
Genetic Evidence of an Isolation Barrier between Flea Subspecies of Citellophilus tesquorum (Wagner, 1898) (Siphonaptera: Ceratophyllidae)


 ,
,
Abstract
:Simple Summary
Abstract

1. Introduction
2. Materials and Methods
2.1. Sample Collection
2.2. DNA Extraction, Amplification and Sequencing
2.3. Evolutionary Analysis
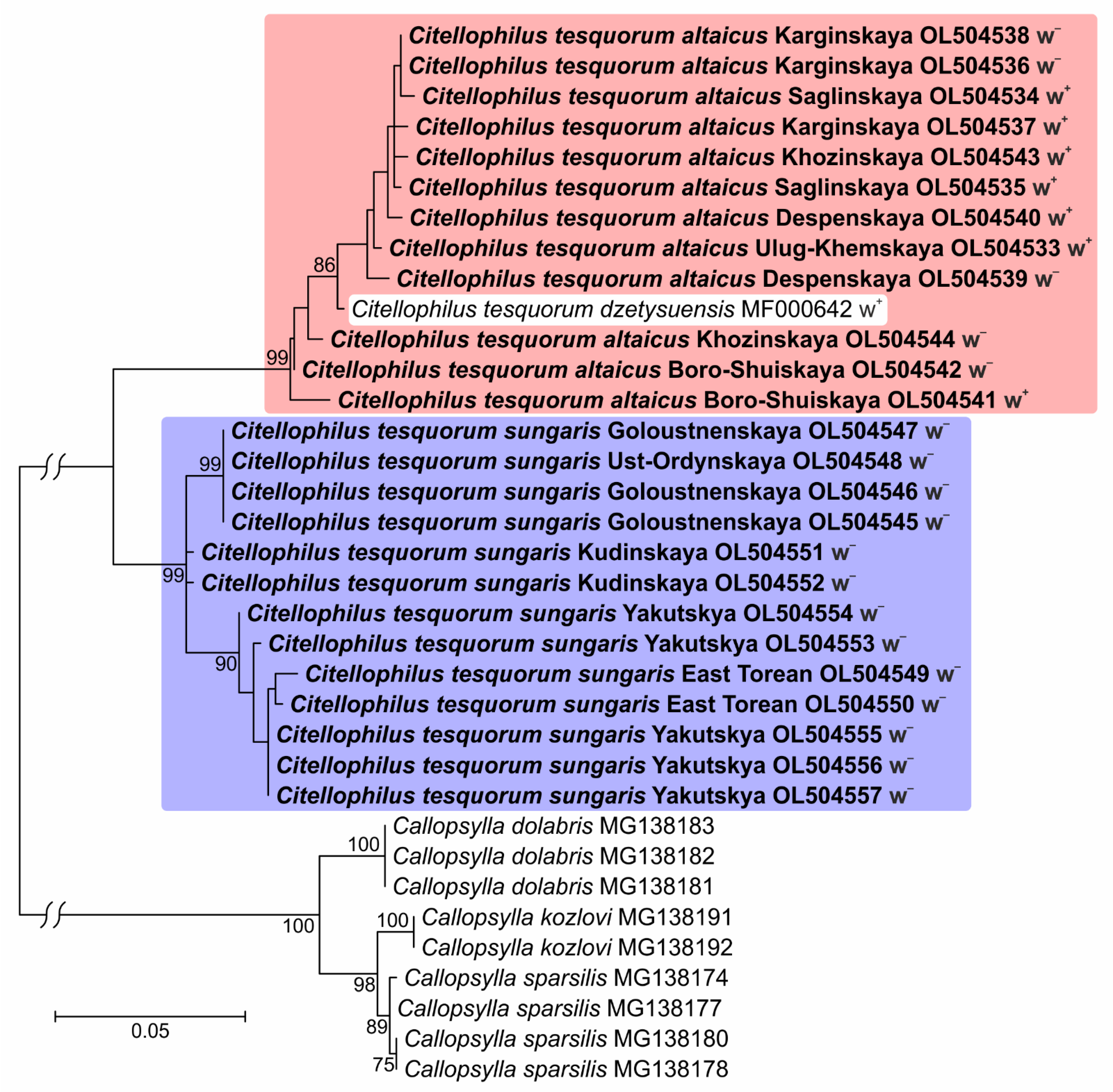
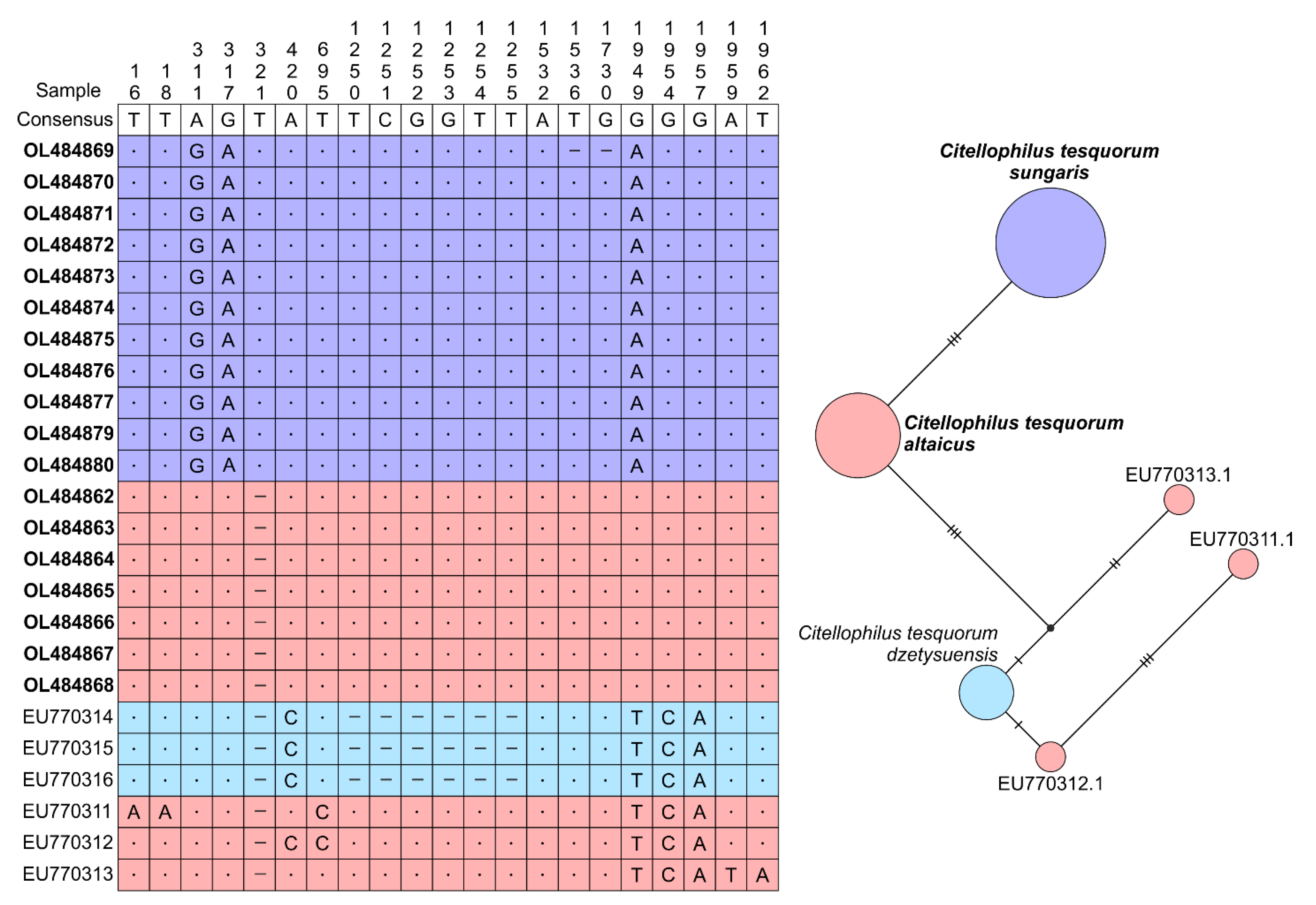
3. Results
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Medvedev, S.G.; Lobanov, A.L.; Lyanguzov, I.A. World Database of Fleas (Nov 2004 version). In Species—2000 & ITIS Catalogue of Life—2005 Annual Checklist [CD-ROM]; Bisby, F.A., Ruggiero, M.A., Wilson, K.L., Cachuela-Palacio, M., Kimani, S.W., Roskov, Y.R., Soulier-Perkins, A., van Hertum, J., Eds.; Species 2000: Reading, UK, 2005. [Google Scholar]
- Bitam, I.; Dittmar, K.; Parola, P.; Whiting, M.F.; Raoult, D. Fleas and flea-borne diseases. Int. J. Infect. Dis. 2010, 14, e667–e676. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zangwill, K.M. Cat Scratch Disease and Bartonellaceae: The Known, the Unknown and the Curious. Pediatric Infect. Dis. J. 2021, 40, S11–S15. [Google Scholar] [CrossRef] [PubMed]
- Glatter, K.A.; Finkelman, P. History of the plague: An ancient pandemic for the age of COVID-19. Am. J. Med. 2021, 134, 176–181. [Google Scholar] [CrossRef] [PubMed]
- Dubyanskiy, V.M.; Yeszhanov, A.B. Ecology of Yersinia pestis and the epidemiology of plague. In Yersinia Pestis: Retrospective and Perspective; Advances in Experimental Medicine and Biology; Yang, R., Anisimov, A., Eds.; Springer: Berlin, Germany, 2016; Volume 918, pp. 101–170. [Google Scholar]
- Medvedev, S.G.; Kotti, B.K.; Verzhutsky, D.B. Diversity of Fleas (Siphonaptera), Vectors of Plague Pathogens: The Flea Citellophilus tesquorum (Wagner, 1898), a Parasite of Ground Squirrels of the Genus Spermophilus. Entomol. Rev. 2019, 99, 565–579. [Google Scholar] [CrossRef]
