
Current Aspects on the Pathophysiology of Bone Metabolic Defects during Progression of Scoliosis in Neurofibromatosis Type 1

 , , , ,
, , , ,  , and
, and
Abstract
:1. Introduction
2. Epidemiological Data and Clinical Characteristics
3. Bone Metabolism in NF1
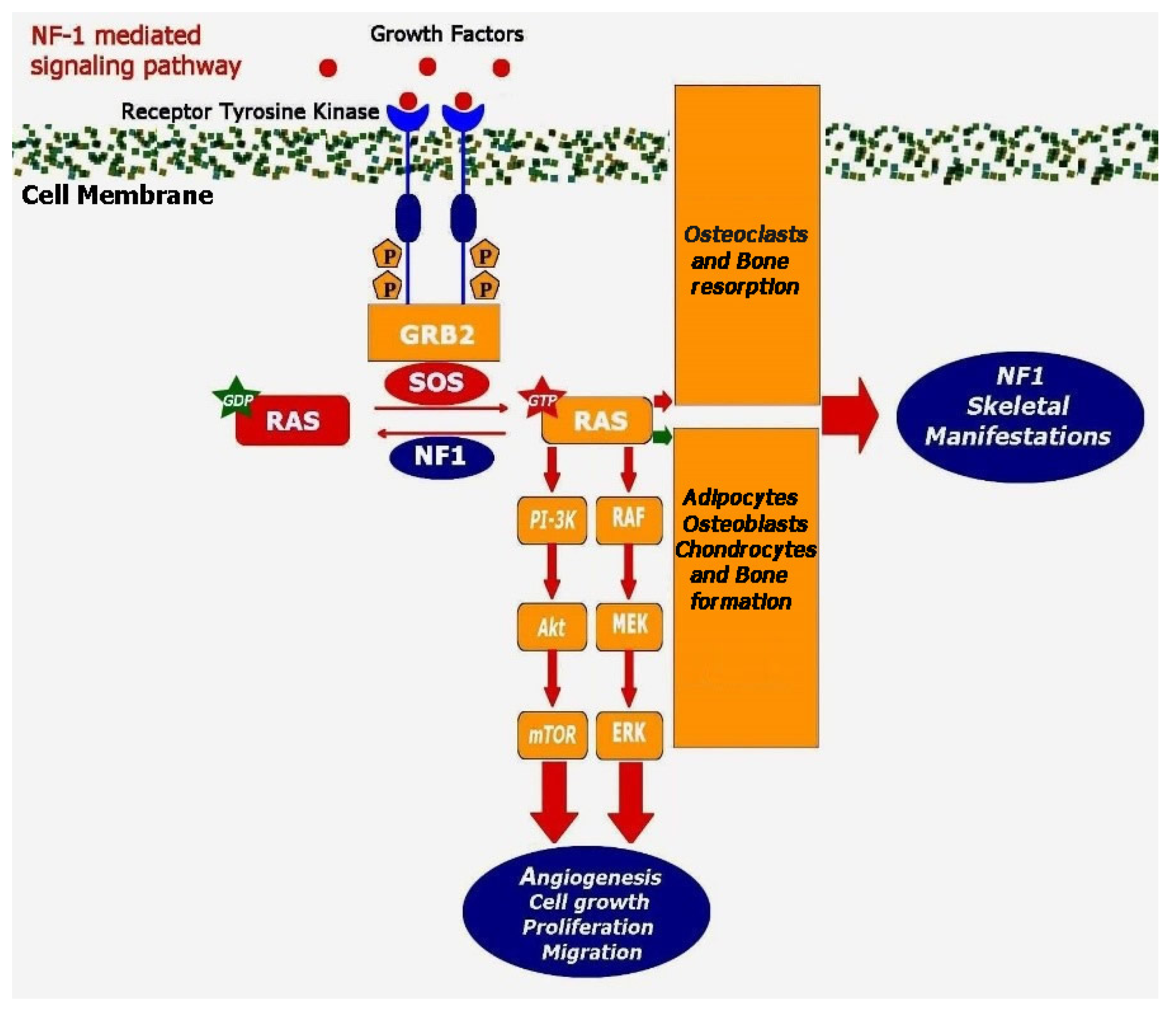
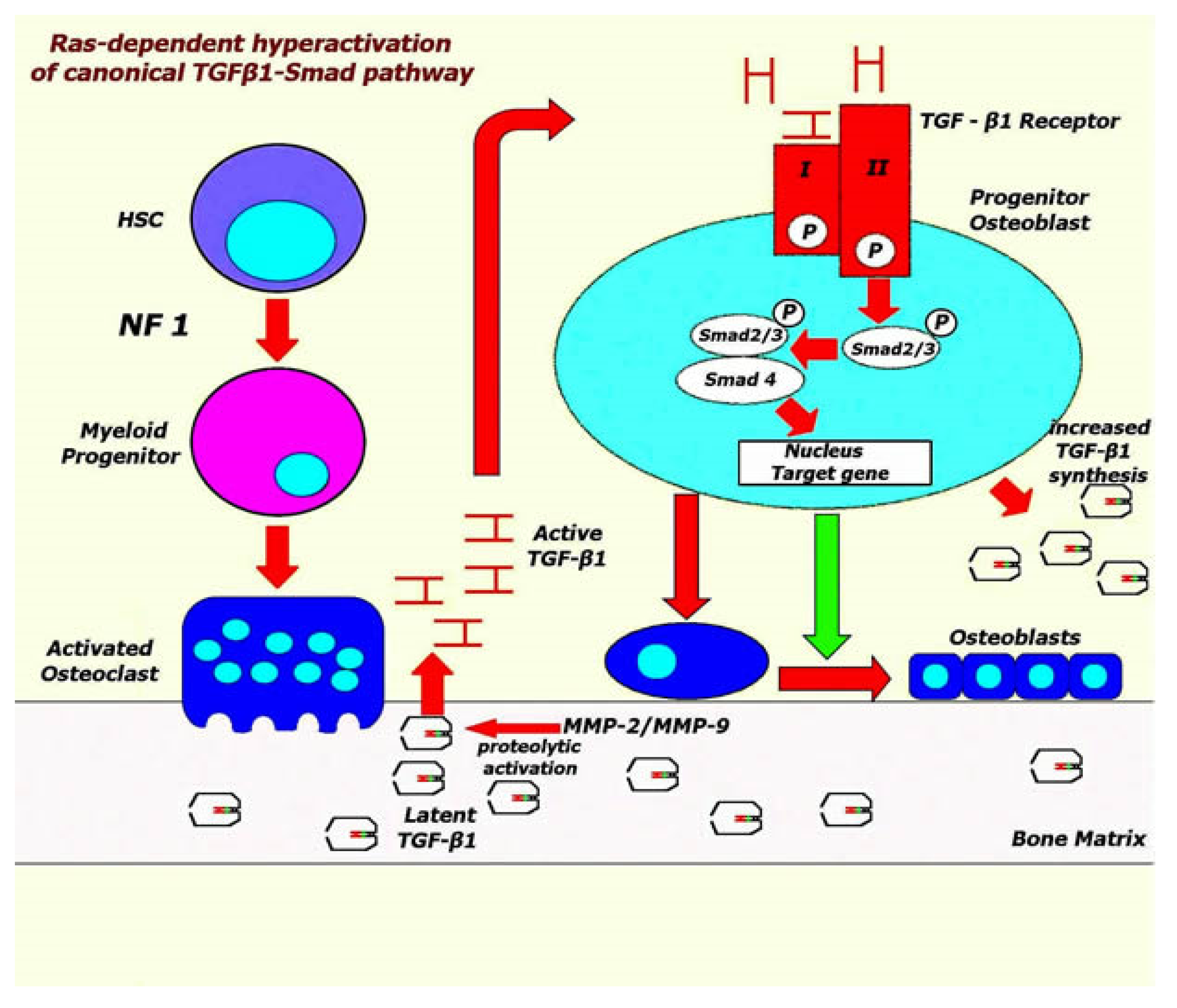
4. Molecular Basis for Skeletal Deformities in NF1
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Lammert, M.; Friedman, J.M.; Kluwe, L.; Mautner, V.F. Prevalence of neurofibromatosis 1 in German children at elementary school enrollment. Arch. Dermatol. 2005, 141, 71–74. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moramarco, A.; Mallone, F.; Sacchetti, M.; Lucchino, L.; Miraglia, E.; Roberti, V.; Lambiase, A.; Giustini, S. Hyperpigmented spots at fundus examination: A new ocular sign in Neurofibromatosis Type I. Orphanet J. Rare Dis. 2021, 16, 147. [Google Scholar] [CrossRef]
- Kallionpää, R.A.; Uusitalo, E.; Leppävirta, J.; Pöyhönen, M.; Peltonen, S.; Peltonen, J. Prevalence of neurofibromatosis type 1 in the Finnish population. Genet. Med. 2018, 20, 1082–1086. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jett, K.; Friedman, J.M. Clinical and genetic aspects of neurofibromatosis 1. Genet. Med. 2010, 12, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Easton, D.F.; Ponder, M.A.; Huson, S.M.; Ponder, B.A. An analysis of variation in expression of neurofibromatosis (NF) type 1 (NF1): Evidence for modifying genes. Am. J. Hum. Genet. 1993, 53, 305–313. [Google Scholar]
- Hannan, F.; Ho, I.; Tong, J.J.; Zhu, Y.; Nurnberg, P.; Zhong, Y. Effect of neurofibromatosis type I mutations on a novel pathway for adenylyl cyclase activation requiring neurofibromin and Ras. Hum. Mol. Genet. 2006, 15, 1087–1098. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ratner, N.; Miller, S.J. A RASopathy gene commonly mutated in cancer: The neurofibromatosis type 1 tumour suppressor. Nat. Rev. Cancer 2015, 15, 290–301. [Google Scholar] [CrossRef]
- Yap, Y.S.; McPherson, J.R.; Ong, C.K.; Rozen, S.G.; Teh, B.T.; Lee, A.S.; Callen, D.F. The NF1 gene revisited—From bench to bedside. Oncotarget 2014, 5, 5873–5892. [Google Scholar] [CrossRef] [Green Version]
- Stumpf, D.A. Neurofibromatosis. Conference statement, National Institute of Health development conference. Arch. Neurol. 1988, 45, 575–578. [Google Scholar]
- Williams, V.C.; Lucas, J.; Babcock, M.A.; Gutmann, D.H.; Korf, B.; Maria, B.L. Neurofibromatosis type 1 revisited. Pediatrics 2009, 123, 124–133. [Google Scholar] [CrossRef] [Green Version]
- Elefteriou, F.; Kolanczyk, M.; Schindeler, A.; Viskochil, D.H.; Hock, J.M.; Schorry, E.K.; Crawford, A.H.; Friedman, J.M.; Little, D.; Peltonen, J.; et al. Skeletal abnormalities in neurofibromatosis type 1: Approaches to therapeutic options. Am. J. Med. Genet. A 2009, 149A, 2327–2338. [Google Scholar] [CrossRef]
