Theoretical Insights into the Aerobic Hydrogenase Activity of Molybdenum–Copper CO Dehydrogenase

 , , and
, , and
Abstract
:1. Introduction
2. Results and Discussion
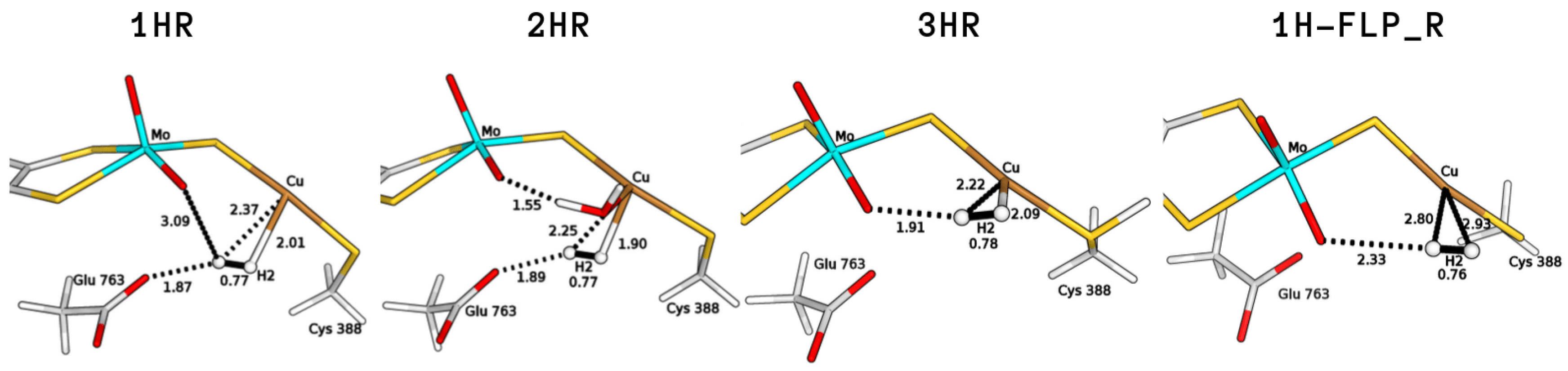
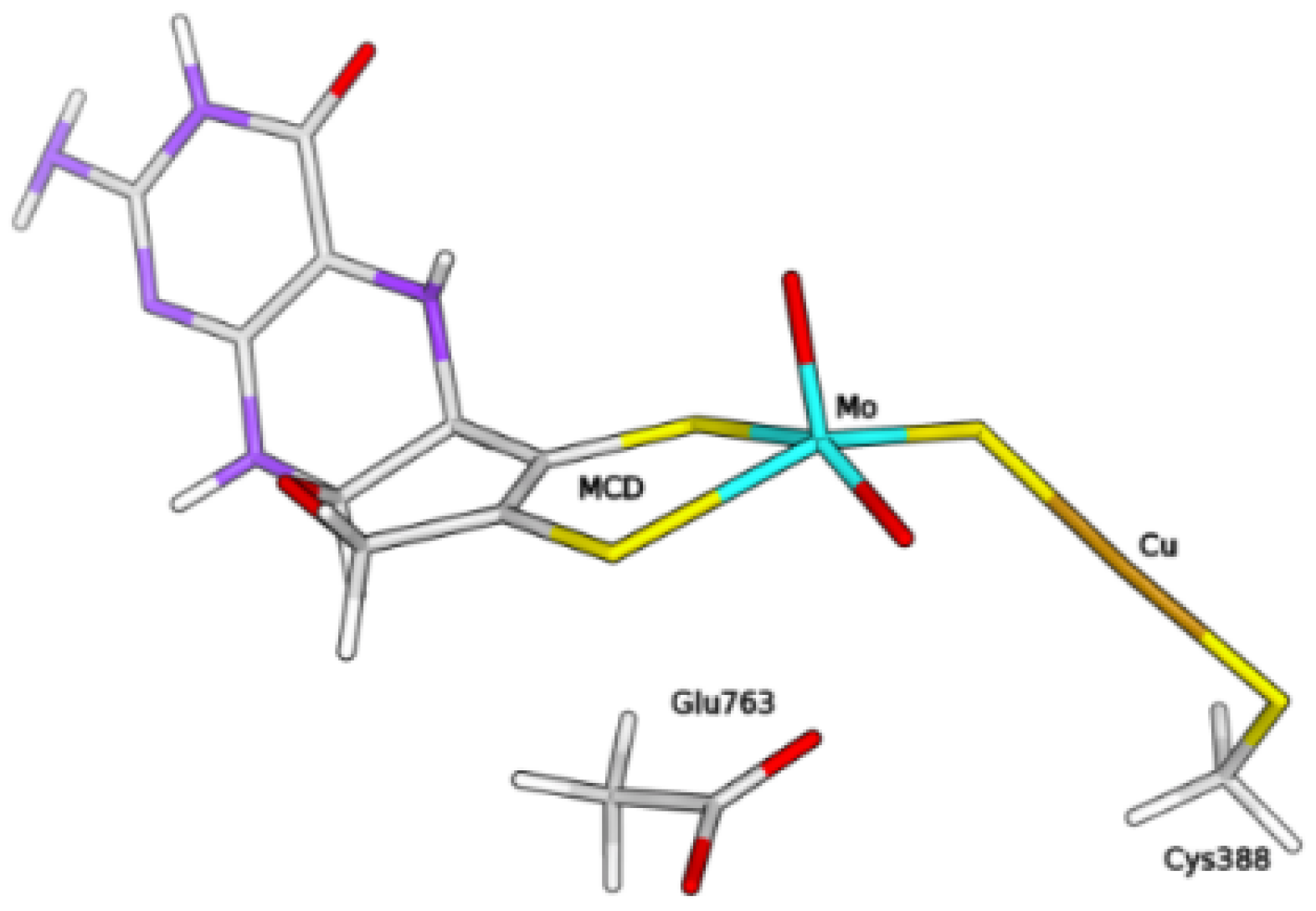
2.1. Study of H Binding Modes to the Copper Centre
2.2. Exploring Basic Residues in the Active Site
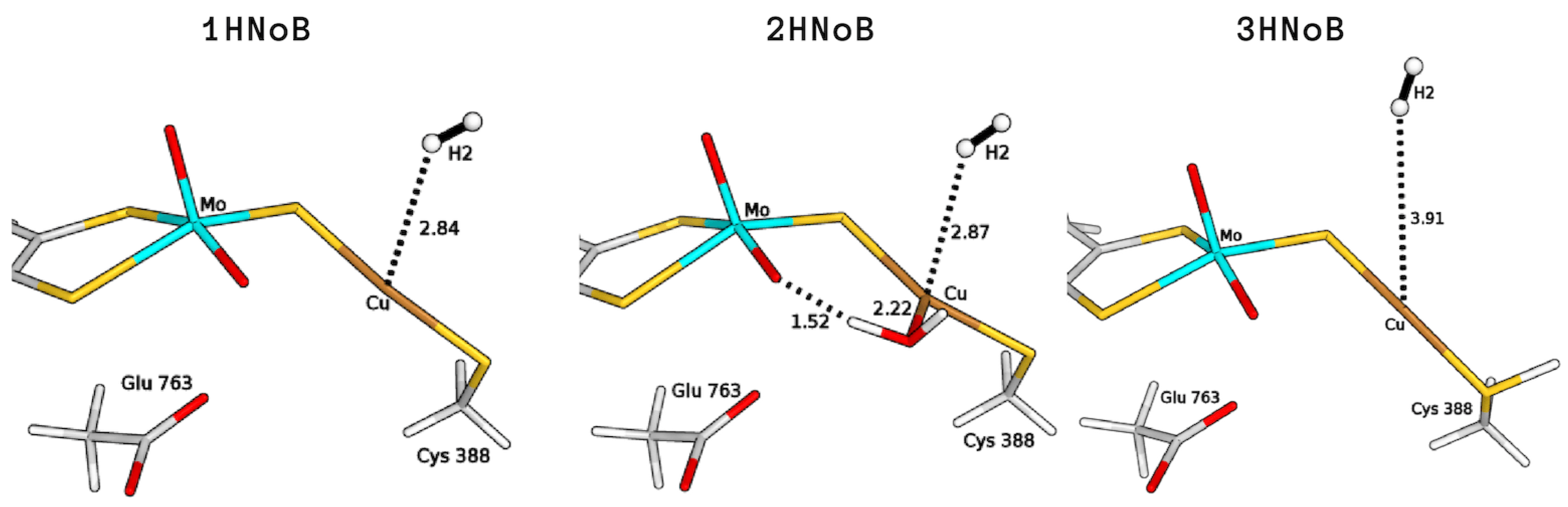
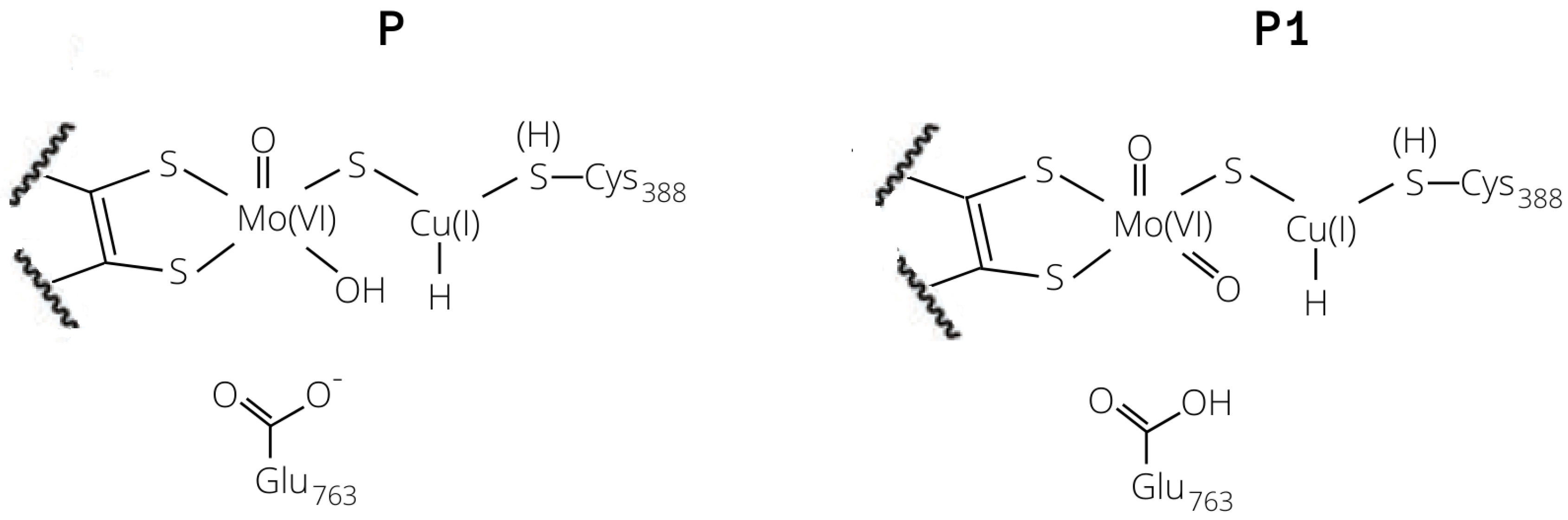
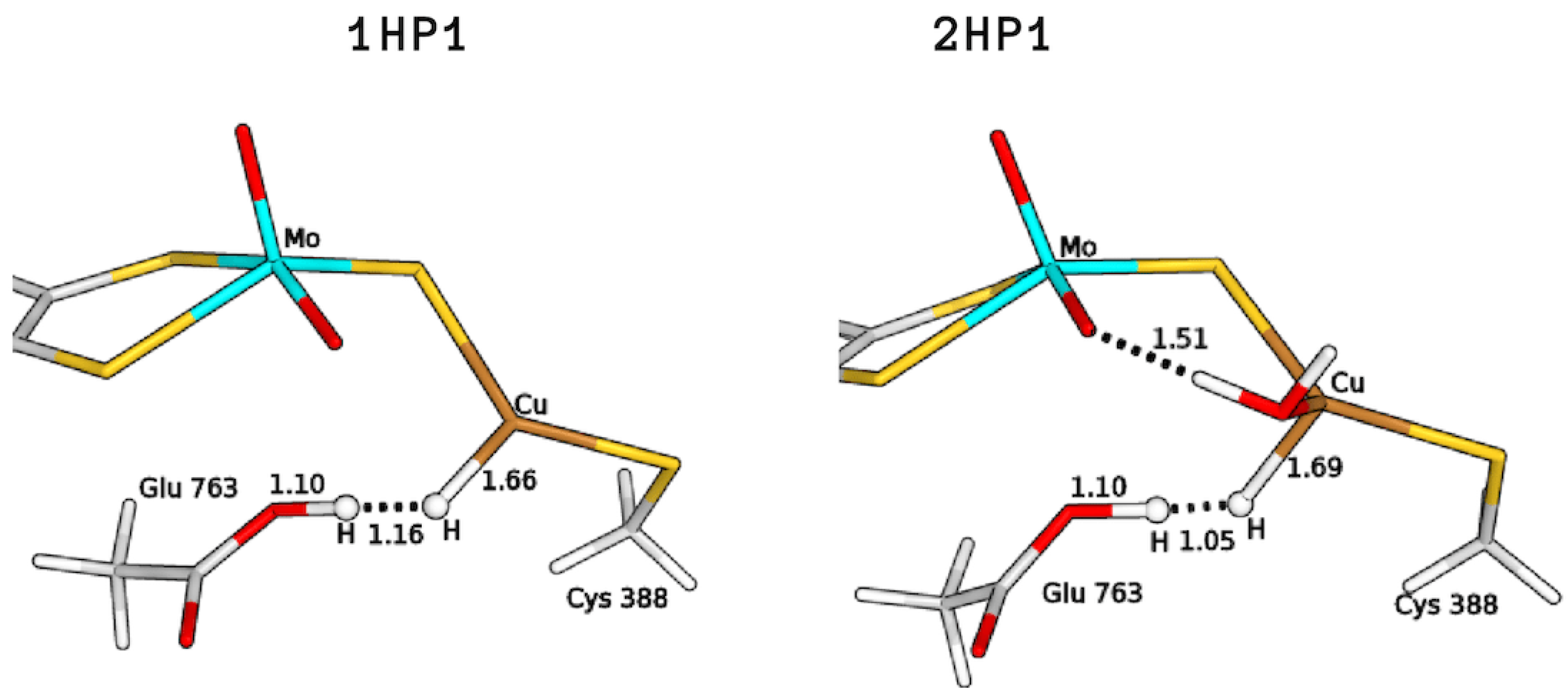
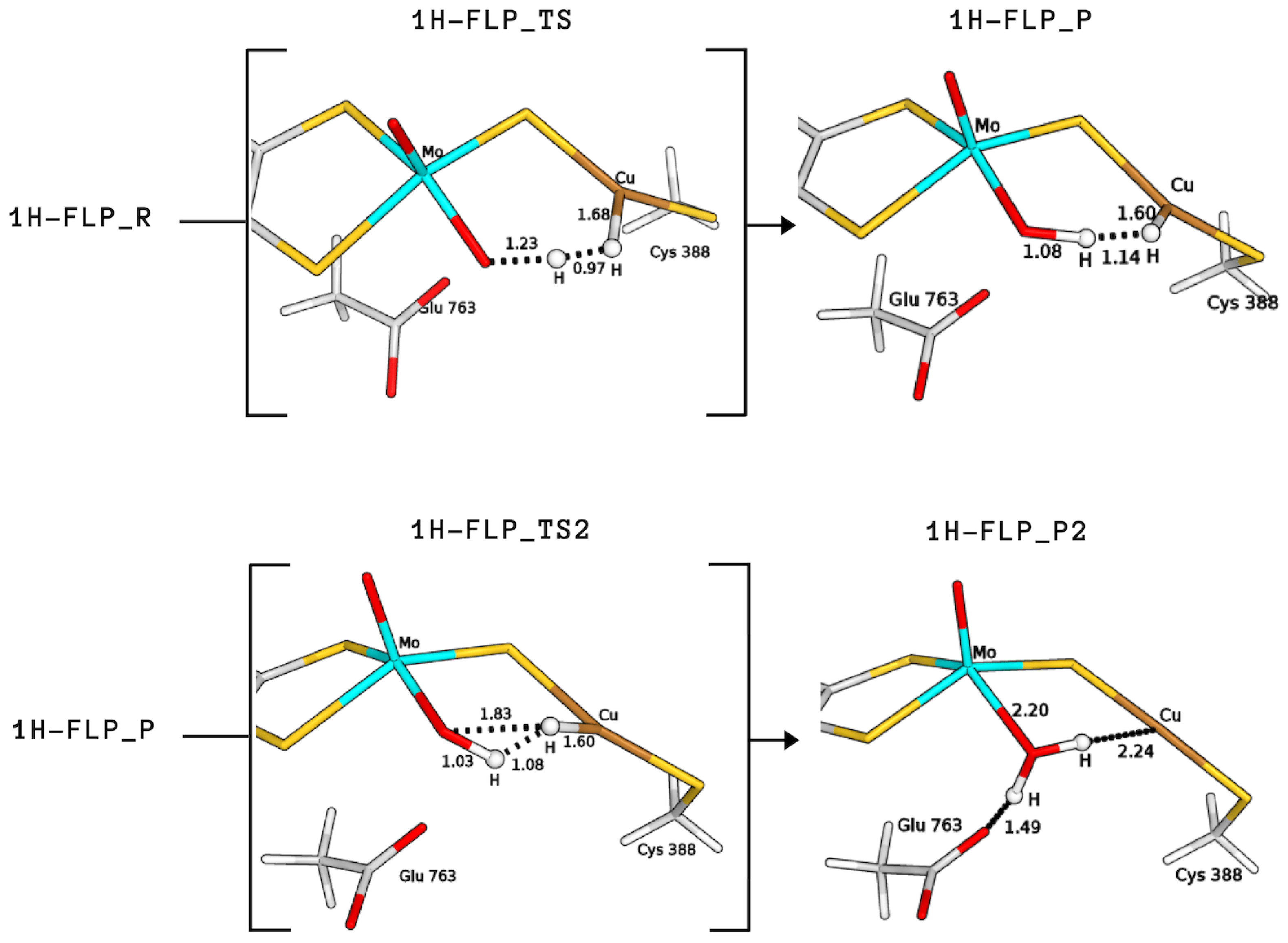
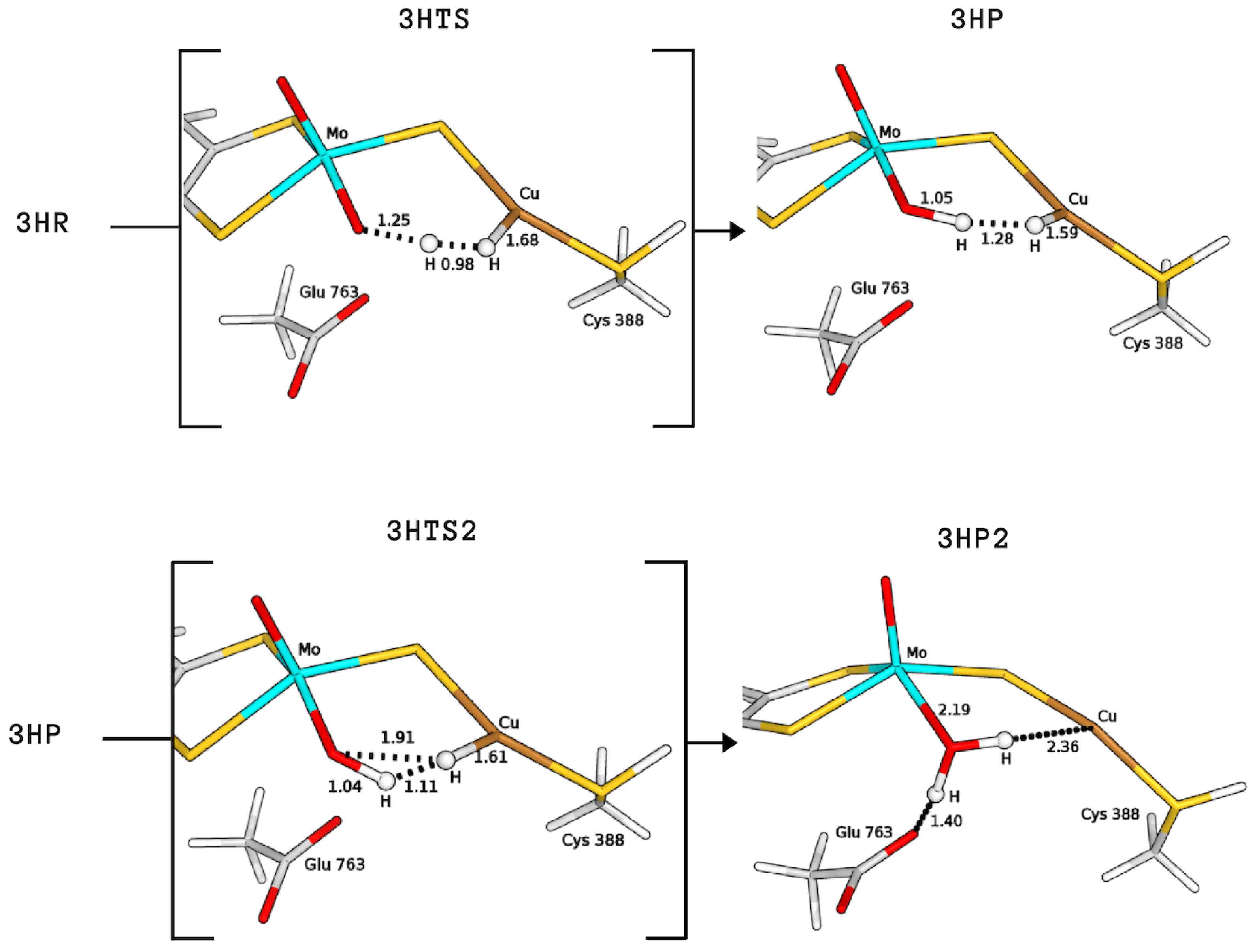
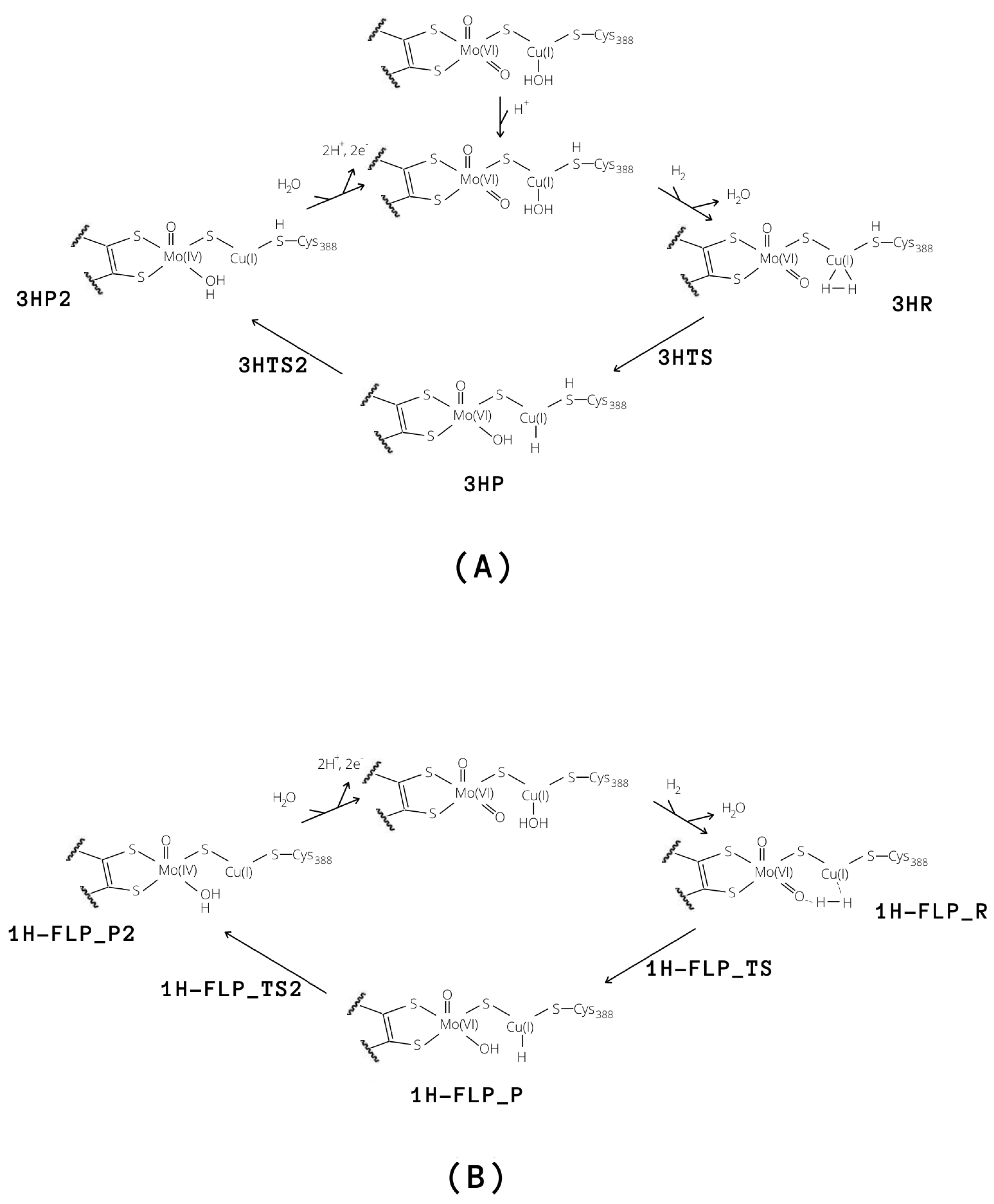
2.3. Plausible Activation Mechanisms for H Splitting
3. Methods
3.1. The Protein
3.2. QM/MM Calculations
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Mörsdorf, G.; Frunzke, K.; Gadkari, D.; Meyer, O. Microbial growth on carbon monoxide. Biodegradation 1992, 3, 61–82. [Google Scholar] [CrossRef]
- Xavier, J.C.; Preiner, M.; Martin, W.F. Something special about CO-dependent CO2 fixation. FEBS J. 2018, 285, 4181–4195. [Google Scholar] [CrossRef] [PubMed]
- Jeoung, J.-H.; Martins, B.M.; Dobbek, H. X-ray Crystallography of Carbon Monoxide Dehydrogenases. In Metalloproteins; Hu, Y., Ed.; Humana Press: New York, NY, USA, 2019; pp. 167–178. [Google Scholar]
- Santiago, B.; Meyer, O. Characterization of hydrogenase activities associated with the molybdenum CO dehydrogenase from Oligotropha carboxidovorans. FEMS Microbiol. Lett. 1996, 136, 157–162. [Google Scholar] [CrossRef]
