Direct Reprogramming of Cardiac Fibroblasts to Repair the Injured Heart


Abstract
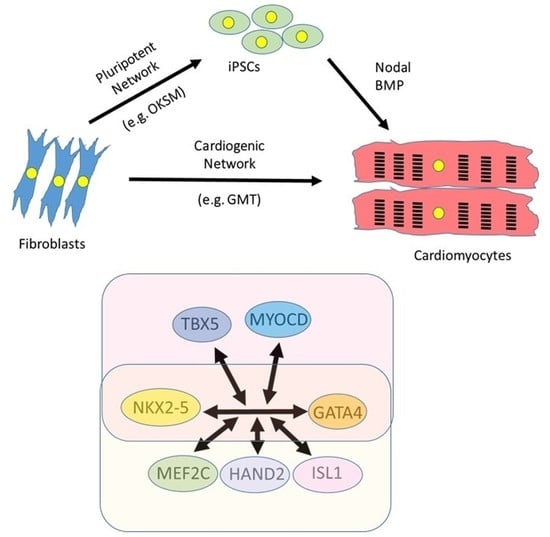
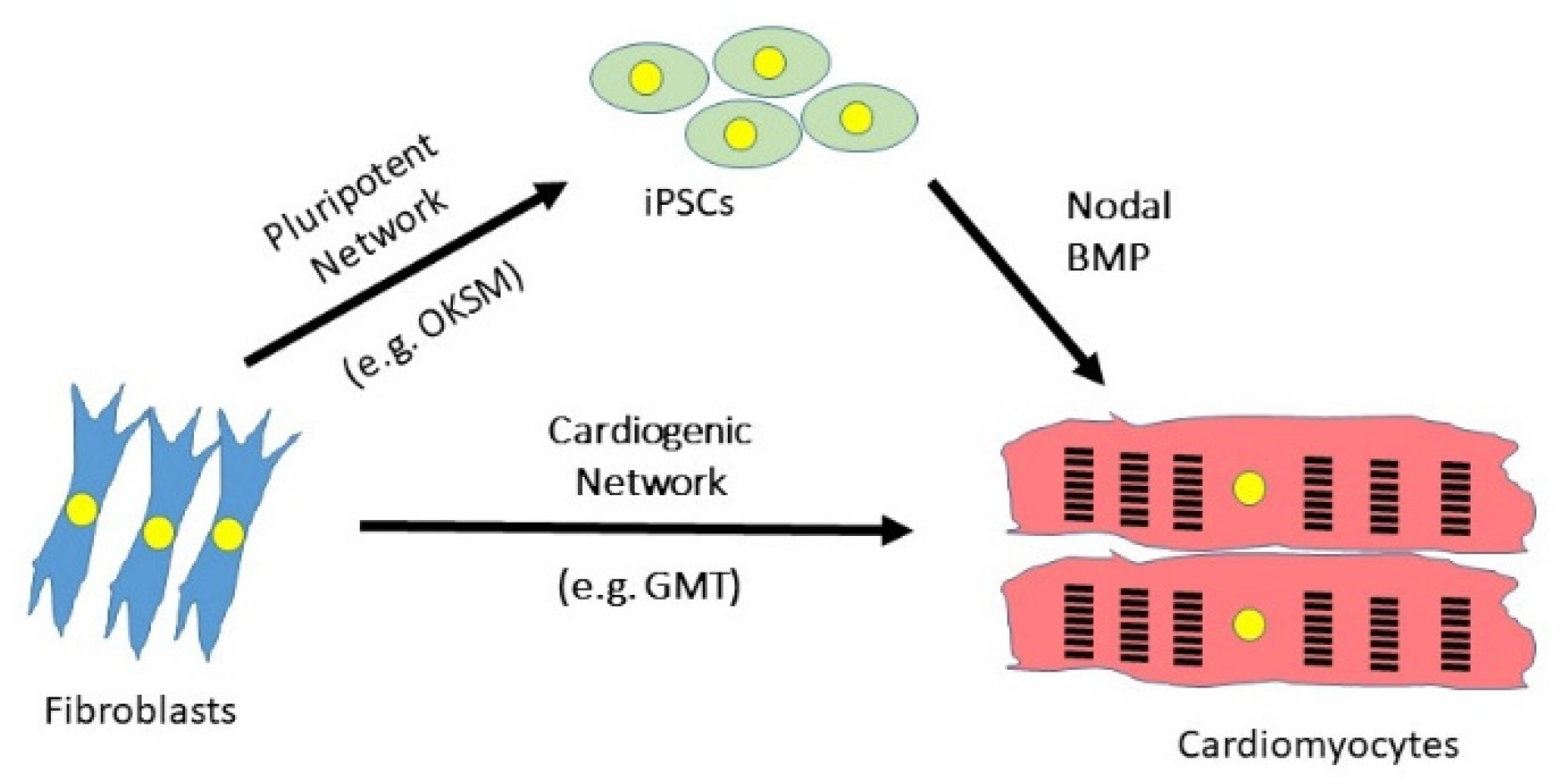
:
1. Introduction
2. Embryonic Development of the Heart
2.1. Cellular Composition of the Adult Heart
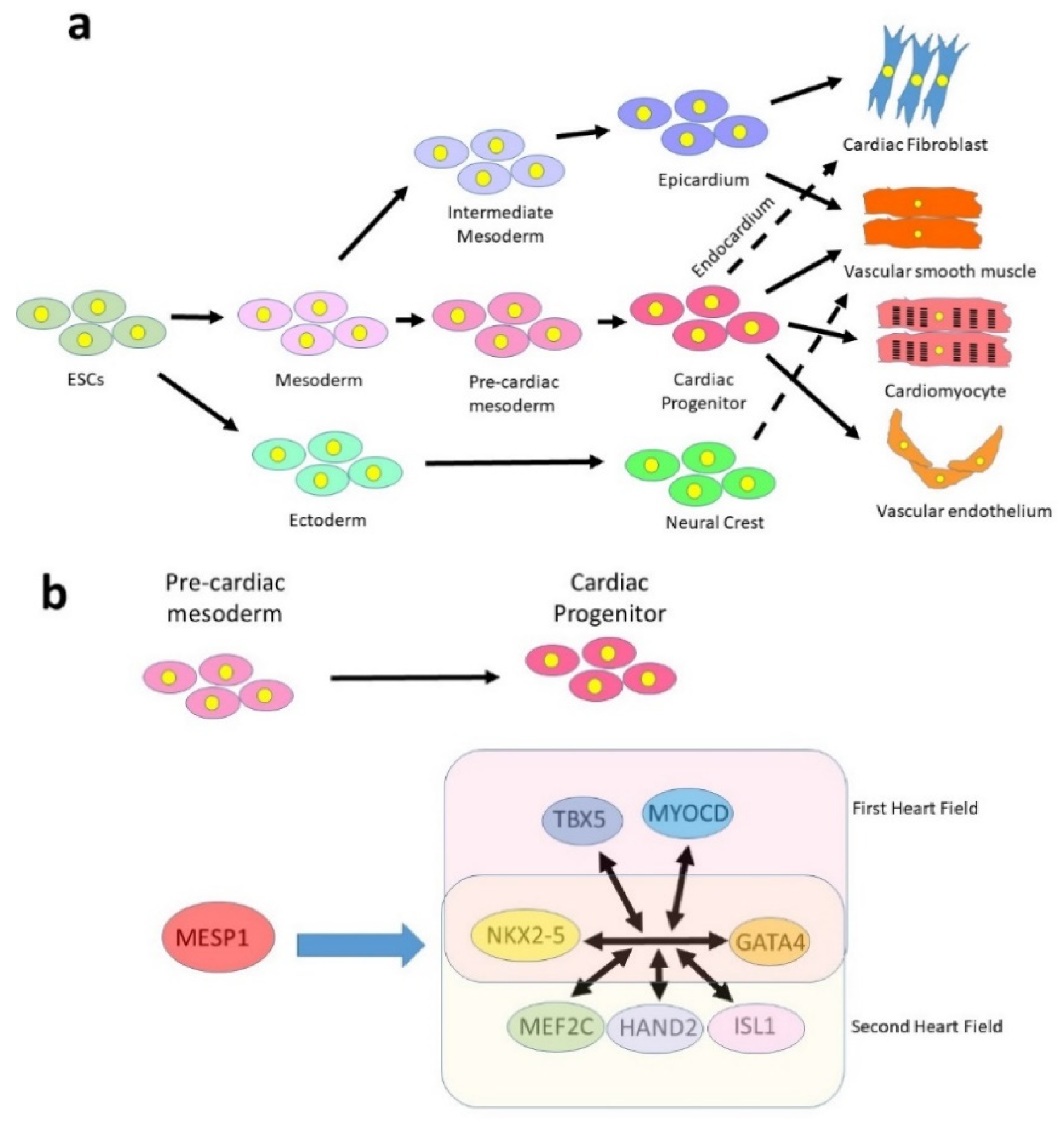
2.2. Fate Specification and Lineage Restriction during Embryonic Heart Development
3. Cellular Events Resulting from Myocardial Infarction
3.1. Initial Inflammatory Response
3.2. Fibrosis
3.3. Progression to Heart Failure
4. Regenerative Medicine
4.1. Induced Pluripotent Technology
4.2. Direct Reprogramming of Cardiac Fibroblasts
4.3. Identification of a Cardiogenic Network in Mouse Models

{kind=link}
{kind=link}
{kind=link}
| Laboratory | Species | Reprogramming Factors | Reference | ||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Transcription Factors | miRNA | Pathway Targeting | |||||||||||||||||||||||||
| GATA4 | MEF2C | TBX5 | MESP1 | MYOCD | HAND2 | NKX2-5 | ZNF281 | ESRRG | ZFPM2 | 1 | 133a | 208 | 499 | PI3/AKT | JAK/STAT | WNT | FGF | VEGF | TGFβ | RhoA-ROCK | Notch | cAMP/PKA | Epigenetic | BECN1 shRNA | |||
| Srivistava | Mouse | X | X | X | Ieda 2010 [81] | ||||||||||||||||||||||
| Mouse | X | X | X | Qian 2102 [98] | |||||||||||||||||||||||
| Human | X | X | X | X | X | X | X | Fu 2013 [99] | |||||||||||||||||||
| Mouse | X | X | X | I | I | Mohamed 2017 [100] | |||||||||||||||||||||
| Olson | Mouse | X | X | X | X | Song 2012 [90] | |||||||||||||||||||||
| Human | X | X | X | X | X | X | Nam 2013 [101] | ||||||||||||||||||||
| Mouse | X | X | X | X | A | Zhou 2015 [92] | |||||||||||||||||||||
| Mouse | X | X | X | X | X | A | Zhou 2017 [93] | ||||||||||||||||||||
| Mouse | X | X | X | X | I | Abad 2017 [91] | |||||||||||||||||||||
| Ieda | Mouse | X | X | X | Inagawa 2012 [102] | ||||||||||||||||||||||
| Human | X | X | X | X | X | Wada 2013 [103] | |||||||||||||||||||||
| Mouse | X | X | X | X | Muraoka 2014 [104] | ||||||||||||||||||||||
| Human | X | X | X | X | X | ||||||||||||||||||||||
| Mouse | X | X | X | A | A | Yamakawa 2015 [105] | |||||||||||||||||||||
| Dzau | Mouse | X | X | X | X | I | Jayawardena 2012 [106] | ||||||||||||||||||||
| Gearhart | Mouse | X | X | X | X | X | Addis 2013 [95] | ||||||||||||||||||||
| Mouse | X | X | X | X | X | I | Ifkovits 2014 [96] | ||||||||||||||||||||
| Song | Mouse | X | X | X | X | X | X | I | I | Zhao 2015 [94] | |||||||||||||||||
| Ravens | Mouse | X | X | X | Protze 2012 [97] | ||||||||||||||||||||||
| Qian | Mouse | X | X | X | Wang 2015 [88] | ||||||||||||||||||||||
| Mouse | X | X | X | X | Wang 2020 [5] | ||||||||||||||||||||||
| Human | X | X | X | X | Garbutt 2020 [107], Zhou 2019 [108] | ||||||||||||||||||||||
| Xie | Mouse | A | I | A | A | Fu 2015 [109] | |||||||||||||||||||||
| Kamp | Mouse | X | X | X | X | A | Lalit 2016 [110] | ||||||||||||||||||||
| Leong | Human | X | X | X | X | X | X | X | Christoforou 2017 [111] | ||||||||||||||||||
| Wu | Mouse | X | X | X | Chen 2012 [112] | ||||||||||||||||||||||
| Laboratory | Species | In vitro/In Vivo | Source Cell | Developmental Stage | Reprogramming Efficiency | Comments | Reference |
|---|---|---|---|---|---|---|---|
| Srivistava | Mouse | In vitro | Cardiac Fibroblast | Postnatal | 20% express MYH6 at 10 days | Although transdifferentiation is rapid, maturation (gain of TNNT2) takes several weeks | Ieda 2010 [81] |
| Mouse | In vivo | Cardiac Fibroblast | Adult | 10–15% | Cells are more mature than those reprogrammed in vitro | Qian 2012 [98] | |
| Human | In vitro | Cardiac Fibroblast | Foetal | 20% express MYH6 | Report that GMT alone cannot reprogramme human cells. | Fu 2013 [99] | |
| Dermal fibroblast | Neonatal | ||||||
| H9 ES-derived fibroblast | n/a | ||||||
| Mouse | In vitro | Cardiac Fibroblast | Neonatal | 30% express MYH6 at 2 weeks | Almost doubles efficiency over GMT alone | Mohamed 2017 [100] | |
| Olson | Mouse | In vitro | Cardiac fibroblast | Adult | 6.8% express both MYH6 and TNNT2 | Efficiency is 1.4% with GMT alone | Song 2012 [90] |
| Tail tip Fibroblast | Adult | 9.2% express both MYH6 and TNNT2 | |||||
| In vivo | Cardiac Fibroblast | Adult | At least 10,000 cells transdifferentiated | Improves heart function following infarction | |||
| Human | In vitro | Cardiac Fibroblast | Adult | 13% express TNNT2 | Sarcomere structures and calcium transients seen at 4–11 weeks | Nam 2013 [101] | |
| Dermal fibroblast | Adult | 9.5% express TNNT2 | |||||
| Mouse | In vitro | MEF | Embryo | ~25% express both MYH6 and TNNT2 | Reprogramming more efficient in embryonic than adult cells. | Zhou 2015 [92] | |
| Tail tip fibroblast | Adult | ~5% express both MYH6 and TNNT2 | |||||
| Cardiac Fibroblast | Adult | ~6% express both MYH6 and TNNT2 | |||||
| Mouse | In vitro | Tail tip fibroblast | Adult | ~28% express both MYH6 and TNNT2 after 7 days | Suppresses inflammatory signalling | Zhou 2017 [93] | |
