
Synthesis and Characterization of Super Bulky β-Diketiminato Group 1 Metal Complexes


School of Chemistry, Monash University, P.O. Box 23, Melbourne, VIC 3800, Australia
*
Author to whom correspondence should be addressed.
Inorganics 2021, 9(9), 72; https://0-doi-org.brum.beds.ac.uk/10.3390/inorganics9090072
Submission received: 25 August 2021
/
Revised: 15 September 2021
/
Accepted: 15 September 2021
/
Published: 16 September 2021
(This article belongs to the Special Issue Cornerstones in Contemporary Inorganic Chemistry)
Abstract
:Sterically bulky β-diketiminate (or Nacnac) ligand systems have recently shown the ability to kinetically stabilize highly reactive low-oxidation state main group complexes. Metal halide precursors to such systems can be formed via salt metathesis reactions involving alkali metal complexes of these large ligand frameworks. Herein, we report the synthesis and characterization of lithium and potassium complexes of the super bulky anionic β-diketiminate ligands, known [TCHPNacnac]− and new [TCHP/DipNacnac]− (ArNacnac = [(ArNCMe)2CH]−) (Ar = 2,4,6-tricyclohexylphenyl (TCHP) or 2,6-diisopropylphenyl (Dip)). The reaction of the proteo-ligands, ArNacnacH, with nBuLi give the lithium etherate compounds, [(TCHPNacnac)Li(OEt2)] and [(TCHP/DipNacnac)Li(OEt2)], which were isolated and characterized by multinuclear NMR spectroscopy and X-ray crystallography. The unsolvated potassium salts, [{K(TCHPNacnac)}2] and [{K(TCHP/DipNacnac)}∞], were also synthesized and characterized in solution by NMR spectroscopy. In the solid state, these highly reactive potassium complexes exhibit differing alkali metal coordination modes, depending on the ligand involved. These group 1 complexes have potential as reagents for the transfer of the bulky ligand fragments to metal halides, and for the subsequent stabilization of low-oxidation state metal complexes.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
The use of sterically demanding β-diketiminate ligand systems for the stabilization of reactive low-oxidation state metal compounds has become commonplace in organometallic chemistry [1,2,3,4,5,6,7,8]. One of the most common β-diketiminate ligands used is [DipNacnac]− ([(DipNCMe)2CH]−, Dip = 2,6-diisopropylphenyl). This has seen widespread use due to its ease of synthesis from commercially available starting materials and the high kinetic stabilization it imparts to the coordinated metal. A particularly successful use of β-diketiminates has been the stabilization of magnesium(I) dimers, e.g., LMg–MgL (L = β-diketiminate), which have subsequently been widely applied as soluble, selective reducing reagents in organic and inorganic synthesis [9,10,11,12]. Attempts to increase the steric profile of β-diketiminate ligands, in the hope that compounds elusive to stabilization by the DipNacnac system can be isolated, are worthwhile pursuits, with notable successes coming from the groups of Piers [13], Holland [14] and Hill [15]. With respect to the stabilization of group 2 compounds, bulky β-diketiminate ligands based on 2,6-bis(diphenylmethyl)-aryl N-substituents, [ArNacnac]− ([(ArNCMe)2CH]−, Ar = 2,6-[C(H)Ph2]2-4-MeC6H2 (Ar*); 2,6-[C(H)Ph2]2-4-iPrC6H2 (Ar†)) were used to isolate the first monomeric magnesium hydrides [15]. More recently, it has been shown that increasing the steric bulk of the Nacnac ligand, by the replacement of the N-bound Dip groups with either DIPeP (DIPeP = 2,6-diisopentylphenyl) or TCHP (TCHP = 2,4,6-tricyclohexylphenyl) groups, allows for the isolation of the magnesium(I) compounds, [{(ArNacnac)Mg}2] (Ar = DIPeP, TCHP), both of which show elongated Mg–Mg bonds compared to previously reported examples [16,17,18].
The activation of β-diketiminate coordinated Mg–Mg bonded fragments by the addition of a sub-stoichiometric amount of a Lewis base is known to generate unsymmetrical magnesium(I) compounds with elongated Mg–Mg bonds. These have been shown to increase the reactivity of the magnesium(I) compound towards inert molecules such as CO and ethylene [19,20]. Recently, we showed that a TCHP-substituted magnesium(I) dimer, upon irradiation, was able to reduce benzene to generate “Birch-like” cyclohexadienyl bridged products, e.g., [{(TCHPNacnac)Mg}2(µ-C6H6)] [21]. Further to this, Harder and co-workers have shown that Nacnac ligands incorporating the DIPeP aryl moiety are able to stabilize molecular magnesium(0) compounds [22,23], as well as calcium complexes, which activate dinitrogen, as in [{(DIPePNacnac)Ca}2(μ-N2)] [24].
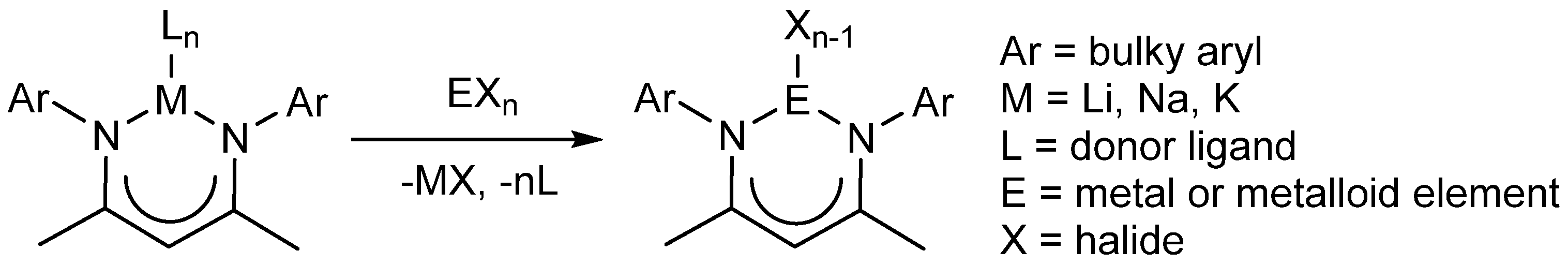
Routes to low-oxidation state metal complexes bearing Nacnac ligands typically proceed via the reduction of β-diketiminato metal halide precursors, which can be synthesized by the reaction of an alkali metal salt of the β-diketiminate ligand with a metal halide (Scheme 1). As such, the synthesis of alkali metal complexes of new very bulky ligands is desirable, with the view to using such complexes as ligand transfer reagents in the preparation of metal halide precursors to highly reactive low-valent metal complexes. Potentially, compounds previously inaccessible using the archetypal DipNacnac ligand system can be realized using this approach. Herein, we describe the synthesis and characterization of a new unsymmetrical, bulky β-diketimine pro-ligand incorporating the TCHP aryl moiety, as well as four alkali metal complexes derived from this, and a previously reported TCHP-substituted β-diketimine.
