Imaging of PDE2- and PDE3-Mediated cGMP-to-cAMP Cross-Talk in Cardiomyocytes


1
Institute of Experimental Cardiovascular Research, University Medical Center Hamburg-Eppendorf, 20246 Hamburg, Germany
2
German Center for Cardiovascular Research (DZHK), Partner Site Hamburg/Kiel/Lübeck, 20246 Hamburg, Germany
*
Author to whom correspondence should be addressed.
J. Cardiovasc. Dev. Dis. 2018, 5(1), 4; https://0-doi-org.brum.beds.ac.uk/10.3390/jcdd5010004
Submission received: 29 December 2017
/
Revised: 16 January 2018
/
Accepted: 17 January 2018
/
Published: 19 January 2018
(This article belongs to the Special Issue Cyclic Nucleotide Signaling and the Cardiovascular System)
{kind=link}
{kind=link}
{kind=link}
Abstract
:Cyclic nucleotides 3′,5′-cyclic adenosine monophosphate (cAMP) and 3′,5′-cyclic guanosine monophosphate (cGMP) are important second messengers that regulate cardiovascular function and disease by acting in discrete subcellular microdomains. Signaling compartmentation at these locations is often regulated by phosphodiesterases (PDEs). Some PDEs are also involved in the cross-talk between the two second messengers. The purpose of this review is to summarize and highlight recent findings about the role of PDE2 and PDE3 in cardiomyocyte cyclic nucleotide compartmentation and visualization of this process using live cell imaging techniques.
1. Introduction
Cyclic nucleotides 3′,5′-cyclic adenosine monophosphate (cAMP) and 3′,5′-cyclic guanosine monophosphate (cGMP) are ubiquitous intracellular second messengers that regulate multiple physiological functions as well as pathological conditions. In cardiomyocytes, there are at least three pathways that normally trigger their production after initial first messenger stimuli: (i) the β-adrenergic pathway for cAMP production, (ii) the nitric oxide, and (iii) the natriuretic peptide (NP) receptor pathways for cGMP synthesis.
1.1. The cAMP and β-Adrenergic Pathway
In healthy cardiomyocytes, sympathetic activation mainly via β-adrenergic receptor (β-AR) signaling leads to the production of cAMP and thereby to increased contractile force (inotropy), heart rate (chronotropy), and cell relaxation (lusitropy) [1]. When a ligand binds to a G protein-coupled receptor (GPCR) located on the plasma membrane, a conformational change occurring in the receptor leads to G-protein activation. Activated G-proteins can in turn, activate or inhibit cAMP-forming enzymes adenylyl cyclases (ACs) which generate cAMP from ATP. Subsequently, cAMP acts in cells via one or more of the following effector proteins:
- (a)
- cAMP-dependent protein kinase (PKA), which is responsible for phosphorylation of several calcium handling proteins involved in cardiac excitation-contraction coupling (ECC) including L-type Ca2+ channel (LTCC) at the plasmalemma, phospholamban, and ryanodine receptors at the sarcoplasmic reticulum (SR), myosin-binding protein C, and troponin I at the myofilaments [1,2]. PKA is the main effector protein in the cAMP cascade, while Ca2+-inhibited AC5 and AC6 are the predominant cAMP generating adenylyl cyclases in adult (AC5 and AC6) and fetal (AC6) ventricular cardiac tissue [3];
- (b)
- (c)
- (d)
1.2. NO/sGC/cGMP Pathway
Biosynthesis of cGMP is catalyzed by two discrete guanylyl cyclase (GC) families, one being activated in the presence of nitric oxide (NO) and called soluble guanylyl cyclase (sGC) and the other acting as membrane receptors for natriuretic peptides (NPs), also called particulate guanylyl cyclase (pGC).
NO, alternatively known as “endothelial-derived relaxant factor” (EDRF) [12,13], is produced for example by endothelial cells after acetylcholine administration. It increases cGMP levels, activates cGMP-dependent protein kinase (PKG), and behaves in a way similar to nitrovasodilators [14,15]. Seminal work on the field [16,17,18] has firmly established that NO is produced by a family of NO biosynthetic enzymes called nitric oxide synthases (NOS). It includes neuronal (NOS-1 or nNOS), inducible (NOS-2 or iNOS), and endothelial nitric oxide synthases (NOS-3 or eNOS) [19], all of which having been detected in heart and vessels [20,21,22,23,24]. iNOS is an inducible biosynthetic enzyme, while eNOS and nNOS are both constitutive and inducible enzymes [25]. NO activates sGC by binding to both heme and non-heme sites [26,27,28], which leads to the production of cGMP [29] and its subsequent downstream effects [25,30,31].
1.3. NP/pGC/cGMP Pathway
Natriuretic peptides (NPs) constitute important cardiovascular regulators of inotropy and blood pressure [32] with atrial (ANP), brain (BNP), and C-type natriuretic peptides (CNP) being the most well-known ligands. In response to neurohumoral (catecholamines or angiotensin II) or mechanical (e.g., increased myocardial stretch or blood pressure) stimuli [33,34], ANP and BNP are produced and released by the atria and the ventricles of the heart, while CNP is produced mainly by endothelial cells of the vasculature [34].
These NPs can bind and activate several pGCs, two of which are expressed in the heart and exert the majority of their physiological effects. NPR1 (also called NPR-A or GC-A) is the receptor that binds both ANP and BNP with relatively high affinity (ANP > BNP) [35,36,37]. After ligand binding at its extracellular domain, pGCs undergo a conformational change upon which its intracellular domain generates cGMP [25]. As a widely distributed receptor in the cardiovascular system (heart, vessels, and kidneys), NPR1 regulates blood pressure, exerts antihypertrophic action, and preserves body homeostasis [35,36,37]. NPR2 (also called NPR-B or GC-B) is the CNP-specific receptor responsible for vascular regeneration and endochondral ossification. It is mainly localized in fibroblasts [38], the sympathetic nervous system [39], and the vascular endothelium and smooth muscle [40] and exerts antihypertrophic effects in cardiomyocytes [41,42].
Both NO/sGC and NP/pGC pathways stimulate cGMP synthesis and participate in the homeostasis of the cardiovascular system via (i) PKG-mediated protein phosphorylation [29,30,31], (ii) the activation of CNGCs, and (iii) the regulation of PDEs [25]. Physiologically, cGMP binds to specific sites in the regulatory domains of PKG, CNGC, or PDE in order to induce conformational changes and downstream effects. Disruption of downstream cascade at any level can initiate pathophysiological effects and may lead to hypertension, atherosclerosis, pulmonary hypertension, hypertrophy, ventricular remodeling, myocardial ischemia, dystrophy-related cardiomyopathies, mitochondrial metabolism, or heart failure [25].
Apart from the classical cyclic nucleotides, cyclic cytidine (cCMP) and cyclic uridine monophosphates (cUMP) have been recently introduced as non-canonical second messengers generated by ACs and GCs [43,44]. However, available published data provides limited information regarding their effector proteins and physiological significance, so further studies are required to fully elucidate their role in the cardiovascular system.
2. Compartmentation of cAMP and cGMP Signaling
The fact that multiple receptor stimuli can trigger diverse intracellular effects generated via the production of just a few second messengers such as cAMP and cGMP led to a currently accepted theory of cyclic nucleotide compartmentation. Compartmentation refers to the mechanisms by which multiple spatially segregated cAMP/PKA and cGMP/PKG signaling pathways exert different or even opposing functional effects in distinct subcellular microdomains of the same cell [9,45]. It appears to be of critical importance for cardiovascular system, since local cyclic nucleotide actions and the interplay of the cAMP and cGMP signaling pathways have been implicated in physiological functions or pathological conditions.
Several proteins [46,47,48,49,50] contribute to cyclic nucleotide compartmentation, which spatially, temporally, and functionally controls the downstream effects of cyclic nucleotides (extensively studied for cAMP) in the cardiovascular system [25,51,52,53]. They include (a) GPCRs located in lipid rafts [54,55], at transverse tubules [56] and in non-caveolar membrane domains [57]; (b) ACs and GCs [58,59]; (c) Scaffold proteins [60,61,62]) such as A-kinase anchoring proteins (AKAPs) [52,63,64] and Calveolin-3 [54,65,66,67]; (d) physical barriers—e.g., mitochondria, cAMP buffering by PKA, cAMP export [68,69] are some of the mechanisms that create locally confined intracellular domains regulating signaling; and (e), the most prominent and extensively studied of all, the PDE-mediated hydrolysis of cyclic nucleotides, which is of high pharmacological and clinical interest [64,70,71].
PDEs can control cAMP and cGMP compartmentation by providing their local hydrolytic degradation and creating spatial second messenger gradients [72]. Although much fewer scientific data are available on cGMP compartmentation, the role of PDEs in local confinement of cGMP pools has recently been elucidated, especially that of PDE2, PDE5, and PDE9 [25,73]. Furthermore, spatial organization of PKG and GCs in distinct subcellular complexes appears to be another important aspect of cGMP microdomain regulation [74]. It still remains to be established whether, for example, myosin, NPR1, and troponin T could act as PKG scaffolding proteins [75].
Among the relevant experimental evidence, studies on knockout mice do also highlight the importance of the crucial role PDEs play in the cAMP/cGMP signaling pathways and their respective crosstalk [76]. The interplay among the β-adrenergic and NO/cGMP/PKG pathways can be interpreted as a network phenomenon arising from the molecular selectivity of PDEs to cAMP and cGMP [77].
3. Phosphodiesterases (PDEs)
PDEs are the hydrolyzing enzymes that terminate intracellular effects of cyclic nucleotides by their hydrolysis to fine-tune the signaling and to prevent continuous activation of the downstream effector proteins. These cyclic nucleotide-degrading enzymes constitute one of the most important mechanisms, by which cyclic nucleotides are spatially, temporally, and functionally compartmentalized in cardiomyocytes and other cells. Of the 12 PDE families [78,79], there are seven, namely PDE1 [80], PDE2 [81], PDE3 [76], PDE4 [82], PDE5 [83], PDE8 [84], and PDE9 [73] that have been reported to be expressed and active in mammalian cardiomyocytes (Figure 1). They are an integral part of the multimolecular signaling/regulatory complexes, i.e., signalosomes [52,64,76,84]. This review will particularly explore the so-called cGMP-regulated PDEs, especially PDE2 and PDE3, which critically regulate cGMP-to-cAMP cross-talk and cyclic nucleotide actions in cardiomyocyte microdomains.
3.1. Phosphodiesterase 2
PDE2 is a dual-substrate enzyme, which hydrolyzes both cAMP and cGMP with similar maximal rates in bovine adrenal and heart tissues [85]. Only one gene (Pde2a) gives rise to three known PDE2A isoforms, which are differentially located in the cytosol, mitochondria, and cellular membranes [58]. It is characteristic of this PDE family that the cGMP-mediated control of cAMP hydrolysis arises, when cGMP binds allosterically to the GAF-B domain of PDE2A, so that cAMP hydrolysis occurs with a 10-fold higher rate [86,87,88]. In this manner, cGMP via PDE2A is able to negatively regulate cAMP levels [51] and therefore to initiate a negative cGMP-to-cAMP cross-talk [89] (Figure 2). Initially cloned from rat brain [90] and purified from bovine or calf tissues (heart, liver adrenal gland, and platelets) [85,91], the PDE2A protein is also found in endothelial cells, macrophages, and brain [92,93]. Platelet aggregation [94], aldosterone secretion [95], and regulation of calcium channels [96] require PDE2A-mediated hydrolysis of cAMP. Recently, a PDE2A isoform regulating the mitochondrial respiratory chain has been detected, discovering a possible new pathway for the drug-induced control of mitochondrial function [97]. Of particular importance are those studies referring to PDE2A expression in isolated cardiomyocytes and myocardium. In cardiomyocytes, PDE2A together with PDE5 is also involved in the degradation of sGC-synthesized cGMP, whereas pGC-synthesized cGMP is preferentially hydrolyzed by PDE2A [72,98,99].
