
Synthesis of N,O-Chelating Hydrazidopalladium Complexes from 1,2-Bis(trifluoroacetyl)hydrazine


Department of Chemical Science and Engineering, Tokyo Institute of Technology, School of Materials and Chemical Technology, 2-12-1-E4-1 O-okayama, Meguro-ku, Tokyo 152-8552, Japan
*
Author to whom correspondence should be addressed.
†
Deceased on 21 April 2017.
Inorganics 2021, 9(10), 76; https://0-doi-org.brum.beds.ac.uk/10.3390/inorganics9100076
Submission received: 10 September 2021
/
Revised: 4 October 2021
/
Accepted: 5 October 2021
/
Published: 7 October 2021
(This article belongs to the Section Organometallic Chemistry)
Abstract
:N,O-chelating dicarbonylhydrazido-palladium complexes were synthesized by treatment of 1,2-bis(trifluoroacetyl)hydrazine with a mixture of a Pd(0) source, [Pd(dba)2] (DBA = dibenzylideneacetone), and four-electron donors including 1,3-bis(diphenylphosphino)propane (DPPP), N,N,N′,N′-tetramethylethylenediamine (TMEDA), and two equivalents of triphenylphosphine. The same products from DPPP and TMEDA could be obtained alternatively by using Pd(OAc)2 through deprotonation of the diacylhydrazine. The five-membered chelate structure was confirmed by NMR spectra and X-ray crystal structure determination. The X-ray structures indicate that the products are formally considered as Pd(II) complexes with a hydrazido(2–) ligand. In the case of the triphenylphosphine-coordinated complex, a fluxional behavior in dichloromethane-d2 was observed by variable temperature NMR experiments, possibly due to structural changes between the square planar and pseudo-tetrahedral geometries.
1. Introduction
Transformation of hydrazine on metals has received much attention with a view to its potential synthetic utility and enzymatic nitrogen fixation mechanism [1,2,3,4]. A number of transition metal complexes exhibiting protean coordination modes of hydrazine and its derivatives have been reported [5]. As for the variable chemical properties of the hydrazine family, substituted hydrazines have been subjected to specific reactions involving N–N and N–H bond cleavages. In light of reductive N–N bond transformations promoted by carbonyl functional groups [6,7,8], acylhydrazines are expected to be eligible for oxidative addition. However, the N–N bond scission has not been observed for low-valent late transition metal complexes [9] with the exception of a highly strained hydrazine analog, specifically di-tert-butyldiaziridinone, convertible to a four-membered oxidative addition product by Pd(0) species [10].
Introduction of acyl substituents into hydrazine also exerts a stimulatory influence on the access to hydrazides [11], as typified by the platinum-catalyzed intramolecular hydrohydrazination of alkenes [12]. Enhanced acidity of hydrazines attached to the electron withdrawing groups facilitates the formation of hydrazido complexes, as seen in widespread studies on the deprotonative conversion of 1,2-diacylhydrazines [13,14,15,16,17,18,19,20]. Dilworth treated 1,2-dibenzoylhydrazine with cis-[PtCl2(PPh3)2] in the presence of NaHCO3 to obtain a mononuclear dibenzoylhydrazido(2–) complex [13]. A non-symmetrical N,O-chelating structure of the hydrazidoplatinum complex was crystallographically determined by Ibers around the same time [14] and analogous platinum complexes were synthesized instead by coordination of dicarbonyldiazenes to [Pt(PPh3)2(C2H4)] [15]. Aside from the luminescence application of dinuclear complexes connected by a diacylhydrazido bridge [21,22], the diacylhydrazine framework has been utilized as a substructure of air-stable and oxidatively degradable high-performance polymers [23], and represents a versatile building block for the synthesis of 1,3,4-oxadiazoles [24,25,26,27] with regard to straightforward methods to construct heterocycles containing the N–N bond [28]. In the context of these attractive aspects, we turned our attention to an activated hydrazine molecule, 1,2-bis(trifluoroacetyl)hydrazine (1), in which two strongly electron-withdrawing carbonyl groups were expected to facilitate N–N and N–H bond transformations [29]. We herein disclose the reactivity of 1 towards Pd(0) species, in conjunction with its deprotonative coordination toward Pd(II) variants.
2. Results and Discussion
2.1. Reaction of 1,2-Bis(trifluoroacetyl)hydrazine in the Presence of Pd(0)
Coordination of the electron-deficient hydrazine 1 was examined by treatment with a versatile Pd(0) source, [Pd(dba)2] [30]. When 1 reacted with an equimolar mixture of [Pd(dba)2] and 1,3-bis(diphenylphosphino)propane (DPPP) in THF under reflux conditions for 19 h, a new air-stable Pd complex (2) was obtained as a yellow solid after removing the solvent under vacuum (Scheme 1). Product 2 could be purified by washing with diethyl ether and following recrystallization by slow diffusion of diethyl ether into a solution in CH2Cl2 at room temperature. The 19F NMR spectrum of 2 in CD2Cl2 exhibited a doublet signal at −68.2 ppm with a small coupling constant of 2 Hz and an independent singlet signal at −68.8 ppm, while no N–H peaks appeared in the 1H NMR spectrum. An unsymmetrical coordination structure of 2 was also suggested by the 31P{1H} NMR spectrum, in which a set of two doublet signals at 15.7 and 9.4 ppm were shown to be coupled to each other with JPP = 48 Hz and the latter resonance was observed with an additional JFP coupling of 2 Hz due to the fluorine nuclei on the trifluoroacetyl substituent. Finally, X-ray crystallography confirmed the atom connectivity of 2, deciphering that the dehydrogenated 1,2-diacylhydrazido unit coordinates with the palladium center non-equivalently through the nitrogen and oxygen atoms to form a five-membered chelate (vide infra).
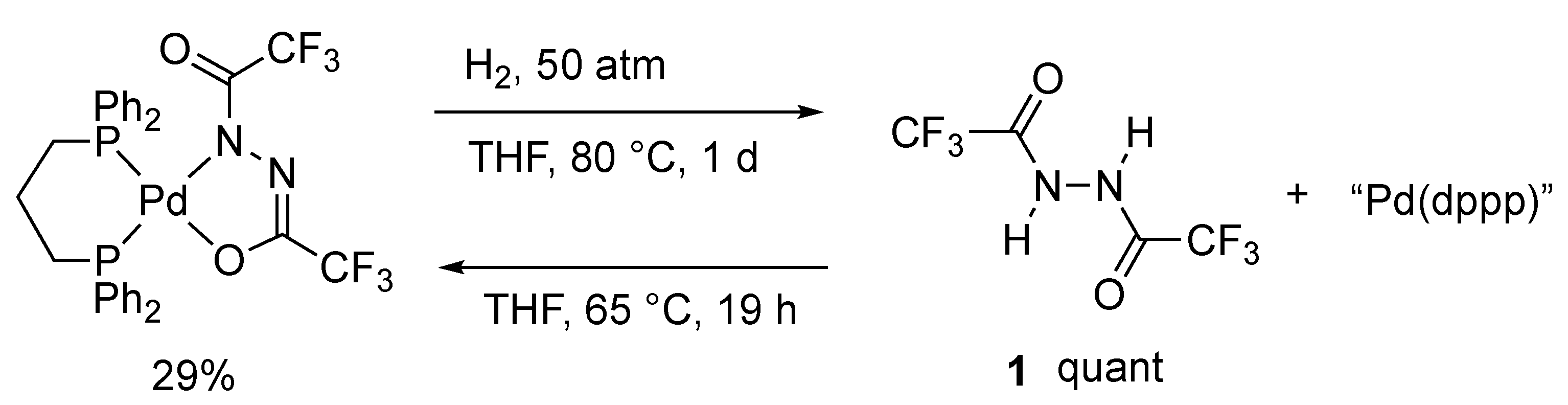
In the monitoring experiment performed in a closed NMR tube containing stoichiometric amounts (10 μmol) of 1, [Pd(dba)2], and DPPP in THF-d8 (0.045 mL) at 65 °C for 19 h, a signal ascribed to H2 was not observed in the 1H NMR spectrum. The concentration was possibly below the detection limit by NMR spectroscopy, even in the case of the quantitative formation of H2, considering the fact that H2 concentration in THF-d8 (0.050 mL) under atmospheric H2 (0.08 mmol) in an NMR tube was estimated to be 19 mM [9]. Oxidative addition of the N–N bond was not observed throughout the reaction, unlike the reported O–O and N–O bond cleavage by Pd(0) [31,32]. Additionally, a quantitative recovery of 1 from 2 was observed by treatment with a pressurized H2 (50 atm) in THF at 80 °C for one day (Scheme 2). It is noteworthy that the subsequent reoxidation of 1 using the resulting mixture proceeded at 65 °C to afford 2 with a moderate conversion of 29%. The reversibility of the hydrogen transfer stood in the way of the dehydrogenative coordination of 1 in the closed reaction system.