- Stevenson, D.A.; Little, D.; Armstrong, L.; Crawford, A.H.; Eastwood, D.; Friedman, J.M.; Greggi, T.; Gutierrez, G.; Hunter-Schaedle, K.; Kendler, D.L.; et al. Approaches to treating NF1 tibial pseudarthrosis: Consensus from the Children’s Tumor Foundation NF1 Bone Abnormalities Consortium. J. Pediatr. Orthop. 2013, 33, 269–275. [Google Scholar] [CrossRef]
- Heervä, E.; Koffert, A.; Jokinen, E.; Kuorilehto, T.; Peltonen, S.; Aro, H.T.; Peltonen, J. A controlled register-based study of 460 neurofibromatosis 1 patients: Increased fracture risk in children and adults over 41 years of age. J. Bone Miner. Res. 2012, 27, 2333–2337. [Google Scholar] [CrossRef]
- Jalabert, M.; Ferkal, S.; Souberbielle, J.C.; Sbidian, E.; Mageau, A.; Eymard, F.; Le Corvoisier, P.; Allanore, L.; Chevalier, X.; Wolkenstein, P.; et al. Bone Status According to Neurofibromatosis Type 1 Phenotype: A Descriptive Study of 60 Women in France. Calcif. Tissue Int. 2021, in press. [Google Scholar] [CrossRef] [PubMed]
- Lammert, M.; Kappler, M.; Mautner, V.F.; Lammert, K.; Störkel, S.; Friedman, J.M.; Atkins, D. Decreased bone mineral density in patients with neurofibromatosis 1. Osteoporos. Int. 2005, 16, 1161–1166. [Google Scholar] [CrossRef] [PubMed]
- Heervä, E.; Leinonen, P.; Kuorilehto, T.; Peltonen, S.; Pöyhönen, M.; Väänänen, K.; Peltonen, J. Neurofibromatosis 1-related osteopenia often progresses to osteoporosis in 12 years. Calcif. Tissue Int. 2013, 92, 23–27. [Google Scholar] [CrossRef]
- Seitz, S.; Schnabel, C.; Busse, B.; Schmidt, H.U.; Beil, F.T.; Friedrich, R.E.; Schinke, T.; Mautner, V.F.; Amling, M. High bone turnover and accumulation of osteoid in patients with neurofibromatosis 1. Osteoporos. Int. 2010, 21, 119–127. [Google Scholar] [CrossRef]
- Yang, F.C.; Chen, S.; Robling, A.G.; Yu, X.; Nebesio, T.D.; Yan, J.; Morgan, T.; Li, X.; Yuan, J.; Hock, J.; et al. Hyperactivation of p21ras and PI3K cooperate to alter murine and human neurofibromatosis type 1-haploinsufficient osteoclast functions. J. Clin. Investig. 2006, 116, 2880–2891. [Google Scholar] [CrossRef] [Green Version]
- Wu, X.; Estwick, S.A.; Chen, S.; Yu, M.; Ming, W.; Nebesio, T.D.; Li, Y.; Yuan, J.; Kapur, R.; Ingram, D.; et al. Neurofibromin plays a critical role in modulating osteoblast differentiation of mesenchymal stem/progenitor cells. Hum. Mol. Genet. 2006, 15, 2837–2845. [Google Scholar] [CrossRef] [Green Version]
- Rhodes, S.D.; Wu, X.; He, Y.; Chen, S.; Yang, H.; Staser, K.W.; Wang, J.; Zhang, P.; Jiang, C.; Yokota, H.; et al. Hyperactive transforming growth factor-β1 signaling potentiates skeletal defects in a neurofibromatosis type 1 mouse model. J. Bone Miner. Res. 2013, 28, 2476–2489. [Google Scholar] [CrossRef] [PubMed]
- Toro, G.; Santoro, C.; Ambrosio, D.; Landi, G.; Scilipoti, M.; Moretti, A.; Paoletta, M.; Liguori, S.; Schiavone Panni, A.; Picariello, S.; et al. Natural History of Scoliosis in Children with NF1: An Observation Study. Healthcare 2021, 9, 881. [Google Scholar] [CrossRef]