| Mouse | In vitro | MEF | Embryo | Up to 70% express MYH6 and TNNT2 or ACTN2 | Improves efficiency of generation of mature cardiomyocytes by GMHT by 5–6 fold | Abad 2017 [91] | |
| Ieda | Mouse | In vivo | Cardiac Fibroblast | Adult | 3% express MYH6 at 1 week | Reprogramming efficiency is lower than in other mouse in vivo studies | Inagawa 2012 [102] |
| Human | In vitro | Cardiac Fibroblast | Adult | 5% express TNNT2 and ACTN2 at 4 weeks | Report that GMT alone cannot reprogramme human cells. | Wada 2013 [103] | |
| Mouse | In vitro | MEF | Embryo | ~35% express MYH6 at 1 week | Cardiomyocytes mature more quickly than GMT alone | Muraoka 2014 [104] | |
| Human | In vitro | Cardiac Fibroblast | Adult | ~20% express TNNT2 at 1 week | |||
| Mouse | In vitro | MEF | Embryo | ~70% beating cells at 4 weeks | Yamakawa 2015 [105] | ||
| Dzau | Mouse | In vitro | Fibroblast | 1.5–7.7% express MYH6 with miR alone, increasing to 28% with JAK inhibitor | JAK inhibition dramatically improves reprogramming using miR | Jayawardena 2012 [106] | |
| Mouse | In vivo | Cardiac Fibroblast | Adult | Induced cardiomyocytes represent ~1% of total and express TNNT2 | |||
| Gearhart | Mouse | In vitro | MEF | Embryo | ~1.5% show calcium oscillations at 2 weeks | Developed a quantifiable calcium reporter to assay efficiency HNGMT reported to be >50-fold more efficient than GMT alone | Addis 2013 [95] |
| Mouse | In vitro | MEF | Embryo | ~15% show calcium oscillations at 2 weeks | TGFβ inhibition improves efficiency 5 fold over HNGMT alone | Ifkovits 2014 [96] | |
| Song | Mouse | In vitro | MEF | Embryo | Zhao 2015 [94] | ||
| Cardiac Fibroblast | Adult | Up to 18% express TNNT2 at 4 weeks. 2.5% beating at 5 weeks | |||||
| Tail tip fibroblast | Adult | Up to 20% express TNNT2 at 4 weeks. 4% beating at 5 weeks | |||||
| Ravens | Mouse | In vitro | MEF | Embryo | 2.5% express MYH6 | Found that a number of triplet combinations can be used. | Protze 2012 [97] |
| Qian | Mouse | In vitro | Cardiac Fibroblast | Adult | ~10% express MYH6, ~5% express TNNT2 | Demonstrated that expression of GMT as polycistronic MGT improves efficiency | Wang 2015 [88] |
| Mouse | In vitro | MEF | Embryo | ~10% express TNNT2 (GMT) | Use of Sendai virus improves efficiency over retrovirus | Miyamoto 2018 [113] | |
| In vitro | Tail tip fibroblast | Postnatal | ~22% express TNNT2 (GMHT) | ||||
| In vivo | Cardiac Fibroblast | Adult | ~1.5% express TNNT2 (GMT) | ||||
| Human | In vitro | Cardiac Fibroblast | Adult | ~4% express TNNT2 (GMTMM) ~15% express TNNT2 (GMTMM+miR133) | |||
| Mouse | In vitro (In vivo) | Cardiac Fibroblast | Adult | Becn1 shRNA knockdown improves GMT efficiency | Wang 2020 [114] | ||
| Human | In vitro | H9 ES-derived fibroblast | n/a | 40–60% express TNNT2 at 2 weeks | Very efficient streamlined cocktail for human cells | Garbutt 2020 [107] | |
| Human | In vitro | Cardiac Fibroblast | Adult | ~40% express TNNT2 at 2 weeks | Zhou 2019 [108] | ||
| Xie | Mouse | In vitro | MEF | Embryo | 14.5% express ACTN2, 9% MYH6 on day 24 | Fu 2015 [109] | |
| In vitro | Tail tip fibroblast | Adult | |||||
| Kamp | Mouse | In vitro | Cardiac Fibroblast | Adult | ~7.25 colonies/50,000 cells | This method generates proliferating cardiac progenitor cells | Lalit 2016 [110] |
| In vitro | Lung fibroblast | Adult | |||||
| In vitro | Tail tip fibroblast | Adult | |||||
| Leong | Human | In vitro | Dermal fibroblast | Adult | Not stated | Christoforou 2017 [111] | |
| Wu | Mouse | In vitro | Cardiac Fibroblast | 2–3 weeks | No MYH6 expression at 3 weeks | Data suggest GMT reprogramming is inefficient | Chen 2012 [112] |
| In vitro | Tail tip fibroblast | Adult | No MYH6 expression but 35% express TNNT2 at 3 weeks |
4.4. Reprogramming Human Cells
4.5. In Vivo Reprogramming of the Injured Heart
5. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Sulo, G.; Igland, J.; Vollset, S.E.; Nygård, O.; Ebbing, M.; Sulo, E.; Egeland, G.M.; Tell, G.S. Heart failure complicating acute myocardial infarction; burden and timing of occurrence: A nation-wide analysis including 86,771 patients from the Cardiovascular Disease in Norway (CVDNOR) Project. J. Am. Heart Assoc. 2016, 5, e002667. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hung, J.; Teng, T.H.K.; Finn, J.; Knuiman, M.; Briffa, T.; Stewart, S.; Sanfilippo, F.M.; Ridout, S.; Hobbs, M. Trends From 1996 to 2007 in Incidence and Mortality Outcomes of Heart Failure After Acute Myocardial Infarction: A Population-Based Study of 20,812 Patients With First Acute Myocardial Infarction in W estern Australia. J. Am. Heart Assoc. 2013, 2, e000172. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jenča, D.; Melenovský, V.; Stehlik, J.; Staněk, V.; Kettner, J.; Kautzner, J.; Adámková, V.; Wohlfahrt, P. Heart failure after myocardial infarction: Incidence and predictors. ESC Heart Fail. 2021, 8, 222–237. [Google Scholar] [CrossRef]