2. Results and Discussion
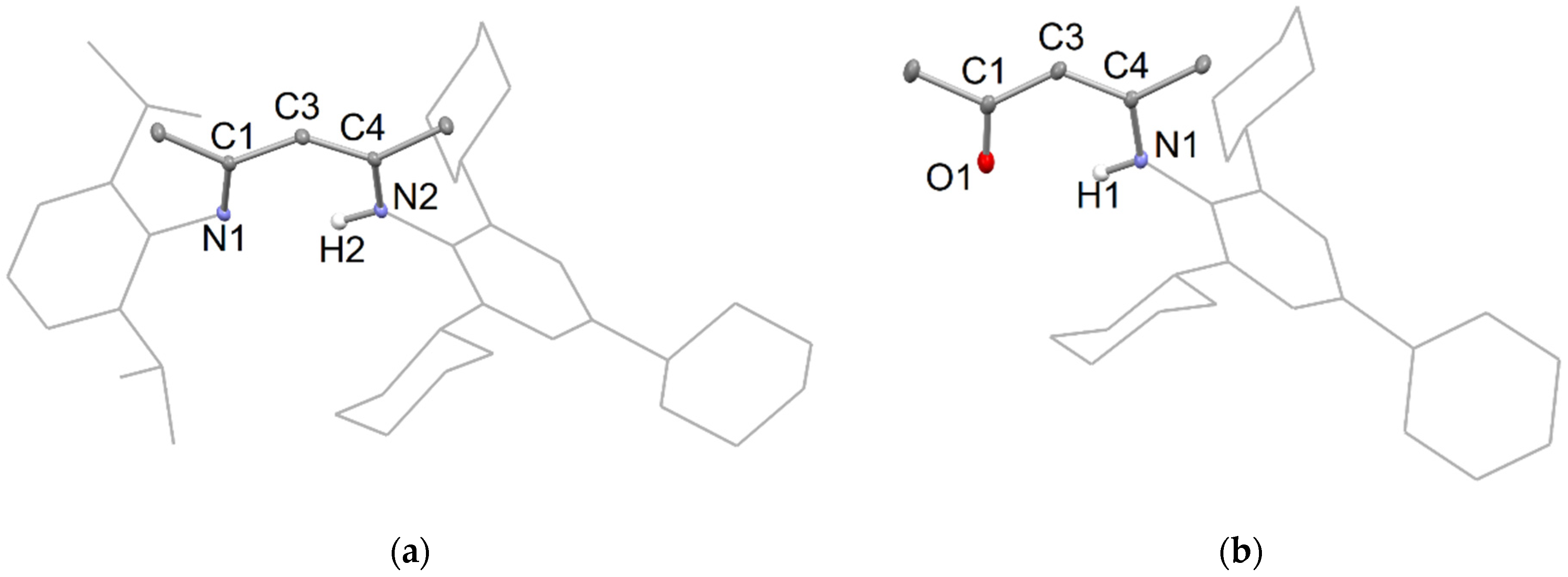
The β-diketimine, TCHPNacnacH, 1, was recently reported to be prepared by a condensation reaction between acacH, H2C(MeC=O)2 and two equivalents of (TCHP)NH2 [17]. Here, the new, unsymmetrical β-diketimine, TCHP/DipNacnacH 2, was similarly synthesized by the condensation of one equivalent of (TCHP)NH2 with DipNacacH, {DipN(H)CMe}CHC(O)Me, which gave 2 in a 43% isolated yield [25]. It is of note that, in the current study, during one preparation of 1, a low yield of the β-ketimine, TCHPNacacH, {(TCHP)N(H)CMe}CHC(O)Me 3, was obtained as a by-product. This likely results from the use of slightly less than two equivalents of (TCHP)NH2 in the synthesis. A rational synthesis of 3 was not attempted, but it was spectroscopically characterized. The molecular structure of 2 is shown in Figure 1a, which reveals it to exist as its ene-imine conjugated tautomer in the solid state, as is typical for β-diketimines. The proton on N2 is hydrogen bonded to N1, which leads to the two double bonds within the C3N2 backbone (viz. N1-C1 and C3-C4) adopting a cis-configuration, relative to each other. A similar structure is adopted by β-ketimine, 3 (Figure 1b), in which the double bonds within the C3NO backbone are C1-O1 and C3-C4.
The bulky β-diketimines, 1 and 2, were deprotonated by treating diethyl ether solutions of the compounds with a slight excess of nBuLi at −78 °C. Upon warming to room temperature and stirring the reaction solutions overnight, they were concentrated and stored at −30 °C to yield colorless crystals of lithium β-diketiminate complexes [(TCHPNacnac)Li(OEt2)], 4, and [(TCHP/DipNacnac)Li(OEt2)], 5, in moderate isolated yields. The solution state NMR spectroscopic data for the compounds are consistent with them retaining their solid-state structures (see below) in solution. However, it is noteworthy that the 13C{1H} NMR spectrum of compound 4 exhibits 10 cyclohexyl carbon signals (δ = 25–45 ppm), which suggests restricted rotation around one of the sterically more encumbered ortho-cyclohexyl rings of the TCHP substituents.
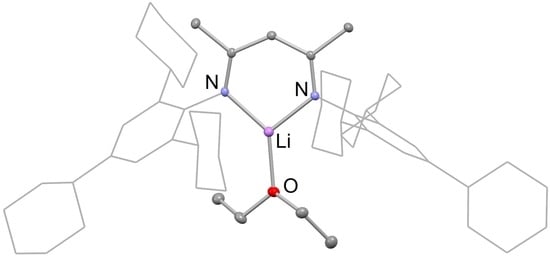
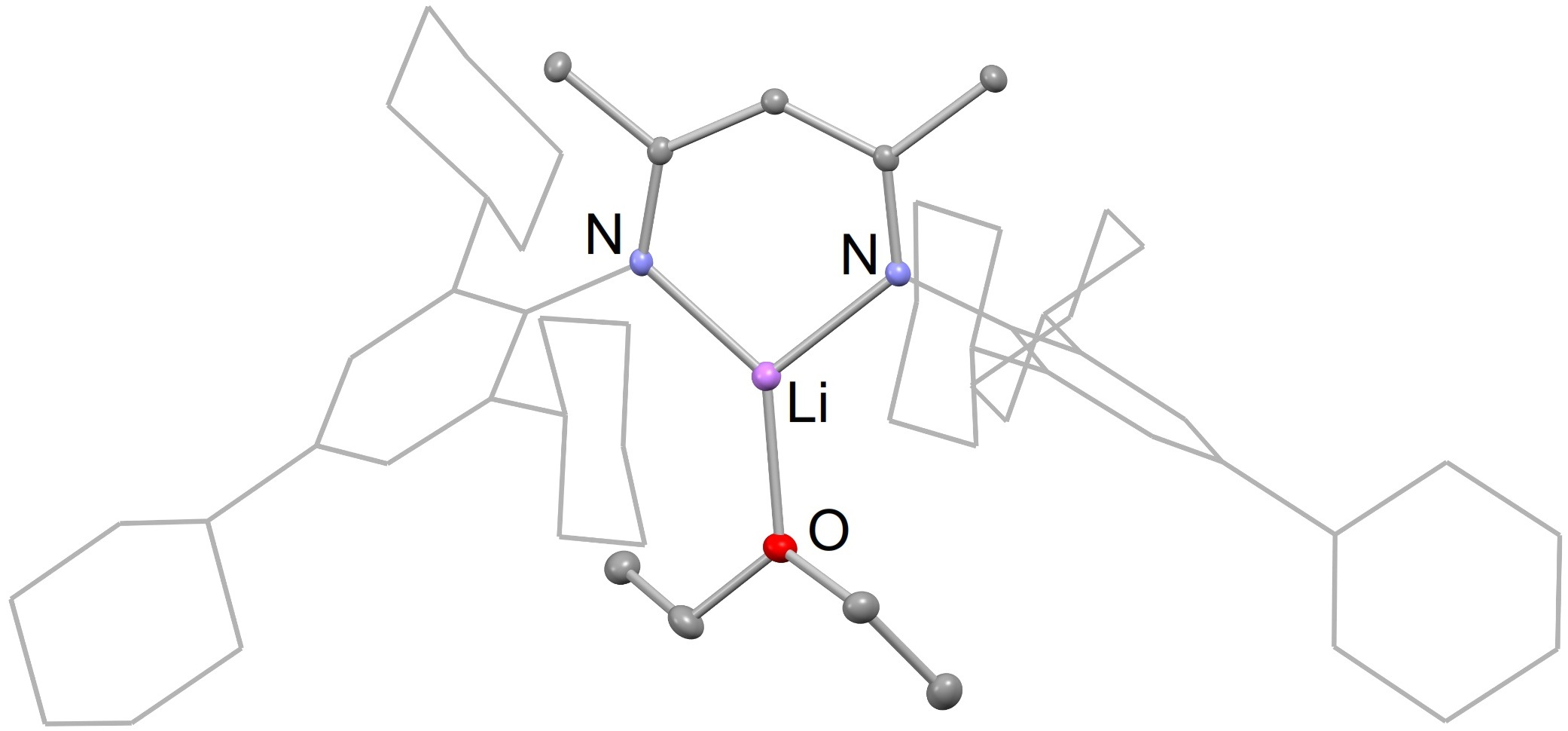
Compounds 4 and 5 have very similar solid-state structures, as is evident from Figure 2. The molecular structure of these compounds are typical of previously reported β-diketiminate lithium etherate complexes, e.g., [(DipNacnac)Li(OEt2)] [26]. That is, they are monomeric and their lithium centers possess trigonal planar coordination geometries with one coordinated ether ligand. The β-diketiminate ligand binds as an N,N-chelate to the lithium, with Li-N bond distances between 1.921 and 1.935 Å, within the normal range for such complexes [26]. The Li-O distances are slightly longer when compared to the previously reported [(DipNacnac)Li(OEt2)] complex (1.953(4) Å for 4 and 1.975(4) Å for 5; cf. 1.911(4) Å for [(DipNacnac)Li(OEt2)]), which is likely caused by steric repulsion from its bulky TCHP aryl groups. This also leads to a slight narrowing of the N-Li-N angle for 4 and 5 (97.67(18)° and 97.79(17)°, respectively) compared to the angle in [(DipNacnac)Li(OEt2)] (99.9(2)°). The bond lengths within the C3N2 backbones of both compounds imply significant electronic delocalizations over those fragments.