3.2. Phosphodiesterase 3
Another important cGMP-regulated PDE is PDE3. This enzyme hydrolyzes both cAMP and cGMP. Often referred to as cGMP-inhibited PDE, PDE3 shows higher catalytic rates for cAMP but relatively high affinity for cGMP, which acts as a competitive inhibitor of cAMP hydrolysis [100,101]. This creates the so-called positive cGMP-to-cAMP cross-talk (Figure 2). PDE3A and PDE3B are the two PDE3 subfamilies, with the former being abundant in cardiomyocytes, oocytes, vascular smooth muscle and platelets and the latter being expressed in the pancreas, liver, and adipose tissue [100]. PDE3A controls myocardial contractility by interacting with the sarcoplasmic/endoplasmic reticulum calcium ATPase (SERCA2a) [102]. By utilizing their direct positive inotropic effects, PDE3 inhibitors are used for acute treatment of end-stage heart failure (HF) [103], albeit presenting increased mortality as well as incidence of arrhythmias and sudden death after chronic use [104,105]. In contrast, PDE3B seems to be more actively engaged in energy metabolism [106,107], but it can also protect the heart from ischemia/reperfusion injury [108]. Knockout models have revealed that PDE3A, but not PDE3B, exerts inotropic and chronotropic effects after treatment with PDE3 inhibitors [109] because PDE3A regulates SERCA2a activity and subsequent SR Ca2+ uptake [102]. By chronically suppressing its expression or action, myocyte apoptosis in vitro [110] or deterioration of ischemia/reperfusion-induced apoptosis and cardiac injury in vivo [111] have been observed. Similarly, disruption of PDE3B interaction with phosphoinositide 3-kinase γ, which can serve as an AKAP, has deleterious effects [112,113,114] such as arrhythmias [114], necrotic cardiac tissue damage, and fibrosis [112].
PDE3 along with other PDEs constitutes an integral part of cAMP degradation. Evidence suggests that it may also controls cGMP levels [108,115], atrial dynamics, and myocyte ANP release, depending on the involved induction mechanism [72,116]. In terms of cGMP and cAMP pathway interactions, cGMP binding to PDE2 enhances the hydrolytic activity of the enzyme and enables the negative cGMP-to-cAMP cross-talk [75]. Conversely, cGMP binding to the catalytic domains of PDE3 reduced the rate of cAMP degradation, thereby mediating the positive cGMP-to-cAMP cross-talk. The previously reported experimental data regarding the affinity, specificity, and enzymatic activity of PDE2 and PDE3 can largely explain their crucial role in cGMP/cAMP crosstalk [75].
4. Visualization of Compartmentalized cAMP and cGMP
Initially, the idea of compartmentalized action of cyclic nucleotides was conceived [117] and revealed by several research groups [118,119,120,121,122] with Buxton and Brunton (1983) [122] using classical biochemical methods to show that prostaglandin induces different PKA activity rates in particulate and soluble fractions of cardiac myocytes after cAMP generation. Later on, Juvericius and Fischmeister (1996) [123], by utilizing a combination of two-barrel microperfusion and whole patch clamp techniques, further confirmed the compartmentation theory in frog ventricular cells, where local application of a β-adrenergic agonist preferentially stimulated the LTCCs close to activated receptors.
To detect cAMP compartmentation in health and disease, multiple techniques have been employed that were only able to detect global concentrations of cyclic nucleotides and required plenty of tissue material [124]. However, biochemical (radio- and enzyme-linked immunoassays) or even electrophysiological approaches (patch-clamp technique), though sensitive and specific, are limited in their capability to record and analyze cyclic nucleotide gradients directly in subcellular microdomains under physiological conditions [124]. Therefore, novel live cell imaging techniques have been developed for the visualization of cyclic nucleotide signaling and its compartmentation in real time with high temporal and spatial resolution [9,124]. Such techniques are mostly based on Förster Resonance Energy Transfer (FRET) biosensors.
FRET biosensors report a non-radiative energy transfer from an excited fluorescent molecule that acts as a donor to a neighboring (located at nm distance) molecule that acts as an acceptor with subsequent fluorescence emission without the direct excitation of the acceptor [125]. Multiple FRET-based biosensors for cGMP [115,126,127,128,129,130] and cAMP [81], and for the activity of the downstream effector proteins such as PKA [131,132,133,134,135,136], Epac [137,138,139,140,141], or CNG channels [142,143,144], have been developed and successfully used to visualize cGMP and cAMP gradients [124,145,146]. They can be further combined with other techniques such as scanning ion conductance microscopy (SICM), which can be used to deliver receptor ligands onto defined membrane structures to targeted distinct cAMP or cGMP pools and to study receptor–microdomain interactions. SICM is a non-optical imaging technique that uses a small glass nanopipette to obtain a highly resolved morphological profile of a living cell membrane based on ion current measurement [147,148,149,150]. It can also be combined with FRET for more accurate and specific detection of microdomain alterations in health and disease [149,151,152].
5. Imaging of cGMP-to-cAMP Crosstalk via PDE2 and PDE3
Employing FRET for live cell imaging, recent studies have revealed strongly remodeled cAMP/cGMP microdomains and subcellular concentration profiles in various cardiac pathologies, leading among other mechanisms to a putatively enhanced involvement of PDE2 in cAMP/cGMP breakdown and crosstalk compared to the other cardiac PDEs.
As mentioned above, the hydrolytic activity of PDE2 can be allosterically stimulated by cGMP to limit cAMP levels, referred to as a negative cGMP-to-cAMP crosstalk. In cardiomyocytes, cGMP can be produced by either pGC after ANP, BNP, and CNP stimulation or by the NO-dependent sGC. Sources for NO include both synthesis in other cell types (e.g., by endothelial cells) and inside cardiomyocytes, e.g., by β3-adrenoreceptor (β3-AR) stimulated pathway, which via inhibitory G-proteins leads to NOS activation (Figure 2). PDE2 hydrolyzes cAMP (e.g., produced in response to the β1/2-adrenergic agonists such as noradrenaline), but its stimulation can be in turn limited by its cGMP hydrolyzing activity, which increases in importance when cGMP concentration rises [51,89]. It has been suggested that PDE2-dependent cAMP hydrolysis might have a more critical effect on cardiomyocyte function, at least under adrenergic overdrive conditions [81,153,154].
The specific role of PDE2 in orchestrating the cyclic nucleotide compartmentation (i.e., cAMP) was supported by experimental evidence coming from a study that demonstrated that, in neonatal rat ventricular myocytes, activation of PDE2 was ineffective in counteracting the forskolin-mediated rise in intracellular cAMP levels [81]. It could also be inferred that, at least in part, stimulation of PDE2-mediated cAMP hydrolysis occurs via a β3-AR/eNOS/sGC pathway (Figure 2). On the contrary, evidence from other studies [155,156,157,158] showed that PDE2 was effective in blocking intracellular increases of cAMP levels mediated by catecholaminergic activation of β-adrenergic receptors or forskolin-mediated AC activation under hypertrophic conditions. By inhibiting the subsequent inotropic effects, these groups were able to argue for a distinct subcellular localization and activity of PDE2 within cardiomyocytes.
More recently, Mehel and colleagues [155] were able to show that myocardial PDE2 is unregulated in human and experimental heart failure and blocks cAMP increase after acute β-AR stimulation. PDE2 upregulation may act as a counterbalance, neutralizing neurohormonal (i.e., β-adrenergic) hyperactivity typically seen in heart failure [155,159]. Furthermore, specific PDE2 inhibition has restored β-AR-mediated signal in diseased cardiomyocytes, while PDE2 overexpression has completely abolished catecholamine effects and hypertrophy without affecting basal contractility [155]. In addition, cAMP hydrolysis via PDE2 mediated the reduction of aldosterone production in adrenal cells, suggesting beneficial synergy between cardiovascular and renal systems [88]. However, for every experimental study, the limitations dictated by the in vitro acquired results might not reflect the in vivo PDE functions, and further experiments in large animal models are required to fully explore the PDE2 role in heart failure pathophysiology. Nevertheless, the overexpressed PDE2 activity may constitute a potential approach to effectively control the deleterious effects of heart failure, e.g., by augmenting its microdomain-specific actions.
On the other hand, there are also studies in which PDE2 may not necessarily exert beneficial effects, but rather contribute to hypertrophy. In cell-based experiments, another pool of cAMP/PDE2 was found to modulate hypertrophic growth of cardiac myocytes by regulating PKA-dependent phosphorylation of nuclear factors of activated T cells (NFAT) [156]. In this study, Zoccarato and colleagues [156] showed that PDE3 and PDE4 inhibition increase cAMP levels and result in hypertrophy, whereas PDE2 inhibition is antihypertrophic despite an increase in cellular cAMP content. Live cell imaging of intact cardiomyocytes revealed that PDE2 inhibition exerted its antihypertrophic effects by generating a locally confined cAMP microdomain, in which PKA type II plays a significant role by phosphorylating NFAT. These are clearly contradicting reports showing remarkable discrepancies especially in the in vivo actions of cardiac PDE2. Further experimental work is required to fully elucidate this question as well as the role of PDE2 in different subcellular cAMP microdomains. It will be especially important to develop and study a tissue-specific knockout mouse model for PDE2.
Another live cell imaging study has developed the first in vivo model expressing a cAMP biosensor targeted to SERCA2a in transgenic mouse cardiomyocytes [157]. Using FRET imaging, it was able to unveil impaired cAMP signal communication between β1-AR located at the membrane and sarcoplasmic reticulum microdomains during early heart failure. By inhibiting PDE2, the authors demonstrated its higher contribution to the regulation of local cAMP levels under pathological conditions [157]. These data suggest that PDE2, when locally or globally upregulated, might potentially contribute to cardioprotective effects in certain microdomains.
Moreover, an elegantly-designed study by Perera et al. [158] proved experimentally for the first time that, in early compensated cardiac hypertrophy preceding heart failure, cGMP-sensitive PDE2 and PDE3 were already physically and functionally rearranged between β1- and β2-AR-associated cAMP microdomains despite unchanged whole cell expression levels and activities. More specifically, the switch of PDEs from PDE3 to PDE2 at the β2-AR, accompanied by a reduction of PDE2 at the β1-AR, led to a turnaround of cAMP cross-talk in a way that, in this pathological setting, the ANP/cGMP signaling pathway by this mechanism could enhance β-AR-mediated cardiac contractility inducing positive inotropic and chronotropic effects following β-AR stimulation (Figure 3). The provided evidence shed light on the poorly understood early microdomain remodeling mechanisms. It has been suggested that, in this way, the heart can compensate for the increased contractility demand under pressure overload [158]. However, our knowledge about microdomain-related contractility mechanisms in early disease is still in its infancy and has to be improved.
The ability of PDE2 to compartmentalize local pools of cAMP has been in part attributed to a much higher speed of cAMP hydrolysis by this PDE as compared to its synthesis by ACs based on FRET imaging in aldosterone producing cells [139]. The most recent finding in regard to cGMP-sensitive cyclic nucleotide compartmentation via PDE2/3 is described in a study using a cardiomyocyte-specific PDE2 transgenic mouse model [159]. In fact, it was shown that endogenous PDE2 contributes to heart rate control under physiological conditions and that PDE2 overexpression protects against arrhythmias and enhances inotropic performance after myocardial infarction [159], providing evidence in support of PDE2 overexpression and highlighting its beneficial role in diseased heart. However, the conclusions from such data obtained from a transgenic mouse model overexpressing this PDE several folds above the endogenous level should be treated with caution since excessive amounts of PDE2 might vanish the boundaries between at least some cAMP microdomains.