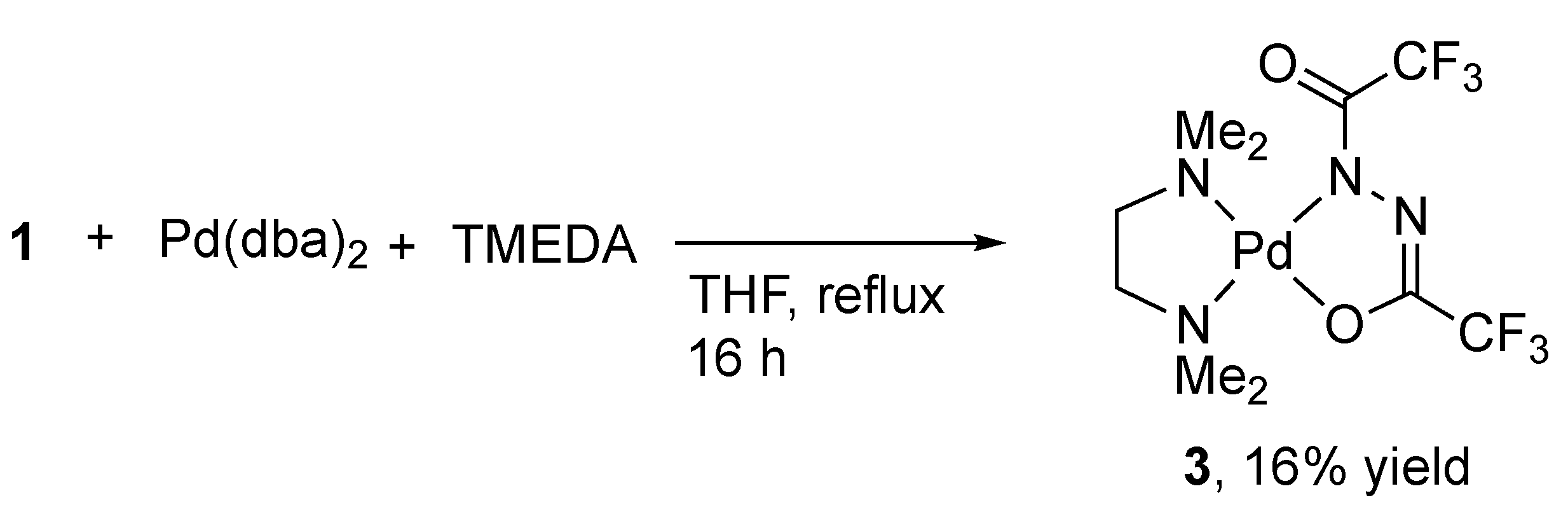
A similar conversion of 1 into an N,O-chelate was investigated by using N,N,N′,N′-tetramethylethylenediamine (TMEDA) in place of DPPP (Scheme 3). After refluxing a mixture of 1, [Pd(dba)2], and TMEDA in THF for 19 h, a yellow solid of 1,2-diacylhydrazido complex 3 was isolated in 12% yield after recrystallization from a mixture of CH2Cl2 and diethyl ether. The 19F NMR spectrum of 3 in CD2Cl2 showed two separate singlet signals at −68.0 and −68.9 ppm. The carbonyl carbon resonances were detected as two quartet signals at 152.9 and 162.4 ppm, accommodating the unsymmetrical N,O-chelating ligand. The X-ray crystal structure and elemental analysis also recognized 3 as an analog of 2.
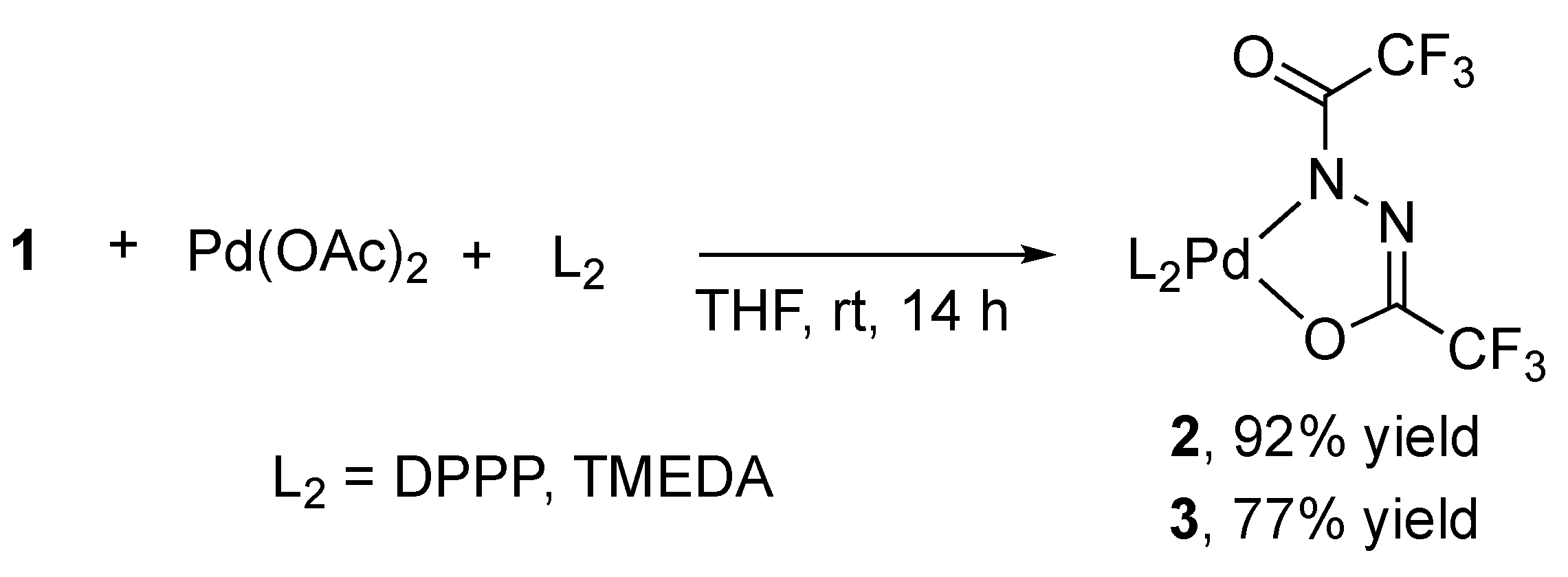
2.2. Alternative Synthesis of Diacylhydrazido Complexes from 1 and Pd(OAc)2
To verify the dihydrazido structure originated from 1, we next tried alternative synthesis of 2 and 3 from Pd(II) species. On the ground of the studies on the Pt congeners prepared from 1,2-diacylhydrazines and [PtCl2(PPh3)2] [13,14,15], 1 was treated with Pd(OAc)2 in the presence of DPPP or TMEDA in THF at room temperature for 14 h. The deprotonation of 1 proceeded without base additives with the aid of the favorable leaving ability of acetate rather than chloride. After removal of acetic acid from the reaction mixture by washing with diethyl ether, 2 and 3 were isolated in the yield of 92% and 77%, respectively (Scheme 4). The identical spectroscopic results of the products from [Pd(dba)2] and Pd(OAc)2 corroborate the dehydrogenative coordination of 1 to the Pd(0) precursor.
