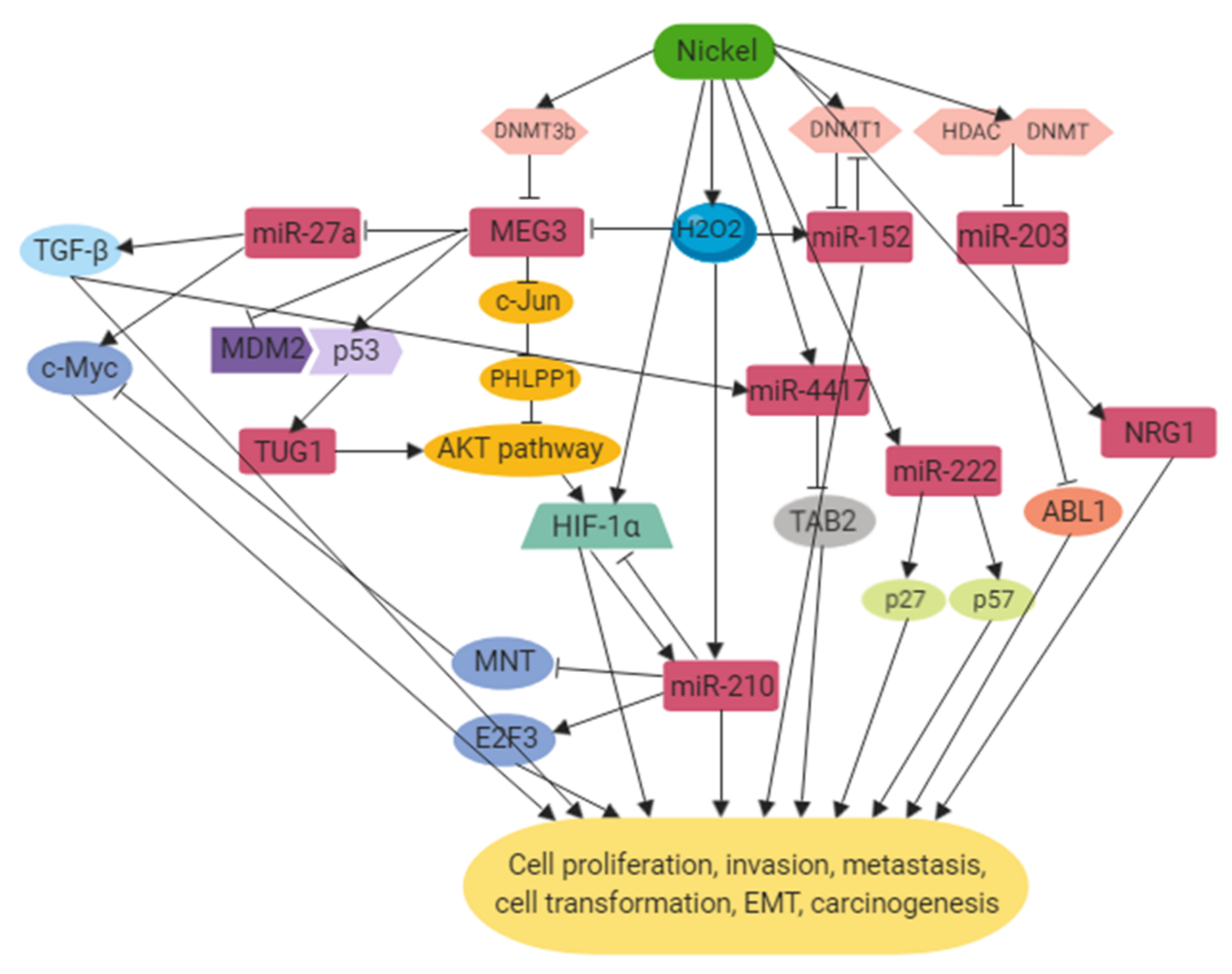
However, only a small amount of non-coding RNAs related to nickel exposure have been characterized so far and their functions and mechanisms in carcinogenesis remain to be elucidated. The mechanisms by which ncRNAs were dysregulated upon nickel exposure were mainly through the increased expression of DNMTs followed by hypermethylation at the promoter region of the ncRNAs, resulting in altered expression levels of their target genes which are involved in cell cycle regulation, proliferation, apoptosis, EMT, metastasis and many aspects of tumorigenesis. Instead of being upstream regulators, some other ncRNAs were found to be regulated by HIF, TGF-β and p53 during the signaling pathways induced by nickel. Figure 1 provides a schematic diagram of the network that ncRNAs are involved in in nickel-induced carcinogenesis. Future studies are warranted to elucidate the functions of these ncRNAs as potential therapeutic targets, diagnostic markers and prognosis predictions for lung cancer induced by nickel.