- Nishino, T.; Okamoto, K.; Leimkühler, S. Enzymes of the Xanthine Oxidase Family. In Molybdenum and Tungsten Enzymes: Biochemistry; Hille, R., Schulzke, C., Kirk, M.L., Eds.; The Royal Society of Chemistry: London, UK, 2016; pp. 192–239. [Google Scholar]
- Hille, R.; Hall, J.; Basu, P. The mononuclear molybdenum enzymes. Chem. Rev. 2014, 114, 3963–4038. [Google Scholar] [CrossRef] [PubMed]
- Dobbek, H.; Gremer, L.; Kiefersauer, R.; Huber, R.; Meyer, O. Catalysis at a dinuclear [CuSMo(=O)OH] cluster in a CO dehydrogenase resolved at 1.1-Å resolution. Proc. Natl. Acad. Sci. USA 2002, 99, 15971–15976. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Hemann, C.F.; Hille, R. Kinetic and spectroscopic studies of the molybdenum-copper CO dehydrogenase from Oligotropha carboxidovorans. J. Biol. Chem. 2010, 285, 12571–12578. [Google Scholar] [CrossRef] [PubMed]
- Wilcoxen, J.; Hille, R. The Hydrogenase Activity of the Molybdenum/Copper-containing Carbon Monoxide Dehydrogenase of Oligotropha carboxidovorans. J. Biol. Chem. 2013, 288, 36052–36060. [Google Scholar] [CrossRef] [PubMed]
- Gourlay, C.; Nielsen, D.J.; White, J.M.; Knottenbelt, S.Z.; Kirk, M.L.; Young, C.G. Paramagnetic active site models for the molybdenum–copper carbon monoxide dehydrogenase. J. Am. Chem. Soc. 2006, 128, 2164–2165. [Google Scholar] [CrossRef] [PubMed]
- Kaufmann, P.; Duffus, B.R.; Teutloff, C.; Leimkühler, S. Functional Studies on Oligotropha carboxidovorans Molybdenum-Copper CO Dehydrogenase Produced in Escherichia coli. Biochemistry 2018, 57, 2889–2901. [Google Scholar] [CrossRef] [PubMed]
- Hille, R.; Dingwall, S.; Wilcoxen, J. The aerobic CO dehydrogenase from Oligotropha carboxidovorans. J. Biol. Inorg. Chem. JBIC 2015, 20, 243–251. [Google Scholar] [CrossRef] [PubMed]