2.3. Crystal Structure of Diacylhydrazido Complexes 2 and 3
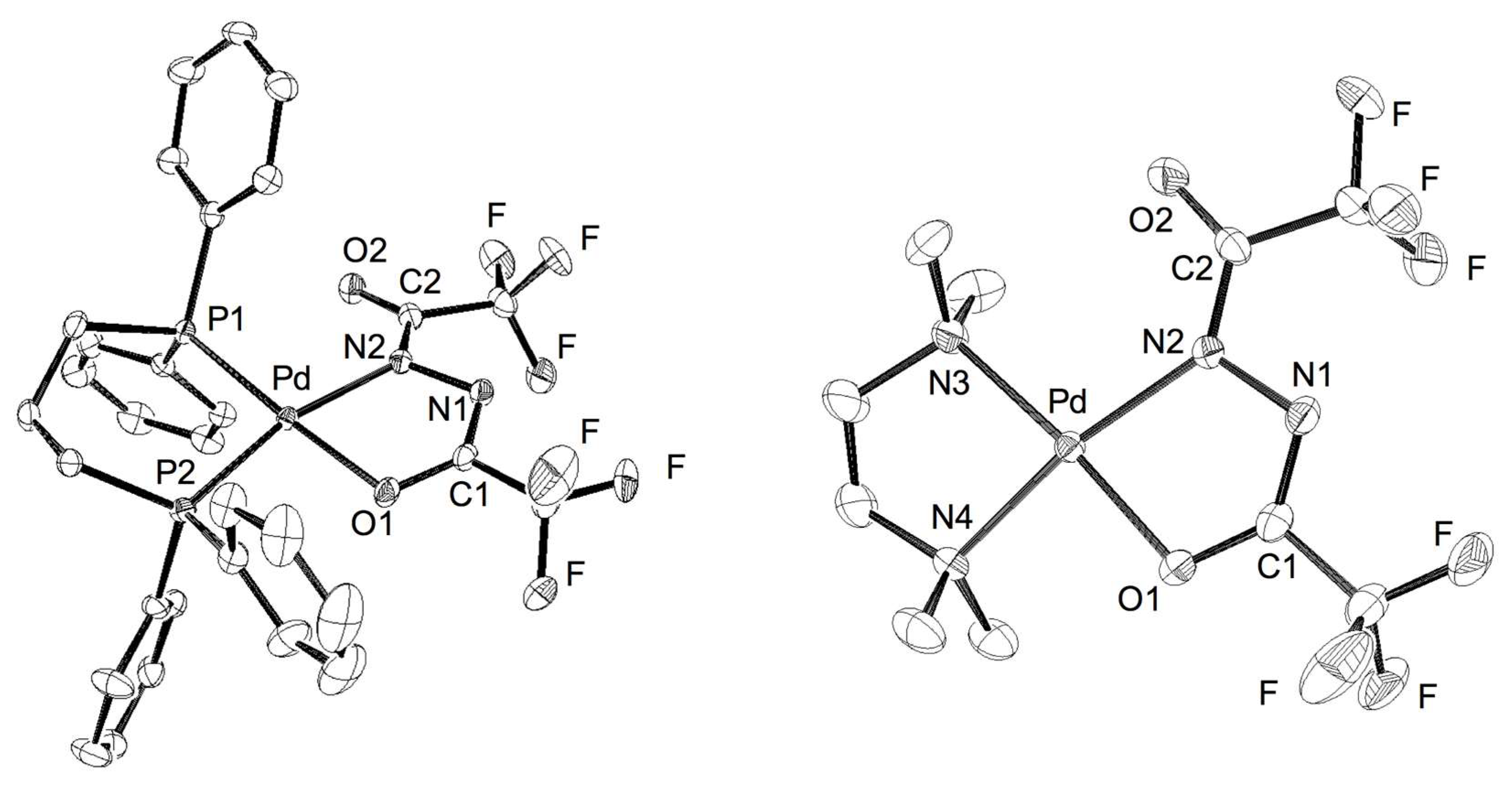
The molecular structures of 2 and 3 were identified by X-ray crystallographic analysis. The ORTEP drawings are depicted in Figure 1 and both the relevant bond lengths and angles are listed in Table 1 and Table 2. Both complexes featured a square planar arrangement of the Pd center coordinated with a diphosphine or diamine ligand and a N,O-chelate originated from 1.
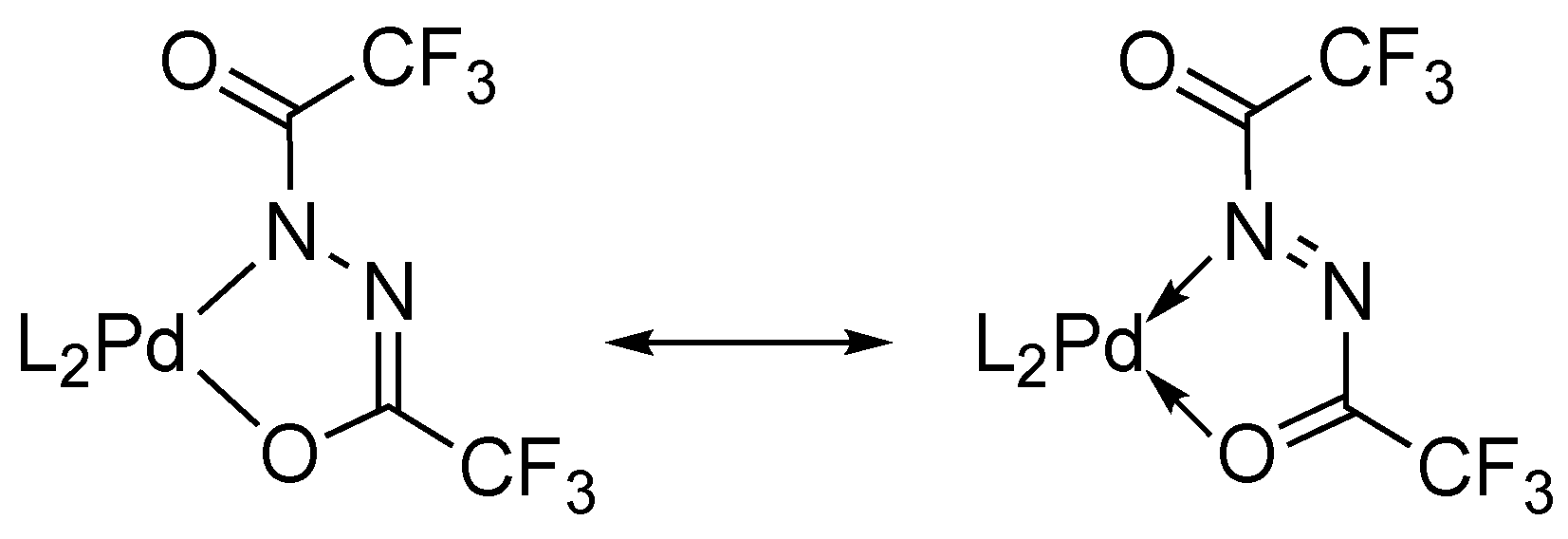
The five-membered ring containing Pd, O1, C1, N1, and N2 atoms is nearly planar. The structures of 2 and 3 were compatible with the geometric attributes observed in the reported Pt complexes with diacylhydrazido(2–) skeletons [13,14,15]. Both palladium complexes involve elongated C1–O1 bonds (1.297(6) and 1.295(5) Å) relative to exocyclic carbonyl C2–O2 bonds (1.228(6) and 1.227(5) Å), along with a specific intermediate N1–N2 bond length (1.425(6) and 1.418(5) Å) between single and double bonds. Relatively short bond lengths of N1–C1 (1.281(7) and 1.289(5) Å) indicate a nature of double bonds. These structural data explicitly support the potent contribution of a divalent Pd center on 2 and 3 in preference to another resonance form of diazene-Pd(0), as shown in Scheme 5. In accordance with the formation of hydrazido(2–)-based Pd(II) species from the Pd(0) precursor, the oxidation of 1 can be accounted for by a plausible pathway involving oxidative addition of the N–H bond to generate a hydrido(hydrazido)palladium intermediate and the following tautomerization to an enol form, which is accessible to the hydrazido(2–) moiety with releasing H2 gas via intramolecular protonation of the hydrido ligand.
