The Aging Vasculature: Glucose Tolerance, Hypoglycemia and the Role of the Serum Response Factor

{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
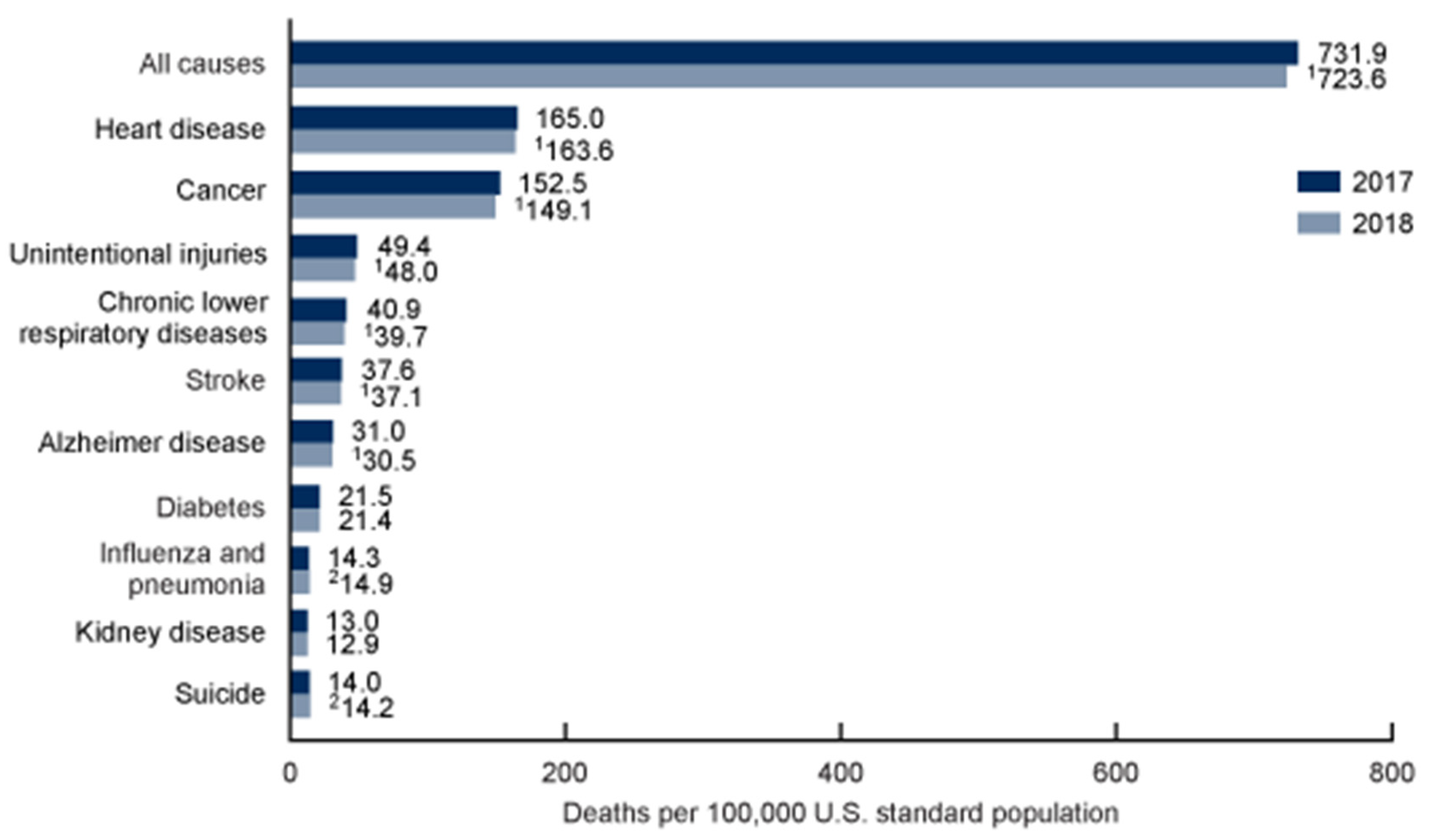
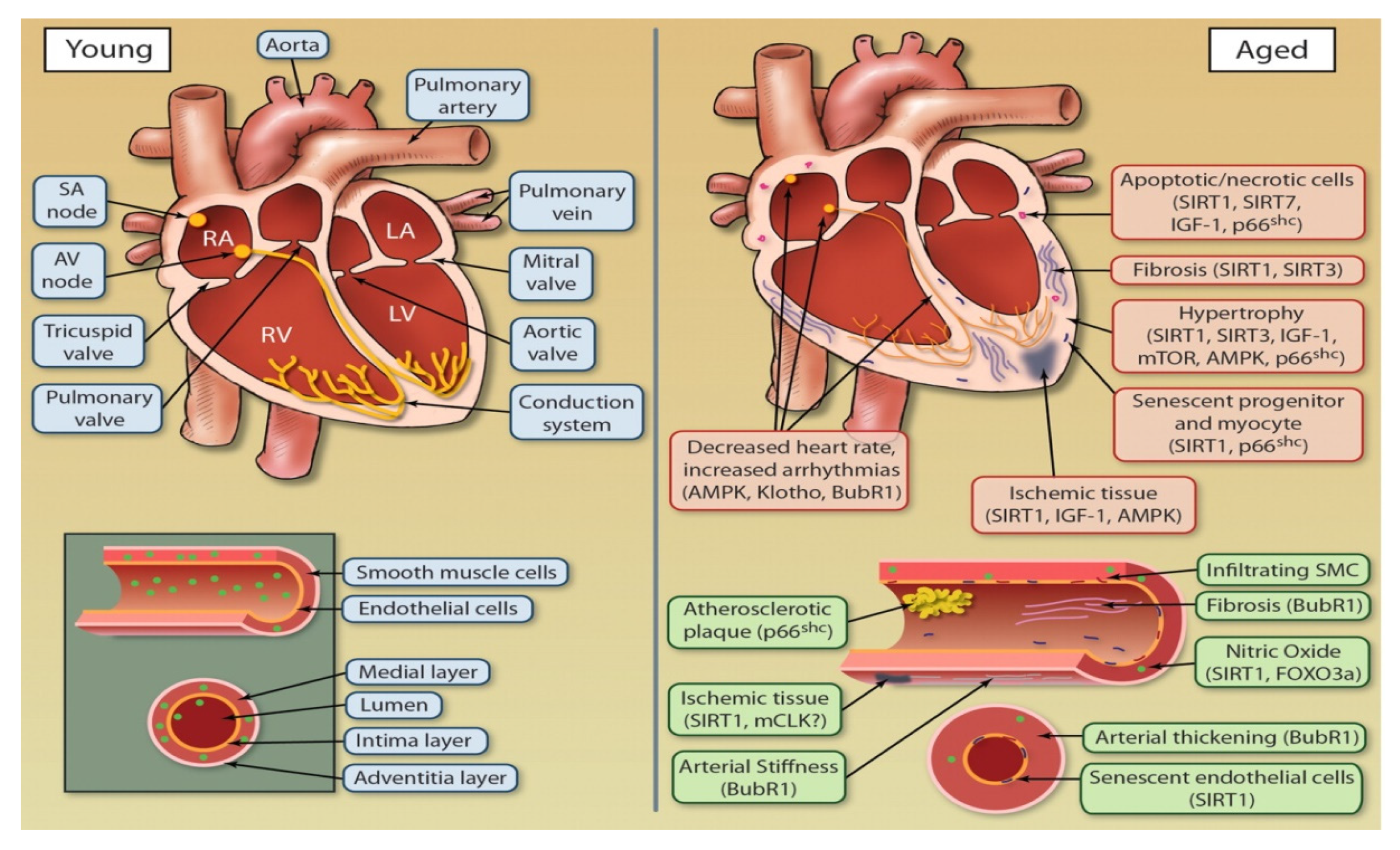
1.1. The Aging Heart
1.2. The Aging Vasculature
1.3. The Aging Vasculature: Endothelium
1.4. Endothelial Cellular Senescence
1.5. Glucose Tolerance and Aging
1.6. Hyperglycemia, Diabetes, and Aging
1.7. Hypoglycemia and Aging
1.8. Serum Response Factor (SRF)
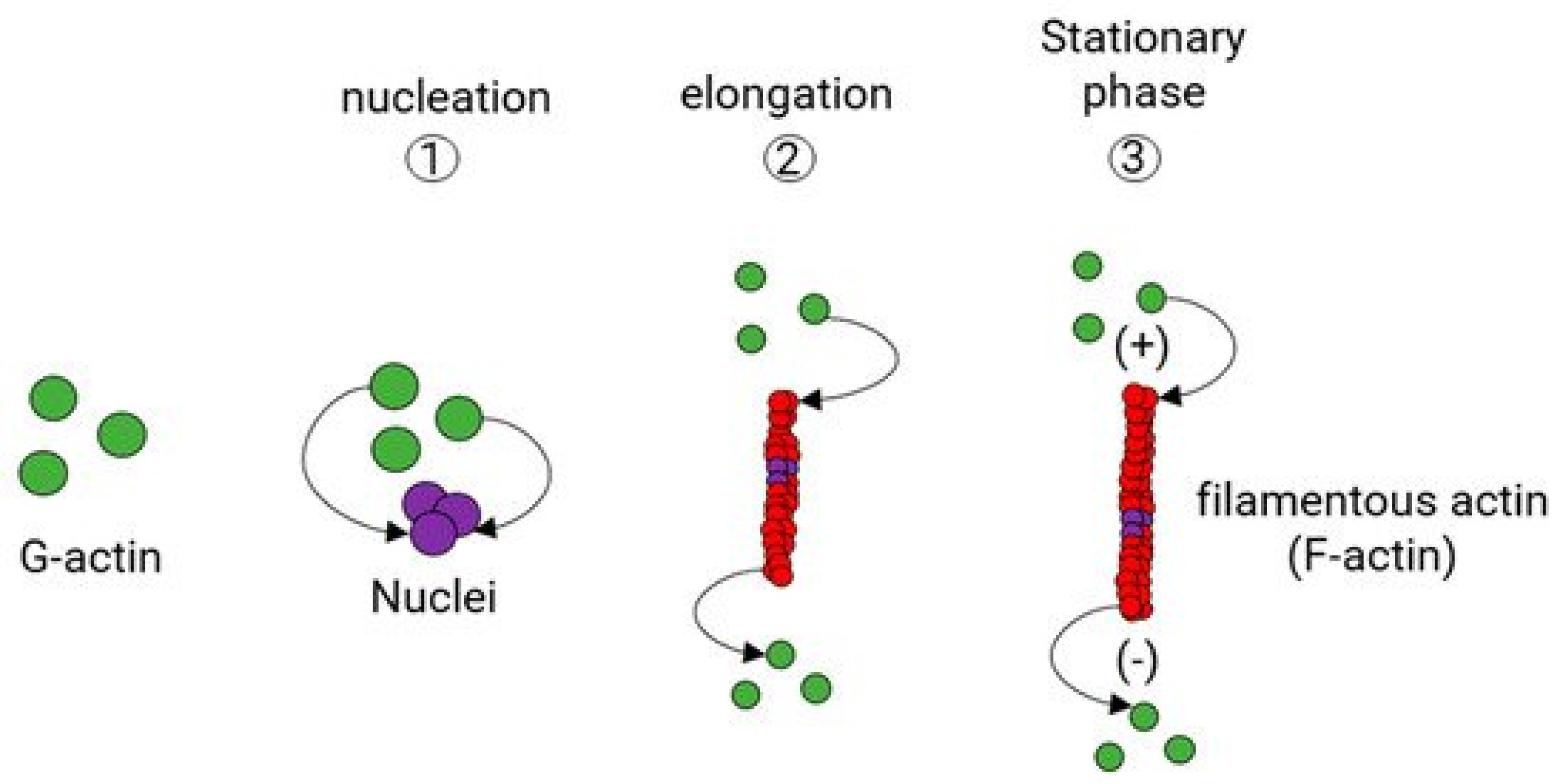
2. The Aging Cytoskeleton: Glucose Sensitivity
3. SRF: Glucose Sensitivity
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- U.S. Census Bureau. Population Division, Annual Estimates of the Resident Population for Selected Age Groups by Sex for the United States, States, Counties, and Puerto Rico Commonwealth and Municipios: 1 April 2010 to 1 July 2013. Release Date: June 2014; 2010 Census Special Reports, Centenarians: 2010, C2010SR-03; National Vital Statistics Reports; U.S. Census Bureau: Suitland-Silver Hill, MD, USA, 2012.