- Rokhsana, D.; Large, T.A.G.; Dienst, M.C.; Retegan, M.; Neese, F. A realistic in silico model for structure/function studies of molybdenum–copper CO dehydrogenase. J. Biol. Inorg. Chem. JBIC 2016, 21, 491–499. [Google Scholar] [CrossRef] [PubMed]
- Breglia, R.; Bruschi, M.; Cosentino, U.; De Gioia, L.; Greco, C.; Miyake, T.; Moro, G. A theoretical study on the reactivity of the Mo/Cu-containing carbon monoxide dehydrogenase with dihydrogen. Protein Eng. Des. Sel. 2017, 30, 169–174. [Google Scholar] [CrossRef] [PubMed]
- Plitt, H.S.; Bär, M.R.; Ahlrichs, R.; Schnöckel, H. [Cu(η2-H2)Cl], a model compound for H2 complexes. Ab initio calculations and identification by IR spectroscopy. Angew. Chem. Int. Ed. 1991, 30, 832–834. [Google Scholar] [CrossRef]
- Frohman, D.J.; Grubbs, G.S., II; Yu, Z.; Novick, S.E. Probing the Chemical Nature of Dihydrogen Complexation to Transition Metals, a Gas Phase Case Study: H2–CuF. Inorg. Chem. 2013, 52, 816–822. [Google Scholar] [CrossRef] [PubMed]
- Serykh, A.I.; Kazansky, V.B. Unusually strong adsorption of molecular hydrogen on Cu+ sites in copper-modified ZSM-5. Phys. Chem. Chem. Phys. 2004, 6, 5250–5255. [Google Scholar] [CrossRef]
- Spoto, G.; Gribov, E.; Bordiga, S.; Lamberti, C.; Ricchiardi, G.; Scarano, D.; Zecchina, A. Cu+(H2) and Na+(H2) adducts in exchanged ZSM-5 zeolites. Chem. Commun. 2004, 2768–2769. [Google Scholar] [CrossRef] [PubMed]