2.4. Synthesis and NMR Analyses of PPh3-Coordinated Diacylhydrazido Complex 4
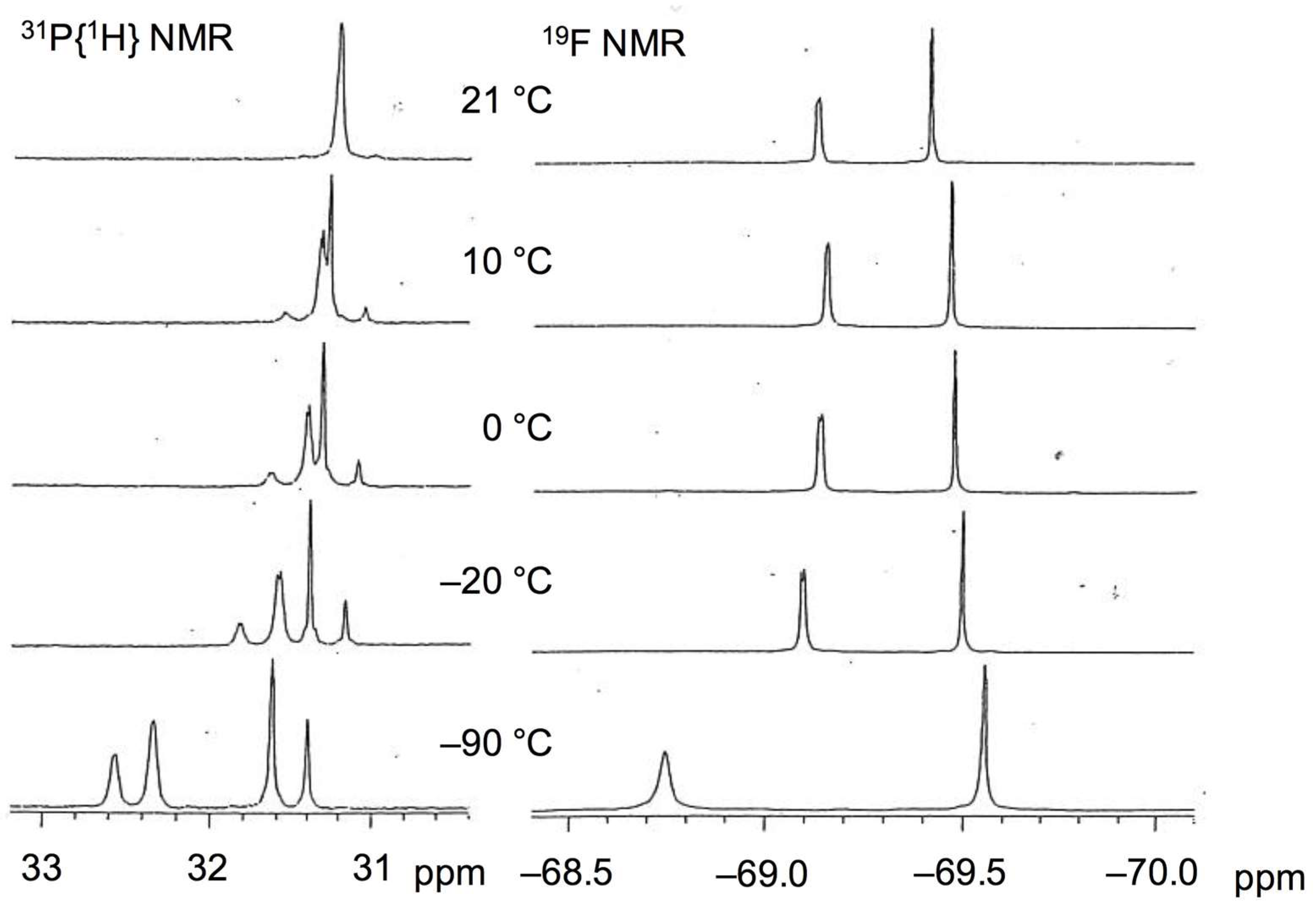
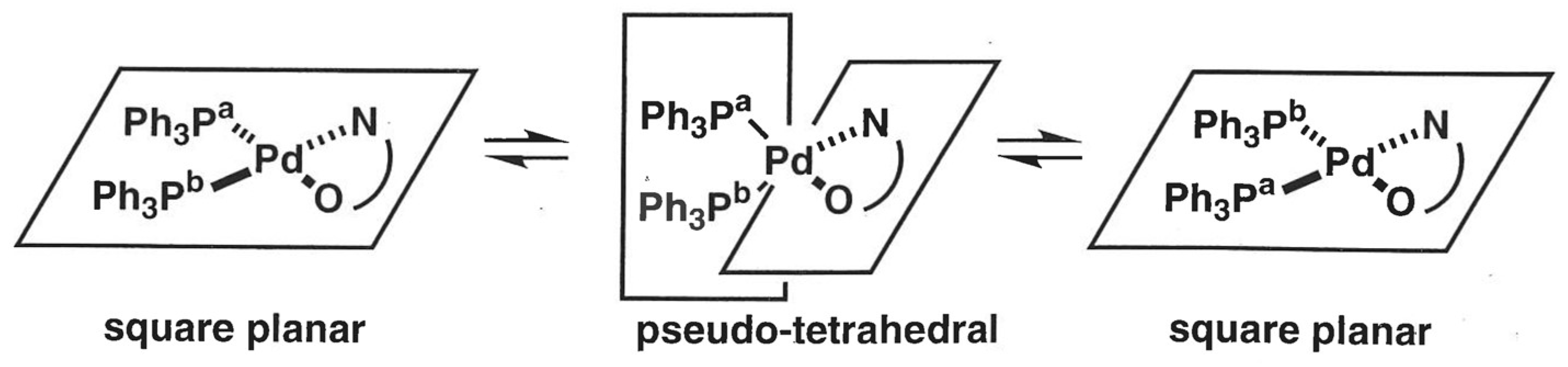
Following a similar procedure as for the DPPP and TMEDA complexes, 1 could be transformed dehydrogenatively into a monodentate phosphine-ligated palladium complex (4) (Scheme 6). After refluxing a mixture of 1, [Pd(dba)2], and two molar amounts of PPh3 in THF for 19 h, an orange-red solid of bis(triphenylphosphine)palladium 4 was isolated in 27% yield. Inequivalent N,O-coordination of 1,2-bis(trifluoroacetyl)hydrazido in 4 was corroborated by two independent 19F NMR signals at −69.2 and −69.5 ppm in CD2Cl2. Interestingly, the 31P{1H} NMR spectrum of 3 displayed only a singlet signal at 31.2 ppm at room temperature, distinct from that of the DPPP-coordinated complex 2. Lowering the temperature below −90 °C resulted in a peak separation as an AB quartet, as shown in Figure 2.