- Takahashi, K.; Yamanaka, S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 2006, 126, 663–676. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Yang, Y.; Liu, J.; Qian, L. Direct cell reprogramming: Approaches, mechanisms and progress. Nat. Rev. Mol. Cell Biol. 2021, 22, 410–424. [Google Scholar] [CrossRef]
- Werner, J.H.; Rosenberg, J.H.; Um, J.Y.; Moulton, M.J.; Agrawal, D.K. Molecular discoveries and treatment strategies by direct reprogramming in cardiac regeneration. Transl. Res. 2019, 203, 73–87. [Google Scholar] [CrossRef]
- Kurotsu, S.; Suzuki, T.; Ieda, M. Direct reprogramming, epigenetics, and cardiac regeneration. J. Card. Fail. 2017, 23, 552–557. [Google Scholar] [CrossRef]
- Tucker, N.R.; Chaffin, M.; Fleming, S.J.; Hall, A.W.; Parsons, V.A.; Bedi, K.C.; Akkad, A.-D.; Herndon, C.N.; Arduini, A.; Papangeli, I.; et al. Transcriptional and Cellular Diversity of the Human Heart. Circulation 2020, 142, 466–482. [Google Scholar] [CrossRef] [PubMed]
- Dykes, I.M.; van Bueren, K.L.; Scambler, P.J. HIC2 regulates isoform switching during maturation of the cardiovascular system. J. Mol. Cell. Cardiol. 2018, 114, 29–37. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yin, Z.; Ren, J.; Guo, W. Sarcomeric protein isoform transitions in cardiac muscle: A journey to heart failure. Biochim. Biophys. Acta 2015, 1852, 47–52. [Google Scholar] [CrossRef] [Green Version]
- England, J.; Loughna, S. Heavy and light roles: Myosin in the morphogenesis of the heart. Cell. Mol. Life Sci. CMLS 2013, 70, 1221–1239. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paik, D.T.; Cho, S.; Tian, L.; Chang, H.Y.; Wu, J.C. Single-Cell RNA sequencing in cardiovascular development, disease and medicine. Nat. Rev. Cardiol. 2020, 17, 457–473. [Google Scholar] [CrossRef]
- Ackers-Johnson, M.; Tan, W.L.W.; Foo, R.S.-Y. Following hearts, one cell at a time: Recent applications of single-cell RNA sequencing to the understanding of heart disease. Nat. Commun. 2018, 9, 4434. [Google Scholar] [CrossRef] [Green Version]
- Zhou, P.; Pu, W.T. Recounting Cardiac Cellular Composition. Circ. Res. 2016, 118, 368–370. [Google Scholar] [CrossRef]
- Weber, K.T.; Sun, Y.; Bhattacharya, S.K.; Ahokas, R.A.; Gerling, I.C. Myofibroblast-mediated mechanisms of pathological remodelling of the heart. Nat. Rev. Cardiol. 2013, 10, 15. [Google Scholar] [CrossRef]
- Tallquist, M.D.; Molkentin, J.D. Redefining the identity of cardiac fibroblasts. Nat. Rev. Cardiol. 2017, 14, 484–491. [Google Scholar] [CrossRef]
- Furtado, M.B.; Costa, M.W.; Pranoto, E.A.; Salimova, E.; Pinto, A.R.; Lam, N.T.; Park, A.; Snider, P.; Chandran, A.; Harvey, R.P. Cardiogenic genes expressed in cardiac fibroblasts contribute to heart development and repair. Circ. Res. 2014, 114, 1422–1434. [Google Scholar] [CrossRef] [PubMed]
- Swirski, F.K.; Nahrendorf, M. Cardioimmunology: The immune system in cardiac homeostasis and disease. Nat. Rev. Immunol. 2018, 18, 733–744. [Google Scholar] [CrossRef] [PubMed]
- Greenberg, M.V.; Bourc’his, D. The diverse roles of DNA methylation in mammalian development and disease. Nat. Rev. Mol. Cell Biol. 2019, 20, 590–607. [Google Scholar] [CrossRef] [PubMed]
- Klemm, S.L.; Shipony, Z.; Greenleaf, W.J. Chromatin accessibility and the regulatory epigenome. Nat. Rev. Genet. 2019, 20, 207–220. [Google Scholar] [CrossRef]
- Prummel, K.D.; Nieuwenhuize, S.; Mosimann, C. The lateral plate mesoderm. Development 2020, 147, dev175059. [Google Scholar] [CrossRef]
- Chabab, S.; Lescroart, F.; Rulands, S.; Mathiah, N.; Simons, B.D.; Blanpain, C. Uncovering the Number and Clonal Dynamics of Mesp1 Progenitors during Heart Morphogenesis. Cell Rep. 2016, 14, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chan, S.S.; Shi, X.; Toyama, A.; Arpke, R.W.; Dandapat, A.; Iacovino, M.; Kang, J.; Le, G.; Hagen, H.R.; Garry, D.J.; et al. Mesp1 patterns mesoderm into cardiac, hematopoietic, or skeletal myogenic progenitors in a context-dependent manner. Cell Stem Cell 2013, 12, 587–601. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soibam, B.; Benham, A.; Kim, J.; Weng, K.-C.; Yang, L.; Xu, X.; Robertson, M.; Azares, A.; Cooney, A.J.; Schwartz, R.J.; et al. Genome-Wide Identification of MESP1 Targets Demonstrates Primary Regulation Over Mesendoderm Gene Activity. Stem Cells 2015, 33, 3254–3265. [Google Scholar] [CrossRef]
- Bondue, A.; Lapouge, G.; Paulissen, C.; Semeraro, C.; Iacovino, M.; Kyba, M.; Blanpain, C. Mesp1 Acts as a Master Regulator of Multipotent Cardiovascular Progenitor Specification. Cell Stem Cell 2008, 3, 69–84. [Google Scholar] [CrossRef]
- Saga, Y.; Miyagawa-Tomita, S.; Takagi, A.; Kitajima, S.; Miyazaki, J.; Inoue, T. MesP1 is expressed in the heart precursor cells and required for the formation of a single heart tube. Development 1999, 126, 3437–3447. [Google Scholar] [CrossRef]