- Brunetti-Pierri, N.; Doty, S.B.; Hicks, J.; Phan, K.; Mendoza-Londono, R.; Blazo, M.; Tran, A.; Carter, S.; Lewis, R.A.; Plon, S.E.; et al. Generalized metabolic bone disease in Neurofibromatosis type I. Mol. Genet. Metab. 2008, 94, 105–111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Riccardi, C.; Perrone, L.; Napolitano, F.; Sampaolo, S.; Melone, M.A.B. Understanding the Biological Activities of Vitamin D in Type 1 Neurofibromatosis: New Insights into Disease Pathogenesis and Therapeutic Design. Cancers 2020, 12, 2965. [Google Scholar] [CrossRef]
- Durrani, A.A.; Crawford, A.H.; Chouhdry, S.N.; Saifuddin, A.; Morley, T.R. Modulation of spinal deformities in patients with neurofibromatosis type 1. Spine 2000, 25, 69–75. [Google Scholar] [CrossRef] [PubMed]
- Akbarnia, B.A.; Gabriel, K.R.; Beckman, E.; Chalk, D. Prevalence of scoliosis in neurofibromatosis. Spine 1992, 17 (Suppl. 8), S244–S248. [Google Scholar] [CrossRef]
- Lykissas, M.G.; Schorry, E.K.; Crawford, A.H.; Gaines, S.; Rieley, M.; Jain, V.V. Does the presence of dystrophic features in patients with type 1 neurofibromatosis and spinal deformities increase the risk of surgery? Spine 2013, 38, 1595–1601. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, Y.; Ehara, Y.; Koga, M.; Imafuku, S.; Yamamoto, O. Epidemiological Analysis of Major Complications Requiring Medical Intervention in Patients with Neurofibromatosis 1. Acta Derm. Venereol. 2018, 98, 753–756. [Google Scholar] [CrossRef] [Green Version]
- Well, L.; Careddu, A.; Stark, M.; Farschtschi, S.; Bannas, P.; Adam, G.; Mautner, V.F.; Salamon, J. Phenotyping spinal abnormalities in patients with Neurofibromatosis type 1 using whole-body MRI. Sci. Rep. 2021, 11, 16889. [Google Scholar] [CrossRef] [PubMed]
- Karmazyn, B.; Cohen, M.D.; Jennings, S.G.; Robertson, K.A. Marrow signal changes observed in follow-up whole-body MRI studies in children and young adults with neurofibromatosis type 1 treated with imatinib mesylate (Gleevec) for plexiform neurofibromas. Pediatr. Radiol. 2012, 42, 1218–1222. [Google Scholar] [CrossRef] [PubMed]
- Delucia, T.A.; Yohay, K.; Widmann, R.F. Orthopaedic aspects of neurofibromatosis: Update. Curr. Opin. Pediatr. 2011, 23, 46–52. [Google Scholar] [CrossRef] [PubMed]
- Petramala, L.; Giustini, S.; Zinnamosca, L.; Marinelli, C.; Colangelo, L.; Cilenti, G.; Formicuccia, M.C.; D’Erasmo, E.; Calvieri, S.; Letizia, C. Bone mineral metabolism in patients with neurofibromatosis type 1 (von Recklingausen disease). Arch. Dermatol. Res. 2012, 304, 325–331. [Google Scholar] [CrossRef]
- Kuorilehto, T.; Pöyhönen, M.; Bloigu, R.; Heikkinen, J.; Väänänen, K.; Peltonen, J. Decreased bone mineral density and content in neurofibromatosis type 1: Lowest local values are located in the load-carrying parts of the body. Osteoporos. Int. 2005, 16, 928–936. [Google Scholar] [CrossRef]
- Illés, T.; Halmai, V.; de Jonge, T.; Dubousset, J. Decreased bone mineral density in neurofibromatosis-1 patients with spinal deformities. Osteoporos. Int. 2001, 12, 823–827. [Google Scholar] [CrossRef] [PubMed]
- Fowlkes, J.L.; Thrailkill, K.M.; Bunn, R.C. RASopathies: The musculoskeletal consequences and their etiology and pathogenesis. Bone 2021, 152, 116060. [Google Scholar] [CrossRef]