- Hinnebusch, B.J.; Chouikha, I.; Sun, Y.C. Ecological opportunity, evolution, and the emergence of flea-borne plague. Infect. Immun. 2016, 84, 1932–1940. [Google Scholar] [CrossRef] [Green Version]
- Lawrence, A.L.; Brown, G.K.; Peters, B.; Spielman, D.S.; Morin-Adeline, V.; Šlapeta, J. High phylogenetic diversity of the cat flea (Ctenocephalides felis) at two mitochondrial DNA markers. Med. Vet. Entomol. 2014, 28, 330–336. [Google Scholar] [CrossRef]
- Lawrence, A.L.; Hii, S.; Jirsová, D.; Panáková, L.; Ionică, A.M.; Gilchrist, K.; Modrý, D.; Mihalca, A.D.; Webb, C.E.; Traub, R.J.; et al. Integrated morphological and molecular identification of cat fleas (Ctenocephalides felis) and dog fleas (Ctenocephalides canis) vectoring Rickettsia felis in central Europe. Vet. Parasitol. 2015, 210, 215–223. [Google Scholar] [CrossRef]
- Zhao, F.; Zhang, T.; Su, J.; Huang, Z.; Wu, A.; Lin, G. Genetic differentiation of the oriental rat flea, Xenopsylla cheopis, from two sympatric host species. Parasites Vectors 2018, 11, 1–6. [Google Scholar] [CrossRef]
- Zurita, A.; Callejón, R.; García-sánchez, Á.M.; Urdapilleta, M.; Lareschi, M.; Cutillas, C. Origin, evolution, phylogeny and taxonomy of Pulex irritans. Med. Vet. Entomol. 2019, 33, 296–311. [Google Scholar] [CrossRef]
- Wagner, J. Aphanipterologische Studien. III. Ueber die Gattung Pulex und Beschreibungneuer Arten der Gattungen Ceratophyllus, Ctenopsylla, Ceratopsylla und Typhlopsylla. Trudy Russk. Ent. Obshch. 1898, 31, 555–594. [Google Scholar]
- Tan, J.A.; Shen, E.L. The Atlas of Plague and Its Environment in the People’s Republic of China; Science Press: Beijing, China, 2000. [Google Scholar]
- Traub, R.E.; Rothschild, M.; Haddow, J.F. The Ceratophyllidae: Key to the genera and host relationships, with notes on their evolution, zoogeography and medical importance; Cambridge University Press: Cambridge, UK, 1983; p. 288. [Google Scholar]