Unsolvated potassium salts of the TCHP-substituted β-diketiminates could also be synthesized using a similar methodology to that previously reported for the synthesis of [K(DipNacnac)] [27]. That is, β-diketimine, 1, was combined with benzyl potassium in toluene, pro-ligand 2 was combined with potassium bis(trimethylsilyl)amide (KHMDS) in toluene and both reactions were monitored by NMR spectroscopy. No reaction could be observed after 16 h at room temperature for either pro-ligand, contrary to the synthesis of [K(DipNacnac)], which showed good conversion under these conditions. This can be attributed to the increased steric bulk from the TCHP aryl group, perturbing deprotonation of 1 and 2 by the base. However, heating these solutions at 60 °C overnight led to clean conversion to the potassium salts [{K(TCHPNacnac)}2], 6, and [{K(TCHP/DipNacnac)}∞], 7, as determined by 1H NMR spectroscopy. It should be noted that compound 6 could also be formed using KHMDS as the base, though this route gave variable yields of product. Attempts to isolate compound 6 repeatedly led to a degree of decomposition of the complex to 1 upon manipulation of the reaction mixture. Moreover, in the presence of silicone grease, compound 6 decomposed to unknown products in solution over time. This suggests that compound 6 is highly reactive, and as such could only be characterized by NMR spectroscopy after formation in situ. As monitoring the formation of 6 from 1 by NMR spectroscopy showed complete conversion, we believe this compound could be used as a ligand transfer reagent after its formation in situ, and in the absence of silicone grease.
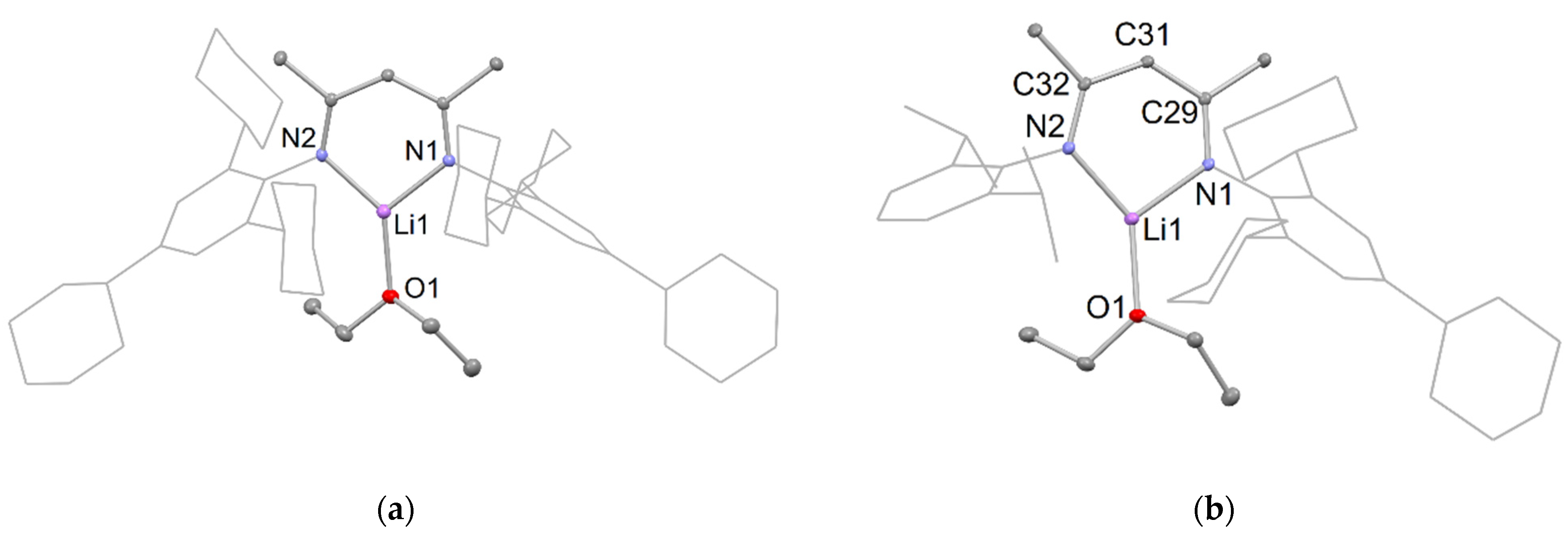
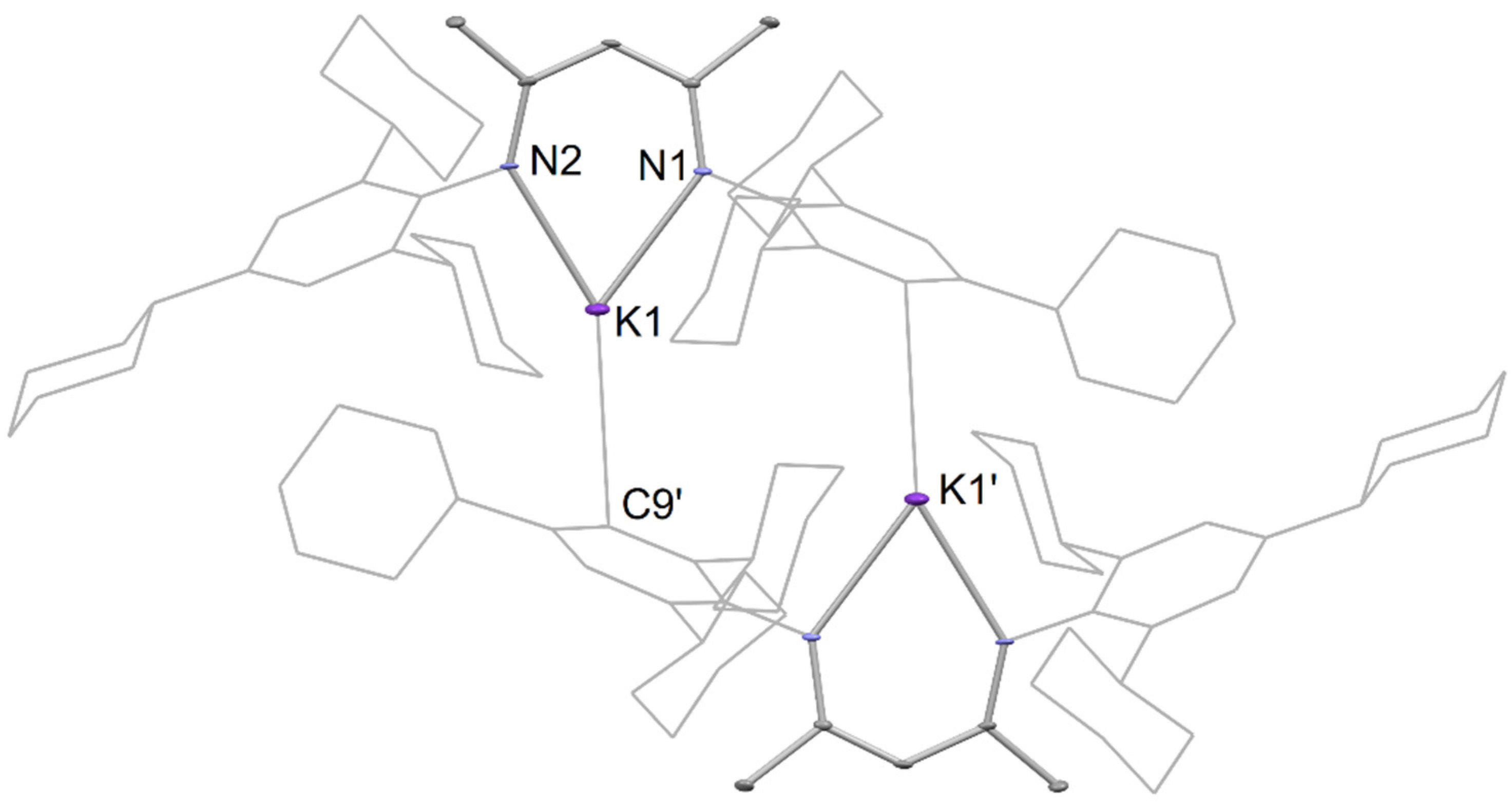
Crystals of 6 suitable for an X-ray diffraction study were formed by the addition of n-hexane to a concentrated toluene solution of 6 at room temperature. The molecular structure (Figure 3) shows the compound crystallizing as a dimer, with the potassium centers having trigonal planar geometries, being N,N-chelated by a β-diketiminate, as well as possessing an intermolecular C∙∙∙K interaction with a meta-carbon of an opposing TCHP group of the second [K(TCHPNacnac)] monomer unit. The N-K distances (2.633(3) and 2.619(3) Å) are in the expected range and correspond well to those previously reported for [K(DipNacnac)] (2.681 Å mean) [27]. The C∙∙∙K distance of 3.309(3) Å is outside the sum of the covalent radii for the two elements (2.71 Å [28]), but well within the sum of their van der Waals radii (4.45 Å [29]).