- Filopanti, M.; Verga, U.; Ulivieri, F.M.; Giavoli, C.; Rodari, G.; Arosio, M.; Natacci, F.; Spada, A. Trabecular Bone Score (TBS) and Bone Metabolism in Patients Affected with Type 1 Neurofibromatosis (NF1). Calcif. Tissue Int. 2019, 104, 207–213. [Google Scholar] [CrossRef] [Green Version]
- Yilmaz, K.; Ozmen, M.; Bora Goksan, S.; Eskiyurt, N. Bone mineral density in children with neurofibromatosis 1. Acta Paediatr. 2007, 96, 1220–1222. [Google Scholar] [CrossRef] [PubMed]
- Dulai, S.; Briody, J.; Schindeler, A.; North, K.N.; Cowell, C.T.; Little, D.G. Decreased bone mineral density in neurofibromatosis type 1: Results from a pediatric cohort. J. Pediatr. Orthop. 2007, 27, 472–475. [Google Scholar] [CrossRef]
- Lodish, M.B.; Dagalakis, U.; Sinaii, N.; Bornstein, E.; Kim, A.; Lokie, K.B.; Baldwin, A.M.; Reynolds, J.C.; Dombi, E.; Stratakis, C.A.; et al. Bone mineral density in children and young adults with neurofibromatosis type 1. Endocr. Relat. Cancer 2012, 19, 817–825. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duman, O.; Ozdem, S.; Turkkahraman, D.; Olgac, N.D.; Gungor, F.; Haspolat, S. Bone metabolism markers and bone mineral density in children with neurofibromatosis type-1. Brain Dev. 2008, 30, 584–588. [Google Scholar] [CrossRef]
- Eelloo, J.; Ward, K.; Huson, S.M.; Adams, J.E.; Russell, S.; Wright, N.; Evans, G.; Mugha, M.Z. Longitudinal assessment of spinal bone mineral density in children with neurofibromatosis type 1 using dual energy absorptiometry and quantitative computed tomography. Bone Abstr. 2013, 2, P172. [Google Scholar]
- Stevenson, D.A.; Moyer-Mileur, L.J.; Murray, M.; Slater, H.; Sheng, X.; Carey, J.C.; Dube, B.; Viskochil, D.H. Bone mineral density in children and adolescents with neurofibromatosis type 1. J. Pediatr. 2007, 150, 83–88. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weinstein, R.S.; Harris, R.L. Hypercalcemic hyperparathyroidism and hypophosphatemic osteomalacia complicating neurofibromatosis. Calcif. Tissue Int. 1990, 46, 361–366. [Google Scholar] [CrossRef]
- Schnabel, C.; Dahm, S.; Streichert, T.; Thierfelder, W.; Kluwe, L.; Mautner, V.F. Differences of 25-hydroxyvitamin D3 concentrations in children and adults with neurofibromatosis type 1. Clin. Biochem. 2014, 47, 560–563. [Google Scholar] [CrossRef]
- Poyrazoğlu, H.G.; Baş, V.N.; Arslan, A.; Bastug, F.; Canpolat, M.; Per, H.; Gümüs, H.; Kumandas, S. Bone mineral density and bone metabolic markers’ status in children with neurofibromatosis type 1. J. Pediatr. Endocrinol. Metab. 2017, 30, 175–180. [Google Scholar] [CrossRef]
- Kluwe, L.; Hagel, C.; Friedrich, R.E.; Schnabel, C.; Schön, G.; Mautner, V. Vitamin D receptor expression and serum 25(OH)D concentration inversely associates with burden of neurofibromas. Eur. J. Cancer Prev. 2019, 28, 220–224. [Google Scholar] [CrossRef]