- Lewis, R.E. The Ceratophyllidae: Currently accepted valid taxa (Insecta: Siphonaptera). In Koenigstein, Koeltz Scientific Books; Koeltz: Koenigstein, Germany, 1990; p. 267. [Google Scholar]
- Lewis, R.E. Siphonaptera, 15th ed.; Iowa State University: Ames, IA, USA, 2003; p. 62. [Google Scholar]
- Jordan, K. On fleas collected by Dr. H. M. Jettmar in Mongolia and Manchuria in 1927 and 1928. Novit. Zool. 1929, 34, 155–164. [Google Scholar]
- Cyprich, D.; Kiefer, M.; Krumpal, M. Revision of the genus Citellophilus (Siphonaptera) from Mongolia. Acta Fac. Ferum Nat. Univ. Comen. Zool. 1985, 28, 33–56. [Google Scholar]
- Verzhutsky, D.B.; Verzhutskaya, J.u.A.; Kholin, A.V.; Medvedev, S.G. The boundary of the areas of two subspecies of fleas—Parasites of Ground squirrels (Citellophilus tesquorum sungaris and Citellophilus tesquorum altaicus). Baikalskij Zool. Žurnal 2021, 1, 116–120. [Google Scholar]
- Folmer, O.; Black, M.; Hoeh, W.; Lutz, R.; Vrijenhoek, R. DNA primers for amplification of mitochondrial cytochrome C oxidase subunit I from diverse metazoan invertebrates. Mol. Mar. Biol. 1994, 3, 294–299. [Google Scholar]
- Bykov, R.; Kerchev, I.; Demenkova, M.; Ryabinin, A.; Ilinsky, Y.Y. Sex-Specific Wolbachia Infection Patterns in Populations of Polygraphus proximus Blandford (Coleoptera; Curculionidae: Scolytinae). Insects 2020, 11, 547. [Google Scholar] [CrossRef]
- Ilinsky, Y. Coevolution of Drosophila melanogaster mtDNA and Wolbachia genotypes. PLoS ONE 2013, 8, e54373. [Google Scholar] [CrossRef] [Green Version]
- White, T.J.; Bruns, T.; Lee, S.J.; Taylor, J. Amplification and direct sequencing of fungal ribosomal RNA genes for phylogenetics. In PCR Protocols: A Guide to Methods and Applications; Academic Press: Cambridge, MA, USA, 1990; Volume 18, pp. 315–322. [Google Scholar]