Funding
This research was funded by National Institutes Health (NIH) grants number ES000260, ES022935, ES023174, ES026138.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Schmidt, M.; Goebeler, M. Nickel allergies: Paying the Toll for innate immunity. J. Mol. Med. 2011, 89, 961–970. [Google Scholar] [CrossRef] [PubMed]
- Cempel, M.; Nikel, G. Nickel: A Review of Its Sources and Environmental Toxicology. Pol. J. Environ. Stud. 2006, 15, 375–382. [Google Scholar]
- Zambelli, B.; Uversky, V.N.; Ciurli, S. Nickel impact on human health: An intrinsic disorder perspective. Biochim. Biophys. Acta 2016, 1864, 1714–1731. [Google Scholar] [CrossRef] [PubMed]
- Coogan, T.P.; Latta, D.M.; Snow, E.T.; Costa, M. Toxicity and carcinogenicity of nickel compounds. Crit. Rev. Toxicol. 1989, 19, 341–384. [Google Scholar] [CrossRef] [PubMed]
- Barceloux, D.G. Nickel. J. Toxicol. Clin. Toxicol. 1999, 37, 239–258. [Google Scholar] [CrossRef] [PubMed]
- Rezuke, W.N.; Knight, J.A.; Sunderman, F.W., Jr. Reference values for nickel concentrations in human tissues and bile. Am. J. Ind. Med. 1987, 11, 419–426. [Google Scholar] [CrossRef] [PubMed]
- Duda-Chodak, A.D.A.; Błaszczyk, U. The impact of nickel on human health. J. Elementol. 2008, 13, 685–696. [Google Scholar]
- Morgan, L.G.; Usher, V. Health problems associated with nickel refining and use. Ann. Occup. Hyg. 1994, 38, 189–198. [Google Scholar]
- Berge, S.R.; Skyberg, K. Radiographic evidence of pulmonary fibrosis and possible etiologic factors at a nickel refinery in Norway. J. Environ. Monit. 2003, 5, 681–688. [Google Scholar] [CrossRef]
- Oller, A.R.; Costa, M.; Oberdorster, G. Carcinogenicity assessment of selected nickel compounds. Toxicol. Appl. Pharmacol. 1997, 143, 152–166. [Google Scholar] [CrossRef]
- Doll, R. Report of the International Committee on Nickel Carcinogenesis in Man; Final Report (No. PB-91-109801/XAB); Program Resources, Inc.: Research Triangle Park, NC, USA, 1990; p. 84. [Google Scholar]
- Huvinen, M.; Pukkala, E. Cancer incidence among Finnish ferrochromium and stainless steel production workers in 1967–2011: A cohort study. BMJ Open 2013, 3, e003819. [Google Scholar] [CrossRef] [PubMed]
- Grimsrud, T.K.; Andersen, A. Evidence of carcinogenicity in humans of water-soluble nickel salts. J. Occup. Med. Toxicol. 2010, 5, 7. [Google Scholar] [CrossRef] [PubMed]
- Andersen, A.; Berge, S.R.; Engeland, A.; Norseth, T. Exposure to nickel compounds and smoking in relation to incidence of lung and nasal cancer among nickel refinery workers. Occup. Environ. Med. 1996, 53, 708–713. [Google Scholar] [CrossRef] [PubMed]
- Seilkop, S.K.; Oller, A.R. Respiratory cancer risks associated with low-level nickel exposure: An integrated assessment based on animal, epidemiological, and mechanistic data. Regul. Toxicol. Pharmacol. 2003, 37, 173–190. [Google Scholar] [CrossRef]
- Moulin, J.J.; Clavel, T.; Roy, D.; Dananche, B.; Marquis, N.; Fevotte, J.; Fontana, J.M. Risk of lung cancer in workers producing stainless steel and metallic alloys. Int. Arch. Occup. Environ. Health 2000, 73, 171–180. [Google Scholar] [CrossRef]
- Anttila, A.; Pukkala, E.; Aitio, A.; Rantanen, T.; Karjalainen, S. Update of cancer incidence among workers at a copper/nickel smelter and nickel refinery. Int. Arch. Occup. Environ. Health 1998, 71, 245–250. [Google Scholar] [CrossRef]
- Carninci, P.; Kasukawa, T.; Katayama, S.; Gough, J.; Frith, M.C.; Maeda, N.; Oyama, R.; Ravasi, T.; Lenhard, B.; Wells, C.; et al. The transcriptional landscape of the mammalian genome. Science 2005, 309, 1559–1563. [Google Scholar]
- Zhao, Y.; Srivastava, D. A developmental view of microRNA function. Trends Biochem. Sci. 2007, 32, 189–197. [Google Scholar] [CrossRef]
- Di Leva, G.; Garofalo, M.; Croce, C.M. MicroRNAs in cancer. Annu. Rev. Pathol. 2014, 9, 287–314. [Google Scholar] [CrossRef]
- Uddin, A.; Chakraborty, S. Role of miRNAs in lung cancer. J. Cell. Physiol. 2018. [Google Scholar] [CrossRef]
- Ofir, M.; Hacohen, D.; Ginsberg, D. MiR-15 and miR-16 are direct transcriptional targets of E2F1 that limit E2F-induced proliferation by targeting cyclin E. Mol. Cancer Res. 2011, 9, 440–447. [Google Scholar] [CrossRef]