- Islas, J.F.; Liu, Y.; Weng, K.C.; Robertson, M.J.; Zhang, S.; Prejusa, A.; Harger, J.; Tikhomirova, D.; Chopra, M.; Iyer, D.; et al. Transcription factors ETS2 and MESP1 transdifferentiate human dermal fibroblasts into cardiac progenitors. Proc. Natl. Acad. Sci. USA 2012, 109, 13016–13021. [Google Scholar] [CrossRef] [Green Version]
- Olson, E.N. Gene Regulatory Networks in the Evolution and Development of the Heart. Science 2006, 313, 1922–1927. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bruneau, B.G. Signaling and transcriptional networks in heart development and regeneration. Cold Spring Harb. Perspect. Biol. 2013, 5, a008292. [Google Scholar] [CrossRef] [Green Version]
- Hiroi, Y.; Kudoh, S.; Monzen, K.; Ikeda, Y.; Yazaki, Y.; Nagai, R.; Komuro, I. Tbx5 associates with Nkx2-5 and synergistically promotes cardiomyocyte differentiation. Nat. Genet. 2001, 28, 276–280. [Google Scholar] [CrossRef] [PubMed]
- Durocher, D.; Charron, F.; Warren, R.; Schwartz, R.J.; Nemer, M. The cardiac transcription factors Nkx2-5 and GATA-4 are mutual cofactors. EMBO J. 1997, 16, 5687–5696. [Google Scholar] [CrossRef] [Green Version]
- Lescroart, F.; Chabab, S.; Lin, X.; Rulands, S.; Paulissen, C.; Rodolosse, A.; Auer, H.; Achouri, Y.; Dubois, C.; Bondue, A. Early lineage restriction in temporally distinct populations of Mesp1 progenitors during mammalian heart development. Nat. Cell Biol. 2014, 16, 829. [Google Scholar] [CrossRef]
- Moses, K.A.; DeMayo, F.; Braun, R.M.; Reecy, J.L.; Schwartz, R.J. Embryonic expression of an Nkx2-5/Cre gene using ROSA26 reporter mice. Genesis 2001, 31, 176–180. [Google Scholar] [CrossRef] [PubMed]
- Molkentin, J.D.; Lin, Q.; Duncan, S.A.; Olson, E.N. Requirement of the transcription factor GATA4 for heart tube formation and ventral morphogenesis. Genes Dev. 1997, 11, 1061–1072. [Google Scholar] [CrossRef] [Green Version]
- Bruneau, B.G.; Nemer, G.; Schmitt, J.P.; Charron, F.; Robitaille, L.; Caron, S.; Conner, D.A.; Gessler, M.; Nemer, M.; Seidman, C.E. A murine model of Holt-Oram syndrome defines roles of the T-box transcription factor Tbx5 in cardiogenesis and disease. Cell 2001, 106, 709–721. [Google Scholar] [CrossRef] [Green Version]
- Lin, Q.; Schwarz, J.; Bucana, C.; Olson, E.N. Control of mouse cardiac morphogenesis and myogenesis by transcription factor MEF2C. Science 1997, 276, 1404–1407. [Google Scholar] [CrossRef] [Green Version]
- Cai, C.L.; Liang, X.; Shi, Y.; Chu, P.H.; Pfaff, S.L.; Chen, J.; Evans, S. Isl1 identifies a cardiac progenitor population that proliferates prior to differentiation and contributes a majority of cells to the heart. Dev. Cell 2003, 5, 877–889. [Google Scholar] [CrossRef] [Green Version]
- Ma, Q.; Zhou, B.; Pu, W.T. Reassessment of Isl1 and Nkx2-5 cardiac fate maps using a Gata4-based reporter of Cre activity. Dev. Biol. 2008, 323, 98–104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dodou, E.; Verzi, M.P.; Anderson, J.P.; Xu, S.-M.; Black, B.L. Mef2c is a direct transcriptional target of ISL1 and GATA factors in the anterior heart field during mouse embryonic development. Development 2004, 131, 3931–3942. [Google Scholar] [CrossRef] [Green Version]
- Miano, J.M. Myocardin in biology and disease. J. Biomed. Res. 2015, 29, 3–19. [Google Scholar]
- Wystub, K.; Besser, J.; Bachmann, A.; Boettger, T.; Braun, T. miR-1/133a Clusters Cooperatively Specify the Cardiomyogenic Lineage by Adjustment of Myocardin Levels during Embryonic Heart Development. PLoS Genet. 2013, 9, e1003793. [Google Scholar] [CrossRef] [PubMed]
- Furtado, M.B.; Nim, H.T.; Boyd, S.E.; Rosenthal, N.A. View from the heart: Cardiac fibroblasts in development, scarring and regeneration. Development 2016, 143, 387–397. [Google Scholar] [CrossRef] [Green Version]
- Moore-Morris, T.; Guimarães-Camboa, N.; Banerjee, I.; Zambon, A.C.; Kisseleva, T.; Velayoudon, A.; Stallcup, W.B.; Gu, Y.; Dalton, N.D.; Cedenilla, M. Resident fibroblast lineages mediate pressure overload–induced cardiac fibrosis. J. Clin. Investig. 2014, 124, 2921–2934. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ali, S.R.; Ranjbarvaziri, S.; Talkhabi, M.; Zhao, P.; Subat, A.; Hojjat, A.; Kamran, P.; Müller, A.M.; Volz, K.S.; Tang, Z. Developmental heterogeneity of cardiac fibroblasts does not predict pathological proliferation and activation. Circ. Res. 2014, 115, 625–635. [Google Scholar] [CrossRef] [Green Version]
- Simões, F.C.; Riley, P.R. The ontogeny, activation and function of the epicardium during heart development and regeneration. Development 2018, 145, dev155994. [Google Scholar] [CrossRef] [Green Version]
- Dyer, L.A.; Patterson, C. Development of the endothelium: An emphasis on heterogeneity. Semin. Thromb. Hemost. 2010, 36, 227–235. [Google Scholar] [CrossRef] [Green Version]
- Qiu, J.; Hirschi, K.K. Endothelial Cell Development and Its Application to Regenerative Medicine. Circ. Res. 2019, 125, 489–501. [Google Scholar] [CrossRef] [PubMed]