The dynamic behavior is possibly due to a rapid formal rearrangement of PPh3 ligands via intramolecular twist-rotation involving transient pseudo-tetrahedral species, as depicted in Scheme 7. A similar fluxional motion relevant to the square planar/tetrahedral interconversion has been investigated in group 9 and 10 metal complexes, with and without the changing of spin states [33,34,35,36,37,38,39]. Based on the fact that the variable temperature 31P{1H} NMR spectra of 4 were not altered by the addition of an extra molar equivalent of PPh3, associative and dissociative processes other than the unimolecular motion can be excluded [40]. In line with the two-sided character of the N–O chelate, the Pd(0) character arising from a carbonyldiazene ligand will contribute to reducing barriers on the path to the pseudo-tetrahedral structure, allowing for the facile twist-rotation.
3. Materials and Methods
3.1. General Information
All manipulations of oxygen and moisture-sensitive materials were performed under argon atmosphere using standard Schlenk techniques. Solvents were purchased from Kanto Chemical Co., Inc. (Tokyo, Japan), dried by refluxing over sodium benzophenone ketyl (THF and diethyl ether) or P2O5 (CH3CN and CH2Cl2), and distilled under argon before use. Dichloromethane-d2 was degassed by three freeze-pump-thaw cycles and purified by trap-to-trap distillation after being dried with P2O5. Tetrahydrofuran-d8 was analogously degassed after trap-to-trap distillation by using CaH2 as a drying agent. The other reagents were purchased from Sigma-Aldrich Co. LLC. (St. Louis, MO, USA); Tokyo Chemical Industry Co., Ltd. (Tokyo, Japan); and Kanto Chemical Co., Ltd. (Tokyo, Japan), and used as delivered. Pd2(dba)4 [30] and 1,2-bis(trifluoroacetyl)hydrazine [29] were prepared according to the procedures described in the literature with modifications. 1H, 19F, 13C{1H}, and 31P{1H} NMR spectra were recorded on JEOL JNM-LA300 and JNM-ECX400 spectrometers (JEOL Ltd., Tokyo, Japan) at around 25 °C unless otherwise noted. The chemical shifts were referenced to an external tetramethylsilane signal (0.0 ppm) by using the signals of residual proton impurities in the deuterated solvents for 1H NMR and by using the solvent resonance for 13C{1H} NMR. The 19F and 31P{1H} chemical shifts were referenced to external CF3COOH (−76.5 ppm) and H3PO4 (0.0 ppm) signals, respectively. Abbreviations for NMR are as follows: s = singlet, d = doublet, t = triplet, q = quartet, m = multiplet, or br = broad. IR spectra were recorded on a JASCO FT/IR-610 spectrometer (JASCO Corporation, Tokyo, Japan). Elemental analyses were carried out using a PE2400 Series II CHNS/O analyzer (PerkinElmer, Inc., Waltham, MA, USA).
3.2. Reaction of 1 with Pd(0) Species
3.2.1. Formation of 2 by Treatment of 1 with [Pd(dba)2] and DPPP
A THF (10 mL) solution of 1 (0.13 g, 0.58 mmol) was added to a mixture of [Pd(dba)2] (0.33 g, 0.58 mmol) and DPPP (0.15 g, 0.58 mmol) in THF (15 mL) at room temperature. After stirring at a refluxing temperature for 19 h, the reaction mixture was filtered through a pad of activated charcoal on Celite. The solvent was removed under reduced pressure, and the resulting residue was washed with diethyl ether. Evaporation to dryness gave a yellow powder of 2 (0.34 g, 45% yield). Single crystals suitable for X-ray diffraction analysis were obtained by slow diffusion of diethyl ether into the solution in CH2Cl2.
Mp. 211–216 °C (decomp.). 1H NMR (300.5 MHz, tetrahydrofuran-d8, RT): δ 1.88–2.09 (m, 2H), 2.20–2.41 (m, 4H), 7.38–7.57 (m, 12H), 7.67–7.73 (m, 4H), 7.80–7.88 (m, 4H). 19F NMR (282.8 MHz, CD2Cl2, RT): δ –68.8 (s) –68.2 (d, 5JFP = 2 Hz). 31P{1H} NMR (121.7 MHz, CD2Cl2, RT): δ 9.4 (dq, 2JPP = 48 Hz, 5JFP = 2 Hz) 15.7 (d, 2JPP = 48 Hz). 13C{1H} NMR (100.5 MHz, CD2Cl2, RT): δ 18.4, 23.0 (dd, JCP = 33 and 7 Hz), 28.5 (dd, JCP = 35 and 14 Hz), 117.4 (q, 1JCF = 292 Hz, CF3), 119.8 (q, 1JCF = 277 Hz, CF3), 128.3 (d, JCP = 13 Hz), 128.6 (d, JCP = 118 Hz), 129.2 (d, JCP = 126 Hz), 129.3 (d, JCP = 12 Hz), 131.2 (d, JCP = 3 Hz), 131.8 (d, JCP = 3 Hz), 132.9 (d, JCP = 12 Hz), 133.2 (d, JCP = 11 Hz), 154.0 (q, 2JCF = 34 Hz), 157.7 (q, 2JCF = 31 Hz). IR (cm–1, KBr): 1638, 1437, 1369, 1201, 1168, 1141, 1101. Anal. Calcd for C31H26N2O2P2F6Pd: C 50.25, H 3.54 N 3.78. Found: C 50.41, H 3.73, N 3.85.
3.2.2. Formation of 3 by Treatment of 1 with [Pd(dba)2] and TMEDA
A THF (10 mL) solution of 1 (97 mg, 0.43 mmol) was added to a mixture of [Pd(dba)2] (0.25 g, 0.44 mmol) and TMEDA (6.8 × 10–2 mL, 0.45 mmol) in THF (10 mL) at room temperature. After stirring at a refluxing temperature for 16 h, the reaction mixture was filtered through a pad of activated charcoal on Celite. The solvent was removed under reduced pressure, and the resulting residue was washed with diethyl ether. Evaporation to dryness gave an orange powder of 3 (32 mg, 16% yield). Single crystals suitable for X-ray diffraction analysis were obtained by slow diffusion of diethyl ether into the solution in CH2Cl2.
Mp. 246–248 °C (decomp.). 1H NMR (399.8 MHz, CD2Cl2, RT): δ 2.67 (s, 4H), 2.75 (s, 6H), 3.06 (s, 6H). 19F NMR (376.2 MHz, CD2Cl2, RT): δ –68.9 (s) –68.0 (s). 13C{1H} NMR (100.5 MHz, CD2Cl2, RT): δ 51.2, 51.5, 61.1, 64.9, 117.0 (q, 1JCF = 276 Hz, CF3), 117.4 (q, 1JCF = 291 Hz, CF3), 152.9 (q, 2JCF = 35 Hz), 162.4 (q, 2JCF = 30 Hz). IR (cm–1, KBr): 1644, 1469, 1437, 1364, 1156. Anal. Calcd for C31H26N2O2P2F6Pd: C 27.01, H 3.63 N 12.60. Found: C 27.05, H 3.65, N 12.48.
3.2.3. Formation of 4 by Treatment of 1 with [Pd(dba)2] and PPh3
A THF (10 mL) solution of 1 (0.16 g, 0.71 mmol) was added to a mixture of [Pd(dba)2] (0.40 g, 0.70 mmol) and PPh3 (0.37 g, 1.4 mmol) in THF (15 mL) at room temperature. After stirring at a refluxing temperature for 19 h, the reaction mixture was filtered through a pad of activated charcoal on Celite. The solvent was removed under reduced pressure and the resulting residue was washed with diethyl ether. Evaporation to dryness gave a red powder of 4 (0.16 g, 27% yield). Single crystals suitable for X-ray diffraction analysis were obtained by slow diffusion of diethyl ether into the solution in CH2Cl2.