- Gamerschlag, S.; Mehlhorn, H.; Heukelbach, J.; Feldmeier, H.; D’Haese, J. Repetitive sequences in the ITS1 region of the ribosomal DNA of Tunga penetrans and other flea species (Insecta, Siphonaptera). Parasitol. Res. 2008, 102, 193–199. [Google Scholar] [CrossRef]
- Navajas, M.; Gutierrez, J.; Bonato, O.; Bolland, H.R.; Mapangou-Divassa, S. Intraspecific diversity of the cassava green mite Mononychellus progresivus (Acari: Tetranychidae) using comparisons of mitochondrial and nuclear ribosomal DNA sequences and cross-breeding. Exp. Appl. Acarol. 1994, 18, 351–360. [Google Scholar] [CrossRef] [Green Version]
- Vobis, M. Flöhe und Ihre Molekularbiologische Charakterisier-ung. Diploma Thesis, Heinrich-Heine-University, Düsseldorf, Germany, 2002. [Google Scholar]
- Lo, N.; Casiraghi, M.; Salati, E.; Bazzocchi, C.; Bandi, C. How many Wolbachia supergroups exist? Mol. Biol. Evol. 2002, 19, 341–346. [Google Scholar] [CrossRef] [Green Version]
- Baldo, L.; Hotopp, J.C.D.; Jolley, K.A.; Bordenstein, S.R.; Biber, S.A.; Choudhury, R.R.; Hayashi, C.; Maiden, M.C.; Tettelin, H.; Werren, J.H. Multilocus sequence typing system for the endosymbiont Wolbachia pipientis. Appl. Environ. Microbiol. 2006, 72, 7098–7110. [Google Scholar] [CrossRef] [Green Version]
- Edgar, R.C. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Librado, P.; Rozas, J. DnaSP v5: A software for comprehensive analysis of DNA polymorphism data. Bioinformatics 2009, 25, 1451–1452. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tamura, K.; Stecher, G.; Peterson, D.; Filipski, A.; Kumar, S. MEGA6: Molecular evolutionary genetics analysis version 6.0. Mol. Biol. Evol. 2013, 30, 2725–2729. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clement, M.; Posada, D.; Crandall, K.A. TCS: A computer program to estimate gene genealogies. Mol. Ecol. 2000, 9, 1657–1659. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leigh, J.W.; Bryant, D. PopART: Full-feature software for haplotype network construction. Methods Ecol Evol. 2015, 6, 1110–1116. [Google Scholar] [CrossRef]