Trying to further elucidate the NO/cGMP or NPs/cGMP and cAMP crosstalk, which was also demonstrated in CMs, additional studies utilizing previously developed targeted FRET biosensors [160] and live cell imaging techniques uncovered that the interconnection between cGMP and cAMP in CMs is closely linked to the intracellular locus of regulation [161]. Depending on the recruited cyclase (soluble or particulate) and the associated PDE, cGMP can either augment or inhibit the cAMP levels after catecholamine stimulation and further affect downstream phosphorylation of PKA and contractility. In fact, cGMP can inhibit PDE3 as a competitive substrate for cAMP and allosterically stimulate PDE2A-mediated cAMP hydrolysis [72] locally without largely affecting global cAMP levels in the cell [161]. Induction of cGMP by catecholamine stimulation was found to differentially regulate intracellular cAMP pools that either activate PKA-RI/PDE3- or PKA-RII/PDE2-associated compartments and provoke opposing effects on local cAMP signals [161]. Low basal cGMP levels (~10–50 nmol/L) which could be detected by FRET in adult cardiomyocytes [115] can even facilitate cGMP hydrolyzing activity of PDE3, while higher (between 200 and 500 nmol/L) cGMP levels can activate PDE2A and inhibit PDE3 towards cAMP hydrolysis [162]. Similarly, NO donors via sGC affect both the PKA-RI and PKA-RII compartments, whereas ANP via pGC limits cGMP action to the PKA-RII compartment only [161]. This evidence supports that cGMP exerts local but not global cAMP control in the cardiomyocyte in neonatal rat ventricular myocytes (NRVMs) when isoproterenol is administered in a microdomain-specific manner [161]. For instance, cGMP diminishes cAMP gradients by PDE2 activity when sGC and ANP/pGC mediate its production, while it augments cAMP gradients by inhibiting PDE3 when sGC does so. It is evident that PDE2 exerts cardioprotective regulation against excessive adrenergic stimulation by interconnecting β1/2-AR/cAMP and β3-AR/cGMP pathways [163] and paves the way for further experimental exploration.
Recently, Li and colleagues (2015) [164] showed that PDE2A overexpression blunted BNP-mediated effects by decreasing cGMP production and negatively affecting downstream effectors such as calcium current, intracellular calcium transient, and neurotransmitter release. PDE2A inhibition was also sufficient to reverse the abrogated BNP response. It was also observed that the stellate neurons of the prohypertensive rats express higher PDE2A levels as compared to the normotensive control. These data again underpin the importance of PDE2A upregulation in preventing the BNP-mediated inhibition of sympathetic transmission with subsequent maladaptive changes. Nevertheless, further experimental evidence is required to support whether the BNP-cGMP-PDE2A pathway is actually impaired in hypertensive and heart failure models.
More recently, Meier and colleagues [165] demonstrated the beneficial effect of CNP on β1- and β2-adrenoceptor signaling in rat hearts through cGMP-cAMP crosstalk, when PDE3 is inhibited by cGMP. The CNP-mediated interplay of the signaling pathways was unaffected both in healthy and failing hearts, while BNP was not able to regulate similar cAMP-mediated effects in any experimental group. This study analyzed mechanisms of cyclic nucleotide crosstalk, trying to explain the lack of long-term positive effects of natriuretic peptide in therapeutic schemes for heart failure.
In general, the use of family-selective PDE inhibitors and of genetic knock-down or knock-out models is another way to assess the contribution of individual PDE families in the compartmentalization of cAMP signaling pathways in cardiac myocytes [81,134,156,161,166,167]. It would be interesting to generate a tissue-specific PDE2 knock-out mouse line and explore the role of this particular phosphodiesterase in cardiovascular disease. This goal has remained unattainable due to perinatal lethality of global PDE2 knockout mice. The effect of PDE2 overexpression or upregulation e.g., by inflammation, remains to be clarified as to whether it counterbalances or further deteriorates cardiovascular disease in response to pathologic stimuli.
6. Conclusions
In conclusion, cAMP and cGMP signaling pathways as well as their crosstalk offer a high level of intracellular organization and constitute an interesting pharmacological topic in health and disease. The positive or negative cGMP-mediated regulation of cAMP response that occurs in intracellularly confined loci controlled by distinct PDE isoenzymes could potentially pave the way for novel pharmacological approaches in heart failure treatment. Despite the encouraging evidence, there is still a long way to go before we can fully decipher and understand the exact mechanisms by which these distinct molecular effectors maintain homeostasis and induce maladaptive changes in the heart.
Acknowledgments
We thank all members of our group for their support and helpful discussions. The work of our laboratory was supported by the Deutsche Forschungsgemeinschaft (grant NI 1301/3-2 to V.O.N.), the European Research Area Network on Cardiovascular Diseases (ERA-CVD), and the Gertraud und Heinz-Rose Stiftung.
Author Contributions
N.P. and V.O.N. developed the concept and drafted the manuscript.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Bers, D.M. Calcium cycling and signaling in cardiac myocytes. Annu. Rev. Physiol. 2008, 70, 23–49. [Google Scholar] [CrossRef] [PubMed]
- Bers, D.M. Cardiac excitation-contraction coupling. Nature 2002, 415, 198–205. [Google Scholar] [CrossRef] [PubMed]
- Defer, N.; Best-Belpomme, M.; Hanoune, J. Tissue specificity and physiological relevance of various isoforms of adenylyl cyclase. Am. J. Physiol. Ren. Physiol. 2000, 279, F400–F416. [Google Scholar] [CrossRef] [PubMed]
- De Rooij, J.; Zwartkruis, F.J.; Verheijen, M.H.; Cool, R.H.; Nijman, S.M.; Wittinghofer, A.; Bos, J.L. Epac is a Rap1 guanine-nucleotide-exchange factor directly activated by cyclic AMP. Nature 1998, 396, 474–477. [Google Scholar] [CrossRef] [PubMed]
- Morel, E.; Marcantoni, A.; Gastineau, M.; Birkedal, R.; Rochais, F.; Garnier, A.; Lompre, A.M.; Vandecasteele, G.; Lezoualc’h, F. cAMP-binding protein Epac induces cardiomyocyte hypertrophy. Circ. Res. 2005, 97, 1296–1304. [Google Scholar] [CrossRef] [PubMed]
- Métrich, M.; Lucas, A.; Gastineau, M.; Samuel, J.L.; Heymes, C.; Morel, E.; Lezoualc’h, F. Epac mediates β-adrenergic receptor–induced cardiomyocyte hypertrophy. Circ. Res. 2008, 102, 959–965. [Google Scholar] [CrossRef] [PubMed]
- Wit, A.L.; Rosen, M.R. Pathophysiologic mechanisms of cardiac arrhythmias. Am. Heart J. 1983, 106, 798–811. [Google Scholar] [CrossRef]
- Larsson, H.P. How is the heart rate regulated in the sinoatrial node? Another piece to the puzzle. J. Gen. Physiol. 2010, 88, 96–107. [Google Scholar] [CrossRef] [PubMed]
- Zoccarato, A.; Zaccolo, M. cAMP compartmentalisation and hypertrophy of the heart: “Good” pools of cAMP and “Bad” pools of cAMP coexist in the same cardiac myocyte. In Microdomains in the Cardiovascular System; Nikolaev, V.O., Zaccolo, M., Eds.; Springer Nature: Cham, Switzerland, 2017. [Google Scholar]
- Froese, A.; Breher, S.S.; Waldeyer, C.; Schindler, R.F.; Nikolaev, V.O.; Rinné, S.; Vauti, F. Popeye domain containing proteins are essential for stress-mediated modulation of cardiac pacemaking in mice. J. Clin. Investig. 2012, 122. [Google Scholar] [CrossRef] [PubMed]
- Schindler, R.F.R.; Brand, T. The Popeye domain containing protein family-A novel class of cAMP effectors with important functions in multiple tissues. Prog. Biophys. Mol. Biol. 2016, 120, 28–36. [Google Scholar] [CrossRef] [PubMed]
- Furchgott, R.F.; Zawadzki, J.V. The obligatory role of endothelial cells in the relaxation of arterial smooth muscle by acetylcholine. Nature 1980, 288, 373–376. [Google Scholar] [CrossRef] [PubMed]
- Cherry, P.D.; Furchgott, R.F.; Zawadski, J.V.; Jothianandan, D. Role of endothelial cells in relaxation of isolated arteries by bradykinin. Proc. Natl. Acad. Sci. USA 1982, 79, 2106–2110. [Google Scholar] [CrossRef] [PubMed]
- Rapoport, R.M.; Draznin, M.B.; Murad, F. Endothelium-dependent relaxation in rat aorta may be mediated through cyclic GMP-dependent protein phosphorylation. Nature 1983, 306, 174–176. [Google Scholar] [CrossRef] [PubMed]
- Rapoport, R.M.; Murad, F. Agonist-induced endothelium-dependent relaxation in rat thoracic aorta may be mediated through cGMP. Circ. Res. 1983, 52, 352–357. [Google Scholar] [CrossRef] [PubMed]
- Furchgott, R.F.; Carvalho, M.H.; Khan, M.T.; Matsunaga, K. Evidence for endothelium-dependent vasodilation of resistance vessels by acetylcholine. J. Vasc. Res. 1987, 24, 145–149. [Google Scholar] [CrossRef]
- Ignarro, L.J.; Buga, G.M.; Wood, K.S.; Byrns, R.E.; Chaudhuri, G. Endothelium-derived relaxing factor produced and released from artery and vein is nitric oxide. Proc. Natl. Acad. Sci. USA 1987, 84, 9265–9269. [Google Scholar] [CrossRef] [PubMed]
- Ignarro, L.J.; Byrns, R.E.; Buga, G.M.; Wood, K.S. Endothelium-derived relaxing factor from pulmonary artery and vein possesses pharmacologic and chemical properties identical to those of nitric oxide radical. Circ. Res. 1987, 61, 866–879. [Google Scholar] [CrossRef] [PubMed]