Mp. 162–166 °C (decomp.); 1H NMR (300.5 MHz, CD2Cl2, RT): δ 7.06–7.36 (m, 30H); 19F NMR (282.8 MHz, CD2Cl2, RT): δ –69.5 (s) –69.2 (s); 31P{1H} NMR (121.7 MHz, CD2Cl2, RT): δ 31.2; IR (cm–1, KBr): 1654, 1438, 1355, 1177, 1122, and 1096; Anal. Calcd for C40H30N2O2P2F6Pd: C 56.32, H 3.54 N 3.28; and found: C 55.80, H 3.70, N 3.18.
3.3. Alternative Synthesis of 2 and 3 Derived from a Pd(II) Precursor
A mixture of Pd(OAc)2 (228 mg, 1.0 mmol) in THF (20 mL) was added a solution of DPPP or TMEDA (1.0 mmol) in THF (10 mL). Subsequently, a solution of 1 (0.23 g, 1.0 mmol) in THF (10 mL) added to the reaction mixture. After stirring at room temperature for 14 h, the mixture was concentrated to ca. 10 mL under reduced pressure. The resulting precipitate was collected by filtration and washed with diethyl ether (10 mL). Evaporation to dryness gave the product 2 or 3 as a yellow powder in the yield of 92% and 77%, respectively.
3.4. NMR Monitoring on the Formation of 2
An NMR tube equipped with a J-Young valve was loaded with equimolar amounts (1.0 × 10−2 mmol) of 1, [Pd(dba)2], and DPPP, and was dissolved in THF-d8 (0.045 mL) at room temperature. The resulting solution was degassed by three freeze-pump-thaw cycles. After heating the mixture at 65 °C for 19 h, the products were analyzed by 1H NMR spectroscopy at room temperature.
3.5. Reaction of 2 with Pressurized H2 and Subsequent Dehydrogenation
A 50 mL stainless steel autoclave equipped with a pressure gauge and a magnetic stirrer was loaded with 2 (0.14 mmol) in THF (15 mL) under an argon atmosphere. The autoclave was flushed with H2 and then pressurized to 50 atm. The solution was stirred at 80 °C for 26.5 h. After carefully venting hydrogen, a sample of the reaction mixture was diluted in CD2Cl2 under argon and 19F NMR was recorded. Subsequently, the resulting mixture was transferred to a 20 mL Schlenk tube and stirred at 65 °C for 16 h. The recovery of 2 from the hydrogenolysis products was confirmed by 19F NMR.
3.6. X-ray Crystal Structure Determination
All measurements were made on a Rigaku Saturn 70 (Rigaku Corporation, Tokyo, Japan) using graphite-monochromated Mo-Kα radiation (λ = 0.71075 Å) under nitrogen stream at 193 K. Single crystals suitable for X-ray analyses were mounted on glass fibers. The crystal-to-detector distance was 45.0 mm. Data were collected to a maximum 2θ value of 55.0°. A total of 720 oscillation images were collected. A sweep of the data was carried out by using ω scans from −110.0 to 70.0° in 0.5° steps at χ = 45.0° and ϕ = 0.0°. A second sweep was performed by using ω scans from −110.0 to 70.0° in 0.5° steps at χ = 45.0° and ϕ = 90.0°. Intensity data were collected for Lorentz-polarization effects and absorption. Details of crystal and data collection parameters for the complexes 2 and 3 are summarized in Tables S1 and S2. Structure solution and refinements were performed with the CrystalStructure program package [41]. The heavy atom positions were determined by a direct program method (SIR92) [42] and the remaining non-hydrogen atoms were found by subsequent Fourier syntheses as well as refined by full-matrix least-squares techniques against F2 using the SHELXL-2014/7 program [43]. The hydrogen atoms were placed at calculated positions and were refined with a riding model.
4. Conclusions
In summary, new mononuclear N,O-chelating hydrazidopalladium complexes 2–4 were prepared by dehydrogenative coordination of 1 to Pd(0) complexes, having relevance to the oxidative transformation of hydrazine into diazene. In consort with the alternative synthesis of 2 and 3 by spontaneous deprotonation of 1 using Pd(II) acetate, the hydrazido(2–) component derived from 1 was structurally verified by X-ray crystallography. Another contribution of the diazene-Pd(0) character was possibly actualized in solution, leading to dynamic behavior due to the unimolecular twist-rotation of the square-planar Pd(II) complex. These experimental results indicate the potential of related acylhydrazido/acyldiazene units to serve as redox-active ligands in analogy with well-known α-diimine systems.
Supplementary Materials
The following are available online at https://0-www-mdpi-com.brum.beds.ac.uk/article/10.3390/inorganics9100076/s1, copies of NMR spectra of 2–4.
Author Contributions
Conceptualization, Y.K.; methodology, Y.K.; validation, Y.K.; formal analysis, Y.K. and T.H.; investigation, Y.K. and T.H.; data curation, Y.K.; writing, Y.K.; project administration, Y.K.; funding acquisition, T.I.; supervision, T.I. All authors except T.I. have read and agreed to the published version of the manuscript.
Funding
This study received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
The data presented in this study are available in the Supplementary Materials.