- Ortman, J.M.; Hogan, H. An Aging Nation: The older population in the United States. Population estimates and projections. In Current Population Reports; National Vital Statistics Reports; 2014; Available online: https://www.giaging.org/resources/an-aging-nation-the-older-population-in-the-united-states (accessed on 15 May 2021).
- Berrío, M.I. Envejecimiento de la población: Un reto para la salud pública. Rev. Colomb. Anestesiol. 2012, 40, 192–194. [Google Scholar] [CrossRef] [Green Version]
- Morens, D.M. Predominant role of bacterial pneumonia as a cause of death in pandemic influenza: Implications for pandemic influenza preparedness. J. Infect. Dis. 2008, 198, 962–970. [Google Scholar] [CrossRef] [PubMed]
- Murphy, S.L. Deaths: Final Data for 2010. National Vital Statistics Reports; National Center for Health Statistics: Hyattsville, MD, USA, 2013; Volume 64, p. 4.
- Available online: https://www.cdc.gov/nchs/products/databriefs/db355.htm (accessed on 3 January 2021).
- Heidenreich, P.A. Forecasting the future of cardiovascular disease in the United States: A policy statement from the American Heart Association. Circulation 2011, 123, 933–944. [Google Scholar] [CrossRef] [Green Version]
- Available online: https://www.physio-pedia.com/index.php?title=Cardiovascular_Considerations_in_the_Older_Patient&veaction=edit§ion=8 (accessed on 1 March 2021).
- North, B.J. The Intersection between Aging and Cardiovascular Disease. Circ. Res. 2012, 110, 1097–1108. [Google Scholar] [CrossRef] [PubMed]
- Lakatta, E.G. Arterial and cardiac aging: Major shareholders in cardiovascular disease enterprises, part I: Aging arteries: A “set up” for vascular disease. Circulation 2003, 107, 139–146. [Google Scholar] [CrossRef] [Green Version]
- Olivetti, G. Cardiomyopathy of the aging human heart: Myocyte loss and reactive cellular hypertrophy. Circ. Res. 1991, 68, 1560–1568. [Google Scholar] [CrossRef] [Green Version]
- Goldspink, D.F. Cardiomyocyte death and the ageing and failing heart. Exp. Physiol. 2003, 88, 447–458. [Google Scholar] [CrossRef]
- Chimenti, C. Senescence and death of primitive cells and myocytes lead to premature cardiac aging and heart failure. Circ. Res. 2003, 93, 604–613. [Google Scholar] [CrossRef]
- Anversa, P. Life and death of cardiac stem cells: A paradigm shift in cardiac biology. Circulation 2006, 113, 1451–1463. [Google Scholar] [CrossRef] [Green Version]
- Nadal-Ginard, B. Myocyte death, growth, and regeneration in cardiac hypertrophy and failure. Circ. Res. 2003, 92, 139–150. [Google Scholar] [CrossRef] [Green Version]
- Anversa, P. Myocyte growth and cardiac repair. J. Mol. Cell Cardiol. 2002, 34, 91–105. [Google Scholar] [CrossRef]
- Lakatta, E.G. Arterial and cardiac aging: Major shareholders in cardiovascular disease enterprises, part II: The aging heart in health: Links to heart disease. Circulation 2003, 107, 346–354. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Antelmi, I. Influence of age, gender, body mass index, and functional capacity on heart rate variability in a cohort of subjects without heart disease. Am. J. Cardiol. 2004, 93, 381–385. [Google Scholar] [CrossRef]
- Stern, S. Cardiology patient pages. Aging and diseases of the heart. Circulation 2003, 108, e99–e101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fleg, J.L. Impact of age on the cardiovascular response to dynamic upright exercise in healthy men and women. J. Appl. Physiol. 1995, 78, 890–900. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schulman, S.P. Age-related decline in left ventricular filling at rest and exercise. Am. J. Physiol. 1992, 263, H1932–H1938. [Google Scholar] [CrossRef] [PubMed]
- Levy, D. Echocardiographically detected left ventricular hypertrophy: Prevalence and risk factors. The Framingham Heart Study. Ann. Intern. Med. 1988, 108, 7–13. [Google Scholar] [CrossRef] [PubMed]
- Akazawa, H. Roles of cardiac transcription factors in cardiac hypertrophy. Circ. Res. 2003, 92, 1079–1088. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Angeli, F. Hypertension, inflammation and atrial fibrillation. J. Hypertens. 2014, 32, 480. [Google Scholar] [CrossRef] [PubMed]
- Izzo, J.L. Arterial stiffness: Clinical relevance, measurement, and treatment. Rev. Cardiovasc. Med. 2001, 2, 29–34. [Google Scholar]
- Sandoo, A. The endothelium and its role in regulating vascular tone. Open Cardiovasc. Med. J. 2010, 4, 302–312. [Google Scholar] [CrossRef] [PubMed]
- Sumpio, B.E. Cells in focus: Endothelial cell. Int. J. Biochem. Cell Biol. 2002, 34, 1508–1512. [Google Scholar] [CrossRef]
- Rajendran, P. The vascular endothelium and human diseases. Int. J. Biol. Sci. 2013, 9, 1057–1069. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hong, Y.M. Atherosclerotic cardiovascular disease beginning in childhood. Korean Circ. J. 2010, 40, 1–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Napoli, C. Fatty streak formation occurs in human fetal aortas and is greatly enhanced by maternal hypercholesterolemia. Intimal accumulation of low density lipoprotein and its oxidation precede monocyte recruitment into early atherosclerotic lesions. J. Clin. Investig. 1997, 100, 2680–2690. [Google Scholar] [CrossRef]
- Napoli, C. Primary prevention of atherosclerosis: A clinical challenge for the reversal of epigenetic mechanisms? Circulation 2012, 125, 2363–2373. [Google Scholar] [CrossRef] [Green Version]