- Georgiev, P.A.; Albinati, A.; Mojet, B.L.; Ollivier, J.; Eckert, J. Observation of Exceptionally Strong Binding of Molecular Hydrogen in a Porous Material: Formation of an η2-H2 Complex in a Cu-Exchanged ZSM-5 Zeolite. J. Am. Chem. Soc. 2007, 129, 8086–8087. [Google Scholar] [CrossRef] [PubMed]
- Reguera, L.; Krap, C.P.; Balmaseda, J.; Reguera, E. Hydrogen storage in copper prussian blue analogues: Evidence of H2 coordination to the copper atom. J. Phys. Chem. C 2008, 112, 15893–15899. [Google Scholar] [CrossRef]
- Stephan, D.W. “Frustrated Lewis pairs”: A concept for new reactivity and catalysis. Org. Biomol. Chem. 2008, 6, 1535–1539. [Google Scholar] [CrossRef] [PubMed]
- Carr, S.B.; Evans, R.M.; Brooke, E.J.; Wehlin, S.A.M.; Nomerotskaia, E.; Sargent, F.; Armstrong, F.A.; Phillips, S.E.V. Hydrogen activation by [NiFe]-hydrogenases. Biochem. Soc. Trans. 2016, 44, 863–868. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miyake, T.; Bruschi, M.; Cosentino, U.; Baffert, C.; Fourmond, V.; Legér, C.; Moro, G.; De Gioia, L.; Greco, C. Does the environment around the H-cluster allow coordination of the pendant amine to the catalytic iron center in [FeFe] hydrogenases? Answers from theory. J. Biol. Inorg. Chem. JBIC 2013, 18, 693–700. [Google Scholar] [CrossRef] [PubMed]
- Greco, C.; Silakov, A.; Bruschi, M.; Ryde, U.; De Gioia, L.; Lubitz, W. Magnetic Properties of [FeFe]-Hydrogenases: A Theoretical Investigation Based on Extended QM and QM/MM Models of the H-Cluster and Its Surroundings. Eur. J. Inorg. Chem. 2011, 1043–1049. [Google Scholar] [CrossRef]