- Hatley, M.E.; Patrick, D.M.; Garcia, M.R.; Richardson, J.A.; Bassel-Duby, R.; van Rooij, E.; Olson, E.N. Modulation of K-Ras-dependent lung tumorigenesis by MicroRNA-21. Cancer Cell 2010, 18, 282–293. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Sempere, L.F.; Ouyang, H.; Memoli, V.A.; Andrew, A.S.; Luo, Y.; Demidenko, E.; Korc, M.; Shi, W.; Preis, M.; et al. MicroRNA-31 functions as an oncogenic microRNA in mouse and human lung cancer cells by repressing specific tumor suppressors. J. Clin. Investig. 2010, 120, 1298–1309. [Google Scholar] [CrossRef] [PubMed]
- Luo, L.; Zhang, T.; Liu, H.; Lv, T.; Yuan, D.; Yao, Y.; Lv, Y.; Song, Y. MiR-101 and Mcl-1 in non-small-cell lung cancer: Expression profile and clinical significance. Med. Oncol. 2012, 29, 1681–1686. [Google Scholar] [CrossRef]
- Wapinski, O.; Chang, H.Y. Long noncoding RNAs and human disease. Trends Cell Biol. 2011, 21, 354–361. [Google Scholar] [CrossRef] [PubMed]
- Poliseno, L.; Salmena, L.; Zhang, J.; Carver, B.; Haveman, W.J.; Pandolfi, P.P. A coding-independent function of gene and pseudogene mRNAs regulates tumour biology. Nature 2010, 465, 1033–1038. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ling, H.; Fabbri, M.; Calin, G.A. MicroRNAs and other non-coding RNAs as targets for anticancer drug development. Nat. Rev. Drug Discov. 2013, 12, 847–865. [Google Scholar] [CrossRef] [Green Version]
- Sullenger, B.A.; Nair, S. From the RNA world to the clinic. Science 2016, 352, 1417–1420. [Google Scholar] [CrossRef] [Green Version]
- Schmitt, A.M.; Chang, H.Y. Long Noncoding RNAs in Cancer Pathways. Cancer Cell 2016, 29, 452–463. [Google Scholar] [CrossRef] [Green Version]
- Schmitz, S.U.; Grote, P.; Herrmann, B.G. Mechanisms of long noncoding RNA function in development and disease. Cell. Mol. Life Sci. 2016, 73, 2491–2509. [Google Scholar] [CrossRef] [Green Version]
- Wei, M.M.; Zhou, G.B. Long Non-coding RNAs and Their Roles in Non-small-cell Lung Cancer. Genom. Proteom. Bioinform. 2016, 14, 280–288. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shen, L.; Chen, L.; Wang, Y.; Jiang, X.; Xia, H.; Zhuang, Z. Long noncoding RNA MALAT1 promotes brain metastasis by inducing epithelial-mesenchymal transition in lung cancer. J. Neurooncol. 2015, 121, 101–108. [Google Scholar] [CrossRef] [PubMed]
- Loewen, G.; Jayawickramarajah, J.; Zhuo, Y.; Shan, B. Functions of lncRNA HOTAIR in lung cancer. J. Hematol. Oncol. 2014, 7, 90. [Google Scholar] [CrossRef] [PubMed]
- Zhou, C.; Huang, C.; Wang, J.; Huang, H.; Li, J.; Xie, Q.; Liu, Y.; Zhu, J.; Li, Y.; Zhang, D.; et al. LncRNA MEG3 downregulation mediated by DNMT3b contributes to nickel malignant transformation of human bronchial epithelial cells via modulating PHLPP1 transcription and HIF-1α translation. Oncogene 2017, 36, 3878–3889. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Zhou, Y.; Wu, Y.; Ma, L.; Fan, Y.; Kang, X.; Shi, H.; Zhang, J. Isolation and characterization of a novel noncoding RNA from nickel-induced lung cancer. Biol. Trace. Elem. Res. 2012, 150, 258–263. [Google Scholar] [CrossRef] [PubMed]
- Cameron, K.S.; Buchner, V.; Tchounwou, P.B. Exploring the molecular mechanisms of nickel-induced genotoxicity and carcinogenicity: A literature review. Rev. Environ. Health 2011, 26, 81–92. [Google Scholar] [CrossRef] [PubMed]
- Sakai, T.; Toguchida, J.; Ohtani, N.; Yandell, D.W.; Rapaport, J.M.; Dryja, T.P. Allele-specific hypermethylation of the retinoblastoma tumor-suppressor gene. Am. J. Hum. Genet. 1991, 48, 880–888. [Google Scholar] [PubMed]
- Lee, Y.W.; Klein, C.B.; Kargacin, B.; Salnikow, K.; Kitahara, J.; Dowjat, K.; Zhitkovich, A.; Christie, N.T.; Costa, M. Carcinogenic nickel silences gene expression by chromatin condensation and DNA methylation: A new model for epigenetic carcinogens. Mol. Cell. Biol. 1995, 15, 2547–2557. [Google Scholar] [CrossRef] [PubMed]
- Ohtani-Fujita, N.; Fujita, T.; Aoike, A.; Osifchin, N.E.; Robbins, P.D.; Sakai, T. CpG methylation inactivates the promoter activity of the human retinoblastoma tumor-suppressor gene. Oncogene 1993, 8, 1063–1067. [Google Scholar] [PubMed]