- Jia, G.; Aroor, A.R.; Jia, C.; Sowers, J.R. Endothelial cell senescence in aging-related vascular dysfunction. Biochim. Biophys. Acta (BBA) Mol. Basis Dis. 2019, 1865, 1802–1809. [Google Scholar] [CrossRef]
- Egashira, K. Effects of age on endothelium-dependent vasodilation of resistance coronary artery by acetylcholine in humans. Circulation 1993, 88, 77–81. [Google Scholar] [CrossRef] [Green Version]
- Taddei, S. Aging and endothelial function in normotensive subjects and patients with essential hypertension. Circulation 1995, 91, 1981–1987. [Google Scholar] [CrossRef]
- Hatake, K. Effect of aging on endothelium-dependent vascular relaxation of isolated human basilar artery to thrombin and bradykinin. Stroke 1995, 21, 1039–1043. [Google Scholar] [CrossRef] [Green Version]
- Singh, N. Ageing is associated with impairment of nitric oxide and prostanoid dilator pathways in the human forearm. Clin. Sci. 2002, 102, 595–600. [Google Scholar] [CrossRef]
- Lyons, D. Impaired nitric oxide-mediated vasodilatation and total body nitric oxide production in healthy old age. Clin. Sci. 1997, 93, 519–525. [Google Scholar] [CrossRef] [PubMed]
- Singh, R.B. Pathogenesis of atherosclerosis: A multifactorial process. Exp. Clin. Cardiol. 2002, 7, 40–53. [Google Scholar] [PubMed]
- Buerk, D.G. Nitric oxide signaling in the microcirculation. Crit. Rev. Biomed. Eng. 2011, 39, 397–433. [Google Scholar] [CrossRef] [Green Version]
- Förstermann, U. Nitric oxide synthases: Regulation and function. Eur. Heart J. 2012, 33, 829–837. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moncada, S. The discovery of nitric oxide and its role in vascular biology. Br. J. Pharmacol. 2006, 147, S193–S201. [Google Scholar] [CrossRef] [Green Version]
- Napoli, C. Effects of nitric oxide on cell proliferation: Novel insights. J. Am. Coll. Cardiol. 2013, 62, 89–95. [Google Scholar] [CrossRef]
- Cai, H. Endothelial dysfunction in cardiovascular diseases: The role of oxidant stress. Circ. Res. 2000, 87, 840–844. [Google Scholar] [CrossRef] [Green Version]
- Csiszar, A. Aging-induced phenotypic changes and oxidative stress impair coronary arteriolar function. Circ. Res. 2002, 90, 1159–1166. [Google Scholar] [CrossRef] [Green Version]
- Barton, M. Anatomic heterogeneity of vascular aging: Role of nitric oxide and endothelin. Hypertension 1997, 30, 817–824. [Google Scholar] [CrossRef] [PubMed]
- Tanabe, T. Exercise training improves ageing-induced decrease in eNOS expression of the aorta. Acta Physiol. Scand. 2003, 178, 3–10. [Google Scholar] [CrossRef] [PubMed]
- van der Loo, B. Enhanced peroxynitrite formation is associated with vascular aging. J. Exp. Med. 2000, 192, 1731–1744. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goettsch, W. Increased expression of endothelin-1 and inducible nitric oxide synthase isoform II in aging arteries in vivo: Implications for atherosclerosis. Biochem. Biophys. Res. Commun. 2001, 280, 908–913. [Google Scholar] [CrossRef] [PubMed]
- Hamilton, C.A. Superoxide excess in hypertension and aging: A common cause of endothelial dysfunction. Hypertension 2001, 37, 529–534. [Google Scholar] [CrossRef]
- Carlsson, L.M.; Marklund, S.L.; Edlund, T. The Rat Extracellular Superoxide Dismutase Dimer is Converted to a Tetramer by the Exchange of a Single Amino Acid. Proc. Natl. Acad. Sci. USA 1996, 93, 5219–5222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ranjan, V. Constitutive NOS expression in cultured endothelial cells is elevated by fluid shear stress. Am. J. Physiol. 1995, 269, H550–H555. [Google Scholar] [CrossRef] [Green Version]
- Davis, M.E. Shear stress regulates endothelial nitric oxide synthase expression through c-Src by divergent signaling pathways. Circ. Res. 2001, 89, 1073–1080. [Google Scholar] [CrossRef] [Green Version]
- Pierce, G.L. Reduced vascular tetrahydrobiopterin (BH4) and endothelial function with ageing: Is it time for a chronic BH4 supplementation trial in middle-aged and older adults? J. Physiol. 2008, 586 Pt 11, 2673–2674. [Google Scholar] [CrossRef]
- Foreman, K.E. Molecular mechanisms of replicative senescence in endothelial cells. Exp. Gerontol. 2003, 38, 1251–1257. [Google Scholar] [CrossRef]
- Campisi, J. Aging, cellular senescence, and cancer. Annu. Rev. Physiol. 2013, 75, 685–705. [Google Scholar] [CrossRef] [Green Version]
- Ambrose, C.T. Pro-Angiogenesis Therapy and Aging: A Mini-Review. Gerontology 2017, 63, 393–400. [Google Scholar] [CrossRef] [Green Version]
- Minamino, T. Endothelial cell senescence in human atherosclerosis: Role of telomere in endothelial dysfunction. Circulation 2002, 105, 1541–1544. [Google Scholar] [CrossRef] [Green Version]
- Buys, C.H. Telomeres, telomerase, and cancer. N. Engl. J. Med. 2002, 342, 1282–1283. [Google Scholar] [CrossRef]
- Chang, E. Telomere length and replicative aging in human vascular tissues. Proc. Natl. Acad. Sci. USA 1995, 92, 11190–11194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murasawa, S. Constitutive human telomerase reverse transcriptase expression enhances regenerative properties of endothelial progenitor cells. Circulation 2002, 106, 1133–1139. [Google Scholar] [CrossRef] [Green Version]