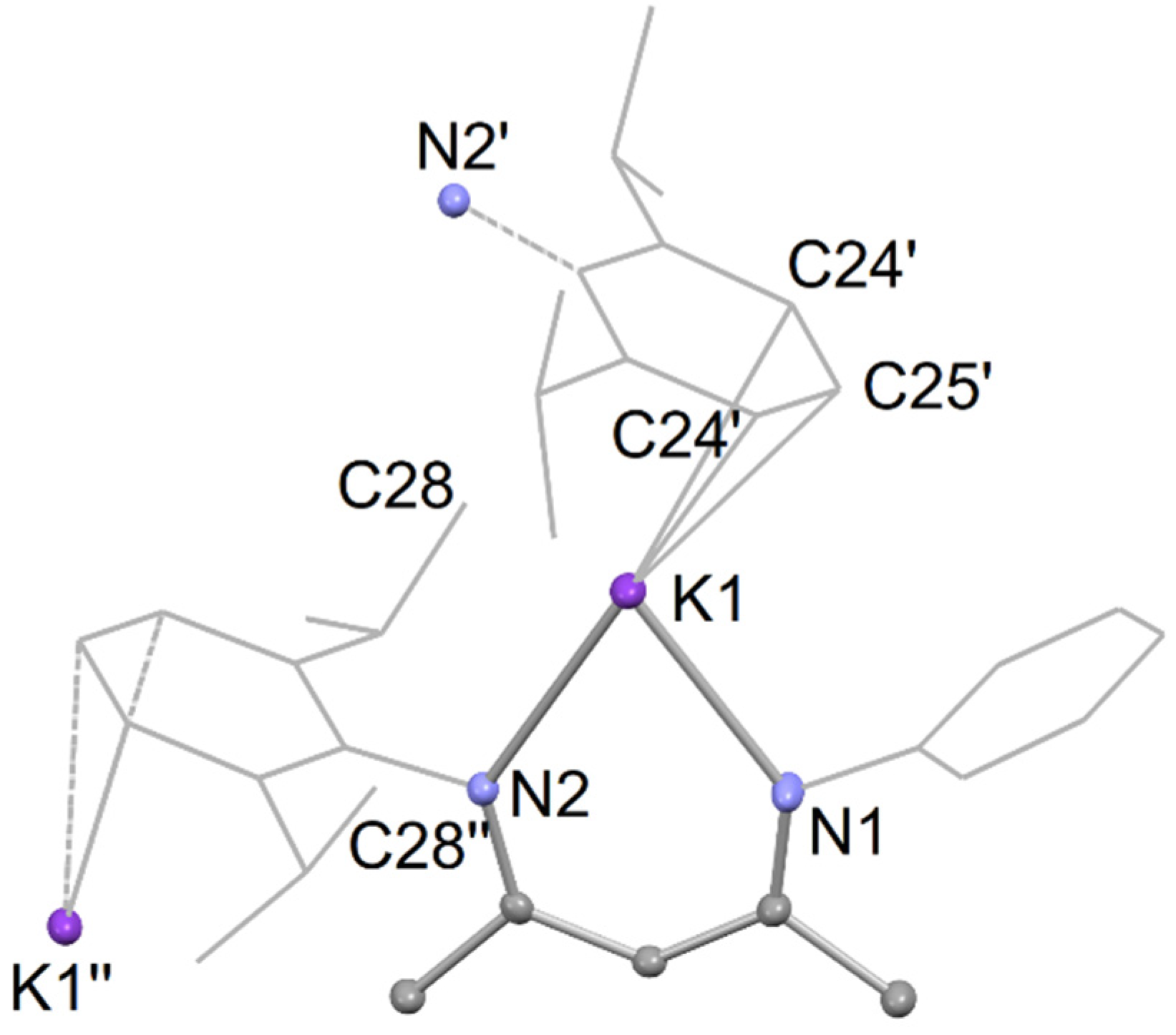
Compound 7 crystallized as a colorless material from the toluene reaction solution, upon cooling it to room temperature from 60 °C. However, it should be noted that, like 6, compound 7 undergoes decomposition to the proteo-ligand 2 in the presence of silicone grease. Crystals of 7 suitable for an X-ray diffraction were obtained from a saturated benzene solution, and its molecular structure is shown in Figure 4. It is polymeric, similar to previously reported [K(DipNacnac)] [27]. The polymer is propagated via intermolecular C∙∙∙K interactions between the potassium centers and para- and meta-carbons of a Dip group on an adjacent monomer unit. The K∙∙∙C bond distances are 3.178(4) Å for K–Cpara and 3.254(3) Å for K–Cmeta. These separations are slightly shorter than the equivalent C∙∙∙K interactions in [K(DipNacnac)], which are 3.214(3) Å for K–Cpara, and 3.351(3) Å and 3.276(3) Å for K–Cmeta interactions. The K-N distances in the compound are comparable to those in 6.
3. Materials and Methods
3.1. General Considerations
All manipulations were carried out using standard Schlenk and glove box techniques under an atmosphere of high purity dinitrogen. Toluene and hexane were distilled over molten potassium, while diethyl ether was distilled over 1:1 Na/K alloy. Benzene-d6 was stored over a mirror of sodium and degassed three times via freeze-pump-thawing before use. The 1H, 13C{1H} and 7Li NMR spectra were recorded on a Bruker AvanceIII 400 spectrometer and were referenced to the residual resonances of the solvent used or external 1 M LiCl. Mass spectra were collected using an Agilent Technologies 5975D inert MSD with a solid-state probe. FTIR spectra were collected as Nujol mulls on an Agilent Cary 630 attenuated total reflectance (ATR) spectrometer. Microanalyses were carried out at the Science Centre, London Metropolitan University or using a PerkinElmer- 2400 CHNS/O Series II System. Melting points were determined in sealed glass capillaries under dinitrogen and are uncorrected. The starting materials, TCHPNacnacH 1 [17], DipNacacH [30] (TCHP)NH2 [31] and benzyl potassium [32], were prepared by literature procedures. All other reagents were used as received.