- Sutcliffe, J.S.; Nelson, D.L.; Zhang, F.; Pieretti, M.; Caskey, C.T.; Saxe, D.; Warren, S.T. DNA methylation represses FMR-1 transcription in fragile X syndrome. Hum. Mol. Genet. 1992, 1, 397–400. [Google Scholar] [CrossRef] [PubMed]
- Greger, V.; Debus, N.; Lohmann, D.; Hopping, W.; Passarge, E.; Horsthemke, B. Frequency and parental origin of hypermethylated RB1 alleles in retinoblastoma. Hum. Genet. 1994, 94, 491–496. [Google Scholar] [CrossRef] [PubMed]
- Hansen, R.S.; Gartler, S.M.; Scott, C.R.; Chen, S.H.; Laird, C.D. Methylation analysis of CGG sites in the CpG island of the human FMR1 gene. Hum. Mol. Genet. 1992, 1, 571–578. [Google Scholar] [CrossRef] [PubMed]
- Yasaei, H.; Gilham, E.; Pickles, J.C.; Roberts, T.P.; O’Donovan, M.; Newbold, R.F. Carcinogen-specific mutational and epigenetic alterations in INK4A, INK4B and p53 tumour-suppressor genes drive induced senescence bypass in normal diploid mammalian cells. Oncogene 2013, 32, 171–179. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.W.; Broday, L.; Costa, M. Effects of nickel on DNA methyltransferase activity and genomic DNA methylation levels. Mutagen. Res. 1998, 415, 213–218. [Google Scholar] [CrossRef]
- Zingg, J.M.; Jones, P.A. Genetic and epigenetic aspects of DNA methylation on genome expression, evolution, mutation and carcinogenesis. Carcinogenesis 1997, 18, 869–882. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.H.; Tang, S.C.; Wang, P.H.; Lee, H.; Ko, J.L. Nickel-induced epithelial-mesenchymal transition by reactive oxygen species generation and E-cadherin promoter hypermethylation. J. Biol. Chem. 2012, 287, 25292–25302. [Google Scholar] [CrossRef] [PubMed]
- Pecina-Slaus, N. Tumor suppressor gene E-cadherin and its role in normal and malignant cells. Cancer Cell Int. 2003, 3, 17. [Google Scholar] [CrossRef] [PubMed]
- Sato, F.; Ono, T.; Kawahara, A.; Kawaguchi, T.; Tanaka, H.; Shimamatsu, K.; Kakuma, T.; Akiba, J.; Umeno, H.; Yano, H. Prognostic impact of p16 and PD-L1 expression in patients with oropharyngeal squamous cell carcinoma receiving a definitive treatment. J. Clin. Pathol. 2019. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.Y.; DesMarais, T.; Costa, M. Metals and Mechanisms of Carcinogenesis. Annu. Rev. Pharmacol. Toxicol. 2019, 59, 537–554. [Google Scholar] [CrossRef]
- Lee, Y.W.; Pons, C.; Tummolo, D.M.; Klein, C.B.; Rossman, T.G.; Christie, N.T. Mutagenicity of soluble and insoluble nickel compounds at the gpt locus in G12 Chinese hamster cells. Environ. Mol. Mutagen. 1993, 21, 365–371. [Google Scholar] [CrossRef] [PubMed]
- Ellen, T.P.; Kluz, T.; Harder, M.E.; Xiong, J.; Costa, M. Heterochromatinization as a potential mechanism of nickel-induced carcinogenesis. Biochemistry 2009, 48, 4626–4632. [Google Scholar] [CrossRef] [PubMed]
- Carone, D.M.; Lawrence, J.B. Heterochromatin instability in cancer: From the Barr body to satellites and the nuclear periphery. Semin. Cancer Biol. 2013, 23, 99–108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morgan, M.A.; Shilatifard, A. Chromatin signatures of cancer. Genes Dev. 2015, 29, 238–249. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miyoshi, N.; Wagatsuma, H.; Wakana, S.; Shiroishi, T.; Nomura, M.; Aisaka, K.; Kohda, T.; Surani, M.A.; Kaneko-Ishino, T.; Ishino, F. Identification of an imprinted gene, Meg3/Gtl2 and its human homologue MEG3, first mapped on mouse distal chromosome 12 and human chromosome 14q. Genes Cells 2000, 5, 211–220. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Zhang, X.; Klibanski, A. MEG3 noncoding RNA: A tumor suppressor. J. Mol. Endocrinol. 2012, 48, R45–R53. [Google Scholar] [CrossRef] [PubMed]
- Benetatos, L.; Vartholomatos, G.; Hatzimichael, E. MEG3 imprinted gene contribution in tumorigenesis. Int. J. Cancer 2011, 129, 773–779. [Google Scholar] [CrossRef] [PubMed]
- Ji, W.; Yang, L.; Yu, L.; Yuan, J.; Hu, D.; Zhang, W.; Yang, J.; Pang, Y.; Li, W.; Lu, J.; et al. Epigenetic silencing of O6-methylguanine DNA methyltransferase gene in NiS-transformed cells. Carcinogenesis 2008, 29, 1267–1275. [Google Scholar] [CrossRef] [PubMed]