- Vasa, M. Nitric oxide activates telomerase and delays endothelial cell senescence. Circ. Res. 2000, 87, 540–542. [Google Scholar] [CrossRef] [Green Version]
- Haendeler, J. Hydrogen peroxide triggers nuclear export of telomerase reverse transcriptase via Src kinase family-dependent phosphorylation of tyrosine 707. Mol. Cell. Biol. 2003, 23, 4598–4610. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haendeler, J. Antioxidants inhibit nuclear export of telomerase reverse transcriptase and delay replicative senescence of endothelial cells. Circ. Res. 2004, 94, 768–775. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matsushita, H. eNOS activity is reduced in senescent human endothelial cells: Preservation by hTERT immortalization. Circ. Res. 2001, 89, 793–798. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, J. Aging enhances the sensitivity of endothelial cells toward apoptotic stimuli: Important role of nitric oxide. Circ. Res. 2001, 89, 709–715. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dimmeler, S. Regulation of endothelial cell apoptosis in atherothrombosis. Curr. Opin. Lipidol. 2002, 13, 531–536. [Google Scholar] [CrossRef] [PubMed]
- Weinsaft, J.W. Aging-associated changes in vascular activity: A potential link to geriatric cardiovascular disease. Am. J. Geriatr. Cardiol. 2001, 10, 348–354. [Google Scholar] [CrossRef] [PubMed]
- Edelberg, J.M. Young adult bone marrow-derived endothelial precursor cells restore aging-impaired cardiac angiogenic function. Circ. Res. 2002, 90, E89–E93. [Google Scholar] [CrossRef] [Green Version]
- Scheubel, R.J. Age-dependent depression in circulating endothelial progenitor cells in patients undergoing coronary artery bypass grafting. J. Am. Coll. Cardiol. 2003, 42, 2073–2080. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Azuma, H. Regeneration of endothelial cells after balloon denudation of the rabbit carotid artery and changes in responsiveness. Jpn. J. Pharmacol. 1990, 52, 541–552. [Google Scholar] [CrossRef]
- Weidinger, F.F. Persistent dysfunction of regenerated endothelium after balloon angioplasty of rabbit iliac artery. Circulation 1990, 81, 1667–1679. [Google Scholar] [CrossRef] [Green Version]
- Fournet-Bourguignon, M.P. Phenotypic and functional changes in regenerated porcine coronary endothelial cells: Increased uptake of modified LDL and reduced production of, N.O. Circ. Res. 2000, 86, 854–861. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, A.R. Vascular endothelial dysfunction in aging: Loss of Akt-dependent endothelial nitric oxide synthase phosphorylation and partial restoration by (R)-alpha-lipoic acid. Biochem. Soc. Trans. 2003, 31, 1447–1449. [Google Scholar] [CrossRef] [PubMed]
- Rivard, A. Age-dependent impairment of angiogenesis. Circulation 1999, 99, 111–120. [Google Scholar] [CrossRef] [Green Version]
- Sakai, Y. Involvement of increased arginase activity in impaired cavernous relaxation with aging in the rabbit. J. Urol. 2004, 172, 369–373. [Google Scholar] [CrossRef] [PubMed]
- Pili, R. Altered angiogenesis underlying age-dependent changes in tumor growth. J. Natl. Cancer Inst. 1994, 86, 1303–1314. [Google Scholar] [CrossRef] [PubMed]
- Ashcroft, G.S. Aging alters the inflammatory and endothelial cell adhesion molecule profiles during human cutaneous wound healing. Lab. Investig. 1998, 78, 47–58. [Google Scholar]
- Koike, T. Inhibited angiogenesis in aging: A role for TIMP-2. J. Gerontol. A Biol. Sci. Med. Sci. 2003, 58, B798–B805. [Google Scholar] [CrossRef]
- Sadoun, E. Impaired angiogenesis in aging is associated with alterations in vessel density, matrix composition, inflammatory response, and growth factor expression. J. Histochem. Cytochem. 2003, 51, 1119–1130. [Google Scholar] [CrossRef] [Green Version]
- Crewe, C.; An, Y.A.; Scherer, P.E. The ominous triad of adipose tissue dysfunction: Inflammation, fibrosis, and impaired angiogenesis. J. Clin. Investig. 2017, 127, 74–82. [Google Scholar] [CrossRef] [Green Version]
- Volpert, O.V. Inhibition of angiogenesis by thrombospondin-2. Biochem. Biophys. Res. Commun. 1995, 217, 326–332. [Google Scholar] [CrossRef]
- Armstrong, L.C. Thrombospondin 2 inhibits microvascular endothelial cell proliferation by a caspase-independent mechanism. Mol. Biol. Cell 2002, 13, 1893–1905. [Google Scholar] [CrossRef] [Green Version]
- Barzilay, J.I. Cardiovascular disease in older adults with glucose disorders: Comparison of American Diabetes Association criteria for diabetes mellitus with WHO criteria. Lancet 1995, 354, 622–625. [Google Scholar] [CrossRef]
- Lerman-Garber, I. Changes in glucose tolerance in elderly. Rev. Investig. Clin. 2010, 62, 312–317. [Google Scholar]
- Singer, D.E. Diabetic myocardial infarction: Interaction of diabetes with other pre-infarction risk factors. Diabetes 1989, 38, 350–357. [Google Scholar] [CrossRef]
- Buttar, H.S. Prevention of cardiovascular diseases: Role of exercise, dietary interventions, obesity and smoking cessation. Exp. Clin. Cardiol. 2005, 10, 229–249. [Google Scholar]
- Geiss, L.S.; National Diabetes Data Group. Diabetes in America; National Institutes of Health, National Institute of Diabetes and Digestive and Kidney Diseases: Bethesda, MD, USA, 1995; pp. 233–257. [Google Scholar]