- Greco, C. H2 Binding and Splitting on a New-Generation [FeFe]-Hydrogenase Model Featuring a Redox-Active Decamethylferrocenyl Phosphine Ligand: A Theoretical Investigation. Inorg. Chem. 2013, 52, 1901–1908. [Google Scholar] [CrossRef] [PubMed]
- Siegbahn, P.E.M.; Shestakov, A.F. Quantum chemical modeling of CO oxidation by the active site of molybdenum CO dehydrogenase. J. Comput. Chem. 2005, 26, 888–898. [Google Scholar] [CrossRef] [PubMed]
- Gnida, M.; Ferner, R.; Gremer, L.; Meyer, O.; Meyer-Klaucke, W. A novel binuclear [CuSMo] cluster at the active site of carbon monoxide dehydrogenase: Characterization by X-ray absorption spectroscopy. Biochemistry 2003, 42, 222–230. [Google Scholar] [CrossRef] [PubMed]
- Rovaletti, A.; Bruschi, M.; Moro, G.; Cosentino, U.; Ryde, U.; Greco, C. A thiocarbonate sink on the enzymatic energy landscape of aerobic CO oxidation? Answers from DFT and QM/MM models of MoCu CO-dehydrogenases. J. Catal. 2019, 372, 201–205. [Google Scholar] [CrossRef]
- Olsson, M.H.M.; Søndergaard, C.R.; Rostkowski, M.; Jensen, J.H. PROPKA3: Consistent treatment of internal and surface residues in empirical pKa Predictions. J. Chem. Theory Comput. 2011, 7, 525–537. [Google Scholar] [CrossRef] [PubMed]
- Ryde, U. The coordination of the catalytic zinc ion in alcohol dehydrogenase studied by combined quantum-chemical and molecular mechanics calculations. J. Comput.-Aided Mol. Des. 1996, 10, 153–164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ryde, U.; Olsson, M.H.M. Structure, strain, and reorganization energy of blue copper models in the protein. Int. J. Quantum Chem. 2001, 81, 335–347. [Google Scholar] [CrossRef]