- Alderton, W.K.; Cooper, C.E.; Knowles, R.G. Nitric oxide synthases: Structure, function and inhibition. Biochem. J. 2001, 357, 593–615. [Google Scholar] [CrossRef] [PubMed]
- Koide, M.; Kawahara, Y.; Nakayama, I.; Tsuda, T.; Yokoyama, M. Cyclic AMP-elevating agents induce an inducible type of nitric oxide synthase in cultured vascular smooth muscle cells. Synergism with the induction elicited by inflammatory cytokines. J. Biol. Chem. 1993, 268, 24959–24966. [Google Scholar] [PubMed]
- Balligand, J.L.; Kobzik, L.; Han, X.; Kaye, D.M.; Belhassen, L.; O’Hara, D.S.; Kelly, R.A.; Smith, T.W.; Michel, T. Nitric oxide-dependent parasympathetic signaling is due to activation of constitutive endothelial (type III) nitric oxide synthase in cardiac myocytes. J. Biol. Chem. 1995, 270, 14582–14586. [Google Scholar] [CrossRef] [PubMed]
- Kurihara, N.; Alfie, M.E.; Sigmon, D.H.; Rhaleb, N.E.; Shesely, E.G.; Carretero, O.A. Role of nNOS in blood pressure regulation in eNOS null mutant mice. Hypertension 1998, 32, 856–861. [Google Scholar] [CrossRef] [PubMed]
- Gyurko, R.; Kuhlencordt, P.; Fishman, M.C.; Huang, P.L. Modulation of mouse cardiac function in vivo by eNOS and ANP. Am. J. Physiol. Heart Circ. Physiol. 2000, 278, H971–H981. [Google Scholar] [CrossRef] [PubMed]
- MacNaul, K.L.; Hutchinson, N.I. Differential expression of iNOS and cNOS mRNA in human vascular smooth muscle cells and endothelial cells under normal and inflammatory conditions. Biochem. Biophys. Res. Commun. 1993, 196, 1330–1334. [Google Scholar] [CrossRef] [PubMed]
- Tsai, E.J.; Kass, D.A. Cyclic GMP signaling in cardiovascular pathophysiology and therapeutics. Pharmacol. Ther. 2009, 122, 216–238. [Google Scholar] [CrossRef] [PubMed]
- Yuen, P.S.; Potter, L.R.; Garbers, D.L. A new form of guanylyl cyclase is preferentially expressed in rat kidney. Biochemistry 1990, 29, 10872–10878. [Google Scholar] [CrossRef] [PubMed]
- Harteneck, C.; Wedel, B.; Koesling, D.; Bo, E. Molecular cloning and expression of a new α-subunit of soluble guanylyl cyclase. Interchangeability of the α-subunits of the enzyme. FEBS Lett. 1991, 292, 217–222. [Google Scholar] [PubMed]
- Behrends, S.; Harteneck, C.; Schultz, G.; Koesling, D. A variant of the α2 subunit of soluble guanylyl cyclase contains an insert homologous to a region within adenylyl cyclases and functions as a dominant negative protein. J. Biol. Chem. 1995, 270, 21109–21113. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.I.; Kass, D.A. Phosphodiesterases and cyclic GMP regulation in heart muscle. Physiology 2012, 27, 248–258. [Google Scholar] [CrossRef] [PubMed]
- Zhao, C.Y.; Greenstein, J.L.; Winslow, R.L. Interaction between phosphodiesterases in the regulation of the cardiac β-adrenergic pathway. J. Mol. Cell. Cardiol. 2015, 88, 29–38. [Google Scholar] [CrossRef] [PubMed]
- Zhao, C.Y.; Greenstein, J.L.; Winslow, R.L. Roles of phosphodiesterases in the regulation of the cardiac cyclic nucleotide cross-talk signaling network. J. Mol. Cell. Cardiol. 2016, 91, 215–227. [Google Scholar] [CrossRef] [PubMed]
- D’souza, S.P.; Davis, M.; Baxter, G.F. Autocrine and paracrine actions of natriuretic peptides in the heart. Pharmacol. Ther. 2004, 101, 113–129. [Google Scholar] [CrossRef] [PubMed]
- De Bold, A.J.; Ma, K.K.Y.; Zhang, Y.; de Bold, M.L.K.; Bensimon, M.; Khoshbaten, A. The physiological and pathophysiological modulation of the endocrine function of the heart. Can. J. Physiol. Pharmacol. 2001, 79, 705–714. [Google Scholar] [CrossRef] [PubMed]
- Kuhn, M. Molecular physiology of membrane guanylyl cyclase receptors. Physiol. Rev. 2016, 96, 751–804. [Google Scholar] [CrossRef] [PubMed]
- Kambayashi, Y.; Nakao, K.; Mukoyama, M.; Saito, Y.; Ogawa, Y.; Shiono, S.; Inouye, K.; Yoshida, N.; Imura, H. Isolation and sequence determination of human brain natriuretic peptide in human atrium. FEBS Lett. 1990, 259, 341–345. [Google Scholar] [CrossRef]
- Mukoyama, M.; Nakao, K.; Saito, Y.; Ogawa, Y.; Hosoda, K.; Suga, S.I.; Shirakami, G.; Jougasaki, M.; Imura, H. Human brain natriuretic peptide, a novel cardiac hormone. Lancet 1990, 335, 801–802. [Google Scholar] [CrossRef]
- Nakao, K.; Itoh, H.; Kambayashi, Y.; Hosoda, K.; Saito, Y.; Yamada, T.; Mukoyama, M.; Arai, H.; Shirakami, G.; Suga, S.I. Rat brain natriuretic peptide. Isolation from rat heart and tissue distribution. Hypertension 1990, 15, 774–778. [Google Scholar] [CrossRef] [PubMed]
- Doyle, D.D.; Upshaw-Earley, J.; Bell, E.L.; Palfrey, H.C. Natriuretic peptide receptor-B in adult rat ventricle is predominantly confined to the nonmyocyte population. Am. J. Physiol. Heart Circ. Physiol. 2002, 282, H2117–H2123. [Google Scholar] [CrossRef] [PubMed]
- Buttgereit, J.; Shanks, J.; Li, D.; Hao, G.; Athwal, A.; Langenickel, T.H.; Wright, H.; da Costa Goncalves, A.C.; Monti, J.; Plehm, R.; et al. C-type natriuretic peptide and natriuretic peptide receptor B signalling inhibits cardiac sympathetic neurotransmission and autonomic function. Cardiovasc. Res. 2016, 112, 637–644. [Google Scholar] [CrossRef] [PubMed]
- Hutchinson, H.G.; Trindade, P.T.; Cunanan, D.B.; Wu, C.F.; Pratt, R.E. Mechanisms of natriuretic-peptide-induced growth inhibition of vascular smooth muscle cells. Cardiovasc. Res. 1997, 35, 158–167. [Google Scholar] [CrossRef]
- Rosenkranz, A.C.; Woods, R.L.; Dusting, G.J.; Ritchie, R.H. Antihypertrophic actions of the natriuretic peptides in adult rat cardiomyocytes: Importance of cyclic GMP. Cardiovasc. Res. 2003, 57, 515–522. [Google Scholar] [CrossRef]
- Tokudome, T.; Horio, T.; Soeki, T.; Mori, K.; Kishimoto, I.; Suga, S.I.; Yoshihara, F.; Kawano, Y.; Kohno, M.; Kangawa, K. Inhibitory effect of C-type natriuretic peptide (CNP) on cultured cardiac myocyte hypertrophy: Interference between CNP and endothelin-1 signaling pathways. Endocrinology 2004, 145, 2131–2140. [Google Scholar] [CrossRef] [PubMed]
- Seifert, R. cCMP and cUMP: Emerging second messengers. Trends Biochem. Sci. 2015, 40, 8–15. [Google Scholar] [CrossRef] [PubMed]
- Seifert, R.; Schneider, E.H.; Bähre, H. From canonical to non-canonical cyclic nucleotides as second messengers: Pharmacological implications. Pharmacol. Ther. 2015, 148, 154–184. [Google Scholar] [CrossRef] [PubMed]
- Zaccolo, M. Spatial control of cAMP signalling in health and disease. Curr. Opin. Pharmacol. 2011, 11, 649–655. [Google Scholar] [CrossRef] [PubMed]
- Saucerman, J.J.; Greenwald, E.C.; Polanowska-Grabowska, R. Mechanisms of cyclic AMP compartmentation revealed by computational models. J. Gen. Physiol. 2014, 143, 39–48. [Google Scholar] [CrossRef] [PubMed]
- Mika, D.; Leroy, J.; Vandecasteele, G.; Fischmeister, R. PDEs create local domains of cAMP signaling. J. Mol. Cell. Cardiol. 2012, 52, 323–329. [Google Scholar] [CrossRef] [PubMed]
- Ziolo, M.T.; Kohr, M.J.; Wang, H. Nitric oxide signaling and the regulation of myocardial function. J. Mol. Cell. Cardiol. 2008, 45, 625–632. [Google Scholar] [CrossRef] [PubMed]
- Fischmeister, R.; Castro, L.R.; Abi-Gerges, A.; Rochais, F.; Jurevičius, J.; Leroy, J.; Vandecasteele, G. Compartmentation of cyclic nucleotide signaling in the heart. Circ. Res. 2006, 99, 816–828. [Google Scholar] [CrossRef] [PubMed]
- Vandecasteele, G.; Rochais, F.; Abi-Gerges, A.; Fischmeister, R. Functional localization of cAMP signalling in cardiac myocytes. Biochem. Soc. Trans. 2006, 34, 484–488. [Google Scholar] [CrossRef] [PubMed]
- Bender, A.T.; Beavo, J.A. Cyclic nucleotide phosphodiesterases: Molecular regulation to clinical use. Pharmacol. Rev. 2006, 58, 488–520. [Google Scholar] [CrossRef] [PubMed]
- Francis, S.H.; Blount, M.A.; Corbin, J.D. Mammalian cyclic nucleotide phosphodiesterases: Molecular mechanisms and physiological functions. Physiol. Rev. 2011, 91, 651–690. [Google Scholar] [CrossRef] [PubMed]
- Keravis, T.; Lugnier, C. Cyclic nucleotide phosphodiesterase (PDE) isozymes as targets of the intracellular signalling network: Benefits of PDE inhibitors in various diseases and perspectives for future therapeutic developments. Br. J. Pharmacol. 2012, 165, 1288–1305. [Google Scholar] [CrossRef] [PubMed]
- Balijepalli, R.C.; Foell, J.D.; Hall, D.D.; Hell, J.W.; Kamp, T.J. Localization of cardiac L-type Ca2+ channels to a caveolar macromolecular signaling complex is required for β2-adrenergic regulation. Proc. Natl. Acad. Sci. USA 2006, 103, 7500–7505. [Google Scholar] [CrossRef] [PubMed]
- Pani, B.; Singh, B.B. Lipid rafts/caveolae as microdomains of calcium signalling. Cell Calcium 2009, 45, 625–633. [Google Scholar] [CrossRef] [PubMed]
- Kamp, T.J.; Hell, J.W. Regulation of cardiac L-type calcium channels by protein kinase A and protein kinase C. Circ. Res. 2000, 87, 1095–1102. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, S.R.; Ostrom, R.S.; Harvey, R.D. Membrane Microdomains and cAMP Compartmentation in Cardiac Myocytes. In Microdomains in the Cardiovascular System; Nikolaev, V.O., Zaccolo, M., Eds.; Springer: Cham, Switzerland, 2017; pp. 17–35. [Google Scholar]
- Guellich, A.; Mehel, H.; Fischmeister, R. Cyclic AMP synthesis and hydrolysis in the normal and failing heart. Pflügers Arch. 2014, 466, 1163–1175. [Google Scholar] [CrossRef] [PubMed]
- Timofeyev, V.; Myers, R.E.; Kim, H.J.; Woltz, R.L.; Sirish, P.; Heiserman, J.P.; Li, N.; Singapuri, A.; Tang, T.; Yarov-Yarovoy, V.; et al. Adenylyl cyclase subtype-specific compartmentalization: Differential regulation of L-type Ca2+ current in ventricular myocytes. Circ. Res. 2013, 112, 1567–1576. [Google Scholar] [CrossRef] [PubMed]