Acknowledgments
We are grateful for the support of the GCOE Program.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Lundgren, R.J.; Stradiotto, M. Palladium-Catalyzed Cross-Coupling of Aryl Chlorides and Tosylates with Hydrazine. Angew. Chem. Int. Ed. 2010, 49, 8686–8690. [Google Scholar] [CrossRef]
- Kinjo, R.; Donnadieu, B.; Bertrand, G. Gold-Catalyzed Hydroamination of Alkynes and Allenes with Parent Hydrazine. Angew. Chem. Int. Ed. 2011, 50, 5560–5563. [Google Scholar] [CrossRef] [PubMed]
- Umehara, K.; Kuwata, S.; Ikariya, T. N–N Bond Cleavage of Hydrazines with a Multi-Proton Responsive Pincer-Type Iron Complex. J. Am. Chem. Soc. 2010, 135, 6754–6757. [Google Scholar] [CrossRef] [PubMed]
- Kiernicki, J.J.; Zeller, M.; Szymczak, N.K. Hydrazine Capture and N–N Bond Cleavage at Iron Enabled by Flexible Appended Lewis Acids. J. Am. Chem. Soc. 2017, 139, 18194–18197. [Google Scholar] [CrossRef]
- Heaton, B.T.; Jacob, C.; Page, P. Transition metal complexes containing hydrazine and substituted hydrazines. Coord. Chem. Rev. 1996, 154, 193–229. [Google Scholar] [CrossRef]
- Mellor, J.M.; Smith, N.M. Reductive cleavage of the nitrogen-nitrogen bond in hydrazine derivatives. J. Chem. Soc. Perkin Trans. 1984, 1, 2927–2931. [Google Scholar] [CrossRef]
- Ding, H.; Friestad, G.K. Trifluoroacetyl-Activated Nitrogen–Nitrogen Bond Cleavage of Hydrazines by Samarium(II) Iodide. Org. Lett. 2004, 6, 637–640. [Google Scholar] [CrossRef]
- Dey, S.; Gadakh, S.K.; Ahuja, B.B.; Kamble, S.P.; Sudalai, A. Pd-catalyzed reductive cleavage of N–N bond in dibenzyl-1-alkylhydrazine-1,2-dicarboxylates with PMHS: Application to a formal enantioselective synthesis of (R)-sitagliptin. Tetrahedron Lett. 2016, 57, 684–687. [Google Scholar] [CrossRef]
- Hoover, J.M.; Freudenthal, J.; Michael, F.E.; Mayer, J.M. Reactivity of Low-Valent Iridium, Rhodium, and Platinum Complexes with Di- and Tetrasubstituted Hydrazines. Organometallics 2008, 27, 2238–2245. [Google Scholar] [CrossRef]
- Zhao, B.; Du, H.; Cui, S.; Shi, Y. Synthetic and Mechanistic Studies on Pd(0)-Catalyzed Diamination of Conjugated Dienes. J. Am. Chem. Soc. 2010, 132, 3523–3532. [Google Scholar] [CrossRef] [Green Version]
- Ligandro, E.; Perdicchia, D. N-Acylhydrazines: Future Perspectives Offered by New Syntheses and Chemistry. Eur. J. Org. Chem. 2004, 2004, 665–675. [Google Scholar] [CrossRef]
- Hoover, J.M.; DiPasquale, A.; Mayer, J.M.; Michael, F.E. Platinum-Catalyzed Intramolecular Hydrohydrazination: Evidence for Alkene Insertion into a Pt–N Bond. J. Am. Chem. Soc. 2010, 132, 5043–5053. [Google Scholar] [CrossRef] [PubMed]
- Dilworth, J.R.; Kasenally, A.S. Dibenzoyl- and diacetylhydrazido(2–) complexes of platinum. J. Organomet. Chem. 1973, 60, 203–207. [Google Scholar] [CrossRef]
- Ittel, S.D.; Ibers, J.A. The Structure of Bis(triphenylphosphine)(dibenzoylhydrazido)platinum, Pt[P(C6H5)3]2[C6H5CONNCOC6H5]·C2H5OH. Inorg. Chem. 1973, 12, 2290–2295. [Google Scholar] [CrossRef]
- Chan, D.; Cronin, L.; Duckett, S.B.; Hupfield, P.; Perutz, R.N. Synthesis, structure and reactivity of N,O-metallacyclic (dicarbonyldiazene) platinum complexes. New. J. Chem. 1998, 22, 511–516. [Google Scholar] [CrossRef]
- Kasenally, A.S.; Hussein, F.M. Hydrazido complexes of the transition metals. Synthesis and reactions of dibenzoyl- and diacetylhydrazido(2–) (N,N′,O,O′)-bis(carbonyltriphenylphosphine)-rhodium(I) and –iridium(I). J. Organomet. Chem. 1976, 111, 355–359. [Google Scholar] [CrossRef]
- Zambrano, C.H.; Sharp, P.R.; Barnes, C.L. Synthesis and X-ray crystal structure of an iridium N,O-chelating hydrazido complex. Polyhedron 1996, 15, 3653–3657. [Google Scholar] [CrossRef]
- Fujisawa, K.; Sugiyama, M.; Miyashita, Y.; Okamoto, K.-i. Structure and chemical properties of a copper(II) hydrazido complex: [{Cu(HB(3,5-iPr2pz)3)}2(μ-NCOOEt)2]. Inorg. Chem. Commun. 2009, 12, 246–248. [Google Scholar] [CrossRef]
- Mondal, S.; Filippou, V.; Bubrin, M.; Schwederski, B.; Fiedler, J.; Kaim, W. Metallo with 1,2-Dipivaloylhydrazido Ligands: Electron Transfer and Alkylation/Protonation Effects. Eur. J. Inorg. Chem. 2019, 2019, 2639–2647. [Google Scholar] [CrossRef]
- Jana, R.; Sarkar, B.; Bubrin, D.; Fiedler, J.; Kaim, W. Structure electrochemistry and spectroscopy of a new diacylhydrazido-bridged diruthenium complex with a strongly near-infrared absorbing RuIIIRuII intermediate. Inorg. Chem. Commun. 2010, 13, 1160–1162. [Google Scholar] [CrossRef]
- Congrave, D.G.; Hsu, Y.-T.; Batsanov, A.S.; Beeby, A.; Bryce, M.R. Sky-blue emitting bridged diiridium complexes: Beneficial effects of intramolecular π–π stacking. Dalton Trans. 2018, 47, 2086–2098. [Google Scholar] [CrossRef] [Green Version]
- Congrave, D.G.; Batsanov, A.S.; Bryce, M.R. Highly luminescent 2-phenylpyridine-free diiridium complexes with bulky 1,2-diarylimidazole cyclometalating ligands. Dalton Trans. 2018, 47, 16524–16533. [Google Scholar] [CrossRef] [Green Version]
- Nagashima, K.; Kihara, N.; Iino, Y. Oxidative Coupling Polymerization of Bishydrazide for the Synthesis of Poly(diacylhydrazine): Oxidative Preparation of Oxidatively Degradable Polymer. J. Polym. Sci. Part A: Polym. Chem. 2012, 50, 4230–4238. [Google Scholar] [CrossRef]
- De Oliveira, C.S.; Lira, B.F.; Barbosa-Filho, J.M.; Lorenzo, J.G.F.; De Athayde-Filho, P.F. Synthetic Approaches and Pharmacological Activity of 1,3,4-Oxadiazoles: A Review of the Literature from 2000–2012. Molecules 2012, 17, 10192–10231. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herrero, M.A.; Wannberg, J.; Larhed, M. Direct Microwave Synthesis of N,N′-Diacylhydrazines and Boc-protected Hydrazines by in situ Carbonylations under Air. Synlett 2004, 2004, 2335–2338. [Google Scholar]
- Andersen, T.L.; Caneschi, W.; Ayoub, A.; Lindhardt, A.T.; Couri, M.R.C.; Skrydstrup, T. 1,2,4- and 1,3,4-Oxadiazole Synthesis by Palladium-Catalyzed Carbonylative Assembly of Aryl Bromides with Amidoximes or Hydrazides. Adv. Synth. Catal. 2014, 356, 3074–3082. [Google Scholar] [CrossRef]
- Mloston, G.; Obijalska, E.; Zurawik, A.; Heimgartner, H. Efficient synthesis of tri- and difluoroacetyl hydrazides as useful building blocks for non-symmetrically substituted, fluoroalkylated 1,3,4-oxadiazoles. Chem. Heterocycl. Compd. 2016, 52, 133–139. [Google Scholar] [CrossRef] [Green Version]