- Xue, Y.; Fleet, J.C. Intestinal vitamin D receptor is required for normal calcium and bone metabolism in mice. Gastroenterology 2009, 136, 1317–1327. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodari, G.; Scuvera, G.; Ulivieri, F.M.; Profka, E.; Menni, F.; Saletti, V.; Esposito, S.; Bergamaschi, S.; Ferrante, E.; Eller-Vainicher, C.; et al. Progressive bone impairment with age and pubertal development in neurofibromatosis type I. Arch. Osteoporos. 2018, 13, 93. [Google Scholar] [CrossRef] [PubMed]
- Tucker, T.; Schnabel, C.; Hartmann, M.; Friedrich, R.E.; Frieling, I.; Kruse, H.P.; Mautner, V.F.; Friedman, J.M. Bone health and fracture rate in individuals with neurofibromatosis 1 (NF1). J. Med. Genet. 2009, 46, 259–265. [Google Scholar] [CrossRef] [PubMed]
- Stevenson, D.A.; Schwarz, E.L.; Viskochil, D.H.; Moyer-Mileur, L.J.; Murray, M.; Firth, S.D.; D’Astous, J.L.; Carey, J.C.; Pasquali, M. Evidence of increased bone resorption in neurofibromatosis type 1 using urinary pyridinium crosslink analysis. Pediatr. Res. 2008, 63, 697–701. [Google Scholar] [CrossRef] [Green Version]
- Ryan, E.A.; Reiss, E. Oncogenous osteomalacia. Review of the world literature of 42 cases and report of two new cases. Am. J. Med. 1984, 77, 501–512. [Google Scholar] [CrossRef]
- Chadha, M.; Singh, A.P.; Singh, A.P. Hypophosphataemic osteomalacia in neurofibromatosis. Acta Orthop. Belg. 2009, 75, 847–850. [Google Scholar] [PubMed]
- Sahoo, S.K.; Kushwaha, P.; Bharti, N.; Khedgikar, V.; Trivedi, R.; Agrawal, V.; Ahmad, N.; Zaidi, G.; Pal, L.; Ito, N.; et al. Elevated FGF23 in a patient with hypophosphatemic osteomalacia associated with neurofibromatosis type 1. Bone 2019, 129, 115055. [Google Scholar] [CrossRef]
- Soveid, M. Tumor associated osteomalacia in neurofibromatosis: Case report and literature review. Med. J. Islam Repub. Iran. 2003, 16, 227–230. [Google Scholar]
- Wattiaux, M.J.; De Vernejoul, M.C.; Bletry, O.; Ulmann, A.; Rondier, J.; Godeau, P. Maladie de Recklinghausen avec hypophosphoremie et osteomalacie. Rev. Med. Interne 1985, 6, 495–502. [Google Scholar] [CrossRef]
- Konishi, K.; Nakamura, M.; Yamakawa, H.; Suzuki, H.; Saruta, T.; Hanaoka, H.; Davatchi, F. Hypophosphatemic osteomalacia in von Recklinghausen neurofibromatosis. Am. J. Med. Sci. 1991, 301, 322–328. [Google Scholar] [CrossRef]
- Fukumoto, S. FGF23 and Bone and Mineral Metabolism. Handb. Exp. Pharmacol. 2020, 262, 281–308. [Google Scholar]
- Kamiya, N.; Yamaguchi, R.; Aruwajoye, O.; Kim, A.J.; Kuroyanagi, G.; Phipps, M.; Adapala, N.S.; Feng, J.Q.; Kim, H.K. Targeted Disruption of NF1 in Osteocytes Increases FGF23 and Osteoid with Osteomalacia-like Bone Phenotype. J. Bone Miner. Res. 2017, 32, 1716–1726. [Google Scholar] [CrossRef] [PubMed]
- Schindeler, A.; Little, D.G. Recent insights into bone development, homeostasis, and repair in type 1 neurofibromatosis (NF1). Bone 2008, 42, 616–622. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.; Chen, S.; Potter, O.L.; Murthy, S.M.; Li, J.; Pulcini, J.M.; Ohashi, N.; Winata, T.; Everett, E.T.; Ingram, D.; et al. Neurofibromin and its inactivation of Ras are prerequisites for osteoblast functioning. Bone 2005, 36, 793–802. [Google Scholar] [CrossRef]