3.2. Syntehsis of TCHP/DipNacnacH 2 and Data for TCHPNacacH 3
Preparation of TCHP/DipNacnacH, 2: 2,4,6-Tricyclohexylaniline (4.57 g, 13.5 mmol), DipNacacH (3.50 g, 13.5 mmol) and p-toluenesulfonic acid (2.32 g, 13.5 mmol) were dissolved in toluene (100 mL), and the mixture was heated at reflux for 3 days in a Dean–Stark apparatus. Volatiles were then removed in vacuo, the resultant oil was dissolved in dichloromethane (100 mL), the extract washed with a saturated Na2CO3 solution (100 mL, 1 M) and the organic layer was separated. The organic layer was dried over MgSO4 and volatiles were removed in vacuo. Methanol (5 mL) was added to the oily residue, which was then sonicated and filtered to yield compound 2 as a crude white powder. This was dried and recrystallized from hot ethyl acetate to yield pure TCHP/DipNacnacH (2.25 g). A further crop of the product could be obtained by concentration of the mother liquor, and letting it stand at room temperature (1.00 g). Total yield: 3.25 g, 42%. X-ray quality crystals could be grown from the slow evaporation of a diethyl ether solution of TCHP/DipNacnacH. M.p: 224–227 °C; 1H NMR (400 MHz, C6D6, 298 K): δ = 1.24 (d, 3JHH = 8 Hz, 6H, CH(CH3)2), 1.28 (d, 3JHH = 8 Hz, 6H, CH(CH3)2), 1.31–1.68 (m, 17H, Cyc-CH2), 1.70 (s, 3H, NCCH3), 1.72 (s, 3H, NCCH3), 1.74–2.06 (m, 13H, Cyc-CH2), 2.52–2.60 (m, 1H, p-Cyc-H), 2.95–3.03 (m, 2H, o-Cyc-H), 3.29–3.38 (sept, 3JHH = 8 Hz, 2H, CH(CH3)2), 4.89 (s, 1H, NCCH), 7.18 (s, 2H, ArH), 7.20 (m, 3H, ArH), 12.29 (s, 1H, NH). 13C{1H} NMR (101 MHz, C6D6, 298 K): δ = 20.6 (NCCH3), 21.4 (NCCH3), 23.8, 24.3 (CH(CH3)2), 26.6, 26.8, 27.4, 2 × 27.7 (Cyc-C), 28.6 (CH(CH3)2), 34.7, 35.3 (Cyc-C), 39.6 (o-Cyc-CH), 45.3 (p-Cyc-CH), 94.1 (NCCH), 123.0, 123.5, 125.0, 137.3, 141.2, 143.3, 143.7, 145.8 (ArC), 159.7 (NCCH3), 163.8 (NCCH3). IR ν/cm−1 (ATR): 2920 (s), 2847 (s), 1618 (s), 1545 (s), 1442 (s), 1359 (w), 1270 (m), 1175 (m). Acc. Mass/ESI m/z: calc. 581.4835 found: 581.4841 [M+H]+.
N.B. During one synthesis of 1, β-ketimine 3 was obtained as a low yield by-product.
Data for TCHPNacacH 3. M.p 183–185 °C. 1H NMR (400 MHz, C6D6, 298 K): N.B. integration of resonances for cyclohexyl groups are estimated due to complex overlapping signals, δ = 1.12–1.44 (m, 10H, Cyc-CH2), 1.46 (s, 3H, NCCH3), 1.48–1.98 (m, 20H, Cyc-CH2), 2.06 (s, 3H, OCCH3), 2.46–2.52 (m, 1H, p-Cyc-CH), 2.78–2.86 (m, 2H, o-Cyc-CH), 5.14 (s, 1H, NCCH), 7.17 (s, 2H, ArH), 12.67 (br, 1H, NH). 13C{1H} NMR (101 MHz, C6D6, 298 K): δ = 19.1 (NCCH3), 2 × 26.6, 27.3, 2 × 27.4 (Cyc-C), 29.1 (OCCH3), 33.5, 35.1, 35.6, 40.0, 45.2 (Cyc-C), 95.9 (NCCH), 122.9, 132.5, 145.5, 147.8 (ArC), 162.9 (NCCH3), 195.6 (OCCH3). IR ν/cm−1 (ATR): 2921 (s), 2848 (s), 1611 (s), 1567 (s), 1492 (w), 1445(s), 1351 (m), 1274 (s), 1235 (w), 1185 (w), 1119 (w), 1014 (w), 998 (w), 951 (w), 919 (w), 893 (w), 865 (m), 774 (w), 741 (m), 674 (w). Acc. Mass/ESI m/z: calc. for [M+H]+ 422.3417 found: 422.3422.
3.3. Syntehsis of Complexes 4–7
Preparation of [(TCHPNacnac)Li(OEt2)], 4. TCHPNacnacH (1.15 g, 1.5 mmol) was dissolved in diethyl ether (20 mL) and the solution cooled to −78 °C. nBuLi (1.05 mL, 1.6 M in hexanes, 1.7 mmol) was added dropwise via syringe and the resultant solution was allowed to warm to room temperature and stirred for 2 h. The solution was concentrated in vacuo and stored at –30 °C overnight, yielding large colorless blocks of 4 (0.62 g) suitable for X-ray diffraction. Further concentration of the mother liquor and storage at −30 °C gave a second crop of crystals (0.14 g). Total yield: 0.76 g, 60%. M.p 112–114 °C (decomp): 1H NMR (400 MHz, C6D6, 298 K) N.B. integration for cyclohexyl groups are estimated due to complex overlapping signals: δ = 0.67 (br, 6H, (CH3CH2)2O), 1.27–1.84 (m, 50H, Cyc-CH2), 1.90 (s, 6H, NCCH3), 1.98–2.03 (m, 10H, Cyc-CH2), 2.52–2.58 (m, 2H, p-Cyc-CH), 2.96–3.04 (br m, 8H, overlapping o-Cyc-CH & (CH3CH2)2O), 4.88 (s, 1H, NCCH), 7.13 (s, 4H, ArH). 13C{1H} NMR (101 MHz, C6D6, 298 K): δ = 14.3 ((CH3CH2)2O), 23.6 (NCCH3), 26.8, 27.0, 27.6, 27.9, 28.1, 34.8, 35.1, 35.6, 39.4, 45.2 (Cyc-C), 65.2 ((CH3CH2)2O), 92.5 (NCCH), 122.4, 139.8, 141.8, 147.9 (ArC), 164.3 (NCCH3). 7Li NMR (155 MHz, C6D6, 298 K): δ = 1.91. IR ν/cm−1 (ATR): 2920 (s), 2849 (m), 1601 (m), 1550 (m), 1438 (m), 1401 (s), 1348 (m), 1282 (m), 1250 (w), 1187 (w), 1116 (m), 1070 (m), 1024 (w), 994 (w), 950 (w), 921 (w), 890 (w), 862 (m), 835 (w), 779 (w), 724 (w), 696 (m). EI/MS (70 eV) m/z (%): 83.1 (Cyc+, 26), 364.3 (Cyc)3C6H2NCMe+, 100), 659.6 (TCHPNacnacH–Cyc+, 10), 727.8 (TCHPNacnacH-CH3+, 17), 742.8 (TCHPNacnacH+, 13), 748.6 (TCHPNacnacLi+, 8). Elemental analysis calculated for C57H87LiN2O: C 83.16%, H 10.65%, Ν 3.40%, found: C 83.02%, H 10.40%, Ν 3.32%.
Preparation of [(TCHP/DipNacnac)Li(OEt2)], 5. TCHP/DipNacnacH (500 mg, 0.86 mmol) was dissolved in diethyl ether (20 mL) and the solution cooled to −78 °C. nBuLi (0.56 mL, 1.6 M in hexanes, 0.90 mmol) was added dropwise via syringe and the resultant solution was allowed to warm to room temperature and stirred overnight. The solution was concentrated in vacuo and stored at −30 °C overnight, yielding large colorless blocks of 5 (258 mg, 45%) suitable for X-ray diffraction. M.p 134–136 °C (decomp): 1H NMR (400 MHz, C6D6, 298 K) N.B. integration for cyclohexyl groups are estimated due to complex overlapping signals: δ = 0.47–0.50 (t, 3JHH = 7.0 Hz, 6H, (CH3CH2)2O), 1.26–1.27 (d, 3JHH = 6.9 Hz, 6H, CH(CH3)2), 1.32–1.33 (d, 3JHH = 6.9 Hz, 6H, CH(CH3)2), 1.37–1.83 (m, 25H, Cyc-CH2), 1.88 (s, 3H, NCCH3), 1.93 (s, 3H, NCCH3), 1.95–1.98 (m, 5H, Cyc-CH2), 2.50–2.58 (m, 1H, p-Cyc-CH), 2.75–2.81 (q, 3JHH = 7.0 Hz, 4H, (CH3CH2)2O), 2.94–3.00 (m, 2H, o-Cyc-CH), 3.40 (sept, 3JHH = 6.9 Hz, 2H, CH(CH3)2) 4.96 (s, 1H, NCCH), 7.08–7.20 (m, 5H, overlapping TCHP-ArH & Dip-ArH). 13C{1H} NMR (101 MHz, C6D6, 298 K): δ = 13.7 ((CH3CH2)2O), 23.4, 23.5 (NCCH3), 24.0, 24.3 (CH(CH3)2), 26.7, 27.0, 27.6, 27.7, 27.9, (Cyc-C), 28.3 (CH(CH3)2), 34.2, 35.2, 35.6, 39.5, 45.3 (Cyc-C), 63.7 ((CH3CH2)2O), 92.8 (NCCH), 122.3, 123.1, 123.4,`139.5, 140.9, 141.9, 147.9, 150.1 (ArC), 163.7, 164.0 (NCCH3). 7Li NMR (155 MHz, C6D6, 298 K): δ = 1.88. IR ν/cm−1 (ATR): 621 (w), 1549 (s), 1518 (m), 1148 (w), 1093 (w), 1058 (w), 1016 (w), 953 (w), 926 (w), 863 (m), 831 (w), 788 (w), 758 (s), 731 (s). A reproducible elemental analysis could not be obtained for this compound as it consistently co-crystallized with small amounts of TCHP/DipNacnacH, which could not be separated by repeated recrystallizations.