- Reuter, N.; Dejaegere, A.; Maigret, B.; Karplus, M. Frontier bonds in QM/MM methods: A comparison of different approaches. J. Phys. Chem. A 2000, 104, 1720–1735. [Google Scholar] [CrossRef]
- Cao, L.; Ryde, U. On the difference between additive and subtractive QM/MM calculations. Front. Chem. 2018, 6, 89. [Google Scholar] [CrossRef] [PubMed]
- TURBOMOLE V6.3 2011, a Development of University of Karlsruhe and Forschungszentrum Karlsruhe GmbH, 1989–2007; TURBOMOLE GmbH, Since 2007. Available online: http://www.turbomole.com (accessed on 8 November 2019).
- Becke, A. Density-functional exchange-energy approximation with correct asymptotic behavior. Phys. Rev. A 1988, 38, 3098–3100. [Google Scholar] [CrossRef] [PubMed]
- Perdew, J.P. Density-functional approximation for the correlation energy of the inhomogeneous electron gas. Phys. Rev. B 1986, 33, 8822–8824. [Google Scholar] [CrossRef] [PubMed]
- Weigend, F.; Ahlrichs, R. Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy. Phys. Chem. Chem. Phys. 2005, 7, 3297–3305. [Google Scholar] [CrossRef] [PubMed]
- Grimme, S.; Ehrlich, S.; Goerigk, L. Effect of the damping function in dispersion corrected density functional theory. J. Comput. Chem. 2011, 32, 1456–1465. [Google Scholar] [CrossRef] [PubMed]
- Eichkorn, K.; Weigend, F.; Treutler, O.; Ahlrichs, R. Auxiliary basis sets for main row atoms and transition metals and their use to approximate Coulomb potentials. Theor. Chem. Acc. 1997, 97, 119–124. [Google Scholar] [CrossRef]