- Stone, P.H. The effect of diabetes mellitus on prognosis and serial left ventricular function after acute myocardial infarction: Contribution of both coronary disease and diastolic left ventricular dysfunction to the adverse prognosis. J. Am. Coll. Cardiol. 1989, 14, 49–57. [Google Scholar] [CrossRef] [Green Version]
- Grund, S.M.; Small, L.D.L. Atherogenic dyslipidemia, and the metabolic syndrome. Circulation 1997, 95, 1–4. [Google Scholar] [CrossRef]
- Austin, M.A. Atherogenic lipoprotein phenotype: A proposed genetic marker for coronary heart disease risk. Circulation 1990, 82, 495–506. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Austin, M.A. Small, dense low density lipoproteins, the insulin resistance syndrome and noninsulin-dependent diabetes. Curr. Opin. Lipidol. 1996, 7, 167–171. [Google Scholar] [CrossRef] [PubMed]
- Arsenault, B.J. Cholesterol levels in small LDL particles predict the risk of coronary heart disease in the EPIC-Norfolk prospective population study. Eur. Heart J. 2007, 28, 2770–2777. [Google Scholar] [CrossRef] [Green Version]
- Chan, A.C. Vitamin E and atherosclerosis. J. Nutr. 1998, 128, 1593–1596. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Napoli, C. Glycosylation enhances oxygen radical-induced modifications and decreases acetylhydrolase activity of human low density lipoprotein. Basic Res. Cardiol. 1997, 92, 96–105. [Google Scholar] [CrossRef]
- Bucala, R. Lipid advanced glycosylated pathway for lipid oxidation in vivo. Proc. Natl. Acad. Sci. USA 1993, 91, 9441–9445. [Google Scholar] [CrossRef] [Green Version]
- Shen, G.X. Lipid disorders in diabetes mellitus and current management. Curr. Pharm. Anal. 2007, 3, 17–24. [Google Scholar] [CrossRef]
- Bokken, B. The pathophysiology of cardiovascular disease and diabetes: Beyond blood pressure and lipids diabetes spectrum. Diabetes Spectr. 2008, 21, 160–165. [Google Scholar]
- Lewis, D.H. The effect of trauma on the regulation of the microcirculation. Pathophysiology 1998, 5 (Suppl. 1), 191. [Google Scholar] [CrossRef]
- Smith, S.E. Cardiac autonomic dysfunction in patients with diabetic retinopathy. Diabetologia 1981, 21, 525–528. [Google Scholar] [PubMed]
- Ewing, D.J. The natural history of diabetic autonomic neuropathy. QIM Int. J. Med. 1980, 49, 95–108. [Google Scholar]
- Taskiran, M. Decreased myocardial perfusion reserve in diabetic autonomic neuropathy. Diabetes 2002, 51, 3306–3310. [Google Scholar] [CrossRef] [PubMed]
- Koh, K.K. Inflammatory markers and the metabolic syndrome. J. Am. Coll. Cardiol. 2005, 46, 1978–1985. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brownlee, M. Biochemistry and molecular cell biology of diabetic complications. Nature 2001, 414, 813–820. [Google Scholar] [CrossRef]
- Williams, S.B. Acute hyperglycemia attenuates endothelium-dependent vasodilation in humans in vivo. Circulation 1998, 97, 1695–1701. [Google Scholar] [CrossRef] [Green Version]
- Hayden, M.R. The central role of vascular extracellular matrix and basement membrane remodeling in metabolic syndrome and type 2 diabetes: The matrix preloaded. Cardiovasc. Diabetol. 2005, 4, 9–29. [Google Scholar] [CrossRef] [Green Version]
- Bauer, T.A. Skeletal muscle deoxygenation after the onset of moderate exercise suggests slowed microvascular blood flow kinetics in type 2 diabetes. Diabetes Care 2007, 30, 2880–2885. [Google Scholar] [CrossRef] [Green Version]
- Weir, M. Microalbuminuria and cardiovascular disease. Clin. J. Am. Soc. Nephrol. 2007, 2, 581–590. [Google Scholar] [CrossRef] [PubMed]
- Sprague, J.E. Glucose counter-regulatory responses to hypoglycemia. Pediatric Endocrinol. Rev. PER 2011, 9, 463–475. [Google Scholar] [PubMed]
- Avila-Fematt, F.M. Hypoglycemia in the elderly with diabetes mellitus. Rev. Invest. Clin. 2015, 62, 366–374. [Google Scholar]
- Rehni, A.K.; Dave, K.R. Impact of Hypoglycemia on Brain Metabolism During Diabetes. Mol. Neurobiol. 2018, 55, 9075–9088. [Google Scholar] [CrossRef] [PubMed]
- Abdelhafiz, A.H. Hypoglycemia in older people—A less well recognized risk factor for frailty. Aging Dis. 2015, 6, 156–167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abdelhafiz, A.H. Hypoglycemia in residential care homes. Br. J. Gen. Pract. 2009, 59, 49–50. [Google Scholar] [CrossRef]
- Schwartz, N. Glycemic thresholds for activation of glucose counter-regulatory systems are higher than the threshold for symptoms. J. Clin. Investig. 1987, 79, 777–781. [Google Scholar] [CrossRef] [Green Version]
- Mitrakou, A. Hierarchy of glycemic thresholds for counter-regulatory hormone secretion, symptoms, and cerebral dysfunction. Am. J. Physiol. 1991, 260, E67–E74. [Google Scholar]
- Cooperberg, B.A. Insulin reciprocally regulates glucagon secretion in humans. Diabetes 2010, 59, 2936–2940. [Google Scholar] [CrossRef] [Green Version]
- Exton, J.H. Mechanisms of hormonal regulation of hepatic glucose metabolism. Diabetes Metab. Rev. 1987, 3, 163–183. [Google Scholar] [CrossRef]
- Cryer, P.E. Exercise-related hypoglycemia-associated autonomic failure in diabetes. Diabetes 2009, 58, 1951–1952. [Google Scholar] [CrossRef] [Green Version]
- Rendell, M. The role of sulphonylureas in the management of type 2 diabetes mellitus. Drugs 2004, 64, 1339–1358. [Google Scholar] [CrossRef]
- United Kingdom Prospective Diabetes Study Group. A 6 year randomised controlled trial comparing sulfonylurea, insulin and metformin therapy in patients with newly diagnosed type 2 diabetes that could not be controlled with diet therapy (UKPDS 24). Ann. Intern. Med. 1998, 128, 165–175. [Google Scholar] [CrossRef] [PubMed]