- Redden, J.M.; Dodge-Kafka, K.L.; Kapiloff, M.S. Function to Failure: Compartmentalization of Cardiomyocyte Signaling by A-Kinase-Anchoring Proteins. In Microdomains in the Cardiovascular System; Nikolaev, V.O., Zaccolo, M., Eds.; Springer: Cham, Switzerland, 2017; pp. 37–57. [Google Scholar]
- Schrade, K.; Klussmann, E. Pharmacological approaches for delineating functions of AKAP-based signalling complexes and finding therapeutic targets. In Microdomains in the Cardiovascular System; Nikolaev, V.O., Zaccolo, M., Eds.; Springer: Cham, Switzerland, 2017; pp. 59–83. [Google Scholar]
- Ghigo, A.; Pirozzi, F.; Li, M.; Hirsch, E. Chatting Second Messengers: PIP3 and cAMP. In Microdomains in the Cardiovascular System; Nikolaev, V.O., Zaccolo, M., Eds.; Springer: Cham, Switzerland, 2017; pp. 85–95. [Google Scholar]
- Dodge-Kafka, K.L.; Langeberg, L.; Scott, J.D. Compartmentation of cyclic nucleotide signaling in the heart: The role of A-kinase anchoring proteins. Circ. Res. 2006, 98, 993–1001. [Google Scholar] [CrossRef] [PubMed]
- Maurice, D.H.; Ke, H.; Ahmad, F.; Wang, Y.; Chung, J.; Manganiello, V.C. Advances in targeting cyclic nucleotide phosphodiesterases. Nat. Rev. Drug Discov. 2014, 13, 290–314. [Google Scholar] [CrossRef] [PubMed]
- Balijepalli, R.C.; Kamp, T.J. Caveolae, ion channels and cardiac arrhythmias. Prog. Biophys. Mol. Biol. 2008, 98, 149–160. [Google Scholar] [CrossRef] [PubMed]
- Carnegie, G.K.; Means, C.K.; Scott, J.D. A-kinase anchoring proteins: From protein complexes to physiology and disease. IUBMB Life 2009, 61, 394–406. [Google Scholar] [CrossRef] [PubMed]
- Nichols, C.B.; Rossow, C.F.; Navedo, M.F.; Westenbroek, R.E.; Catterall, W.A.; Santana, L.F.; McKnight, G.S. Sympathetic stimulation of adult cardiomyocytes requires association of akap5 with a subpopulation of l-type calcium channels novelty and significance. Circ. Res. 2010, 107, 747–756. [Google Scholar] [CrossRef] [PubMed]
- Cheepala, S.; Hulot, J.S.; Morgan, J.A.; Sassi, Y.; Zhang, W.; Naren, A.P.; Schuetz, J.D. Cyclic nucleotide compartmentalization: Contributions of phosphodiesterases and ATP-binding cassette transporters. Annu. Rev. Pharmacol. Toxicol. 2013, 53, 231–253. [Google Scholar] [CrossRef] [PubMed]
- Sassi, Y.; Abi-Gerges, A.; Fauconnier, J.; Mougenot, N.; Reiken, S.; Haghighi, K.; Kranias, E.G.; Marks, A.R.; Lacampagne, A.; Engelhardt, S.; et al. Regulation of cAMP homeostasis by the efflux protein MRP4 in cardiac myocytes. FASEB J. 2012, 26, 1009–1017. [Google Scholar] [CrossRef] [PubMed]
- Houslay, M.D. Underpinning compartmentalised cAMP signalling through targeted cAMP breakdown. Trends Biochem. Sci. 2010, 35, 91–100. [Google Scholar] [CrossRef] [PubMed]
- Conti, M.; Mika, D.; Richter, W. Cyclic AMP compartments and signaling specificity: Role of cyclic nucleotide phosphodiesterases. J. Gen. Physiol. 2014, 143, 29–38. [Google Scholar] [CrossRef] [PubMed]
- Zaccolo, M.; Movsesian, M.A. cAMP and cGMP signaling cross-talk: Role of phosphodiesterases and implications for cardiac pathophysiology. Circ. Res. 2007, 100, 1569–1578. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.I.; Zhu, G.; Sasaki, T.; Cho, G.S.; Hamdani, N.; Holewinski, R.; Jo, S.H.; Danner, T.; Zhang, M.; Rainer, P.P.; et al. Phosphodiesterase 9A controls nitric-oxide-independent cGMP and hypertrophic heart disease. Nature 2015, 519, 472–476. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Kass, D.A. Phosphodiesterases and cardiac cGMP: Evolving roles and controversies. Trends Pharmacol. Sci. 2011, 32, 360–365. [Google Scholar] [CrossRef] [PubMed]
- Brescia, M.; Zaccolo, M. Modulation of compartmentalised cyclic nucleotide signalling via local inhibition of phosphodiesterase activity. Int. J. Mol. Sci. 2016, 17. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, F.; Muratal, T.; Shimizul, K.; Degerma, E.; Maurice, D.; Manganiello, V. Cyclic nucleotide phosphodiesterases: Important signaling modulators and therapeutic targets. Oral Dis. 2015, 21, e25–e50. [Google Scholar] [CrossRef] [PubMed]
- Zhao, C.Y. Computational modeling of cyclic nucleotide signaling mechanisms in cardiac myocytes. In Microdomains in the Cardiovascular System; Nikolaev, V.O., Zaccolo, M., Eds.; Springer Nature: Cham, Switzerland, 2017; pp. 175–213. [Google Scholar]
- Conti, M.; Beavo, J. Biochemistry and physiology of cyclic nucleotide phosphodiesterases: Essential components in cyclic nucleotide signaling. Annu. Rev. Biochem. 2007, 76, 481–511. [Google Scholar] [CrossRef] [PubMed]
- Wood, E.R.; Bledsoe, R.; Chai, J.; Daka, P.; Deng, H.; Ding, Y.; Harris-Gurley, S.; Kryn, L.H.; Nartey, E.; Nichols, J.; et al. The role of phosphodiesterase 12 (PDE12) as a negative regulator of the innate immune response and the discovery of antiviral inhibitors. J. Biol. Chem. 2015, 290, 19681–19696. [Google Scholar] [CrossRef] [PubMed]
- Vandeput, F.; Wolda, S.L.; Krall, J.; Hambleton, R.; Uher, L.; McCaw, K.N.; Radwanski, P.B.; Florio, V.; Movsesian, M.A. Cyclic nucleotide phosphodiesterase PDE1C1 in human cardiac myocytes. J. Biol. Chem. 2007, 282, 32749–32757. [Google Scholar] [CrossRef] [PubMed]
- Mongillo, M.; Tocchetti, C.G.; Terrin, A.; Lissandron, V.; Cheung, Y.F.; Dostmann, W.R.; Pozzan, T.; Kass, D.A.; Paolocci, N.; Houslay, M.D.; et al. Compartmentalized phosphodiesterase-2 activity blunts beta-adrenergic cardiac inotropy via an NO/cGMP-dependent pathway. Circ. Res. 2006, 98, 226–234. [Google Scholar] [CrossRef] [PubMed]
- Richter, W.; Xie, M.; Scheitrum, C.; Krall, J.; Movsesian, M.A.; Conti, M. Conserved expression and functions of PDE4 in rodent and human heart. Basic Res. Cardiol. 2011, 106, 249–262. [Google Scholar] [CrossRef] [PubMed]
- Gamanuma, M.; Yuasa, K.; Sasaki, T.; Sakurai, N.; Kotera, J.; Omori, K. Comparison of enzymatic characterization and gene organization of cyclic nucleotide phosphodiesterase 8 family in humans. Cell Signal. Technol. 2003, 15, 565–574. [Google Scholar] [CrossRef]
- Houslay, M.D.; Baillie, G.S.; Maurice, D.H. cAMP-Specific phosphodiesterase-4 enzymes in the cardiovascular system: A molecular toolbox for generating compartmentalized cAMP signaling. Circ. Res. 2007, 100, 950–966. [Google Scholar] [CrossRef] [PubMed]
- Martins, T.J.; Mumby, M.C.; Beavo, J.A. Purification and characterization of a cyclic GMP-stimulated cyclic nucleotide phosphodiesterase from bovine tissues. J. Biol. Chem. 1982, 257, 1973–1979. [Google Scholar] [PubMed]
- Mumby, M.C.; Martins, T.J.; Chang, M.L.; Beavo, J.A. Identification of cGMP-stimulated cyclic nucleotide phosphodiesterase in lung tissue with monoclonal antibodies. J. Biol. Chem. 1982, 257, 13283–13290. [Google Scholar] [PubMed]
- Martinez, S.E.; Wu, A.Y.; Glavas, N.A.; Tang, X.B.; Turley, S.; Hol, W.G.; Beavo, J.A. The two GAF domains in phosphodiesterase 2A have distinct roles in dimerization and in cGMP binding. Proc. Natl. Acad. Sci. USA 2002, 99, 13260–13265. [Google Scholar] [CrossRef] [PubMed]
- Rosman, G.J.; Martins, T.J.; Sonnenburg, W.K.; Beavo, J.A.; Ferguson, K.; Loughney, K. Isolation and characterization of human cDNAs encoding a cGMP-stimulated 3′, 5′-cyclic nucleotide phosphodiesterase. Gene 1997, 191, 89–95. [Google Scholar] [CrossRef]
- Lugnier, C. Cyclic nucleotide phosphodiesterase (PDE) superfamily: A new target for the development of specific therapeutic agents. Pharmacol. Ther. 2006, 109, 366–398. [Google Scholar] [CrossRef] [PubMed]
- Yang, Q.; Paskind, M.; Bolger, G.; Thompson, W.J.; Repaske, D.R.; Cutler, L.S.; Epstein, P.M. A novel cyclic GMP stimulated phosphodiesterase from rat brain. Biochem. Biophys. Res. Commun. 1994, 205, 1850–1858. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, T.; Manganiello, V.C.; Vaughan, M. Purification and characterization of cyclic GMP-stimulated cyclic nucleotide phosphodiesterase from calf liver. Effects of divalent cations on activity. J. Biol. Chem. 1983, 258, 12526–12533. [Google Scholar] [PubMed]
- Bender, A.T.; Beavo, J.A. Specific localized expression of cGMP PDEs in Purkinje neurons and macrophages. Neurochem. Int. 2004, 45, 853–857. [Google Scholar] [CrossRef] [PubMed]
- Juilfs, D.M.; Soderling, S.; Burns, F.; Beavo, J.A. Cyclic GMP as substrate and regulator of cyclic nucleotide phosphodiesterases (PDEs). In Reviews of Physiology, Biochemistry and Pharmacology; Springer: Berlin/Heidelberg, Germany, 1999. [Google Scholar]
- Dickinson, N.T.; Haslam, R.J. Activation of cGMP-stimulated phosphodiesterase by nitroprusside limits cAMP accumulation in human platelets: Effects on platelet aggregation. Biochem. J. 1997, 323, 371–377. [Google Scholar] [CrossRef] [PubMed]
- MacFarland, R.T.; Zelus, B.D.; Beavo, J.A. High concentrations of a cGMP-stimulated phosphodiesterase mediate ANP-induced decreases in cAMP and steroidogenesis in adrenal glomerulosa cells. J. Biol. Chem. 1991, 266, 136–142. [Google Scholar] [PubMed]
- Simmons, M.A.; Hartzell, H.C. Role of phosphodiesterase in regulation of calcium current in isolated cardiac myocytes. Mol. Pharmacol. 1988, 33, 664–671. [Google Scholar] [PubMed]