- Ji, W.; Yang, L.; Yuan, J.; Yang, L.; Zhang, M.; Qi, D.; Duan, X.; Xuan, A.; Zhang, W.; Lu, J.; et al. MicroRNA-152 targets DNA methyltransferase 1 in NiS-transformed cells via a feedback mechanism. Carcinogenesis 2013, 34, 446–453. [Google Scholar] [CrossRef]
- Zhang, J.; Zhou, Y.; Wu, Y.J.; Li, M.J.; Wang, R.J.; Huang, S.Q.; Gao, R.R.; Ma, L.; Shi, H.J.; Zhang, J. Hyper-methylated miR-203 dysregulates ABL1 and contributes to the nickel-induced tumorigenesis. Toxicol. Lett. 2013, 223, 42–51. [Google Scholar] [CrossRef]
- Salnikow, K.; Davidson, T.; Zhang, Q.; Chen, L.C.; Su, W.; Costa, M. The involvement of hypoxia-inducible transcription factor-1-dependent pathway in nickel carcinogenesis. Cancer Res 2003, 63, 3524–3530. [Google Scholar]
- Wang, G.L.; Jiang, B.H.; Rue, E.A.; Semenza, G.L. Hypoxia-inducible factor 1 is a basic-helix-loop-helix-PAS heterodimer regulated by cellular O2 tension. Proc. Natl. Acad. Sci. USA 1995, 92, 5510–5514. [Google Scholar] [CrossRef] [PubMed]
- Semenza, G.L. Hypoxia-inducible factor 1: Master regulator of O2 homeostasis. Curr. Opin. Genet. Dev. 1998, 8, 588–594. [Google Scholar] [CrossRef]
- Costa, M.; Davidson, T.L.; Chen, H.; Ke, Q.; Zhang, P.; Yan, Y.; Huang, C.; Kluz, T. Nickel carcinogenesis: Epigenetics and hypoxia signaling. Mutagen. Res. 2005, 592, 79–88. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.W.; Bae, S.H.; Jeong, J.W.; Kim, S.H.; Kim, K.W. Hypoxia-inducible factor (HIF-1)α: Its protein stability and biological functions. Exp. Mol. Med. 2004, 36, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Ke, Q.; Costa, M. Hypoxia-inducible factor-1 (HIF-1). Mol. Pharmacol. 2006, 70, 1469–1480. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Chen, H.; Huang, X.; Costa, M. Effects of 12 metal ions on iron regulatory protein 1 (IRP-1) and hypoxia-inducible factor-1 alpha (HIF-1α) and HIF-regulated genes. Toxicol. Appl. Pharmacol. 2006, 213, 245–255. [Google Scholar] [CrossRef] [PubMed]
- Davidson, T.L.; Chen, H.; Di Toro, D.M.; D’Angelo, G.; Costa, M. Soluble nickel inhibits HIF-prolyl-hydroxylases creating persistent hypoxic signaling in A549 cells. Mol. Carcinog. 2006, 45, 479–489. [Google Scholar] [CrossRef]
- Davidson, T.; Chen, H.; Garrick, M.D.; D’Angelo, G.; Costa, M. Soluble nickel interferes with cellular iron homeostasis. Mol. Cell. Biochem. 2005, 279, 157–162. [Google Scholar] [CrossRef]
- Salnikow, K.; Blagosklonny, M.V.; Ryan, H.; Johnson, R.; Costa, M. Carcinogenic Nickel Induces Genes Involved with Hypoxic Stress. Cancer Res. 2000, 60, 38–41. [Google Scholar]
- Rezvani, H.R.; Mahfouf, W.; Ali, N.; Chemin, C.; Ged, C.; Kim, A.L.; de Verneuil, H.; Taieb, A.; Bickers, D.R.; Mazurier, F. Hypoxia-inducible factor-1alpha regulates the expression of nucleotide excision repair proteins in keratinocytes. Nucleic Acids Res. 2010, 38, 797–809. [Google Scholar] [CrossRef]
- Chan, N.; Ali, M.; McCallum, G.P.; Kumareswaran, R.; Koritzinsky, M.; Wouters, B.G.; Wells, P.G.; Gallinger, S.; Bristow, R.G. Hypoxia provokes base excision repair changes and a repair-deficient, mutator phenotype in colorectal cancer cells. Mol. Cancer Res. 2014, 12, 1407–1415. [Google Scholar] [CrossRef] [PubMed]
- Kulshreshtha, R.; Ferracin, M.; Negrini, M.; Calin, G.A.; Davuluri, R.V.; Ivan, M. Regulation of microRNA expression: The hypoxic component. Cell Cycle 2007, 6, 1426–1431. [Google Scholar] [CrossRef] [PubMed]
- He, M.; Lu, Y.; Xu, S.; Mao, L.; Zhang, L.; Duan, W.; Liu, C.; Pi, H.; Zhang, Y.; Zhong, M.; et al. MiRNA-210 modulates a nickel-induced cellular energy metabolism shift by repressing the iron-sulfur cluster assembly proteins ISCU1/2 in Neuro-2a cells. Cell Death Dis. 2014, 5, e1090. [Google Scholar] [CrossRef] [PubMed]
- Kulshreshtha, R.; Ferracin, M.; Wojcik, S.E.; Garzon, R.; Alder, H.; Agosto-Perez, F.J.; Davuluri, R.; Liu, C.G.; Croce, C.M.; Negrini, M.; et al. A microRNA signature of hypoxia. Mol. Cell. Biol. 2007, 27, 1859–1867. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Flach, H.; Onizawa, M.; Wei, L.; McManus, M.T.; Weiss, A. Negative regulation of Hif1a expression and TH17 differentiation by the hypoxia-regulated microRNA miR-210. Nat. Immunol. 2014, 15, 393–401. [Google Scholar] [CrossRef] [PubMed]