- Ma, Y.; Gross, A.M.; Dombi, E.; Pemov, A.; Choi, K.; Chaney, K.; Rhodes, S.D.; Angus, S.P.; Sciaky, N.; Clapp, D.W.; et al. A molecular basis for neurofibroma-associated skeletal manifestations in NF1. Genet. Med. 2020, 22, 1786–1793. [Google Scholar] [CrossRef]
- Li, Y.; Li, J.; Zhou, Q.; Liu, Y.; Chen, W.; Xu, H. mTORC1 signaling is essential for neurofibromatosis type I gene modulated osteogenic differentiation of BMSCs. J. Cell Biochem. 2019, 120, 2886–2896. [Google Scholar] [CrossRef]
- Li, Y.; Zhu, M.; Lin, X.; Li, J.; Yuan, Z.; Liu, Y.; Xu, H. Autophagy is involved in neurofibromatosis type I gene-modulated osteogenic differentiation in human bone mesenchymal stem cells. Exp. Ther. Med. 2021, 22, 1262. [Google Scholar] [CrossRef]
- Zhou, Z.; Shi, G.; Zheng, X.; Jiang, S.; Jiang, L. Autophagy activation facilitates mechanical stimulation-promoted osteoblast differentiation and ameliorates hindlimb unloading-induced bone loss. Biochem. Biophys. Res. Commun. 2018, 498, 667–673. [Google Scholar] [CrossRef]
- Tan, Q.; Wu, J.Y.; Liu, Y.X.; Liu, K.; Tang, J.; Ye, W.H.; Zhu, G.H.; Mei, H.B.; Yang, G. The neurofibromatosis type I gene promotes autophagy via mTORC1 signalling pathway to enhance new bone formation after fracture. J. Cell Mol. Med. 2020, 24, 11524–11534. [Google Scholar] [CrossRef] [PubMed]
- Miyazaki, T.; Katagiri, H.; Kanegae, Y.; Takayanagi, H.; Sawada, Y.; Yamamoto, A.; Pando, M.P.; Asano, T.; Verma, I.M.; Oda, H.; et al. Reciprocal role of ERK and NF-kappaB pathways in survival and activation of osteoclasts. J. Cell Biol. 2000, 148, 333–342. [Google Scholar] [CrossRef]
- Ogasawara, T.; Katagiri, M.; Yamamoto, A.; Hoshi, K.; Takato, T.; Nakamura, K.; Tanaka, S.; Okayama, H.; Kawaguchi, H. Osteoclast differentiation by RANKL requires NF-kappaB-mediated downregulation of cyclin-dependent kinase 6 (Cdk6). J. Bone Miner. Res. 2004, 19, 1128–1136. [Google Scholar] [CrossRef] [PubMed]
- Lee, Z.H.; Kim, H.H. Signal transduction by receptor activator of nuclear factor kappa B in osteoclasts. Biochem. Biophys. Res. Commun. 2003, 305, 211–214. [Google Scholar] [CrossRef]
- Park, J.H.; Lee, N.K.; Lee, S.Y. Current Understanding of RANK Signaling in Osteoclast Differentiation and Maturation. Mol. Cells 2017, 40, 706–713. [Google Scholar]
- Yang, G.; Yu, H.; Liu, Y.; Ye, W.; Zhu, G.; Yan, A.; Tan, Q.; Mei, H. Serum-derived exosomes from neurofibromatosis type 1 congenital tibial pseudarthrosis impaired bone by promoting osteoclastogenesis and inhibiting osteogenesis. Exp. Biol. Med. 2021, 246, 130–141. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.S.; Ali, M.H.; Wydra, F.; Li, X.; Hamilton, J.L.; An, H.S.; Cs-Szabo, G.; Andrews, S.; Moric, M.; Xiao, G.; et al. Characterization of degenerative human facet joints and facet joint capsular tissues. Osteoarthr. Cartil. 2015, 23, 2242–2251. [Google Scholar] [CrossRef] [Green Version]