- Acin-Perez, R.; Gatti, D.L.; Bai, Y.; Manfredi, G. Protein phosphorylation and prevention of cytochrome oxidase inhibition by ATP: Coupled mechanisms of energy metabolism regulation. Cell Metab. 2011, 13, 712–719. [Google Scholar] [CrossRef] [PubMed]
- Castro, L.R.; Verde, I.; Cooper, D.M.; Fischmeister, R. Cyclic guanosine monophosphate compartmentation in rat cardiac myocytes. Circulation 2006, 113, 2221–2228. [Google Scholar] [CrossRef] [PubMed]
- Weber, S.; Zeller, M.; Guan, K.; Wunder, F.; Wagner, M.; El-Armouche, A. PDE2 at the crossway between cAMP and cGMP signalling in the heart. Cell Signal. 2017, 38, 76–84. [Google Scholar] [CrossRef] [PubMed]
- Shakur, Y.; Holst, L.S.; Landstrom, T.R.; Movsesian, M.; Degerman, E.; Manganiello, V. Regulation and function of the cyclic nucleotide phosphodiesterase (PDE3) gene family. Prog. Nucleic Acid Res. Mol. Biol. 2001, 66, 241–277. [Google Scholar] [PubMed]
- Degerman, E.; Belfrage, P.; Manganiello, V.C. Structure, localization, and regulation of cGMP-inhibited phosphodiesterase (PDE3). J. Biol. Chem. 1997, 272, 6823–6826. [Google Scholar] [CrossRef] [PubMed]
- Beca, S.; Ahmad, F.; Shen, W.; Liu, J.; Makary, S.; Polidovitch, N.; Sun, J.; Hockman, S.; Chung, Y.W.; Movsenian, M.; et al. Phosphodiesterase type 3A regulates basal myocardial contractility through interacting with sarcoplasmic reticulum calcium ATPase type 2a signaling complexes in mouse heart novelty and significance. Circ. Res. 2013, 112, 289–297. [Google Scholar] [CrossRef] [PubMed]
- McMurray, J.J.; Adamopoulos, S.; Anker, S.D.; Auricchio, A.; Böhm, M.; Dickstein, K.; Jaarsma, T. ESC Guidelines for the diagnosis and treatment of acute and chronic heart failure 2012. Eur. J. Heart Fail. 2012, 14, 803–869. [Google Scholar] [CrossRef] [PubMed]
- Landry, Y.; Gies, J.P. Drugs and their molecular targets: An updated overview. Fundam. Clin. Pharmacol. 2008, 22, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Packer, M.; Carver, J.R.; Rodeheffer, R.J.; Ivanhoe, R.J.; DiBianco, R.; Zeldis, S.M.; Mallis, G.I. Effect of oral milrinone on mortality in severe chronic heart failure. N. Engl. J. Med. 1991, 325, 1468–1475. [Google Scholar] [CrossRef] [PubMed]
- Choi, Y.H.; Park, S.; Hockman, S.; Zmuda-Trzebiatowska, E.; Svennelid, F.; Haluzik, M.; Gavrilova, O.; Ahmad, F.; Pepin, L.; Napolitano, M.; et al. Alterations in regulation of energy homeostasis in cyclic nucleotide phosphodiesterase 3B–null mice. J. Clin. Investig. 2006, 116, 3240–3251. [Google Scholar] [CrossRef] [PubMed]
- Degerman, E.; Ahmad, F.; Chung, Y.W.; Guirguis, E.; Omar, B.; Stenson, L.; Manganiello, V. From PDE3B to the regulation of energy homeostasis. Curr. Opin. Pharmacol. 2011, 11, 676–682. [Google Scholar] [CrossRef] [PubMed]
- Chung, Y.W.; Lagranha, C.; Chen, Y.; Sun, J.; Tong, G.; Hockman, S.C.; Ahmad, F.; Esfahani, S.G.; Bae, D.H.; Polidovitch, N.; et al. Targeted disruption of PDE3B, but not PDE3A, protects murine heart from ischemia/reperfusion injury. Proc. Natl. Acad. Sci. USA 2015, 112, E2253–E2262. [Google Scholar] [CrossRef] [PubMed]
- Sun, B.; Li, H.; Shakur, Y.; Hensley, J.; Hockman, S.; Kambayashi, J.; Manganiello, V.M.; Liu, Y. Role of phosphodiesterase type 3A and 3B in regulating platelet and cardiac function using subtype-selective knockout mice. Cell Signal. 2007, 19, 1765–1771. [Google Scholar] [CrossRef] [PubMed]
- Ding, B.; Abe, J.I.; Wei, H.; Xu, H.; Che, W.; Aizawa, T.; Liu, W.; Molina, C.A.; Sadoshima, J.; Blaxall, B.C.; et al. A positive feedback loop of phosphodiesterase 3 (PDE3) and inducible cAMP early repressor (ICER) leads to cardiomyocyte apoptosis. Proc. Natl. Acad. Sci. USA 2005, 102, 14771–14776. [Google Scholar] [CrossRef] [PubMed]
- Oikawa, M.; Wu, M.; Lim, S.; Knight, W.E.; Miller, C.L.; Cai, Y.; Lu, C.; Blaxall, B.C.; Takeishi, Y.; Abe, J.I.; et al. Cyclic nucleotide phosphodiesterase 3A1 protects the heart against ischemia-reperfusion injury. J. Mol. Cell. Cardiol. 2013, 64, 11–19. [Google Scholar] [CrossRef] [PubMed]
- Patrucco, E.; Notte, A.; Barberis, L.; Selvetella, G.; Maffei, A.; Brancaccio, M.; Marengo, S.; Russo, G.; Azzolino, O.; Rybalkin, S.D.; et al. PI3Kgamma modulates the cardiac response to chronic pressure overload by distinct kinase-dependent and -independent effects. Cell 2004, 118, 375–387. [Google Scholar] [CrossRef] [PubMed]
- Perino, A.; Ghigo, A.; Ferrero, E.; Morello, F.; Santulli, G.; Baillie, G.S.; Damilano, F.; Dunlop, A.J.; Pawson, C.; Walser, R.; et al. Integrating cardiac PIP 3 and cAMP signaling through a PKA anchoring function of p110γ. Mol. Cell 2011, 42, 84–95. [Google Scholar] [CrossRef] [PubMed]
- Ghigo, A.; Perino, A.; Mehel, H.; Zahradníková, A.; Morello, F.; Leroy, J.; Nikolaev, V.O.; Damilano, F.; Cimino, J.; De Luca, E.; et al. Phosphoinositide 3-kinase γ protects against catecholamine-induced ventricular arrhythmia through protein kinase a–mediated regulation of distinct phosphodiesterases. Circulation 2012, 126, 2073–2083. [Google Scholar] [CrossRef] [PubMed]
- Götz, K.R.; Sprenger, J.U.; Perera, R.K.; Steinbrecher, J.H.; Lehnart, S.E.; Kuhn, M.; Gorelik, J.; Balligand, J.L.; Nikolaev, V.O. Transgenic mice for real-time visualization of cGMP in intact adult cardiomyocytes. Circ. Res. 2014, 114, 1235–1245. [Google Scholar] [CrossRef] [PubMed]
- Wen, J.F.; Cui, X.; Jin, J.Y.; Kim, S.M.; Kim, S.Z.; Kim, S.H.; Kim, S.H.; Lee, H.S.; Cho, K.W. High and low gain switches for regulation of camp efflux concentration. Circ. Res. 2004, 94, 936–943. [Google Scholar] [CrossRef] [PubMed]
- Hayes, J.S.; Brunton, L.L. Functional compartments in cyclic nucleotide action. J. Cycl. Nucleotide Res. 1982, 8, 1–16. [Google Scholar]
- Corbin, J.D.; Sugden, P.H.; Lincoln, T.M.; Keely, S.L. Compartmentalization of adenosine 3′: 5′-monophosphate and adenosine 3′: 5′-monophosphate-dependent protein kinase in heart tissue. J. Biol. Chem. 1977, 252, 3854–3861. [Google Scholar] [PubMed]
- Hayes, J.S.; Brunton, L.L.; Brown, J.H.; Reese, J.B.; Mayer, S.E. Hormonally specific expression of cardiac protein kinase activity. Proc. Natl. Acad. Sci. USA 1979, 76, 1570–1574. [Google Scholar] [CrossRef] [PubMed]
- Hayes, J.S.; Brunton, L.L.; Mayer, S.E. Selective activation of particulate cAMP-dependent protein kinase by isoproterenol and prostaglandin E1. J. Biol. Chem. 1980, 255, 5113–5119. [Google Scholar] [PubMed]
- Keely, S.L. Activation of cAMP-dependent protein kinase without a corresponding increase in phosphorylase activity. Res. Commun. Chem. Pathol. Pharmacol. 1977, 18, 283–290. [Google Scholar] [PubMed]
- Buxton, I.L.; Brunton, L.L. Compartments of cyclic AMP and protein kinase in mammalian cardiomyocytes. J. Biol. Chem. 1983, 258, 10233–10239. [Google Scholar] [PubMed]
- Jurevicius, J.; Fischmeister, R. cAMP compartmentation is responsible for a local activation of cardiac Ca2+ channels by beta-adrenergic agonists. Proc. Natl. Acad. Sci. USA 1996, 93, 295–299. [Google Scholar] [CrossRef] [PubMed]
- Sprenger, J.U.; Nikolaev, V.O. Biophysical techniques for detection of cAMP and cGMP in Living Cells. Int. J. Mol. Sci. 2013, 14, 8025–8046. [Google Scholar] [CrossRef] [PubMed]
- Förster, T. Zwischenmolekulare energiewanderung und fluoreszenz. Ann. Phys. 1948, 437, 55–75. [Google Scholar] [CrossRef]
- Nikolaev, V.O.; Lohse, M.J. Novel techniques for real-time monitoring of cGMP in living cells. Handb. Exp. Pharmacol. 2009, 191, 229–243. [Google Scholar]
- Honda, A.; Adams, S.R.; Sawyer, C.L.; Lev-Ram, V.; Tsien, R.Y.; Dostmann, W.R. Spatiotemporal dynamics of guanosine 3′, 5′-cyclic monophosphate revealed by a genetically encoded, fluorescent indicator. Proc. Natl. Acad. Sci. USA 2001, 98, 2437–2442. [Google Scholar] [CrossRef] [PubMed]
- Sato, M.; Hida, N.; Ozawa, T.; Umezawa, Y. Fluorescent indicators for cyclic GMP based on cyclic GMP-dependent protein kinase I alpha and green fluorescent proteins. Anal. Chem. 2000, 72, 5918–5924. [Google Scholar] [CrossRef] [PubMed]
- Nikolaev, V.O.; Gambaryan, S.; Lohse, M.J. Fluorescent sensors for rapid monitoring of intracellular cGMP. Nat. Methods 2006, 3, 23–25. [Google Scholar] [CrossRef] [PubMed]
- Niino, Y.; Hotta, K.; Oka, K. Simultaneous live cell imaging using dual FRET sensors with a single excitation light. PLoS ONE 2009, 4. [Google Scholar] [CrossRef] [PubMed]
- Adams, S.R.; Harootunian, A.T.; Buechler, Y.J.; Taylor, S.S.; Tsien, R.Y. Fluorescence ratio imaging of cyclic AMP in single cells. Nature 1991, 349, 694–697. [Google Scholar] [CrossRef] [PubMed]
- Bacskai, B.J.; Hochner, B.; Mahaut-Smith, M.; Adams, S.R.; Kaang, B.K.; Kandel, E.R.; Tsien, R.Y. Spatially resolved dynamics of cAMP and protein kinase A subunits in Aplysia sensory neurons. Science 1993, 260, 222–226. [Google Scholar] [CrossRef] [PubMed]
- Zaccolo, M.; De Giorgi, F.; Cho, C.Y.; Feng, L.; Knapp, T.; Negulescu, P.A.; Taylor, S.S.; Tsien, R.Y.; Pozzan, T. A genetically encoded, fluorescent indicator for cyclic AMP in living cells. Nat. Cell. Biol. 2000, 2, 25–29. [Google Scholar] [CrossRef] [PubMed]
- Zaccolo, M.; Pozzan, T. Discrete microdomains with high concentration of cAMP in stimulated rat neonatal cardiac myocytes. Science 2002, 295, 1711–1715. [Google Scholar] [CrossRef] [PubMed]
- Lehnart, S.E.; Wehrens, X.H.; Reiken, S.; Warrier, S.; Belevych, A.E.; Harvey, R.D.; Richter, W.; Jin, S.L.; Conti, M.; Marks, A.R. Phosphodiesterase 4D deficiency in the ryanodine-receptor complex promotes heart failure and arrhythmias. Cell 2005, 123, 25–35. [Google Scholar] [CrossRef] [PubMed]