- Case, D.A.; Babin, V.; Berryman, J.; Betz, R.M.; Cai, Q.; Cerutti, D.S.; Cheatham, T.E., III; Darden, T.A.; Duke, R.E.; Gohlke, H.; et al. AMBER 14; University of California: San Francisco, CA, USA, 2014. [Google Scholar]
- Maier, J.A.; Martinez, C.; Kasavajhala, K.; Wickstrom, L.; Hauser, K.E.; Simmerling, C. ff14SB: Improving the accuracy of protein side chain and backbone parameters from ff99SB. J. Chem. Theory Comput. 2015, 11, 3696–3713. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Wolf, R.M.; Caldwell, J.W.; Kollman, P.A.; Case, D.A. Development and testing of a general Amber force field. J. Comput. Chem. 2004, 25, 1157–1174. [Google Scholar] [CrossRef] [PubMed]
- Bayly, C.I.; Cieplak, P.; Cornell, W.D.; Kollman, P.A. A Well-Behaved Electrostatic Potential Based Method Using Charge Restraints for Deriving Atomic Charges: The RESP Model. J. Phys. Chem. 1993, 97, 10269–10280. [Google Scholar] [CrossRef]
- Romero, E.A.; Olsen, P.M.; Jazzar, R.; Soleilhavoup, M.; Gembicky, M.; Bertrand, G. Spectroscopic Evidence for a Monomeric Copper(I) Hydride and Crystallographic Characterization of a Monomeric Silver(I) Hydride. Angew. Chem. Int. Ed. 2017, 56, 4024–4027. [Google Scholar] [CrossRef] [PubMed]
- Huang, G.; Wagner, T.; Wodrich, M.D.; Ataka, K.; Bill, E.; Ermler, U.; Hu, X.; Shima, S. The atomic-resolution crystal structure of activated [Fe]-hydrogenase. Nat. Catal. 2019, 2, 537–543. [Google Scholar] [CrossRef]
- Tai, H.; Nishikawa, K.; Higuchi, Y.; Mao, Z.-W.; Hirota, S. Cysteine SH and Glutamate COOH Contributions to [NiFe] Hydrogenase Proton Transfer Revealed by Highly Sensitive FTIR Spectroscopy. Angew. Chem. Int. Ed. 2019, 58, 13285–13290. [Google Scholar] [CrossRef] [PubMed]