- Grivas, T.B.; Vasiliadis, E.S.; Kaspiris, A.; Khaldi, L.; Kletsas, D. Expression of matrix metalloproteinase-1 (MMP-1) in Wistar rat’s intervertebral disc after experimentally induced scoliotic deformity. Scoliosis 2011, 6, 9. [Google Scholar] [CrossRef] [Green Version]
- Vasiliadis, E.S.; Kaspiris, A.; Grivas, T.B.; Khaldi, L.; Lamprou, M.; Pneumaticos, S.G.; Nikolopoulos, K.; Korres, D.S.; Papadimitriou, E. Expression of macrophage elastase (MMP12) in rat tail intervertebral disc and growth plate after asymmetric loading. Bone Jt. Res. 2014, 3, 273–279. [Google Scholar] [CrossRef]
- Kaspiris, A.; Chronopoulos, E.; Grivas, T.B.; Vasiliadis, E.; Khaldi, L.; Lamprou, M.; Lelovas, P.P.; Papaioannou, N.; Dontas, I.A.; Papadimitriou, E. Effects of mechanical loading on the expression of pleiotrophin and its receptor protein tyrosine phosphatase beta/zeta in a rat spinal deformity model. Cytokine 2016, 78, 7–15. [Google Scholar] [CrossRef]
- Mashour, G.A.; Ratner, N.; Khan, G.A.; Wang, H.L.; Martuza, R.L.; Kurtz, A. The angiogenic factor midkine is aberrantly expressed in NF1-deficient Schwann cells and is a mitogen for neurofibroma-derived cells. Oncogene 2001, 20, 97–105. [Google Scholar] [CrossRef] [Green Version]
- Wu, M.; Wallace, M.R.; Muir, D. Nf1 haploinsufficiency augments angiogenesis. Oncogene 2006, 25, 2297–2303. [Google Scholar] [CrossRef] [Green Version]
- Kuorilehto, T.; Kinnunen, P.; Nissinen, M.; Alanne, M.; Leskelä, H.V.; Lehenkari, P.; Peltonen, J. Vasculopathy in two cases of NF1-related congenital pseudarthrosis. Pathol. Res. Pract. 2006, 202, 687–690. [Google Scholar] [CrossRef] [PubMed]
- Hermanns-Sachweh, B.; Senderek, J.; Alfer, J.; Klosterhalfen, B.; Büttner, R.; Füzesi, L.; Weber, M. Vascular changes in the periosteum of congenital pseudarthrosis of the tibia. Pathol. Res. Pract. 2005, 201, 305–312. [Google Scholar] [CrossRef] [PubMed]
- Cuthbert, R.J.; Jones, E.; Sanjurjo-Rodríguez, C.; Lotfy, A.; Ganguly, P.; Churchman, S.M.; Castana, P.; Tan, H.B.; McGonagle, D.; Papadimitriou, E.; et al. Regulation of Angiogenesis Discriminates Tissue Resident MSCs from Effective and Defective Osteogenic Environments. J. Clin. Med. 2020, 9, 1628. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.; Lu, A.; Ran, R.; Aronow, B.J.; Schorry, E.K.; Hopkin, R.J.; Gilbert, D.L.; Glauser, T.A.; Hershey, A.D.; Richtand, N.W.; et al. Human blood genomics: Distinct profiles for gender, age and neurofibromatosis type 1. Brain Res. Mol. Brain Res. 2004, 132, 155–167. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.; Wu, X.; Lei, W.; Pang, L.; Wan, C.; Shi, Z.; Zhao, L.; Nagy, T.R.; Peng, X.; Hu, J.; et al. TGF-beta1-induced migration of bone mesenchymal stem cells couples bone resorption with formation. Nat. Med. 2009, 15, 757–765. [Google Scholar] [CrossRef] [Green Version]
- Qin, Y.; Tang, S.; Zhen, G.; Ding, Q.; Ding, S.; Cao, X. Bone-targeted delivery of TGF-β type 1 receptor inhibitor rescues uncoupled bone remodeling in Camurati-Engelmann disease. Ann. N. Y. Acad. Sci. 2018, 1433, 29–40. [Google Scholar] [CrossRef] [PubMed]