- Yamagishi, H. Cardiac Neural Crest. Cold Spring Harb. Perspect. Biol. 2005, 16, 704–715. [Google Scholar] [CrossRef] [PubMed]
- Konstantinidis, K.; Whelan, R.S.; Kitsis, R.N. Mechanisms of cell death in heart disease. Arter. Thromb. Vasc. Biol. 2012, 32, 1552–1562. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gwechenberger, M.; Mendoza, L.H.; Youker, K.A.; Frangogiannis, N.G.; Smith, C.W.; Michael, L.H.; Entman, M.L. Cardiac myocytes produce interleukin-6 in culture and in viable border zone of reperfused infarctions. Circulation 1999, 99, 546–551. [Google Scholar] [CrossRef] [Green Version]
- Frangogiannis, N.G. Chemokines in the ischemic myocardium: From inflammation to fibrosis. Inflamm. Res. 2004, 53, 585–595. [Google Scholar] [CrossRef]
- Hanna, A.; Frangogiannis, N.G. The Role of the TGF-β Superfamily in Myocardial Infarction. Front. Cardiovasc. Med. 2019, 6, 140. [Google Scholar] [CrossRef] [PubMed]
- Poss, K.D.; Wilson, L.G.; Keating, M.T. Heart regeneration in zebrafish. Science 2002, 298, 2188–2190. [Google Scholar] [CrossRef] [PubMed]
- Sanz-Morejón, A.; Mercader, N. Recent insights into zebrafish cardiac regeneration. Curr. Opin. Genet. Dev. 2020, 64, 37–43. [Google Scholar] [CrossRef]
- Porrello, E.R.; Olson, E.N. A neonatal blueprint for cardiac regeneration. Stem Cell Res. 2014, 13, 556–570. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wynn, T. Cellular and molecular mechanisms of fibrosis. J. Pathol. 2008, 214, 199–210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haudek, S.B.; Xia, Y.; Huebener, P.; Lee, J.M.; Carlson, S.; Crawford, J.R.; Pilling, D.; Gomer, R.H.; Trial, J.; Frangogiannis, N.G. Bone marrow-derived fibroblast precursors mediate ischemic cardiomyopathy in mice. Proc. Natl. Acad. Sci. USA 2006, 103, 18284–18289. [Google Scholar] [CrossRef] [Green Version]
- Van Amerongen, M.; Bou-Gharios, G.; Popa, E.; van Ark, J.; Petersen, A.; van Dam, G.; van Luyn, M.; Harmsen, M. Bone marrow-derived myofibroblasts contribute functionally to scar formation after myocardial infarction. J. Pathol. 2008, 214, 377–386. [Google Scholar] [CrossRef]
- Desmoulière, A.; Geinoz, A.; Gabbiani, F.; Gabbiani, G. Transforming growth factor-beta 1 induces alpha-smooth muscle actin expression in granulation tissue myofibroblasts and in quiescent and growing cultured fibroblasts. J. Cell Biol. 1993, 122, 103–111. [Google Scholar] [CrossRef] [Green Version]
- Davis, J.; Molkentin, J.D. Myofibroblasts: Trust your heart and let fate decide. J. Mol. Cell. Cardiol. 2014, 70, 9–18. [Google Scholar] [CrossRef] [Green Version]
- Tomasek, J.J.; Gabbiani, G.; Hinz, B.; Chaponnier, C.; Brown, R.A. Myofibroblasts and mechano-regulation of connective tissue remodelling. Nat. Rev. Mol. Cell Biol. 2002, 3, 349–363. [Google Scholar] [CrossRef]
- Shinde, A.; Frangogiannis, N. Fibroblasts in myocardial infarction: A role in inflammation and repair. J. Mol. Cell. Cardiol. 2014, 70, 74–82. [Google Scholar] [CrossRef] [Green Version]
- Simões, F.C.; Cahill, T.J.; Kenyon, A.; Gavriouchkina, D.; Vieira, J.M.; Sun, X.; Pezzolla, D.; Ravaud, C.; Masmanian, E.; Weinberger, M.; et al. Macrophages directly contribute collagen to scar formation during zebrafish heart regeneration and mouse heart repair. Nat. Commun. 2020, 11, 600. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Piek, A.; de Boer, R.A.; Silljé, H.H.W. The fibrosis-cell death axis in heart failure. Heart Fail. Rev. 2016, 21, 199–211. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gabriel-Costa, D. The pathophysiology of myocardial infarction-induced heart failure. Pathophysiology 2018, 25, 277–284. [Google Scholar] [CrossRef] [PubMed]
- Cleland, J.; Torabi, A.; Khan, N. Epidemiology and management of heart failure and left ventricular systolic dysfunction in the aftermath of a myocardial infarction. Heart 2005, 91 (Suppl. S2), ii7–ii13. [Google Scholar] [CrossRef] [Green Version]
- Li, M.; Belmonte, J.C.I. Ground rules of the pluripotency gene regulatory network. Nat. Rev. Genet. 2017, 18, 180–191. [Google Scholar] [CrossRef] [Green Version]
- Kinoshita, M.; Smith, A. Pluripotency Deconstructed. Dev. Growth Differ. 2018, 60, 44–52. [Google Scholar] [CrossRef]
- Waddington, C.H. The Strategy of the Genes; Routledge: Oxfordshire, UK, 1957. [Google Scholar]
- Takahashi, K.; Tanabe, K.; Ohnuki, M.; Narita, M.; Ichisaka, T.; Tomoda, K.; Yamanaka, S. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell 2007, 131, 861–872. [Google Scholar] [CrossRef] [Green Version]
- Yu, J.; Vodyanik, M.A.; Smuga-Otto, K.; Antosiewicz-Bourget, J.; Frane, J.L.; Tian, S.; Nie, J.; Jonsdottir, G.A.; Ruotti, V.; Stewart, R.; et al. Induced pluripotent stem cell lines derived from human somatic cells. Science 2007, 318, 1917–1920. [Google Scholar] [CrossRef]
- Tsialikas, J.; Romer-Seibert, J. LIN28: Roles and regulation in development and beyond. Development 2015, 142, 2397–2404. [Google Scholar] [CrossRef] [Green Version]