- Jolley, K.A.; Bray, J.E.; Maiden, M.C. Open-access bacterial population genomics: BIGSdb software, the PubMLST. org website and their applications. Wellcome Open Res. 2018, 3, 124. [Google Scholar] [CrossRef] [PubMed]
- Verzhutsky, D.B. Spatial Organization of the Host Population and Its Ectoparasites; Palmarium Academic Publishing: Saarbrucken, Germany, 2012; p. 360. (In Russian) [Google Scholar]
- Van der Mescht, L.; Matthee, S.; Matthee, C.A. New taxonomic and evolutionary insights relevant to the cat flea, Ctenocephalides felis: A geographic perspective. Mol. Phylogenetics Evol. 2021, 155, 106990. [Google Scholar] [CrossRef]
- Zurita, A.; García-Sánchez, Á.M.; Cutillas, C. Ctenophthalmus baeticus boisseauorum (Beaucournu, 1968) and Ctenophthalmus apertus allani (Smit, 1955) (Siphonaptera: Ctenophthalmidae) as synonymous taxa: Morphometric, phylogenetic, and molecular characterization. Bull. Entomol. Res. 2020, 110, 663–676. [Google Scholar] [CrossRef]
- Yudina, M.A.; Bykov, R.A.; Kotti, B.K.; Vysochina, N.P.; Stakheev, V.V.; Broshkov, A.D.; Zakharov, I.K.; Ilinsky, Y.Y. Wolbachia Infection in Flea Populations (Insecta: Siphonaptera). Biol. Bull. Rev. 2019, 9, 403–411. [Google Scholar] [CrossRef]
- Nikitin, A.Y.; Nechaeva, L.K. Hybridization of flea subspecies as a possible basis for the method of regulating the number of vectors. Organization of plague surveillance and prevention measures. In Proceedings of the Interstate Scientific and Practical Conference, Almaty, Kazakhstan, 1992; Volume 3, pp. 400–403. [Google Scholar]