- Valko, M.; Rhodes, C.J.; Moncol, J.; Izakovic, M.; Mazur, M. Free radicals, metals and antioxidants in oxidative stress-induced cancer. Chem. Biol. Interact. 2006, 160, 1–40. [Google Scholar] [CrossRef] [PubMed]
- Pero, R.W.; Roush, G.C.; Markowitz, M.M.; Miller, D.G. Oxidative stress, DNA repair, and cancer susceptibility. Cancer Detect. Prev. 1990, 14, 555–561. [Google Scholar]
- Sies, H. Oxidative stress: Oxidants and antioxidants. Exp. Physiol. 1997, 82, 291–295. [Google Scholar] [CrossRef]
- Halliwell, B. Reactive oxygen species in living systems: Source, biochemistry, and role in human disease. Am. J. Med. 1991, 91, 14s–22s. [Google Scholar] [CrossRef]
- Lee, J.D.; Cai, Q.; Shu, X.O.; Nechuta, S.J. The Role of Biomarkers of Oxidative Stress in Breast Cancer Risk and Prognosis: A Systematic Review of the Epidemiologic Literature. J. Womens Health 2017, 26, 467–482. [Google Scholar] [CrossRef] [Green Version]
- Shi, X.; Dalal, N.S.; Kasprzak, K.S. Generation of free radicals from lipid hydroperoxides by Ni2+ in the presence of oligopeptides. Arch. Biochem. Biophys. 1992, 299, 154–162. [Google Scholar] [CrossRef]
- Tsao, Y.C.; Gu, P.W.; Liu, S.H.; Tzeng, I.S.; Chen, J.Y.; Luo, J.J. Nickel exposure and plasma levels of biomarkers for assessing oxidative stress in nickel electroplating workers. Biomarkers 2017, 22, 455–460. [Google Scholar] [CrossRef] [PubMed]
- Di Bucchianico, S.; Gliga, A.R.; Åkerlund, E.; Skoglund, S.; Wallinder, I.O.; Fadeel, B.; Karlsson, H.L. Calcium-dependent cyto- and genotoxicity of nickel metal and nickel oxide nanoparticles in human lung cells. Part. Fibre Toxicol. 2018, 15, 32. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Klein, C.B.; Costa, M. Crystalline Ni3S2 specifically enhances the formation of oxidants in the nuclei of CHO cells as detected by dichlorofluorescein. Carcinogenesis 1994, 15, 545–548. [Google Scholar] [CrossRef] [PubMed]
- Lynn, S.; Yew, F.H.; Chen, K.S.; Jan, K.Y. Reactive oxygen species are involved in nickel inhibition of DNA repair. Environ. Mol. Mutagen. 1997, 29, 208–216. [Google Scholar] [CrossRef]
- Scanlon, S.E.; Scanlon, C.D.; Hegan, D.C.; Sulkowski, P.L.; Glazer, P.M. Nickel induces transcriptional down-regulation of DNA repair pathways in tumorigenic and non-tumorigenic lung cells. Carcinogenesis 2017, 38, 627–637. [Google Scholar] [CrossRef] [Green Version]
- Wan, Y.; Cui, R.; Gu, J.; Zhang, X.; Xiang, X.; Liu, C.; Qu, K.; Lin, T. Identification of Four Oxidative Stress-Responsive MicroRNAs, miR-34a-5p, miR-1915-3p, miR-638, and miR-150-3p, in Hepatocellular Carcinoma. Oxid. Med. Cell. Longev. 2017, 2017, 5189138. [Google Scholar] [CrossRef]
- Sun, W.; Zhao, L.; Song, X.; Zhang, J.; Xing, Y.; Liu, N.; Yan, Y.; Li, Z.; Lu, Y.; Wu, J.; et al. MicroRNA-210 Modulates the Cellular Energy Metabolism Shift During H2O2-Induced Oxidative Stress by Repressing ISCU in H9c2 Cardiomyocytes. Cell. Physiol. Biochem. 2017, 43, 383–394. [Google Scholar] [CrossRef]
- Ali, T.; Mushtaq, I.; Maryam, S.; Farhan, A.; Saba, K.; Jan, M.I.; Sultan, A.; Anees, M.; Duygu, B.; Hamera, S.; et al. Interplay of N acetyl cysteine and melatonin in regulating oxidative stress-induced cardiac hypertrophic factors and microRNAs. Arch. Biochem. Biophys. 2019, 661, 56–65. [Google Scholar] [CrossRef]
- Clemens, F.; Verma, R.; Ramnath, J.; Landolph, J.R. Amplification of the Ect2 proto-oncogene and over-expression of Ect2 mRNA and protein in nickel compound and methylcholanthrene-transformed 10T1/2 mouse fibroblast cell lines. Toxicol. Appl. Pharmacol. 2005, 206, 138–149. [Google Scholar] [CrossRef]
- Chiocca, S.M.; Sterner, D.A.; Biggart, N.W.; Murphy, E.C., Jr. Nickel mutagenesis: Alteration of the MuSVts110 thermosensitive splicing phenotype by a nickel-induced duplication of the 3’ splice site. Mol. Carcinog. 1991, 4, 61–71. [Google Scholar] [CrossRef] [PubMed]
- Maehle, L.; Metcalf, R.A.; Ryberg, D.; Bennett, W.P.; Harris, C.C.; Haugen, A. Altered p53 gene structure and expression in human epithelial cells after exposure to nickel. Cancer Res. 1992, 52, 218–221. [Google Scholar] [PubMed]
- Shi, Y.; Lv, C.; Shi, L.; Tu, G. MEG3 inhibits proliferation and invasion and promotes apoptosis of human osteosarcoma cells. Oncol. Lett. 2018, 15, 1917–1923. [Google Scholar] [CrossRef] [PubMed]