Preparation of [{K(TCHPNacnac)}2], 6: TCHPNacnacH (400 mg, 0.54 mmol) and benzyl potassium (72 mg, 0.55 mmol) were dissolved in toluene (10 mL) and the mixture stirred overnight at 60 °C. The solution was then cooled to room temperature and volatiles were removed in vacuo. The subsequent solid was dissolved in n-hexane (5 mL) and the extract left to stand at room temperature, yielding colorless crystals of 6 suitable for X-ray diffraction studies. Analysis of the crystalline material by NMR spectroscopy consistently showed contamination of the product with TCHPNacnacH, which could not be removed, despite repeated recrystallizations.
In situ preparation of 6: TCHPNacnacH (10 mg, 0.013 mmol) and benzyl potassium (3 mg, 0.023 mmol) were dissolved in C6D6 (0.6 mL) in a J. Young’s NMR tube equipped with a Teflon screw cap and the mixture heated overnight at 60 °C. The subsequent reaction solution was analyzed by NMR spectroscopy, showing near complete conversion to compound 6. 1H NMR (400 MHz, C6D6, 298 K) N.B. integration for cyclohexyl groups are estimated due to complex overlapping signals: δ = 1.36–1.87 (m, 55H, Cyc-CH2), 1.89 (s, 6H, NCCH3), 2.02–2.06 (m, 5H, Cyc-CH2), 2.54–2.62 (m, 2H, p-Cyc-CH), 3.03–3.10 (m, 4H, o-Cyc-CH), 4.74 (s, 1H, NCCH), 7.16 (s, 4H, TCHP-ArH). 13C{1H} NMR (101 MHz, C6D6, 298 K): δ = 23.8 (NCCH3), 26.8, 27.0, 27.6, 27.8, 28.2, 35.0, 2 × 35.6, 38.8, 45.2 (Cyc-C), 90.1 (NCCH), 122.8, 138.6, 139.6, 140.0, 149.4, (ArC), 161.2, (NCCH3).
Preparation of [{K(TCHP/DipNacnac)}∞], 7. TCHP/DipNacnacH (516 mg, 0.89 mmol) and KHMDS (195 mg, 0.98 mmol) were dissolved in toluene (8 mL) and the mixture heated overnight at 60 °C. The solution was cooled to room temperature, after which time micro-crystalline 7 was deposited. The suspension was filtered, and the solid was dried in vacuo giving compound 7 as a spectroscopically near pure colorless solid (500 mg, 91%). Crystals suitable for X-ray diffraction were grown by slow cooling a saturated solution of 7 in C6D6 from 60 °C to room temperature. M.p. > 260 °C: 1H NMR (400 MHz, C6D6, 298 K) N.B. integration for cyclohexyl groups are estimated due to complex overlapping signals: δ = 1.14–1.17 (d, 3JHH = 6.9 Hz, 6H, CH(CH3)2), 1.24–1.30 (m, 2H, Cyc-CH2), 1.32–1.34 (d, 3JHH = 6.9 Hz, 6H, CH(CH3)2), 1.37–1.84 (m, 24H, Cyc-CH2), 1.89 (s, 3H, NCCH3), 1.91 (s, 3H, NCCH3), 1.97–2.04 (m, 4H, Cyc-CH2), 2.53–2.59 (m, 1H, p-Cyc-CH), 2.96–3.04 (m, 2H, o-Cyc-CH), 3.39 (sept, 3JHH = 6.9 Hz, 2H, CH(CH3)2), 4.80 (s, 1H, NCCH), 7.01–7.19 (m, 5H, overlapping TCHP-ArH & Dip-ArH). 13C{1H} NMR (101 MHz, C6D6, 298 K): δ = 23.6, 23.7 (NCCH3), 24.0, 24.4 (CH(CH3)2), 26.8, 26.9, 27.6, 27.9 (Cyc-C), 28.0 (CH(CH3)2), 34.5, 35.6, 35.8, 39.1, 45.2 (Cyc-C), 91.2 (NCCH), 121.2, 122.8, 123.7, 138.4, 139.6, 140.0, 149.1, 151.2 (ArC), 160.1, 160.9 (NCCH3). IR ν/cm−1 (ATR): 1566 (m), 1518 (m), 1143 (m), 1016 (w), 923 (w), 890 (w), 862 (m), 819 (w), 792 (m), 770 (m), 727 (s). A reproducible elemental analysis could not be obtained for this compound as it consistently co-crystallized with small amounts of TCHP/DipNacnacH, which could not be separated by repeated recrystallizations.
3.4. Crystallographic Details
Crystals of 2–7 suitable for X-ray structural determination were mounted in silicone oil. Crystallographic measurements were carried out at 123(2) K, and were made using a Rigaku Synergy diffractometer using a graphite monochromator with Cu Kα radiation (λ = 1.54184 Å). The structures were solved by direct methods and refined on F2 by full matrix least squares (SHELX16) [33] using all unique data. All non-hydrogen atoms were anisotropic with hydrogen atoms typically included in calculated positions (riding model). Crystal data, details of the data collection and refinement are given in Table S1 (in Supplementary Materials). Crystallographic data for the structures have been deposited with the Cambridge Crystallographic Data Centre (CCDC no. 2105059-2105064). Copies of this information may be obtained free of charge from The Director, CCDC, 12 Union Road, Cambridge, CB2 1EZ, UK (fax: +44-1223-336033; email: [email protected] or www: http://www.ccdc.cam.ac.uk).
4. Conclusions
In summary, the syntheses of four alkali metal complexes of two super bulky β-diketiminate ligand systems are described. These complexes were characterized by X-ray crystallography and multinuclear NMR spectroscopy. The solid-state structures of the lithium etherate complexes are similar to previously reported examples. The molecular structures of the potassium salts show varied coordination environments in potassium, depending on the accompanying ligand framework. That is, [{K(TCHPNacnac)}2] 6 is dimeric with C∙∙∙K interactions involving a meta-aryl carbon of the opposing monomer unit. In contrast, [{K{(TCHP/DipNacnac)}∞] 7 is polymeric, and shows comparable connectivity between monomer units to that seen in the previously reported complex [K(DipNacnac)]. These alkali metal compounds have significant potential for use as ligand transfer reagents in the synthesis of metal complexes incorporating large TCHP-substituted β-diketiminates. We are currently exploring the use of these ligands for the kinetic stabilization of low-oxidation state metal complexes.
Supplementary Materials
The following are available online at https://0-www-mdpi-com.brum.beds.ac.uk/article/10.3390/inorganics9090072/s1, Table S1: Summary of crystallographic data for 2–7; Figures S1–S14: 1H, 13C{1H}, and 7Li{1H} NMR spectra of 2–7; CIF and CheckCIF files of 2–7.
Author Contributions
Conceptualization, D.D.L.J. and C.J.; methodology, D.D.L.J. and S.W.; validation, D.D.L.J. and S.W.; formal analysis, D.D.L.J., S.W. and C.J.; investigation, D.D.L.J. and S.W.; writing—original draft preparation, D.D.L.J.; writing—review and editing, C.J.; visualization, D.D.L.J. and C.J.; supervision, C.J.; project administration, C.J.; funding acquisition, C.J. All authors have read and agreed to the published version of the manuscript.