- Almpani, K.; Liberton, D.K.; Jani, P.; Keyvanfar, C.; Mishra, R.; Curry, N.; Orzechowski, P.; Frischmeyer-Guerrerio, P.A.; Lee, J.S. Loeys-Dietz and Shprintzen-Goldberg syndromes: Analysis of TGF-β-opathies with craniofacial manifestations using an innovative multimodality method. J. Med. Genet. 2021; in press. [Google Scholar] [CrossRef] [PubMed]
- Uehara, M.; Ito, K.; Kosho, T.; Kuraishi, S.; Oba, H.; Hatakenaka, T.; Ikegami, S.; Takizawa, T.; Munakata, R.; Kubota, M.; et al. Posterior spinal fusion for severe kyphoscoliosis in a Loeys-Dietz syndrome patient with a large syringomyelia. J. Clin. Neurosci. 2020, 76, 211–213. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, Y.; Watanabe, K.; Yagi, M.; Suzuki, S.; Nori, S.; Tsuji, O.; Nagoshi, N.; Okada, E.; Fujita, N.; Nakamura, M.; et al. Early-Onset Scoliosis Associated with Shprintzen-Goldberg Syndrome Treated with Growing Rods and Required Multiple Unplanned Surgeries: A Case Report. Spine Surg. Relat. Res. 2020, 5, 214–217. [Google Scholar] [CrossRef]
- Takano, H.; Yonezawa, I.; Okuda, T.; Kajihara, H.; Kaneko, K. Scoliosis in Shprintzen-Goldberg Syndrome. Case Rep. Orthop. 2020, 2020, 8857463. [Google Scholar] [CrossRef]
- Taniguchi, Y.; Matsubayashi, Y.; Kato, S.; Doi, T.; Takeda, N.; Yagi, H.; Inuzuka, R.; Oshima, Y.; Tanaka, S. Predictive Physical Manifestations for Progression of Scoliosis in Marfan Syndrome. Spine 2021, 46, 1020–1025. [Google Scholar] [CrossRef] [PubMed]
- Pollock, L.; Ridout, A.; Teh, J.; Nnadi, C.; Stavroulias, D.; Pitcher, A.; Blair, E.; Wordsworth, P.; Vincent, T.L. The Musculoskeletal Manifestations of Marfan Syndrome: Diagnosis, Impact, and Management. Curr. Rheumatol. Rep. 2021, 23, 81. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
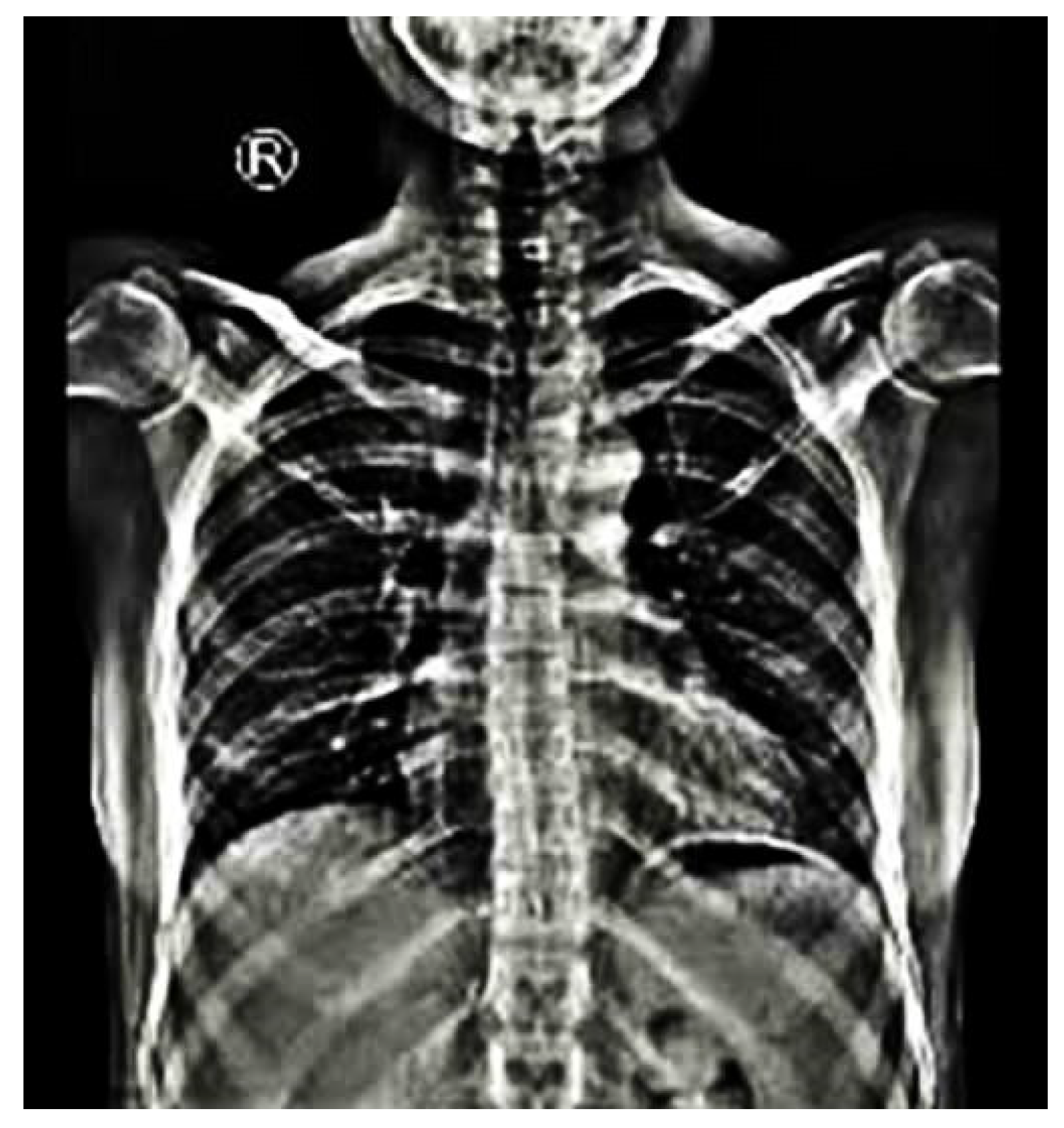
| Scoliotic Vertebral Dystrophic Alterations |
|---|
|
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kaspiris, A.; Savvidou, O.D.; Vasiliadis, E.S.; Hadjimichael, A.C.; Melissaridou, D.; Iliopoulou-Kosmadaki, S.; Iliopoulos, I.D.; Papadimitriou, E.; Chronopoulos, E. Current Aspects on the Pathophysiology of Bone Metabolic Defects during Progression of Scoliosis in Neurofibromatosis Type 1. J. Clin. Med. 2022, 11, 444. https://0-doi-org.brum.beds.ac.uk/10.3390/jcm11020444
Kaspiris A, Savvidou OD, Vasiliadis ES, Hadjimichael AC, Melissaridou D, Iliopoulou-Kosmadaki S, Iliopoulos ID, Papadimitriou E, Chronopoulos E. Current Aspects on the Pathophysiology of Bone Metabolic Defects during Progression of Scoliosis in Neurofibromatosis Type 1. Journal of Clinical Medicine. 2022; 11(2):444. https://0-doi-org.brum.beds.ac.uk/10.3390/jcm11020444
Chicago/Turabian StyleKaspiris, Angelos, Olga D. Savvidou, Elias S. Vasiliadis, Argyris C. Hadjimichael, Dimitra Melissaridou, Stella Iliopoulou-Kosmadaki, Ilias D. Iliopoulos, Evangelia Papadimitriou, and Efstathios Chronopoulos. 2022. "Current Aspects on the Pathophysiology of Bone Metabolic Defects during Progression of Scoliosis in Neurofibromatosis Type 1" Journal of Clinical Medicine 11, no. 2: 444. https://0-doi-org.brum.beds.ac.uk/10.3390/jcm11020444