- Lu, K.H.; Li, W.; Liu, X.H.; Sun, M.; Zhang, M.L.; Wu, W.Q.; Xie, W.P.; Hou, Y.Y. Long non-coding RNA MEG3 inhibits NSCLC cells proliferation and induces apoptosis by affecting p53 expression. BMC Cancer 2013, 13, 461. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Su, C.; Zhao, F.; Tao, J.; Hu, D.; Shi, A.; Pan, J.; Zhang, Y. Paclitaxel promotes lung cancer cell apoptosis via MEG3-P53 pathway activation. Biochem. Biophys. Res. Commun. 2018, 504, 123–128. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Zhou, Y.; Mehta, K.R.; Danila, D.C.; Scolavino, S.; Johnson, S.R.; Klibanski, A. A pituitary-derived MEG3 isoform functions as a growth suppressor in tumor cells. J. Clin. Endocrinol. Metab. 2003, 88, 5119–5126. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Zhong, Y.; Wang, Y.; Zhang, X.; Batista, D.L.; Gejman, R.; Ansell, P.J.; Zhao, J.; Weng, C.; Klibanski, A. Activation of p53 by MEG3 non-coding RNA. J. Biol. Chem. 2007, 282, 24731–24742. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.X.; Deng, W.; Zhou, X.T.; Chen, R.P.; Xiang, M.Q.; Guo, Y.T.; Pu, Z.J.; Li, R.; Wang, G.F.; Wu, L.F. The mechanism of adenosine-mediated activation of lncRNA MEG3 and its antitumor effects in human hepatoma cells. Int. J. Oncol. 2016, 48, 421–429. [Google Scholar] [CrossRef]
- Lv, D.; Sun, R.; Yu, Q.; Zhang, X. The long non-coding RNA maternally expressed gene 3 activates p53 and is downregulated in esophageal squamous cell cancer. Tumour. Biol. 2016, 37, 16259–16267. [Google Scholar] [CrossRef]
- Liu, J.; Wan, L.; Lu, K.; Sun, M.; Pan, X.; Zhang, P.; Lu, B.; Liu, G.; Wang, Z. The Long Noncoding RNA MEG3 Contributes to Cisplatin Resistance of Human Lung Adenocarcinoma. PLoS ONE 2015, 10, e0114586. [Google Scholar] [CrossRef]
- Tay, Y.; Rinn, J.; Pandolfi, P.P. The multilayered complexity of ceRNA crosstalk and competition. Nature 2014, 505, 344–352. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, C.; Liao, X.; Jin, H.; Xie, F.; Zheng, F.; Li, J.; Zhou, C.; Jiang, G.; Wu, X.R.; Huang, C. MEG3, as a Competing Endogenous RNA, Binds with miR-27a to Promote PHLPP2 Protein Translation and Impairs Bladder Cancer Invasion. Mol. Ther. Nucleic Acids 2019, 16, 51–62. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ding, L.; Zhang, S.; Xu, M.; Zhang, R.; Sui, P.; Yang, Q. MicroRNA-27a contributes to the malignant behavior of gastric cancer cells by directly targeting PH domain and leucine-rich repeat protein phosphatase 2. J. Exp. Clin. Cancer Res. 2017, 36, 45. [Google Scholar] [CrossRef] [PubMed]
- Chae, D.K.; Ban, E.; Yoo, Y.S.; Kim, E.E.; Baik, J.H.; Song, E.J. MIR-27a regulates the TGF-β signaling pathway by targeting SMAD2 and SMAD4 in lung cancer. Mol. Carcinog. 2017, 56, 1992–1998. [Google Scholar] [CrossRef]
- Khandelwal, A.; Bacolla, A.; Vasquez, K.M.; Jain, A. Long non-coding RNA: A new paradigm for lung cancer. Mol. Carcinog. 2015, 54, 1235–1251. [Google Scholar] [CrossRef]
- Gao, C.; He, Z.; Li, J.; Li, X.; Bai, Q.; Zhang, Z.; Zhang, X.; Wang, S.; Xiao, X.; Wang, F.; et al. Specific long non-coding RNAs response to occupational PAHs exposure in coke oven workers. Toxicol. Rep. 2016, 3, 160–166. [Google Scholar] [CrossRef] [Green Version]
- Zhang, E.B.; Yin, D.D.; Sun, M.; Kong, R.; Liu, X.H.; You, L.H.; Han, L.; Xia, R.; Wang, K.M.; Yang, J.S.; et al. P53-regulated long non-coding RNA TUG1 affects cell proliferation in human non-small cell lung cancer, partly through epigenetically regulating HOXB7 expression. Cell Death Dis. 2014, 5, e1243. [Google Scholar] [CrossRef]
- Zhang, Z.; Sun, H.; Dai, H.; Walsh, R.M.; Imakura, M.; Schelter, J.; Burchard, J.; Dai, X.; Chang, A.N.; Diaz, R.L.; et al. MicroRNA miR-210 modulates cellular response to hypoxia through the MYC antagonist MNT. Cell Cycle 2009, 8, 2756–2768. [Google Scholar] [CrossRef] [Green Version]
- Giannakakis, A.; Sandaltzopoulos, R.; Greshock, J.; Liang, S.; Huang, J.; Hasegawa, K.; Li, C.; O’Brien-Jenkins, A.; Katsaros, D.; Weber, B.L.; et al. miR-210 links hypoxia with cell cycle regulation and is deleted in human epithelial ovarian cancer. Cancer Biol. Ther. 2008, 7, 255–264. [Google Scholar] [CrossRef] [Green Version]