- Senger, M.; Mebs, S.; Duan, J.; Shulenina, O.; Laun, K.; Kertess, L.; Wittkamp, F.; Apfel, U.P.; Happe, T.; Winkler, M. Protonation/reduction dynamics at the [4Fe–4S] cluster of the hydrogen-forming cofactor in [FeFe]-hydrogenases. Phys. Chem. Chem. Phys. 2018, 20, 3128–3140. [Google Scholar] [CrossRef] [PubMed]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
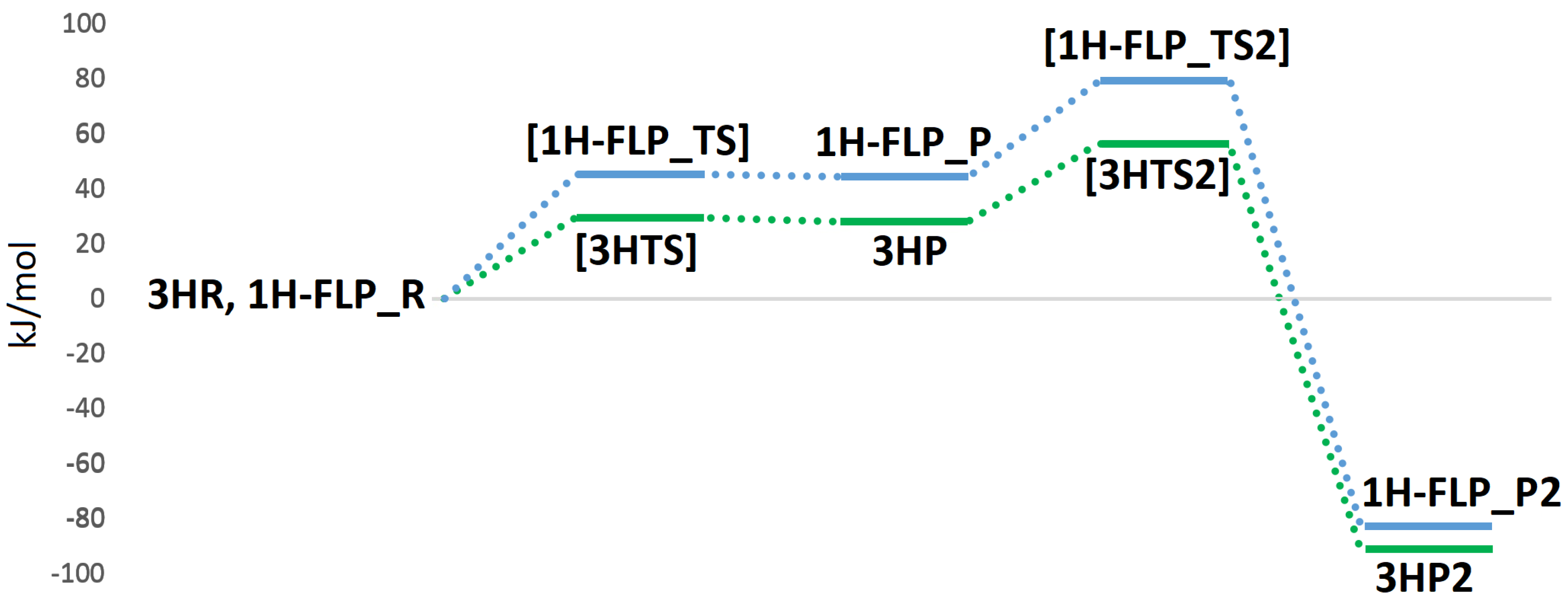{kind=link}
| NoB | R | P1 | TS | P | TS2 | P2 | |
|---|---|---|---|---|---|---|---|
| 1H | −46.9 | 0.0 | 57.7 | −2.9 | 31.8 | −130.1 | |
| 2H | −46.4 | 0.0 | 83.3 | ||||
| 3H | −10.9 | 0.0 | 29.3 | 28.0 | 56.4 | −90.8 | |
| 1H-FLP | 0.8 | 0.0 | 45.2 | 44.4 | 79.1 | −82.8 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rovaletti, A.; Bruschi, M.; Moro, G.; Cosentino, U.; Greco, C.; Ryde, U. Theoretical Insights into the Aerobic Hydrogenase Activity of Molybdenum–Copper CO Dehydrogenase. Inorganics 2019, 7, 135. https://0-doi-org.brum.beds.ac.uk/10.3390/inorganics7110135
Rovaletti A, Bruschi M, Moro G, Cosentino U, Greco C, Ryde U. Theoretical Insights into the Aerobic Hydrogenase Activity of Molybdenum–Copper CO Dehydrogenase. Inorganics. 2019; 7(11):135. https://0-doi-org.brum.beds.ac.uk/10.3390/inorganics7110135
Chicago/Turabian StyleRovaletti, Anna, Maurizio Bruschi, Giorgio Moro, Ugo Cosentino, Claudio Greco, and Ulf Ryde. 2019. "Theoretical Insights into the Aerobic Hydrogenase Activity of Molybdenum–Copper CO Dehydrogenase" Inorganics 7, no. 11: 135. https://0-doi-org.brum.beds.ac.uk/10.3390/inorganics7110135