- Okita, K.; Matsumura, Y.; Sato, Y.; Okada, A.; Morizane, A.; Okamoto, S.; Hong, H.; Nakagawa, M.; Tanabe, K.; Tezuka, K.-I. A more efficient method to generate integration-free human iPS cells. Nat. Methods 2011, 8, 409–412. [Google Scholar] [CrossRef] [Green Version]
- Yu, J.; Hu, K.; Smuga-Otto, K.; Tian, S.; Stewart, R.; Slukvin, I.I.; Thomson, J.A. Human induced pluripotent stem cells free of vector and transgene sequences. Science 2009, 324, 797–801. [Google Scholar] [CrossRef] [Green Version]
- Warren, L.; Manos, P.D.; Ahfeldt, T.; Loh, Y.-H.; Li, H.; Lau, F.; Ebina, W.; Mandal, P.K.; Smith, Z.D.; Meissner, A. Highly efficient reprogramming to pluripotency and directed differentiation of human cells with synthetic modified mRNA. Cell Stem Cell 2010, 7, 618–630. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hou, P.; Li, Y.; Zhang, X.; Liu, C.; Guan, J.; Li, H.; Zhao, T.; Ye, J.; Yang, W.; Liu, K. Pluripotent stem cells induced from mouse somatic cells by small-molecule compounds. Science 2013, 341, 651–654. [Google Scholar] [CrossRef]
- Yang, L.; Soonpaa, M.H.; Adler, E.D.; Roepke, T.K.; Kattman, S.J.; Kennedy, M.; Henckaerts, E.; Bonham, K.; Abbott, G.W.; Linden, R.M.; et al. Human cardiovascular progenitor cells develop from a KDR+ embryonic-stem-cell-derived population. Nature 2008, 453, 524–528. [Google Scholar] [CrossRef]
- Avolio, E.; Caputo, M.; Madeddu, P. Stem cell therapy and tissue engineering for correction of congenital heart disease. Front. Cell Dev. Biol. 2015, 3, 39. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Q.; Yang, H.; Bai, A.; Jiang, W.; Li, X.; Wang, X.; Mao, Y.; Lu, C.; Qian, R.; Guo, F. Functional engineered human cardiac patches prepared from nature’s platform improve heart function after acute myocardial infarction. Biomaterials 2016, 105, 52–65. [Google Scholar] [CrossRef] [PubMed]
- Su, T.; Huang, K.; Daniele, M.A.; Hensley, M.T.; Young, A.T.; Tang, J.; Allen, T.A.; Vandergriff, A.C.; Erb, P.D.; Ligler, F.S. Cardiac stem cell patch integrated with microengineered blood vessels promotes cardiomyocyte proliferation and neovascularization after acute myocardial infarction. ACS Appl. Mater. Interfaces 2018, 10, 33088–33096. [Google Scholar] [CrossRef]
- Ieda, M.; Fu, J.D.; Delgado-Olguin, P.; Vedantham, V.; Hayashi, Y.; Bruneau, B.G.; Srivastava, D. Direct reprogramming of fibroblasts into functional cardiomyocytes by defined factors. Cell 2010, 142, 375–386. [Google Scholar] [CrossRef] [Green Version]
- Franco, D.; Lamers, W.H.; Moorman, A.F.M. Patterns of expression in the developing myocardium: Towards a morphologically integrated transcriptional model. Cardiovasc. Res. 1998, 38, 25–53. [Google Scholar] [CrossRef] [Green Version]
- Wessels, A.; Vermeulen, J.; Virágh, S.; Kalman, F.; Lamers, W.; Moorman, A. Spatial distribution of “tissue-specific” antigens in the developing human heart and skeletal muscle. II. An immunohistochemical analysis of myosin heavy chain isoform expression patterns in the embryonic heart. Anat. Rec. 1991, 229, 355–368. [Google Scholar] [CrossRef] [PubMed]
- Lyons, G.E.; Schiaffino, S.; Sassoon, D.; Barton, P.; Buckingham, M. Developmental regulation of myosin gene expression in mouse cardiac muscle. J. Cell Biol. 1990, 111 Pt 1, 2427–2436. [Google Scholar] [CrossRef] [Green Version]
- Xie, L.; Hoffmann, A.D.; Burnicka-Turek, O.; Friedland-Little, J.M.; Zhang, K.; Moskowitz, I.P. Tbx5-hedgehog molecular networks are essential in the second heart field for atrial septation. Dev. Cell 2012, 23, 280–291. [Google Scholar] [CrossRef] [Green Version]
- Stone, N.R.; Gifford, C.A.; Thomas, R.; Pratt, K.J.B.; Samse-Knapp, K.; Mohamed, T.M.A.; Radzinsky, E.M.; Schricker, A.; Ye, L.; Yu, P.; et al. Context-Specific Transcription Factor Functions Regulate Epigenomic and Transcriptional Dynamics during Cardiac Reprogramming. Cell Stem Cell 2019, 25, 87–102.e9. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto, H.; Wang, Z.; Garry, G.A.; Malladi, V.S.; Botten, G.A.; Ye, W.; Zhou, H.; Osterwalder, M.; Dickel, D.E.; Visel, A.; et al. Cardiac Reprogramming Factors Synergistically Activate Genome-wide Cardiogenic Stage-Specific Enhancers. Cell Stem Cell 2019, 25, 69–86.e5. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Liu, Z.; Yin, C.; Asfour, H.; Chen, O.; Li, Y.; Bursac, N.; Liu, J.; Qian, L. Stoichiometry of Gata4, Mef2c, and Tbx5 Influences the Efficiency and Quality of Induced Cardiac Myocyte Reprogramming. Circ. Res. 2015, 116, 237–244. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, T.K.; Song, F.F.; Packham, E.A.; Buxton, S.; Robinson, T.E.; Ronksley, J.; Self, T.; Bonser, A.J.; Brook, J.D. Physical interaction between TBX5 and MEF2C is required for early heart development. Mol. Cell. Biol. 2009, 29, 2205–2218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, K.; Nam, Y.J.; Luo, X.; Qi, X.; Tan, W.; Huang, G.N.; Acharya, A.; Smith, C.L.; Tallquist, M.D.; Neilson, E.G.; et al. Heart repair by reprogramming non-myocytes with cardiac transcription factors. Nature 2012, 485, 599–604. [Google Scholar] [CrossRef] [Green Version]