- He, R.Q.; Cen, W.L.; Cen, J.M.; Cen, W.N.; Li, J.Y.; Li, M.W.; Gan, T.Q.; Hu, X.H.; Chen, G. Clinical Significance of miR-210 and its Prospective Signaling Pathways in Non-Small Cell Lung Cancer: Evidence from Gene Expression Omnibus and the Cancer Genome Atlas Data Mining with 2763 Samples and Validation via Real-Time Quantitative PCR. Cell. Physiol. Biochem. 2018, 46, 925–952. [Google Scholar] [CrossRef]
- Yang, J.S.; Li, B.J.; Lu, H.W.; Chen, Y.; Lu, C.; Zhu, R.X.; Liu, S.H.; Yi, Q.T.; Li, J.; Song, C.H. Serum miR-152, miR-148a, miR-148b, and miR-21 as novel biomarkers in non-small cell lung cancer screening. Tumour. Biol. 2015, 36, 3035–3042. [Google Scholar] [CrossRef] [PubMed]
- Bueno, M.J.; Perez de Castro, I.; Gomez de Cedron, M.; Santos, J.; Calin, G.A.; Cigudosa, J.C.; Croce, C.M.; Fernandez-Piqueras, J.; Malumbres, M. Genetic and epigenetic silencing of microRNA-203 enhances ABL1 and BCR-ABL1 oncogene expression. Cancer Cell 2008, 13, 496–506. [Google Scholar] [CrossRef] [PubMed]
- Craig, V.J.; Cogliatti, S.B.; Rehrauer, H.; Wundisch, T.; Muller, A. Epigenetic silencing of microRNA-203 dysregulates ABL1 expression and drives Helicobacter-associated gastric lymphomagenesis. Cancer Res. 2011, 71, 3616–3624. [Google Scholar] [CrossRef] [PubMed]
- Furuta, M.; Kozaki, K.I.; Tanaka, S.; Arii, S.; Imoto, I.; Inazawa, J. miR-124 and miR-203 are epigenetically silenced tumor-suppressive microRNAs in hepatocellular carcinoma. Carcinogenesis 2010, 31, 766–776. [Google Scholar] [CrossRef] [PubMed]
- Noguchi, S.; Mori, T.; Otsuka, Y.; Yamada, N.; Yasui, Y.; Iwasaki, J.; Kumazaki, M.; Maruo, K.; Akao, Y. Anti-oncogenic microRNA-203 induces senescence by targeting E2F3 protein in human melanoma cells. J. Biol. Chem. 2012, 287, 11769–11777. [Google Scholar] [CrossRef]
- Yi, R.; Poy, M.N.; Stoffel, M.; Fuchs, E. A skin microRNA promotes differentiation by repressing ‘stemness’. Nature 2008, 452, 225–229. [Google Scholar] [CrossRef]
- Zhang, J.; Zhou, Y.; Ma, L.; Huang, S.; Wang, R.; Gao, R.; Wu, Y.; Shi, H.; Zhang, J. The alteration of miR-222 and its target genes in nickel-induced tumor. Biol. Trace Elem. Res. 2013, 152, 267–274. [Google Scholar] [CrossRef]
- Le Sage, C.; Nagel, R.; Egan, D.A.; Schrier, M.; Mesman, E.; Mangiola, A.; Anile, C.; Maira, G.; Mercatelli, N.; Ciafre, S.A.; et al. Regulation of the p27(Kip1) tumor suppressor by miR-221 and miR-222 promotes cancer cell proliferation. EMBO J. 2007, 26, 3699–3708. [Google Scholar] [CrossRef]
- Fornari, F.; Gramantieri, L.; Ferracin, M.; Veronese, A.; Sabbioni, S.; Calin, G.A.; Grazi, G.L.; Giovannini, C.; Croce, C.M.; Bolondi, L.; et al. MiR-221 controls CDKN1C/p57 and CDKN1B/p27 expression in human hepatocellular carcinoma. Oncogene 2008, 27, 5651–5661. [Google Scholar] [CrossRef] [Green Version]
- Wu, C.H.; Hsiao, Y.M.; Yeh, K.T.; Tsou, T.C.; Chen, C.Y.; Wu, M.F.; Ko, J.L. Upregulation of microRNA-4417 and Its Target Genes Contribute to Nickel Chloride-promoted Lung Epithelial Cell Fibrogenesis and Tumorigenesis. Sci. Rep. 2017, 7, 15320. [Google Scholar] [CrossRef]
Figure 1.
Regulatory networks of non-coding RNAs (ncRNAs) involved in nickel-induced carcinogenesis.
Figure 1.
Regulatory networks of non-coding RNAs (ncRNAs) involved in nickel-induced carcinogenesis.

© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Zhu, Y.; Chen, Q.Y.; Li, A.H.; Costa, M. The Role of Non-Coding RNAs Involved in Nickel-Induced Lung Carcinogenic Mechanisms. Inorganics 2019, 7, 81. https://0-doi-org.brum.beds.ac.uk/10.3390/inorganics7070081
AMA Style
Zhu Y, Chen QY, Li AH, Costa M. The Role of Non-Coding RNAs Involved in Nickel-Induced Lung Carcinogenic Mechanisms. Inorganics. 2019; 7(7):81. https://0-doi-org.brum.beds.ac.uk/10.3390/inorganics7070081
Chicago/Turabian StyleZhu, Yusha, Qiao Yi Chen, Alex Heng Li, and Max Costa. 2019. "The Role of Non-Coding RNAs Involved in Nickel-Induced Lung Carcinogenic Mechanisms" Inorganics 7, no. 7: 81. https://0-doi-org.brum.beds.ac.uk/10.3390/inorganics7070081
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.