- Zammitt, N.N. Hypoglycemia in type 2 diabetes: Pathophysiology, frequency, and effects of different treatment modalities. Diabetes Care 2005, 28, 2948–2961. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shafiee, G. The importance of hypoglycemia in diabetic patients. J. Diabetes Metab. Disord. 2012, 11, 17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wei, M. Low fasting plasma glucose level as a predictor of cardiovascular disease and all-cause mortality. Circulation 2000, 101, 2047–2052. [Google Scholar] [CrossRef] [Green Version]
- Gerstein, H.C. Effects of intensive glucose lowering in type 2 diabetes. N. Engl. J. Med. 2008, 358, 2545–2559. [Google Scholar]
- Duckworth, W. Glucose control and vascular complications in veterans with type 2 diabetes. N. Engl. J. Med. 2009, 360, 129–139. [Google Scholar] [CrossRef] [Green Version]
- Hadaegh, F. Impact of metabolic syndrome, diabetes and prediabetes on cardiovascular events: Tehran lipid and glucose study. Diabetes Res. Clin. Pract. 2010, 87, 342–347. [Google Scholar] [CrossRef]
- Asvold, B.O. Cognitive function in type 1 diabetic adults with early exposure to severe hypoglycemia: A 16-year follow-up study. Diabetes Care 2010, 33, 1945–1947. [Google Scholar] [CrossRef] [Green Version]
- Whitmer, R.A. Hypoglycemic episodes and risk of dementia in older patients with type 2 diabetes mellitus. JAMA 2009, 301, 1565–1572. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chai, J. Serum response factor: Discovery, biochemistry, biological roles and implications for tissue injury healing. J. Physiol. Pharm. 2002, 53, 147–157. [Google Scholar]
- Johansen, F.E. Two pathways for serum regulation of the c-fos serum response element require specific sequence elements and a minimal domain of serum response factor. Mol. Cell. Biol. 1994, 4, 5920–5928. [Google Scholar] [CrossRef] [Green Version]
- Sheng, M. Calcium and growth factor pathways of c-fos transcriptional activation require distinct upstream regulatory sequences. Mol. Cell. Biol. 1988, 8, 2787–2796. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.H. Suppression of Ras transformation by serum response factor. J. Biol. Chem. 1994, 269, 13740–13743. [Google Scholar] [CrossRef]
- Kaplan-Albuquerque, N. Depletion of serum response factor by RNA interference mimics the mitogenic effects of platelet derived growth factor-BB in vascular smooth muscle cells. Circ. Res. 2005, 97, 427–433. [Google Scholar] [CrossRef] [Green Version]
- Olson, E.N. Linking actin dynamics and gene transcription to drive cellular motile functions. Nat. Rev. Mol. Cell Biol. 2010, 11, 353–365. [Google Scholar] [CrossRef] [Green Version]
- Miano, J.M. A transcriptional brake for activated gene expression in the vessel wall? Am. J. Pathol. 1999, 155, 1009–1012. [Google Scholar] [CrossRef] [Green Version]
- Modak, C. Serum response factor: Look into the gut. World J. Gastroenterol. WJG 2010, 16, 2195–2201. [Google Scholar] [CrossRef]
- Tarnawski, A.S. Angiogenesis in gastric mucosa: An important component of gastric erosion and ulcer healing and its impairment in aging. J. Gastroenterol. Hepatol. 2014, 29, 112–123. [Google Scholar] [CrossRef]
- Chen, Y. The yeast Mcm1 protein is regulated post-transcriptionally by the flux of glycolysis. Mol. Cell. Biol. 1995, 15, 4631–4639. [Google Scholar] [CrossRef] [Green Version]
- Belaguli, N.S. Dominant negative murine serum response factor: Alternative splicing within the activation domain inhibits transactivation of serum response factor binding targets. Mol. Cell. Biol. 1999, 19, 4582–4591. [Google Scholar] [CrossRef] [Green Version]
- Charvet, C. New role for serum response factor in postnatal skeletal muscle growth and regeneration via the interleukin 4 and insulin-like growth factor 1 pathways. Mol. Cell. Biol. 2006, 26, 6664–6674. [Google Scholar] [CrossRef] [Green Version]
- Christoforou, N. Transcription factors MYOCD, SRF, Mesp1 and SMARCD3 enhance the cardio-inducing effect of GATA4, TBX5, and MEF2C during direct cellular reprogramming. PLoS ONE 2013, 8, e63577. [Google Scholar] [CrossRef] [Green Version]
- Kitchen, C.M. Expression and promoter analysis of a highly restricted integrin alpha gene in vascular smooth muscle. Gene 2013, 513, 82–89. [Google Scholar] [CrossRef] [Green Version]
- Miano, J.M. Vascular smooth muscle cell differentiation. J. Biomed. Res. 2010, 24, 169–180. [Google Scholar] [CrossRef] [Green Version]
- Huang, J. Modulation of smooth muscle cell phenotype: The other side of the story. Circ. Res. 2012, 111, 659–661. [Google Scholar] [CrossRef] [PubMed]
- Hong, L. The rhoA/rho kinase pathway regulates nuclear localization of serum response factor. Am. J. Respir. Cell Mol. Biol. 2003, 29, 39–47. [Google Scholar]
- Zeng, Y. Formin-like 2 regulates Rho/ROCK pathway to promote actin assembly and cell invasion of colorectal cancer. Cancer Sci. 2015, 106, 1385–1393. [Google Scholar] [CrossRef] [PubMed]
- Minami, T. Reciprocal expression of MRTF-A and myocardin is crucial for pathological vascular remodeling in mice. EMBO J. 2012, 31, 4428–4440. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pomiès, P. The cytoskeleton-associated PDZ-LIM protein, ALP, acts on serum response factor activity to regulate muscle differentiation. Mol. Biol. Cell 2007, 18, 1723–1733. [Google Scholar] [CrossRef] [Green Version]