- Nikitin, A.Y.; Bazanova, L.P.; Nechaeva, L.K.; Korzun, V.M.; Khabarov, A.V.; Kozets, L.I. Experimental study of the ability of hybrids bred from two subspecies of the flea Citellophilus tesquorum to transmit plague bacillus. Med. Parasitol. Parasit. Dis. 1995, 4, 15–17. (In Russian) [Google Scholar]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Subspecies of Citellophilus tesquorum | Population | No of Localities | No of Samples | No of Wolbachia Infected Samples |
|---|---|---|---|---|
| sungaris | Goloustnenskaya | 3 | 7 | 0 |
| sungaris | Ust-Ordynskaya | 2 | 7 | 0 |
| sungaris | East Torean | 2 | 19 | 0 |
| sungaris | Kudinskaya | 1 | 4 | 0 |
| sungaris | Yakutskaya | 1 | 5 | 0 |
| altaicus | Ulug-Khemskaya | 1 | 15 | 15 |
| altaicus | Saglinskaya | 1 | 9 | 7 |
| altaicus | Karginskaya | 2 | 20 | 9 |
| altaicus | Despenskaya | 1 | 6 | 3 |
| altaicus | Boro-Shaiskaya | 1 | 6 | 3 |
| altaicus | Chozinskaya | 1 | 5 | 3 |
| Trait | C. t. altaicus | C. t. sungaris | C. t. mongolicus | C. t. dzetysuensis |
|---|---|---|---|---|
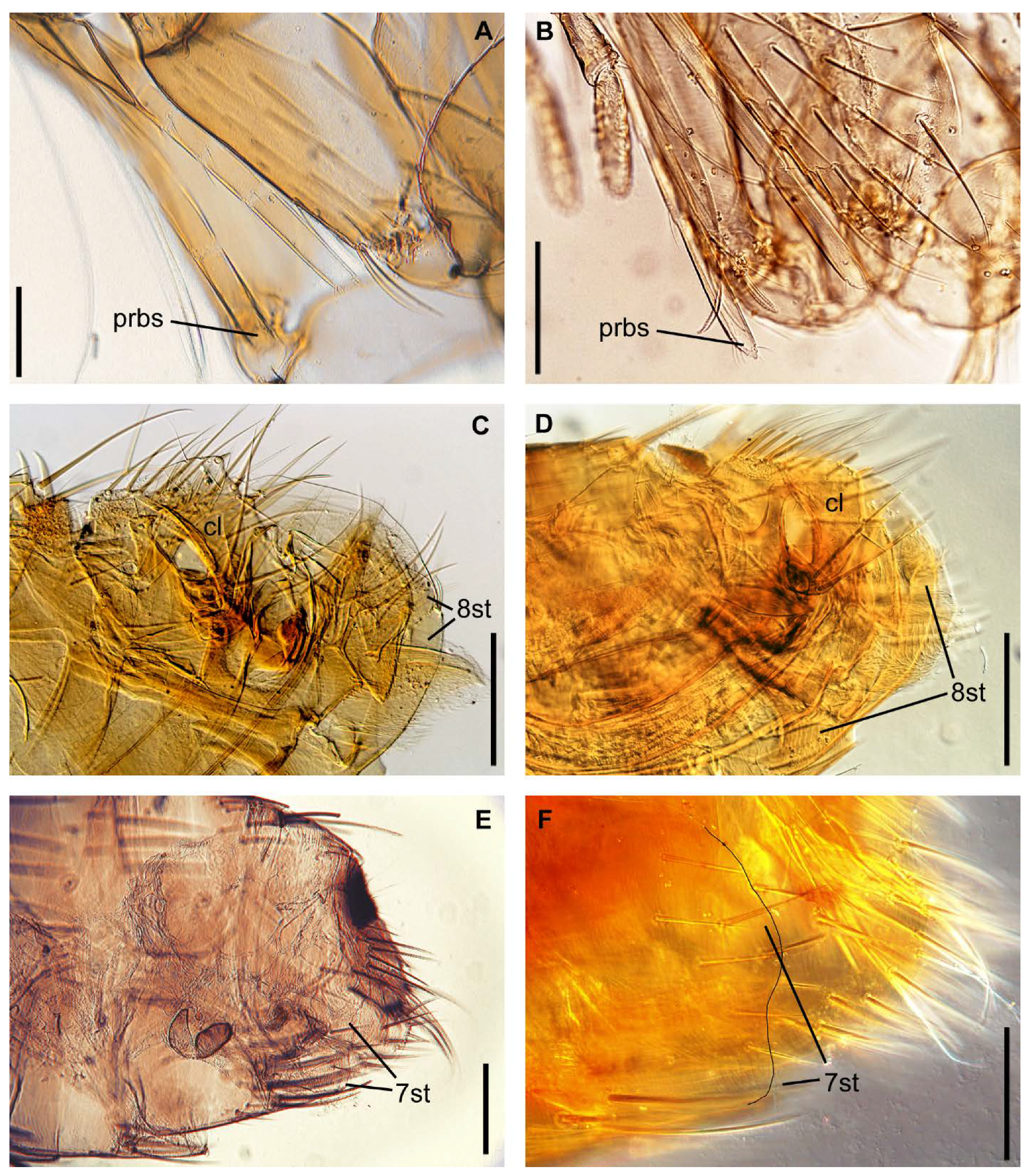
| Head: ratio of proboscis apex to coxa and trochanter | reach apex of coxa, or middle of trochanter (Figure 2A) | reach apex of trochanter (Figure 2B) | reach middle or apex of trochanter | reach middle of trochanter |
| Abdomen: presence of membranous appendage of sternum VIII apical part | present (Figure 2C and Figure S1A) | absent (Figure 2D and Figure S1C) | absent (Figure S1D) | absent (Figure S1B) |
| Abdomen: presence of lateral sinus of posterior margin sternum VIII | absent (Figure 2E and Figure S1E) | present or absent (Figure 2F and Figure S1G) | present (Figure S1H) | no data (Figure S1F) |
| Primer | Target | 5′-3′ Sequence | Reference |
|---|---|---|---|
| ITS5-f1 | ribosomal region | GGAAGTAAAAGTCGTAACAAGG | [23] |
| ITS2-r2 | ribosomal region | CAAGGTTTCCGTAGGTGAACCTG | [24] |
| ITS1ctf2 | ribosomal region | CGCGTACAGGCAGATTATCA | this study |
| ITS1ctr2 | ribosomal region | GCCCGCACTCAAACATTAAA | this study |