- Nikolaev, V.O.; Bünemann, M.; Hein, L.; Hannawacker, A.; Lohse, M.J. Novel single chain cAMP sensors for receptor-induced signal propagation. J. Biol. Chem. 2004, 279, 37215–37218. [Google Scholar] [CrossRef] [PubMed]
- DiPilato, L.M.; Cheng, X.; Zhang, J. Fluorescent indicators of cAMP and Epac activation reveal differential dynamics of cAMP signaling within discrete subcellular compartments. Proc. Natl. Acad. Sci. USA 2004, 101, 16513–16518. [Google Scholar] [CrossRef] [PubMed]
- Ponsioen, B.; Zhao, J.; Riedl, J.; Zwartkruis, F.; van der Krogt, G.; Zaccolo, M.; Moolenaar, W.H.; Bos, J.L.; Jalink, K. Detecting cAMP-induced Epac activation by fluorescence resonance energy transfer: Epac as a novel cAMP indicator. EMBO Rep. 2004, 5, 1176–1180. [Google Scholar] [CrossRef] [PubMed]
- Nikolaev, V.O.; Gambaryan, S.; Engelhardt, S.; Walter, U.; Lohse, M.J. Real-time monitoring of the PDE2 activity of live cells: Hormone-stimulated cAMP hydrolysis is faster than hormone-stimulated cAMP synthesis. J. Biol. Chem. 2005, 280, 1716–1719. [Google Scholar] [CrossRef] [PubMed]
- Calebiro, D.; Nikolaev, V.O.; Gagliani, M.C.; de Filippis, T.; Dees, C.; Tacchetti, C.; Persani, L.; Lohse, M.J. Persistent cAMP-signals triggered by internalized G-protein-coupled receptors. PLoS Biol. 2009, 7. [Google Scholar] [CrossRef] [PubMed]
- Klarenbeek, J.; Goedhart, J.; van Batenburg, A.; Groenewald, D.; Jalink, K. Fourth-generation epac-based FRET sensors for cAMP feature exceptional brightness, photostability and dynamic range: Characterization of dedicated sensors for FLIM, for ratiometry and with high affinity. PLoS ONE 2015, 10. [Google Scholar] [CrossRef] [PubMed]
- Rich, T.C.; Fagan, K.A.; Nakata, H.; Schaack, J.; Cooper, D.M.; Karpen, J.W. Cyclic nucleotide-gated channels colocalize with adenylyl cyclase in regions of restricted cAMP diffusion. J. Gen. Physiol. 2000, 116, 147–161. [Google Scholar] [CrossRef] [PubMed]
- Rich, T.C.; Tse, T.E.; Rohan, J.G.; Schaack, J.; Karpen, J.W. In vivo assessment of local phosphodiesterase activity using tailored cyclic nucleotide-gated channels as cAMP sensors. J. Gen. Physiol. 2001, 118, 63–78. [Google Scholar] [CrossRef] [PubMed]
- Nikolaev, V.O.; Bünemann, M.; Schmitteckert, E.; Lohse, M.J.; Engelhardt, S. Cyclic AMP imaging in adult cardiac myocytes reveals far-reaching beta1-adrenergic but locally confined beta2-adrenergic receptor-mediated signaling. Circ. Res. 2006, 99, 1084–1091. [Google Scholar] [CrossRef] [PubMed]
- Perera, R.K.; Nikolaev, V.O. Compartmentation of cAMP signalling in cardiomyocytes in health and disease. Acta Physiol. 2013, 207, 650–662. [Google Scholar] [CrossRef] [PubMed]
- Bork, N.I.; Nikolaev, V.O. Receptor-cyclic nucleotide microdomains in the heart. In Microdomains in the Cardiovascular System; Nikolaev, V.O., Zaccolo, M., Eds.; Springer Nature: Cham, Switzerland, 2017; pp. 3–15. [Google Scholar]
- Hansma, P.K.; Drake, B.; Marti, O.; Gould, S.A.; Prater, C.B. The scanning ion-conductance microscope. Science 1989, 243, 641–643. [Google Scholar] [CrossRef] [PubMed]
- Korchev, Y.E.; Bashford, C.L.; Milovanovic, M.; Vodyanoy, I.; Lab, M.J. Scanning ion conductance microscopy of living cells. Biophys. J. 1997, 73, 653–658. [Google Scholar] [CrossRef]
- Nikolaev, V.O.; Moshkov, A.; Lyon, A.R.; Miragoli, M.; Novak, P.; Paur, H.; Lohse, M.J.; Korchev, Y.E.; Harding, S.E.; Gorelik, J. Beta2-adrenergic receptor redistribution in heart failure changes cAMP compartmentation. Science 2010, 327, 1653–1657. [Google Scholar] [CrossRef] [PubMed]
- Miragoli, M.; Moshkov, A.; Novak, P.; Shevchuk, A.; Nikolaev, V.O.; El-Hamamsy, I.; Potter, C.M.; Wright, P.; Kadir, S.H.; Lyon, A.R.; et al. Scanning ion conductance microscopy: A convergent high-resolution technology for multi-parametric analysis of living cardiovascular cells. J. R. Soc. Interface 2011, 8, 913–925. [Google Scholar] [CrossRef] [PubMed]
- Froese, A.; Nikolaev, V.O. Imaging alterations of cardiomyocyte cAMP microdomains in disease. Front. Pharmacol. 2015, 6. [Google Scholar] [CrossRef] [PubMed]
- Berisha, F.; Nikolaev, V.O. Cyclic nucleotide imaging and cardiovascular disease. Pharmacol. Ther. 2017, 175, 107–115. [Google Scholar] [CrossRef] [PubMed]
- Fischmeister, R.; Castro, L.; Abi-Gerges, A.; Rochais, F.; Vandecasteele, G. Species-and tissue-dependent effects of NO and cyclic GMP on cardiac ion channels. Comp. Biochem. Physiol. A Mol. Integr. Physiol. 2005, 142, 136–143. [Google Scholar] [CrossRef] [PubMed]
- Vandecasteele, G.; Verde, I.; Rücker-Martin, C.; Donzeau-Gouge, P.; Fischmeister, R. Cyclic GMP regulation of the L-type Ca2+ channel current in human atrial myocytes. J. Physiol. 2001, 533, 329–340. [Google Scholar] [CrossRef] [PubMed]
- Mehel, H.; Emons, J.; Vettel, C.; Wittkopper, K.; Seppelt, D.; Dewenter, M.; Lutz, S.; Sossalla, S.; Maier, L.S.; Lechene, P.; et al. Phosphodiesterase-2 is up-regulated in human failing hearts and blunts beta adrenergic responses in cardiomyocytes. J. Am. Coll. Cardiol. 2013, 62, 1596–1606. [Google Scholar] [CrossRef] [PubMed]
- Zoccarato, A.; Surdo, N.C.; Aronsen, J.M.; Fields, L.A.; Mancuso, L.; Dodoni, G.; Stangherlin, A.; Livie, C.; Jiang, H.; Sin, Y.Y.; et al. Cardiac hypertrophy is inhibited by a local pool of cAMP regulated by phosphodiesterase 2 novelty and significance. Circ. Res. 2015, 117, 707–719. [Google Scholar] [CrossRef] [PubMed]
- Sprenger, J.U.; Perera, R.K.; Steinbrecher, J.H.; Lehnart, S.E.; Maier, L.S.; Hasenfuss, G.; Nikolaev, V.O. In vivo model with targeted cAMP biosensor reveals changes in receptor microdomain communication in cardiac disease. Nat. Commun. 2015, 6. [Google Scholar] [CrossRef] [PubMed]
- Perera, R.K.; Sprenger, J.U.; Steinbrecher, J.H.; Hübscher, D.; Lehnart, S.E.; Abesser, M.; Schuh, K.; El-Armouche, A.; Nikolaev, V.O. Microdomain switch of cGMP-regulated phosphodiesterases leads to ANP-induced augmentation of beta-adrenoceptor-stimulated contractility in early cardiac hypertrophy. Circ. Res. 2015, 116, 1304–1311. [Google Scholar] [CrossRef] [PubMed]
- Vettel, C.; Lindner, M.; Dewenter, M.; Lorenz, K.; Schanbacher, C.; Riedel, M.; Lämmle, S. Phosphodiesterase 2 protects against catecholamine-induced arrhythmia and preserves contractile function after myocardial infarction. Circ. Res. 2017, 120, 120–132. [Google Scholar] [CrossRef] [PubMed]
- Di Benedetto, G.; Zoccarato, A.; Lissandron, V.; Terrin, A.; Li, X.; Houslay, M.D.; Baillie, G.S.; Zaccolo, M. Protein kinase A type I and type II define distinct intracellular signaling compartments. Circ. Res. 2008, 103, 836–844. [Google Scholar] [CrossRef] [PubMed]
- Stangherlin, A.; Gesellchen, F.; Zoccarato, A.; Terrin, A.; Fields, L.A.; Berrera, M.; Surdo, N.C.; Craig, M.A.; Smith, G.; Hamilton, G.; et al. cGMP signals modulate cAMP levels in a compartment-specific manner to regulate catecholamine-dependent signaling in cardiac myocytes. Circ. Res. 2011, 108, 929–939. [Google Scholar] [CrossRef] [PubMed]
- Surapisitchat, J.; Jeon, K.I.; Yan, C.; Beavo, J.A. Differential regulation of endothelial cell permeability by cGMP via phosphodiesterases 2 and 3. Circ. Res. 2007, 101, 811–818. [Google Scholar] [CrossRef] [PubMed]
- Stangherlin, A.; Zaccolo, M. Phosphodiesterases and subcellular compartmentalized cAMP signaling in the cardiovascular system. Am. J. Physiol. Heart Circ. Physiol. 2012, 302, H379–H390. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Lu, C.J.; Hao, G.; Wright, H.; Woodward, L.; Liu, K.; Vergari, E.; Surdo, N.C.; Herring, N.; Zaccolo, M.; et al. Efficacy of B-type natriuretic peptide is coupled to phosphodiesterase 2A in cardiac sympathetic neurons novelty and significance. Hypertension 2015, 66, 190–198. [Google Scholar] [CrossRef] [PubMed]
- Meier, S.; Andressen, K.W.; Aronsen, J.M.; Sjaastad, I.; Hougen, K.; Skomedal, T.; Osnes, J.B.; Qvigstad, E.; Levy, F.O.; Moltzau, L.R. PDE3 inhibition by C-type natriuretic peptide-induced cGMP enhances cAMP-mediated signaling in both non-failing and failing hearts. Eur. J. Pharmacol. 2017, 812, 174–183. [Google Scholar] [CrossRef] [PubMed]
- Mongillo, M.; McSorley, T.; Evellin, S.; Sood, A.; Lissandron, V.; Terrin, A.; Huston, E.; Hannawacker, A.; Lohse, M.J.; Pozzan, T.; et al. Fluorescence resonance energy transfer–based analysis of cAMP dynamics in live neonatal rat cardiac myocytes reveals distinct functions of compartmentalized phosphodiesterases. Circ. Res. 2004, 95, 67–75. [Google Scholar] [CrossRef] [PubMed]
- Rochais, F.; Abi-Gerges, A.; Horner, K.; Lefebvre, F.; Cooper, D.M.; Conti, M.; Fischmeister, R.; Vandecasteele, G. A specific pattern of phosphodiesterases controls the cAMP signals generated by different Gs-coupled receptors in adult rat ventricular myocytes. Circ. Res. 2006, 98, 1081–1088. [Google Scholar] [CrossRef] [PubMed]
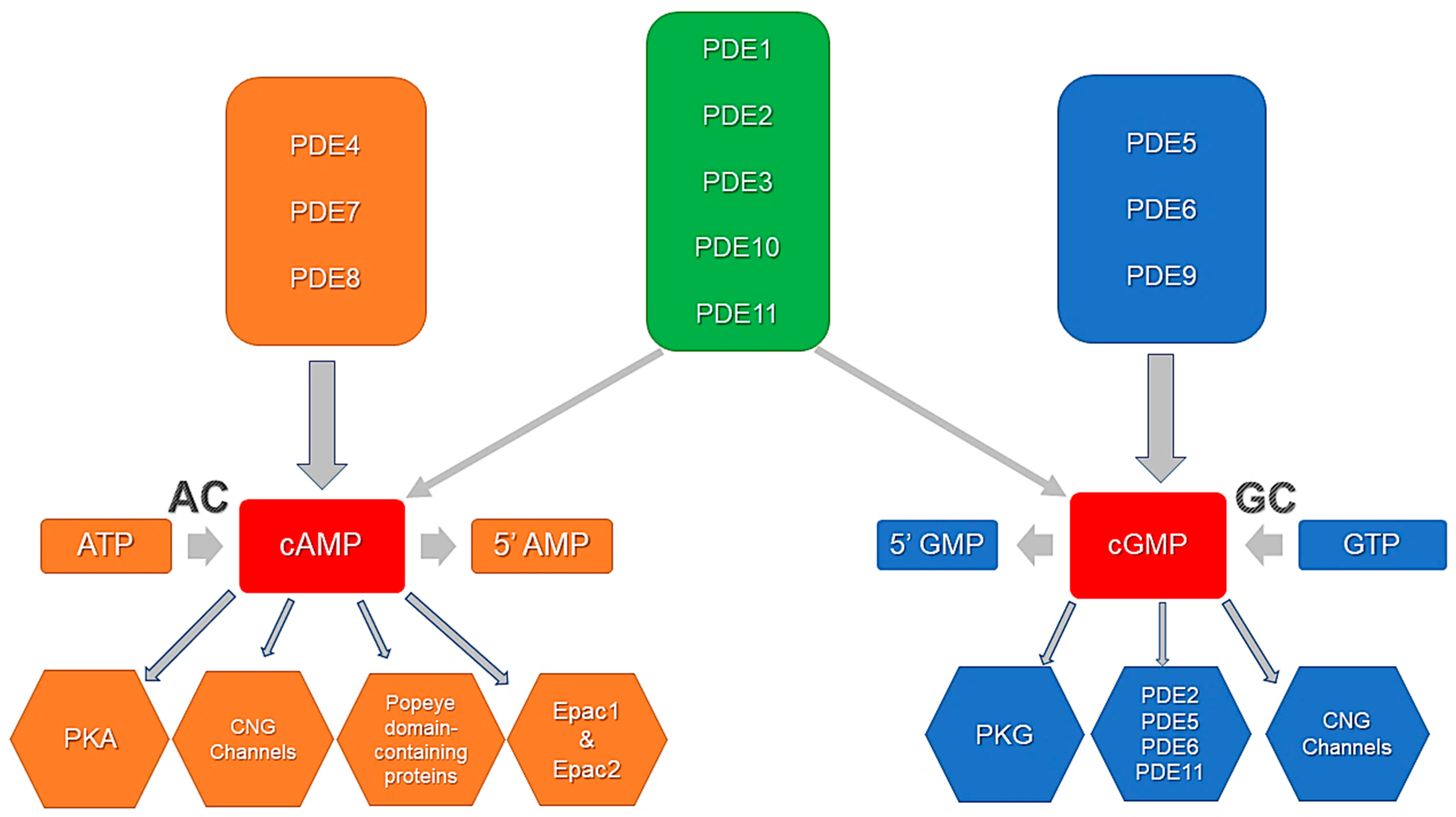
Figure 1.
PDE activity, specificity, and cyclic nucleotide effector proteins. PDE4, PDE7, and PDE8 selectively degrade cAMP. PDE5, PDE6, and PDE9 selectively degrade cGMP. PDE1, PDE2, PDE3, PDE10, and PDE11 are dual-specificity phosphodiesterases that hydrolyze both cAMP and cGMP. Downstream effectors include PKA, PKG, Epacs, PDEs, CNG channels and Popeye domain-containing proteins. Adapted from Ahmad et al., 2015 [76].