- Schratt, G. Serum response factor is crucial for actin cytoskeletal organization and focal adhesion assembly in embryonic stem cells. J. Cell Biol. 2002, 156, 737–750. [Google Scholar] [CrossRef] [Green Version]
- Gutsche-Perelroizen, I. Filament Assembly from Profilin-Actin. J. Biol. Chem. 1999, 274, 6234–6243. [Google Scholar] [CrossRef] [Green Version]
- Rajakylä, E.K. Rho, nuclear actin, and actin-binding proteins in the regulation of transcription and gene expression. Small GTPases 2014, 5, e27539. [Google Scholar] [CrossRef] [Green Version]
- Miano, J.M.; Long, X.; Fujiwara, K. Serum response factor: Master regulator of the actin cytoskeleton and contractile apparatus. Am. J. Physiol. Cell Physiol. 2007, 292, C70–C81. [Google Scholar] [CrossRef] [PubMed]
- Muñoz-Lasso, D.C.; Romá-Mateo, C.; Pallardó, F.V.; Gonzalez-Cabo, P. Much More Than a Scaffold: Cytoskeletal Proteins in Neurological Disorders. Cells 2020, 9, 358. [Google Scholar] [CrossRef] [Green Version]
- Ralf, P.; Brandes, I.; Fleming, R.B. Endothelial aging. Cardiovasc. Res. 2005, 66, 286–294. [Google Scholar]
- King, R.J.; Grant, P.J. Diabetes and cardiovascular disease: Pathophysiology of a life-threatening epidemic. Herz 2016, 41, 184–192. [Google Scholar] [CrossRef]
- Gourlay, C.; Ayscough, K. The actin cytoskeleton: A key regulator of apoptosis and ageing? Nat. Rev. Mol. Cell Biol. 2005, 6, 583–589. [Google Scholar] [CrossRef]
- Pan, M.H.; Sun, M.H.; Li, X.H.; Zhang, Y.; Sun, S.C. RAB7 GTPase regulates actin dynamics for DRP1-mediated mitochondria function and spindle migration in mouse oocyte meiosis. FASEB J. 2020, 34, 9615–9627. [Google Scholar] [CrossRef] [PubMed]
- Olkkonen, V.M.; Koponen, A.; Arora, A. OSBP-related protein 2 (ORP2): Unraveling its functions in cellular lipid/carbohydrate metabolism, signaling and F-actin regulation. J. Steroid Biochem. Mol. Biol. 2019, 192, 105298. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lai, W.F.; Wong, W.T. Roles of the actin cytoskeleton in aging and age-associated diseases. Ageing Res. Rev. 2020, 58, 101021. [Google Scholar] [CrossRef] [PubMed]
- Doshi, B.M.; Hightower, L.E.; Lee, J. HSPB1, actin filament dynamics, and aging cells. Ann. N. Y. Acad. Sci. 2010, 1197, 76–84. [Google Scholar] [CrossRef]
- Feng, X.W.; Huo, L.J.; Yang, M.C.; Wang, J.X.; Shi, X.Z. Thymosins participate in antibacterial immunity of kuruma shrimp, Marsupenaeus japonicus. Fish Shellfish Immunol. 2019, 84, 244–251. [Google Scholar] [CrossRef]
- Liu, L.; Suzuki, K.; Chun, E.; Murashima, A.; Sato, Y.; Nakagata, N.; Fujimori, T.; Yonemura, S.; He, W.; Yamada, G. Androgen Regulates Dimorphic F-Actin Assemblies in the Genital Organogenesis. Sex. Dev. 2017, 11, 190–202. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sasako, T.; Ueki, K. Insulin/IGF-1 signaling and aging. Nihon Rinsho 2016, 74, 1435–1440. [Google Scholar]
- Williams, E.D.; Rogers, S.C.; Zhang, X.; Azhar, G.; Wei, J.Y. p49/STRAP, a Serum Response Factor Binding Protein (SRFBP1), Is Involved in the Redistribution of Cytoskeletal F-Actin Proteins during Glucose Deprivation. J. Nutr. Health Aging 2017, 21, 1142–1150. [Google Scholar] [CrossRef] [PubMed]
- Rogers, S.C.; Zhang, X.; Azhar, G.; Luo, S.; Wei, J.Y. Exposure to high or low glucose levels accelerates the appearance of markers of endothelial cell senescence and induces dysregulation of nitric oxide synthase. J. Gerontol. A Biol. Sci. Med. Sci. 2013, 68, 1469–1481. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Whitson, R.J.; Lee, A.; Urman, N.M.; Mirza, A.; Yao, C.Y.; Brown, A.S.; Li, J.R.; Shankar, G.; Fry, M.A.; Atwood, S.X.; et al. Noncanonical hedgehog pathway activation through SRF-MKL1 promotes drug resistance in basal cell carcinomas. Nat. Med. 2018, 24, 271–281. [Google Scholar] [CrossRef]
- Kong, M.; Chen, X.; Lv, F.; Ren, H.; Fan, Z.; Qin, H.; Yu, L.; Shi, X.; Xu, Y. Serum response factor (SRF) promotes ROS generation and hepatic stellate cell activation by epigenetically stimulating NCF1/2 transcription. Redox Biol. 2019, 26, 101302. [Google Scholar] [CrossRef]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Aberdeen, H.; Battles, K.; Taylor, A.; Garner-Donald, J.; Davis-Wilson, A.; Rogers, B.T.; Cavalier, C.; Williams, E.D. The Aging Vasculature: Glucose Tolerance, Hypoglycemia and the Role of the Serum Response Factor. J. Cardiovasc. Dev. Dis. 2021, 8, 58. https://0-doi-org.brum.beds.ac.uk/10.3390/jcdd8050058
Aberdeen H, Battles K, Taylor A, Garner-Donald J, Davis-Wilson A, Rogers BT, Cavalier C, Williams ED. The Aging Vasculature: Glucose Tolerance, Hypoglycemia and the Role of the Serum Response Factor. Journal of Cardiovascular Development and Disease. 2021; 8(5):58. https://0-doi-org.brum.beds.ac.uk/10.3390/jcdd8050058
Chicago/Turabian StyleAberdeen, Hazel, Kaela Battles, Ariana Taylor, Jeranae Garner-Donald, Ana Davis-Wilson, Bryan T. Rogers, Candice Cavalier, and Emmanuel D. Williams. 2021. "The Aging Vasculature: Glucose Tolerance, Hypoglycemia and the Role of the Serum Response Factor" Journal of Cardiovascular Development and Disease 8, no. 5: 58. https://0-doi-org.brum.beds.ac.uk/10.3390/jcdd8050058