Funding
CJ is grateful to the Australian Research Council for funding part of this work. Moreover, this material is based upon work supported by the Air Force Office of Scientific Research under award number FA2386-21-1-4048.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
NMR spectra and crystal data are given in the supporting information. Crystal data, details of the data collection and refinement are given in Table S1. Crystallographic data for the structures have been deposited with the Cambridge Crystallographic Data Centre (CCDC no. 2105059-2105064). Copies of this information may be obtained free of charge from The Director, CCDC, 12 Union Road, Cambridge, CB2 1EZ, UK (fax: +44-1223-336033; email: [email protected] or http://www.ccdc.cam.ac.uk).
Conflicts of Interest
The authors declare no conflict of interest. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript, or in the decision to publish the results.
References
- Bourget-Merle, L.; Lappert, M.F.; Severn, J.R. The Chemistry of β-Diketiminatometal Complexes. Chem. Rev. 2002, 102, 3031–3066. [Google Scholar] [CrossRef] [PubMed]
- Sarish, S.P.; Nembenna, S.; Nagendran, S.; Roesky, H.W. Chemistry of Soluble β-Diketiminatoalkaline-Earth Metal Complexes with M−X Bonds (M = Mg, Ca, Sr; X = OH, Halides, H). Acc. Chem. Res. 2011, 44, 157–170. [Google Scholar] [CrossRef] [PubMed]
- Yao, S.; Driess, M. Lessons from Isolable Nickel(I) Precursor Complexes for Small Molecule Activation. Acc. Chem. Res. 2012, 45, 276–287. [Google Scholar] [CrossRef]
- Tsai, Y. The chemistry of univalent metal β-diketiminates. Coord. Chem. Rev. 2012, 256, 722–758. [Google Scholar] [CrossRef]
- Zhu, D.; Budzelaar, P.H.M. N-Aryl β-diiminate complexes of the platinum metals. Dalton Trans. 2013, 42, 11343–11354. [Google Scholar] [CrossRef]
- Chen, C.; Bellows, S.M.; Holland, P.L. Tuning steric and electronic effects in transition-metal β-diketiminate complexes. Dalton Trans. 2015, 44, 16654–16670. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hohloch, S.; Kriegel, B.M.; Bergman, R.G.; Arnold, J. Group 5 chemistry supported by β-diketiminate ligands. Dalton Trans. 2016, 45, 15725–15745. [Google Scholar] [CrossRef]
- Camp, C.; Arnold, J. On the non-innocence of “Nacnacs”: Ligand-based reactivity in β-diketiminate supported coordination compounds. Dalton Trans. 2016, 45, 14462–14498. [Google Scholar] [CrossRef]
- Green, S.P.; Jones, C.; Stasch, A. Stable Magnesium(I) Compounds with Mg-Mg Bonds. Science 2007, 318, 1754–1757. [Google Scholar] [CrossRef]
- Stasch, A.; Jones, C. Stable dimeric magnesium(i) compounds: From chemical landmarks to versatile reagents. Dalton Trans. 2011, 40, 5659–5672. [Google Scholar] [CrossRef]
- Jones, C. Dimeric magnesium(I) β-diketiminates: A new class of quasi-universal reducing agent. Nat. Rev. Chem. 2017, 1, 0059. [Google Scholar] [CrossRef]
- Bonyhady, S.; Jones, C.; Nembenna, S.; Stasch, A.; Edwards, A.; McIntyre, G. β-Diketiminate-Stabilized Magnesium(I) Dimers and Magnesium(II) Hydride Complexes: Synthesis, Characterization, Adduct Formation, and Reactivity Studies. Chem. Eur. J. 2010, 16, 938–955. [Google Scholar] [CrossRef]
- Kenward, A.L.; Ross, J.A.; Piers, W.E.; Parvez, M. Metalation-Resistant β-Diketiminato Ligands for Thermally Robust Organoscandium Complexes. Organometallics 2009, 28, 3625–3628. [Google Scholar] [CrossRef]
- Cowley, R.E.; Holland, P.L. Ligand Effects on Hydrogen Atom Transfer from Hydrocarbons to Three-Coordinate Iron Imides. Inorg. Chem. 2012, 51, 8352–8361. [Google Scholar] [CrossRef]
- Arrowsmith, M.; Maitland, B.; Kociok-Köhn, G.; Stasch, A.; Jones, C.; Hill, M.S. Mononuclear Three-Coordinate Magnesium Complexes of a Highly Sterically Encumbered β-Diketiminate Ligand. Inorg. Chem. 2014, 53, 10543–10552. [Google Scholar] [CrossRef] [Green Version]
- Gentner, T.X.; Rösch, B.; Ballmann, G.; Langer, J.; Elsen, H.; Harder, S. Low Valent Magnesium Chemistry with a Super Bulky β-Diketiminate Ligand. Angew. Chem. Int. Ed. 2019, 58, 607–611. [Google Scholar] [CrossRef]
- Yuvaraj, K.; Douair, I.; Jones, D.D.L.; Maron, L.; Jones, C. Sterically controlled reductive oligomerisations of CO by activated magnesium(i) compounds: Deltate vs. ethenediolate formation. Chem. Sci. 2020, 11, 3516–3522. [Google Scholar] [CrossRef] [Green Version]
- Boutland, A.J.; Dange, D.; Stasch, A.; Maron, L.; Jones, C. Two-Coordinate Magnesium(I) Dimers Stabilized by Super Bulky Amido Ligands. Angew. Chem. Int. Ed. 2016, 55, 9239–9243. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yuvaraj, K.; Douair, I.; Paparo, A.; Maron, L.; Jones, C. Reductive Trimerization of CO to the Deltate Dianion Using Activated Magnesium(I) Compounds. J. Am. Chem. Soc. 2019, 141, 8764–8768. [Google Scholar] [CrossRef] [PubMed]
- Yuvaraj, K.; Douair, I.; Maron, L.; Jones, C. Activation of Ethylene by N-Heterocyclic Carbene Coordinated Magnesium(I) Compounds. Chem. Eur. J. 2020, 26, 14665–14670. [Google Scholar] [CrossRef] [PubMed]
- Jones, D.D.L.; Douair, I.; Maron, L.; Jones, C. Photochemically Activated Dimagnesium(I) Compounds: Reagents for the Reduction and Selective C−H Bond Activation of Inert Arenes. Angew. Chem. Int. Ed. 2021, 60, 7087–7092. [Google Scholar] [CrossRef]
- Rösch, B.; Gentner, T.X.; Eyselein, J.; Langer, J.; Elsen, H.; Harder, S. Strongly reducing magnesium(0) complexes. Nature 2021, 592, 717–721. [Google Scholar] [CrossRef] [PubMed]
- Jones, C. Highly reactive form of magnesium stabilized by bulky ligands. Nature 2021, 592, 687–688. [Google Scholar] [CrossRef] [PubMed]
- Rösch, B.; Gentner, T.X.; Langer, J.; Färber, C.; Eyselein, J.; Zhao, L.; Frenking, G.; Harder, S. Dinitrogen complexation and reduction at low-valent calcium. Science 2021, 371, 1125–1128. [Google Scholar] [CrossRef]
- Weerawardhana, E.A.; Pena, A.; Zeller, M.; Lee, W.-T. Synthesis and characterization of iron and cobalt complexes with an asymmetric N-alkyl,N′-aryl-β-diketiminate ligand. Inorg. Chim. Acta 2017, 460, 29–34. [Google Scholar] [CrossRef]
- Stender, M.; Wright, R.J.; Eichler, B.E.; Prust, J.; Olmstead, M.M.; Roesky, H.W.; Power, P.P. The synthesis and structure of lithium derivatives of the sterically encumbered β-diketiminate ligand [{(2,6-Pri2H3C6)N(CH3)C}2CH]−, and a modified synthesis of the aminoimine precursor. J. Chem. Soc. Dalton Trans. 2001, 3465–3469. [Google Scholar] [CrossRef]
- Clegg, W.; Cope, E.K.; Edwards, A.J.; Mair, F.S. Structural Characterization of [(2,6-Pri2C6H3)NC(Me)C(H)C(Me)N(2,6-Pri2C6H3)K·PhCH3]∞: A Heavy Alkali Metal Diazapentadienyl Complex. Inorg. Chem. 1998, 37, 2317–2319. [Google Scholar] [CrossRef]
- Pyykö, P.; Atsumi, M. Molecular Single-Bond Covalent Radii for Elements 1–118. Chem. Eur. J. 2009, 15, 186–197. [Google Scholar] [CrossRef]
- Mantina, M.; Chamberlain, A.C.; Valero, R.; Cramer, C.J.; Truhlar, D.G. Consistent van der Waals Radii for the Whole Main Group. J. Phys. Chem. A 2009, 113, 5806–5812. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dove, A.P.; Gibson, V.C.; Marshall, E.L.; White, A.J.; Williams, D.J. Magnesium and zinc complexes of a potentially tridentate β-diketiminate ligand. Dalton Trans. 2004, 570–578. [Google Scholar] [CrossRef] [PubMed]
- Savka, R.; Plenio, H. Metal Complexes of Very Bulky N,N′-Diarylimidazolylidene N-Heterocyclic Carbene (NHC) Ligands with 2,4,6-Cycloalkyl Substituents. Eur. J. Inorg. Chem. 2014, 2014, 6246–6253. [Google Scholar] [CrossRef]
- Kundu, S.; Sinhababu, S.; Siddiqui, S.S.; Luebben, A.V.; Dittrich, B.; Yang, T.; Frenking, G.; Roesky, H.W. Comparison of Two Phosphinidenes Binding to Silicon(IV)dichloride as well as to Silylene. J. Am. Chem. Soc. 2018, 140, 9409–9412. [Google Scholar] [CrossRef] [PubMed]
- Sheldrick, G.M. SHELX-16. University of Göttingen, 2016. [Google Scholar]
Scheme 1.