Figure 1.
PDE activity, specificity, and cyclic nucleotide effector proteins. PDE4, PDE7, and PDE8 selectively degrade cAMP. PDE5, PDE6, and PDE9 selectively degrade cGMP. PDE1, PDE2, PDE3, PDE10, and PDE11 are dual-specificity phosphodiesterases that hydrolyze both cAMP and cGMP. Downstream effectors include PKA, PKG, Epacs, PDEs, CNG channels and Popeye domain-containing proteins. Adapted from Ahmad et al., 2015 [76].

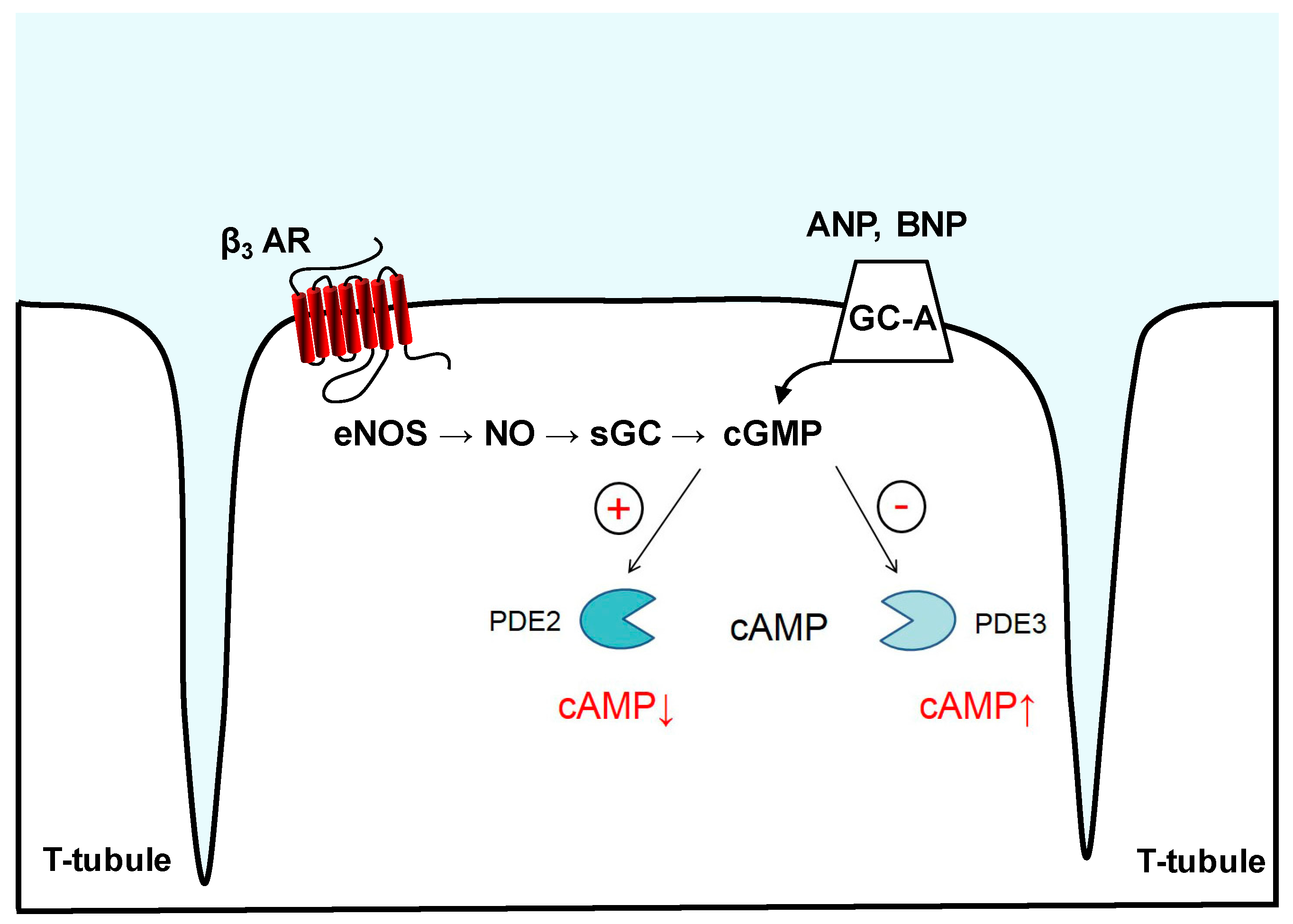
Figure 2.
PDE2- and PDE3-mediated cyclic nucleotide crosstalk. cGMP synthesis occurs by pGCs such as GC-A, which serves as a membrane receptor for ANP and BNP, or by NO-activated sGC, e.g., downstream of eNOS and β3-adrenoreceptor (β3-AR). Binding of cGMP to PDE2 can allosterically increase its hydrolytic activity, lowering cAMP levels in subcellular microdomains. PDE3 is a “cGMP-inhibited” phosphodiesterase that upon cGMP binding and degradation in the catalytic domain shows reduced rates of cAMP hydrolysis, generating a positive cGMP-to-cAMP cross-talk.
Figure 2.
PDE2- and PDE3-mediated cyclic nucleotide crosstalk. cGMP synthesis occurs by pGCs such as GC-A, which serves as a membrane receptor for ANP and BNP, or by NO-activated sGC, e.g., downstream of eNOS and β3-adrenoreceptor (β3-AR). Binding of cGMP to PDE2 can allosterically increase its hydrolytic activity, lowering cAMP levels in subcellular microdomains. PDE3 is a “cGMP-inhibited” phosphodiesterase that upon cGMP binding and degradation in the catalytic domain shows reduced rates of cAMP hydrolysis, generating a positive cGMP-to-cAMP cross-talk.

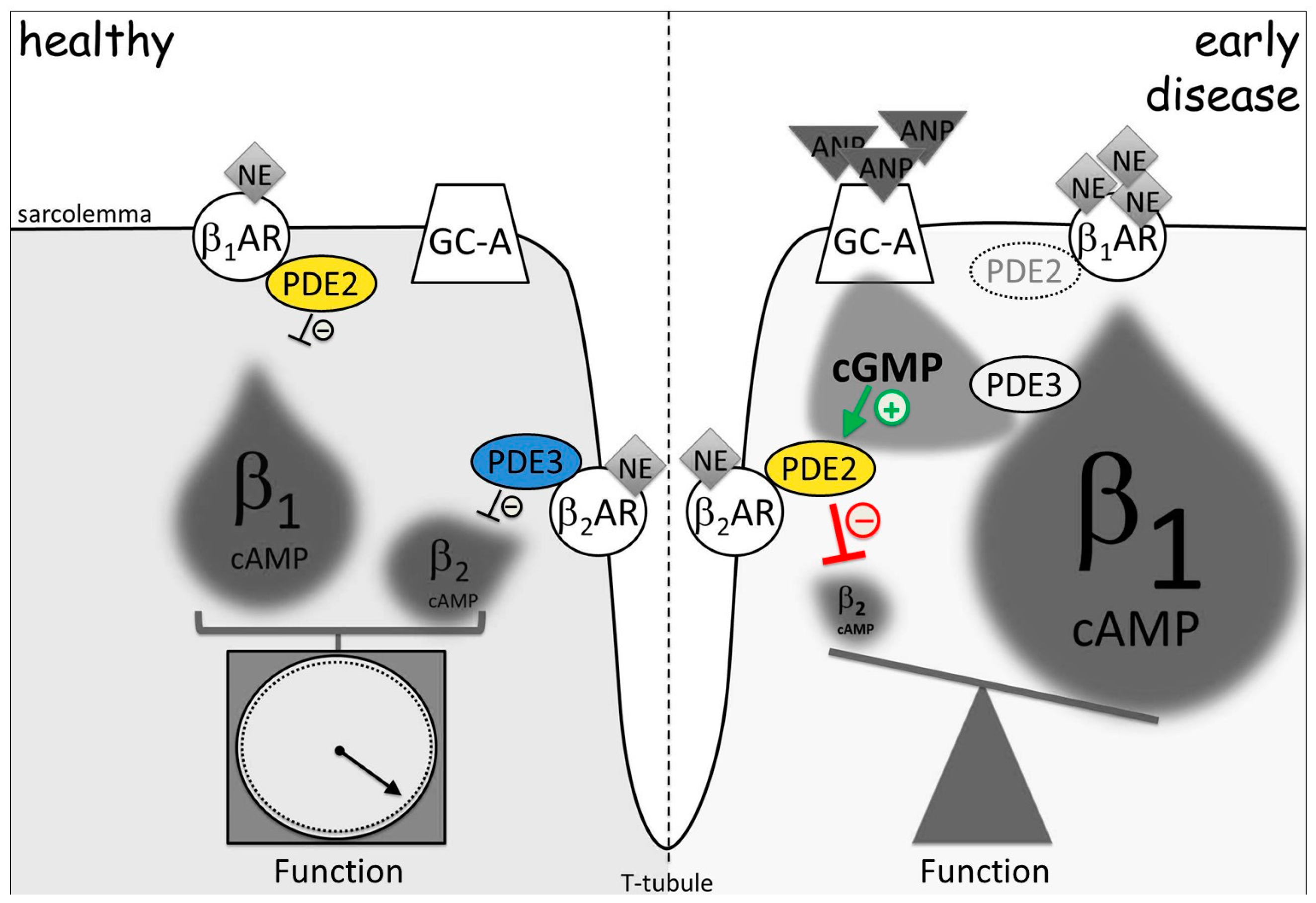
Figure 3.
PDE2 and PDE3 redistribution in early cardiac hypertrophy changes cGMP-to-cAMP cross-talk. β2-AR microdomain is normally controlled by PDE3, while PDE2 is functionally associated with β1-AR. In disease, redistribution of PDE2 from β1- to β2-AR-associated membrane microdomains leads to a decrease of the local β2-AR-cAMP and to an increase of global β1-AR-cAMP pool under elevated ANP and cGMP levels observed in hypertrophy. This relocation of cGMP-regulated PDEs leads to a turnaround of cGMP-to-cAMP cross-talk between both β-AR microdomains. By this mechanism, elevated ANP can augment β-adrenoceptor-stimulated contractile function. NE: norepinephrine, the physiological β-AR agonist. Adapted from [158].
Figure 3.
PDE2 and PDE3 redistribution in early cardiac hypertrophy changes cGMP-to-cAMP cross-talk. β2-AR microdomain is normally controlled by PDE3, while PDE2 is functionally associated with β1-AR. In disease, redistribution of PDE2 from β1- to β2-AR-associated membrane microdomains leads to a decrease of the local β2-AR-cAMP and to an increase of global β1-AR-cAMP pool under elevated ANP and cGMP levels observed in hypertrophy. This relocation of cGMP-regulated PDEs leads to a turnaround of cGMP-to-cAMP cross-talk between both β-AR microdomains. By this mechanism, elevated ANP can augment β-adrenoceptor-stimulated contractile function. NE: norepinephrine, the physiological β-AR agonist. Adapted from [158].

© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Pavlaki, N.; Nikolaev, V.O. Imaging of PDE2- and PDE3-Mediated cGMP-to-cAMP Cross-Talk in Cardiomyocytes. J. Cardiovasc. Dev. Dis. 2018, 5, 4. https://0-doi-org.brum.beds.ac.uk/10.3390/jcdd5010004
AMA Style
Pavlaki N, Nikolaev VO. Imaging of PDE2- and PDE3-Mediated cGMP-to-cAMP Cross-Talk in Cardiomyocytes. Journal of Cardiovascular Development and Disease. 2018; 5(1):4. https://0-doi-org.brum.beds.ac.uk/10.3390/jcdd5010004
Chicago/Turabian StylePavlaki, Nikoleta, and Viacheslav O. Nikolaev. 2018. "Imaging of PDE2- and PDE3-Mediated cGMP-to-cAMP Cross-Talk in Cardiomyocytes" Journal of Cardiovascular Development and Disease 5, no. 1: 4. https://0-doi-org.brum.beds.ac.uk/10.3390/jcdd5010004
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.