- Abad, M.; Hashimoto, H.; Zhou, H.; Morales, M.G.; Chen, B.; Bassel-Duby, R.; Olson, E.N. Notch inhibition enhances cardiac reprogramming by increasing MEF2C transcriptional activity. Stem Cell Rep. 2017, 8, 548–560. [Google Scholar] [CrossRef]
- Zhou, H.; Dickson, M.E.; Kim, M.S.; Bassel-Duby, R.; Olson, E.N. Akt1/protein kinase B enhances transcriptional reprogramming of fibroblasts to functional cardiomyocytes. Proc. Natl. Acad. Sci. USA 2015, 112, 11864–11869. [Google Scholar] [CrossRef] [Green Version]
- Zhou, H.; Morales, M.G.; Hashimoto, H.; Dickson, M.E.; Song, K.; Ye, W.; Kim, M.S.; Niederstrasser, H.; Wang, Z.; Chen, B. ZNF281 enhances cardiac reprogramming by modulating cardiac and inflammatory gene expression. Genes Dev. 2017, 31, 1770–1783. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Y.; Londono, P.; Cao, Y.; Sharpe, E.J.; Proenza, C.; O’Rourke, R.; Jones, K.L.; Jeong, M.Y.; Walker, L.A.; Buttrick, P.M. High-Efficiency reprogramming of fibroblasts into cardiomyocytes requires suppression of pro-fibrotic signalling. Nat. Commun. 2015, 6, 8243. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Addis, R.C.; Ifkovits, J.L.; Pinto, F.; Kellam, L.D.; Esteso, P.; Rentschler, S.; Christoforou, N.; Epstein, J.A.; Gearhart, J.D. Optimization of direct fibroblast reprogramming to cardiomyocytes using calcium activity as a functional measure of success. J. Mol. Cell. Cardiol. 2013, 60, 97–106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ifkovits, J.L.; Addis, R.C.; Epstein, J.A.; Gearhart, J.D. Inhibition of TGFβ signaling increases direct conversion of fibroblasts to induced cardiomyocytes. PLoS ONE 2014, 9, e89678. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Protze, S.; Khattak, S.; Poulet, C.; Lindemann, D.; Tanaka, E.M.; Ravens, U. A new approach to transcription factor screening for reprogramming of fibroblasts to cardiomyocyte-like cells. J. Mol. Cell. Cardiol. 2012, 53, 323–332. [Google Scholar] [CrossRef] [PubMed]
- Qian, L.; Huang, Y.; Spencer, C.I.; Foley, A.; Vedantham, V.; Liu, L.; Conway, S.J.; Fu, J.-D.; Srivastava, D. In vivo reprogramming of murine cardiac fibroblasts into induced cardiomyocytes. Nature 2012, 485, 593–598. [Google Scholar] [CrossRef]
- Fu, J.D.; Stone, N.R.; Liu, L.; Spencer, C.I.; Qian, L.; Hayashi, Y.; Delgado-Olguin, P.; Ding, S.; Bruneau, B.G.; Srivastava, D. Direct reprogramming of human fibroblasts toward a cardiomyocyte-like state. Stem Cell Rep. 2013, 1, 235–247. [Google Scholar] [CrossRef] [Green Version]
- Mohamed, T.M.; Stone, N.R.; Berry, E.C.; Radzinsky, E.; Huang, Y.; Pratt, K.; Ang, Y.-S.; Yu, P.; Wang, H.; Tang, S. Chemical enhancement of in vitro and in vivo direct cardiac reprogramming. Circulation 2017, 135, 978–995. [Google Scholar] [CrossRef]
- Nam, Y.J.; Song, K.; Luo, X.; Daniel, E.; Lambeth, K.; West, K.; Hill, J.A.; DiMaio, J.M.; Baker, L.A.; Bassel-Duby, R.; et al. Reprogramming of human fibroblasts toward a cardiac fate. Proc. Natl. Acad. Sci. USA 2013, 110, 5588–5593. [Google Scholar] [CrossRef] [Green Version]
- Inagawa, K.; Miyamoto, K.; Yamakawa, H.; Muraoka, N.; Sadahiro, T.; Umei, T.; Wada, R.; Katsumata, Y.; Kaneda, R.; Nakade, K.; et al. Induction of cardiomyocyte-like cells in infarct hearts by gene transfer of Gata4, Mef2c, and Tbx5. Circ. Res. 2012, 111, 1147–1156. [Google Scholar] [CrossRef]
- Wada, R.; Muraoka, N.; Inagawa, K.; Yamakawa, H.; Miyamoto, K.; Sadahiro, T.; Umei, T.; Kaneda, R.; Suzuki, T.; Kamiya, K.; et al. Induction of human cardiomyocyte-like cells from fibroblasts by defined factors. Proc. Natl. Acad. Sci. USA 2013, 110, 12667–12672. [Google Scholar] [CrossRef] [Green Version]
- Muraoka, N.; Yamakawa, H.; Miyamoto, K.; Sadahiro, T.; Umei, T.; Isomi, M.; Nakashima, H.; Akiyama, M.; Wada, R.; Inagawa, K.; et al. MiR-133 promotes cardiac reprogramming by directly repressing Snai1 and silencing fibroblast signatures. EMBO J. 2014, 33, 1565–1581. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamakawa, H.; Muraoka, N.; Miyamoto, K.; Sadahiro, T.; Isomi, M.; Haginiwa, S.; Kojima, H.; Umei, T.; Akiyama, M.; Kuishi, Y.; et al. Fibroblast Growth Factors and Vascular Endothelial Growth Factor Promote Cardiac Reprogramming under Defined Conditions. Stem Cell Rep. 2015, 5, 1128–1142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jayawardena, T.M.; Egemnazarov, B.; Finch, E.A.; Zhang, L.; Payne, J.A.; Pandya, K.; Zhang, Z.; Rosenberg, P.; Mirotsou, M.; Dzau, V.J. MicroRNA-mediated in vitro and in vivo direct reprogramming of cardiac fibroblasts to cardiomyocytes. Circ. Res. 2012, 110, 1465–1473. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garbutt, T.A.; Zhou, Y.; Keepers, B.; Liu, J.; Qian, L. An Optimized Protocol for Human Direct Cardiac Reprogramming. STAR Protoc. 2020, 1, 100010. [Google Scholar] [CrossRef]