Generic reaction of a β-diketiminato alkali metal salt with a metal/metalloid halide.

Figure 1.
(a) Partial thermal ellipsoid plot (25% probability surface for displayed ellipsoids) of the molecular structure of 2 (hydrogen atoms, except H2, omitted; Dip and TCHP groups shown as wireframe for clarity). Selected bond lengths (Å) and angles (°): N(1)-C(1) 1.317(2), C(1)-C(3) 1.419(2), C(3)-C(4) 1.391(2), N(2)-C(4) 1.337(2), N(1)-C(1)-C(3) 121.09(14), C(1)-C(3)-C(4) 126.12(14); (b) thermal ellipsoid plot (25% probability surface) of the molecular structure of 3 (hydrogen atoms, except H1, omitted; TCHP group shown as wireframe for clarity). Selected bond lengths (Å) and angles (°): O(1)-C(1) 1.239(2), C(1)-C(3) 1.420(3), C(3)-C(4) 1.381(2), N(1)-C(4) 1.342(2), O(1)-C(1)-C(3) 123.14(17), C(1)-C(3)-C(4) 123.61(17).
Figure 1.
(a) Partial thermal ellipsoid plot (25% probability surface for displayed ellipsoids) of the molecular structure of 2 (hydrogen atoms, except H2, omitted; Dip and TCHP groups shown as wireframe for clarity). Selected bond lengths (Å) and angles (°): N(1)-C(1) 1.317(2), C(1)-C(3) 1.419(2), C(3)-C(4) 1.391(2), N(2)-C(4) 1.337(2), N(1)-C(1)-C(3) 121.09(14), C(1)-C(3)-C(4) 126.12(14); (b) thermal ellipsoid plot (25% probability surface) of the molecular structure of 3 (hydrogen atoms, except H1, omitted; TCHP group shown as wireframe for clarity). Selected bond lengths (Å) and angles (°): O(1)-C(1) 1.239(2), C(1)-C(3) 1.420(3), C(3)-C(4) 1.381(2), N(1)-C(4) 1.342(2), O(1)-C(1)-C(3) 123.14(17), C(1)-C(3)-C(4) 123.61(17).

Figure 2.
(a) Partial thermal ellipsoid plot (25% probability surface for displayed ellipsoids) of the molecular structure of 4 (hydrogen atoms omitted; TCHP groups shown as wireframe for clarity). Selected bond lengths (Å) and angles (°): Li(1)-O(1) 1.953(4), N(1)-Li(1) 1.921(4), N(2)-Li(1) 1.924(4), N(1)-Li(1)-N(2) 97.67(18); (b) thermal ellipsoid plot (25% probability surface) of the molecular structure of 5 (hydrogen atoms omitted; Dip and TCHP groups shown as wireframe for clarity). Selected bond lengths (Å) and angles (°): Li(1)-O(1) 1.975(4), N(1)-Li(1) 1.927(4), N(2)-Li(1) 1.935(4), N(1)-Li(1)-N(2) 97.79(17).
Figure 2.
(a) Partial thermal ellipsoid plot (25% probability surface for displayed ellipsoids) of the molecular structure of 4 (hydrogen atoms omitted; TCHP groups shown as wireframe for clarity). Selected bond lengths (Å) and angles (°): Li(1)-O(1) 1.953(4), N(1)-Li(1) 1.921(4), N(2)-Li(1) 1.924(4), N(1)-Li(1)-N(2) 97.67(18); (b) thermal ellipsoid plot (25% probability surface) of the molecular structure of 5 (hydrogen atoms omitted; Dip and TCHP groups shown as wireframe for clarity). Selected bond lengths (Å) and angles (°): Li(1)-O(1) 1.975(4), N(1)-Li(1) 1.927(4), N(2)-Li(1) 1.935(4), N(1)-Li(1)-N(2) 97.79(17).

Figure 3.
Partial thermal ellipsoid plot (25% probability surface for displayed ellipsoids) of the molecular structure of 6 (hydrogen atoms omitted; TCHP groups shown as wireframe for clarity). Selected bond lengths (Å) and angles (°): K(1)-N(1) 2.633(3), K(1)-N(2) 2.619(3), K(1)-C(9)’ 3.309(3), N(1)-K(1)-N(2) 68.23(10).
Figure 3.
Partial thermal ellipsoid plot (25% probability surface for displayed ellipsoids) of the molecular structure of 6 (hydrogen atoms omitted; TCHP groups shown as wireframe for clarity). Selected bond lengths (Å) and angles (°): K(1)-N(1) 2.633(3), K(1)-N(2) 2.619(3), K(1)-C(9)’ 3.309(3), N(1)-K(1)-N(2) 68.23(10).

Figure 4.
Partial thermal ellipsoid plot (25% probability surface for displayed ellipsoids) of the molecular structure of 7 (hydrogen atoms omitted; Dip groups shown as wireframe, and TCHP group truncated for clarity). Selected bond lengths (Å) and angles (°): K(1)-N(1) 2.634(3), K(1)-N(2) 2.713(3), K(1)-C(24)’ 3.254(3), K(1)-C(25)’ 3.178(4), N(1)-K(1)-N(2) 68.82(9).
Figure 4.
Partial thermal ellipsoid plot (25% probability surface for displayed ellipsoids) of the molecular structure of 7 (hydrogen atoms omitted; Dip groups shown as wireframe, and TCHP group truncated for clarity). Selected bond lengths (Å) and angles (°): K(1)-N(1) 2.634(3), K(1)-N(2) 2.713(3), K(1)-C(24)’ 3.254(3), K(1)-C(25)’ 3.178(4), N(1)-K(1)-N(2) 68.82(9).

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Jones, D.D.L.; Watts, S.; Jones, C. Synthesis and Characterization of Super Bulky β-Diketiminato Group 1 Metal Complexes. Inorganics 2021, 9, 72. https://0-doi-org.brum.beds.ac.uk/10.3390/inorganics9090072
AMA Style
Jones DDL, Watts S, Jones C. Synthesis and Characterization of Super Bulky β-Diketiminato Group 1 Metal Complexes. Inorganics. 2021; 9(9):72. https://0-doi-org.brum.beds.ac.uk/10.3390/inorganics9090072
Chicago/Turabian StyleJones, Dafydd D. L., Samuel Watts, and Cameron Jones. 2021. "Synthesis and Characterization of Super Bulky β-Diketiminato Group 1 Metal Complexes" Inorganics 9, no. 9: 72. https://0-doi-org.brum.beds.ac.uk/10.3390/inorganics9090072
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.