- Tšupova, S.; Mäeorg, U. Hydrazines and azo-compounds in the synthesis of heterocycles comprising N-N bond. Heterocycles 2014, 88, 129–173. [Google Scholar]
- Bredikhin, A.; Tšubrik, O.; Sillard, R.; Mäeorg, U. Increasing the N–H Acidity: Introduction of Highly Electronegative Groups into the Hydrazine Molecule. Synlett 2005, 2005, 1939–1941. [Google Scholar]
- Takahashi, Y.; Ito, T.; Sakai, S.; Ishii, Y.; Bonnet, J.J.; Ibers, J.A. A novel palladium(0) complex; bis(dibenzylideneacetone)palladium(0). J. Chem. Soc. D Chem. Commun. 1970, 17, 1065–1066. [Google Scholar] [CrossRef]
- Unlike BzOOBz: Bird, C.; Booth, B.L.; Haszeldine, R.N.; Neuss, G.R.H.; Smith, M.A.; Flood, A. Reactions Involving Transition Metals. Part 18. Reactions of Transition-metal Complexes with Peroxycarboxylic Acids, Diacyl Peroxides, and t-Butyl Peroxybenzoate. J. Chem. Soc. Dalton Trans. 1982, 6, 1109–1114. [Google Scholar] [CrossRef]
- He, J.; Shigenari, T.; Yu, J.-Q. Palladium(0)/PAr3-Catalyzed Intermolecular Amination of C(sp3)–H Bonds: Synthesis of β-Amino Acids. Angew. Chem. Int. Ed. 2015, 54, 6545–6549. [Google Scholar] [CrossRef]
- Azizian, H.; Dixon, K.R.; Eaborn, C.; Pidcock, A.; Shuaib, N.M.; Vinaixa, J. Dynamic Stereochemistry of cis-[PtH(SiR3)(PPh3)2]. Spontaneous Ligand Interchange with Retention of Nuclear Spin-Spin Correlation. J. Chem. Soc. Chem. Commun. 1982, 17, 1020–1022. [Google Scholar] [CrossRef]
- Wouters, J.M.A.; Klein, R.A.; Elsevier, C.J.; Häming, L.; Stam, C.H. Synthesis of trans-(σ-Allenyl)platinum(II) and -palladium(II) Compounds. X-ray Crystal Structure of trans-[PtBr{C(H)=C=Me2}(PPh3)2] and Highly Diastereoselective trans-cis Isomerization of (σ-Allenyl)palladium(II) Bromides. Organometallics 1994, 13, 4586–4593. [Google Scholar] [CrossRef] [Green Version]
- Obora, Y.; Tsuji, Y.; Nishiyama, K.; Ebihara, M.; Kawamura, T. Structure and Fluxional Behavior of cis-Bis(stannyl)bis(phosphine)platinum: Oxidative Addition of Organodistannane to Platinum(0) Complex. J. Am. Chem. Soc. 1996, 118, 10922–10923. [Google Scholar] [CrossRef]
- Tsuji, Y.; Nishiyama, K.; Hori, S.-i.; Ebihara, M.; Kawamura, T. Structure and Facile Unimolecular Twist Rotation of cis-Bis(silyl)bis(phosphine)platinum and cis-Bis(stannyl)bis(phosphine)palladium Complexes. Organometallics 1998, 17, 507–512. [Google Scholar] [CrossRef]
- Mori, Y.; Shirase, H.; Fukuda, Y. Substituent and Solvent Effects on Square-Planar–Tetrahedral Equilibria of Bis[4-(arylimino)pentan-2-onato]nickel(II) and Bis[1-aryl-3-(phenylimino)butan-1-onato]nickel(II) Complexes. Bull. Chem. Soc. Jpn. 2008, 81, 1108–1115. [Google Scholar] [CrossRef]
- Maganas, D.; Grigoropoulos, A.; Staniland, S.S.; Chatziefthimiou, S.D.; Harrison, A.; Robertson, N.; Kyritsis, P.; Neese, F. Tetrahedral and Aquare Planar Ni[(SPR2)2N]2 Complexes, R = Ph & iPr Revisited: Experimetal and Theoretical Analysis of Interconversion Pathways, Structural Preferences, and Spin Delocalization. Inorg. Chem. 2010, 49, 5079–5093. [Google Scholar]
- Sherbo, R.S.; Bindra, G.S.; Budzelaar, P.H.M. Square-Planar–Tetrahedral Interconversion without Spin Flip in (β-diiminate)Rh(1,3-diene) Complexes. Organometallics 2016, 35, 2039–2048. [Google Scholar] [CrossRef]
- Zhou, X.; Lau, K.-C.; Petro, B.J.; Jordan, R.F. cis/trans Isomerization of o-Phosphino-Arenesulfonate Palladium Methyl Complexes. Organometallics 2014, 33, 7209–7214. [Google Scholar] [CrossRef]
- CrystalStructure 4.1: Crystal Structure Analysis Package; Rigaku Coorporation: Tokyo, Japan, 2015.
- Altomare, A.; Cascarano, G.; Giacovazzo, C.; Guagliardi, A.; Burla, M.; Polidori, G.; Camalli, M. SIR92—A program for automatic solution of crystal structures by direct methods. J. Appl. Cryst. 1994, 27, 435. [Google Scholar] [CrossRef]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Crystallogr. Sect. C 2015, 71, 3–8. [Google Scholar] [CrossRef] [PubMed]
Scheme 1.
Synthesis of 2 from 1 and [Pd(dba)2].

Scheme 2.
Hydrogenolysis of 2 and the reverse dehydrogenation of 1.

Scheme 3.
Synthesis of 3 from 1 and [Pd(dba)2].

Scheme 4.
Alternative synthesis of 2 and 3 from Pd(OAc)2.

Figure 1.
ORTEP drawings of 2 (left) and 3 (right). All hydrogens are omitted for clarity. Thermal ellipsoids are shown at the 30% probability level.
Figure 1.
ORTEP drawings of 2 (left) and 3 (right). All hydrogens are omitted for clarity. Thermal ellipsoids are shown at the 30% probability level.

Scheme 5.
Contribution of hydrazido-Pd(II) and diazene-Pd(0) structures.

Scheme 6.
Synthesis of 4.

Figure 2.
The variable temperature 31P{1H} and 19F NMR spectra of 4 in CD2Cl2.

Scheme 7.
A plausible intramolecular motion.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Selected bond distances (Å) and angles (deg) for 2.
| Pd–O1 | 2.046 (4) | Pd–P1 | 2.2522 (13) |
| Pd–N2 | 2.063 (4) | Pd–P2 | 2.2692 (14) |
| N1–N2 | 1.425 (6) | ||
| N1–C1 | 1.281 (7) | N2–C2 | 1.327 (6) |
| C1–O1 | 1.297 (6) | C2–O2 | 1.228 (6) |
| O1–Pd–N2 | 78.15 (15) | Pd–N2–N1 | 113.5 (3) |
| N2–Pd–P1 | 100.63 (12) | N2–N1–C1 | 109.5 (4) |
| P1–Pd–P2 | 93.60 (5) | N1–C1–O1 | 128.8 (5) |
| P2–Pd–O1 | 87.78 (11) | C1–O1–Pd | 109.5 (3) |
Table 2.
Selected bond distances (Å) and angles (deg) for 3.
| Pd–O1 | 1.997 (3) | Pd–N3 | 2.059 (4) |
| Pd–N2 | 2.034 (3) | Pd–N4 | 2.058 (3) |
| N1–N2 | 1.418 (5) | ||
| N1–C1 | 1.289 (5) | N2–C2 | 1.322 (5) |
| C1–O1 | 1.295 (5) | C2–O2 | 1.227 (5) |
| O1–Pd–N2 | 80.20 (12) | Pd–N2–N1 | 112.3 (2) |
| N2–Pd–N3 | 104.62 (13) | N2–N1–C1 | 109.8 (3) |
| N3–Pd–N4 | 85.16 (13) | N1–C1–O1 | 128.6 (4) |
| N4–Pd–O1 | 90.11 (12) | C1–O1–Pd | 109.0 (2) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Kayaki, Y.; Hayakawa, T.; Ikariya, T. Synthesis of N,O-Chelating Hydrazidopalladium Complexes from 1,2-Bis(trifluoroacetyl)hydrazine. Inorganics 2021, 9, 76. https://0-doi-org.brum.beds.ac.uk/10.3390/inorganics9100076
AMA Style
Kayaki Y, Hayakawa T, Ikariya T. Synthesis of N,O-Chelating Hydrazidopalladium Complexes from 1,2-Bis(trifluoroacetyl)hydrazine. Inorganics. 2021; 9(10):76. https://0-doi-org.brum.beds.ac.uk/10.3390/inorganics9100076
Chicago/Turabian StyleKayaki, Yoshihito, Tomohiro Hayakawa, and Takao Ikariya. 2021. "Synthesis of N,O-Chelating Hydrazidopalladium Complexes from 1,2-Bis(trifluoroacetyl)hydrazine" Inorganics 9, no. 10: 76. https://0-doi-org.brum.beds.ac.uk/10.3390/inorganics9100076
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.