- Zhou, Y.; Liu, Z.; Welch, J.D.; Gao, X.; Wang, L.; Garbutt, T.; Keepers, B.; Ma, H.; Prins, J.F.; Shen, W.; et al. Single-Cell Transcriptomic Analyses of Cell Fate Transitions during Human Cardiac Reprogramming. Cell Stem Cell 2019, 25, 149–164.e9. [Google Scholar] [CrossRef]
- Fu, Y.; Huang, C.; Xu, X.; Gu, H.; Ye, Y.; Jiang, C.; Qiu, Z.; Xie, X. Direct reprogramming of mouse fibroblasts into cardiomyocytes with chemical cocktails. Cell Res. 2015, 25, 1013–1024. [Google Scholar] [CrossRef] [Green Version]
- Lalit, P.A.; Salick, M.R.; Nelson, D.O.; Squirrell, J.M.; Shafer, C.M.; Patel, N.G.; Saeed, I.; Schmuck, E.G.; Markandeya, Y.S.; Wong, R. Lineage reprogramming of fibroblasts into proliferative induced cardiac progenitor cells by defined factors. Cell Stem Cell 2016, 18, 354–367. [Google Scholar] [CrossRef] [Green Version]
- Christoforou, N.; Chakraborty, S.; Kirkton, R.D.; Adler, A.F.; Addis, R.C.; Leong, K.W. Core Transcription Factors, MicroRNAs, and Small Molecules Drive Transdifferentiation of Human Fibroblasts Towards The Cardiac Cell Lineage. Sci. Rep. 2017, 7, 40285. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.X.; Krane, M.; Deutsch, M.A.; Wang, L.; Rav-Acha, M.; Gregoire, S.; Engels, M.C.; Rajarajan, K.; Karra, R.; Abel, E.D.; et al. Inefficient reprogramming of fibroblasts into cardiomyocytes using Gata4, Mef2c, and Tbx5. Circ. Res. 2012, 111, 50–55. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miyamoto, K.; Akiyama, M.; Tamura, F.; Isomi, M.; Yamakawa, H.; Sadahiro, T.; Muraoka, N.; Kojima, H.; Haginiwa, S.; Kurotsu, S.; et al. Direct In Vivo Reprogramming with Sendai Virus Vectors Improves Cardiac Function after Myocardial Infarction. Cell Stem Cell 2018, 22, 91–103.e5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.; Jiang, X.; Zhao, L.; Zuo, S.; Chen, X.; Zhang, L.; Lin, Z.; Zhao, X.; Qin, Y.; Zhou, X. Lineage reprogramming of fibroblasts into induced cardiac progenitor cells by CRISPR/Cas9-based transcriptional activators. Acta Pharm. Sin. B 2020, 10, 313–326. [Google Scholar] [CrossRef] [PubMed]
- Jayawardena, T.M.; Finch, E.A.; Zhang, L.; Zhang, H.; Hodgkinson, C.P.; Pratt, R.E.; Rosenberg, P.B.; Mirotsou, M.; Dzau, V.J. MicroRNA induced cardiac reprogramming in vivo: Evidence for mature cardiac myocytes and improved cardiac function. Circ. Res. 2015, 116, 418–424. [Google Scholar] [CrossRef] [PubMed]
- Chang, Y.; Lee, E.; Kim, J.; Kwon, Y.-W.; Kwon, Y.; Kim, J. Efficient in vivo direct conversion of fibroblasts into cardiomyocytes using a nanoparticle-based gene carrier. Biomaterials 2019, 192, 500–509. [Google Scholar] [CrossRef]
- Ieda, M. Key Regulators of Cardiovascular Differentiation and Regeneration: Harnessing the Potential of Direct Reprogramming to Treat Heart Failure. J. Card. Fail. 2020, 26, 80–84. [Google Scholar] [CrossRef]
- Chen, O.; Qian, L. Direct Cardiac Reprogramming: Advances in Cardiac Regeneration. BioMed. Res. Int. 2015, 2015, 580406. [Google Scholar] [CrossRef]
- Chen, J.X.; Plonowska, K.; Wu, S.M. Somatic Cell Reprogramming into Cardiovascular Lineages. J. Cardiovasc. Pharmacol. Ther. 2014, 19, 340–349. [Google Scholar] [CrossRef] [Green Version]
- Tani, H.; Sadahiro, T.; Ieda, M. Direct Cardiac Reprogramming: A Novel Approach for Heart Regeneration. Int. J. Mol. Sci. 2018, 19, 2629. [Google Scholar] [CrossRef] [Green Version]
- Konermann, S.; Brigham, M.D.; Trevino, A.E.; Joung, J.; Abudayyeh, O.O.; Barcena, C.; Hsu, P.D.; Habib, N.; Gootenberg, J.S.; Nishimasu, H.; et al. Genome-Scale transcriptional activation by an engineered CRISPR-Cas9 complex. Nature 2015, 517, 583–588. [Google Scholar] [CrossRef] [Green Version]
- Dykes, I.M. Exosomes in Cardiovascular Medicine. Cardiol. Ther. 2017, 6, 225–237. [Google Scholar] [CrossRef] [PubMed]
- Murphy, D.E.; de Jong, O.G.; Brouwer, M.; Wood, M.J.; Lavieu, G.; Schiffelers, R.M.; Vader, P. Extracellular vesicle-based therapeutics: Natural versus engineered targeting and trafficking. Exp. Mol. Med. 2019, 51, 1–12. [Google Scholar] [CrossRef] [PubMed]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Adams, E.; McCloy, R.; Jordan, A.; Falconer, K.; Dykes, I.M. Direct Reprogramming of Cardiac Fibroblasts to Repair the Injured Heart. J. Cardiovasc. Dev. Dis. 2021, 8, 72. https://0-doi-org.brum.beds.ac.uk/10.3390/jcdd8070072
Adams E, McCloy R, Jordan A, Falconer K, Dykes IM. Direct Reprogramming of Cardiac Fibroblasts to Repair the Injured Heart. Journal of Cardiovascular Development and Disease. 2021; 8(7):72. https://0-doi-org.brum.beds.ac.uk/10.3390/jcdd8070072
Chicago/Turabian StyleAdams, Emma, Rachel McCloy, Ashley Jordan, Kaitlin Falconer, and Iain M. Dykes. 2021. "Direct Reprogramming of Cardiac Fibroblasts to Repair the Injured Heart" Journal of Cardiovascular Development and Disease 8, no. 7: 72. https://0-doi-org.brum.beds.ac.uk/10.3390/jcdd8070072