| ITS1ctf | ribosomal region | CGTGCTTCGGTGTGTGTTTT | this study |
| ITS1ctr | ribosomal region | GGACAAATTCGCTCTCACGC | this study |
| ITS2-f2 | ribosomal region | GGGTCGATGAAGAACGCAGC | [25] |
| ITS1-r1 | ribosomal region | GCTGCGTTCTTCATCGACCC | [26] |
| ITS2-f3 | ribosomal region | GACCACTCCTGGCTGAGG | this study |
| ITS1-r2 | ribosomal region | CCAGGAGTGGTCCGGGAACAGTATC | this study |
| 28S-r2 | ribosomal region | TAGTTTCTTTTCCTCCGCTTAA | this study |
| 28S-r1 | ribosomal region | GCCGCTACTAAGGGAATCCTA | this study |
| HCO-2198 | COI, mitochondrial gene | TAAACTTCAGGGTGACCAAAAAATCA | [20] |
| LCO-1490 | COI, mitochondrial gene | GGTCAACAAATCATAAAGATATTGG | [20] |
| ftsZuniv1 | Wolbachia symbiont | GG(CT)AA(AG)GGTGC(AG)GCAGAAGA | [27] |
| ftsZuniv2 | Wolbachia symbiont | ATC(AG)AT(AG)CCAGTTGCAAG | [27] |
| ftsZf1 | Wolbachia symbiont | ATYATGGARCATATAAARGATAG | [28] |
| ftsZr1 | Wolbachia symbiont | TCRAGYAATGGATTRGATAT | [28] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ilinsky, Y.; Lapshina, V.; Verzhutsky, D.; Fedorova, Y.; Medvedev, S. Genetic Evidence of an Isolation Barrier between Flea Subspecies of Citellophilus tesquorum (Wagner, 1898) (Siphonaptera: Ceratophyllidae). Insects 2022, 13, 126. https://0-doi-org.brum.beds.ac.uk/10.3390/insects13020126
Ilinsky Y, Lapshina V, Verzhutsky D, Fedorova Y, Medvedev S. Genetic Evidence of an Isolation Barrier between Flea Subspecies of Citellophilus tesquorum (Wagner, 1898) (Siphonaptera: Ceratophyllidae). Insects. 2022; 13(2):126. https://0-doi-org.brum.beds.ac.uk/10.3390/insects13020126
Chicago/Turabian StyleIlinsky, Yury, Vasilina Lapshina, Dmitry Verzhutsky, Yulia Fedorova, and Sergey Medvedev. 2022. "Genetic Evidence of an Isolation Barrier between Flea Subspecies of Citellophilus tesquorum (Wagner, 1898) (Siphonaptera: Ceratophyllidae)" Insects 13, no. 2: 126. https://0-doi-org.brum.beds.ac.uk/10.3390/insects13020126