A Next-Generation Sequencing-Based Approach to Identify Genetic Determinants of Antibiotic Resistance in Cambodian Helicobacter pylori Clinical Isolates

 , ,
, ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Study Design and Sampling Collection
2.2. Ethical Considerations
2.3. Phenotypic Antibiotic Susceptibility Testing
2.4. Genotype Analysis of Antibiotic Susceptibility
2.4.1. DNA Extraction, Library Preparation, and Whole Genome Sequencing
2.4.2. Bioinformatic Analysis to Identify Genetic Determinants of Antibiotic Resistance
Analysis of Drug Susceptibility Genotypes Encoded by Plasmid in H. pylori Isolates
Analysis of Drug Susceptibility Genotypes Encoded by Genomic DNA of H. pylori Isolates
2.5. Statistical Analysis
3. Results
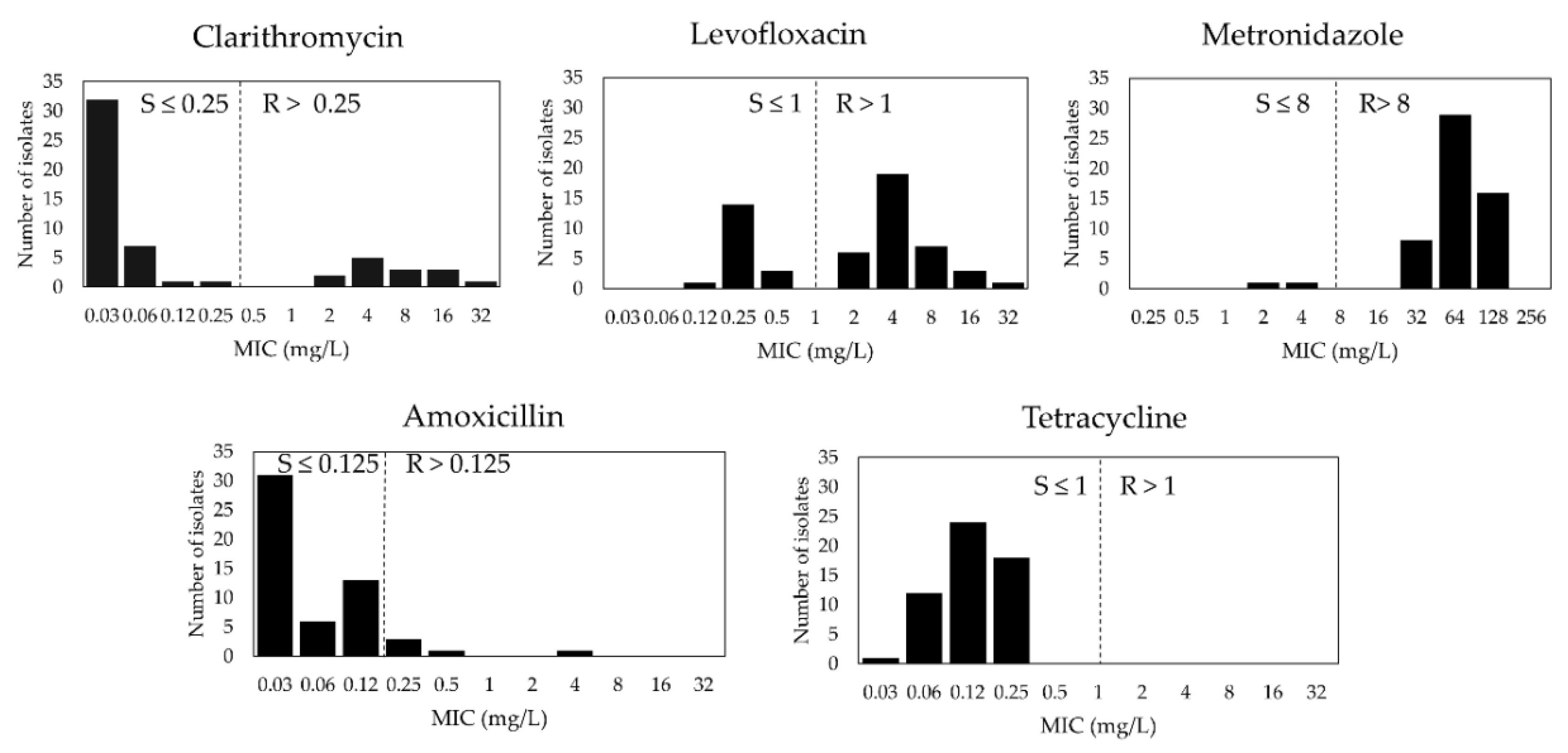
3.1. H. pylori Primary Antibiotic Susceptibility in Cambodia
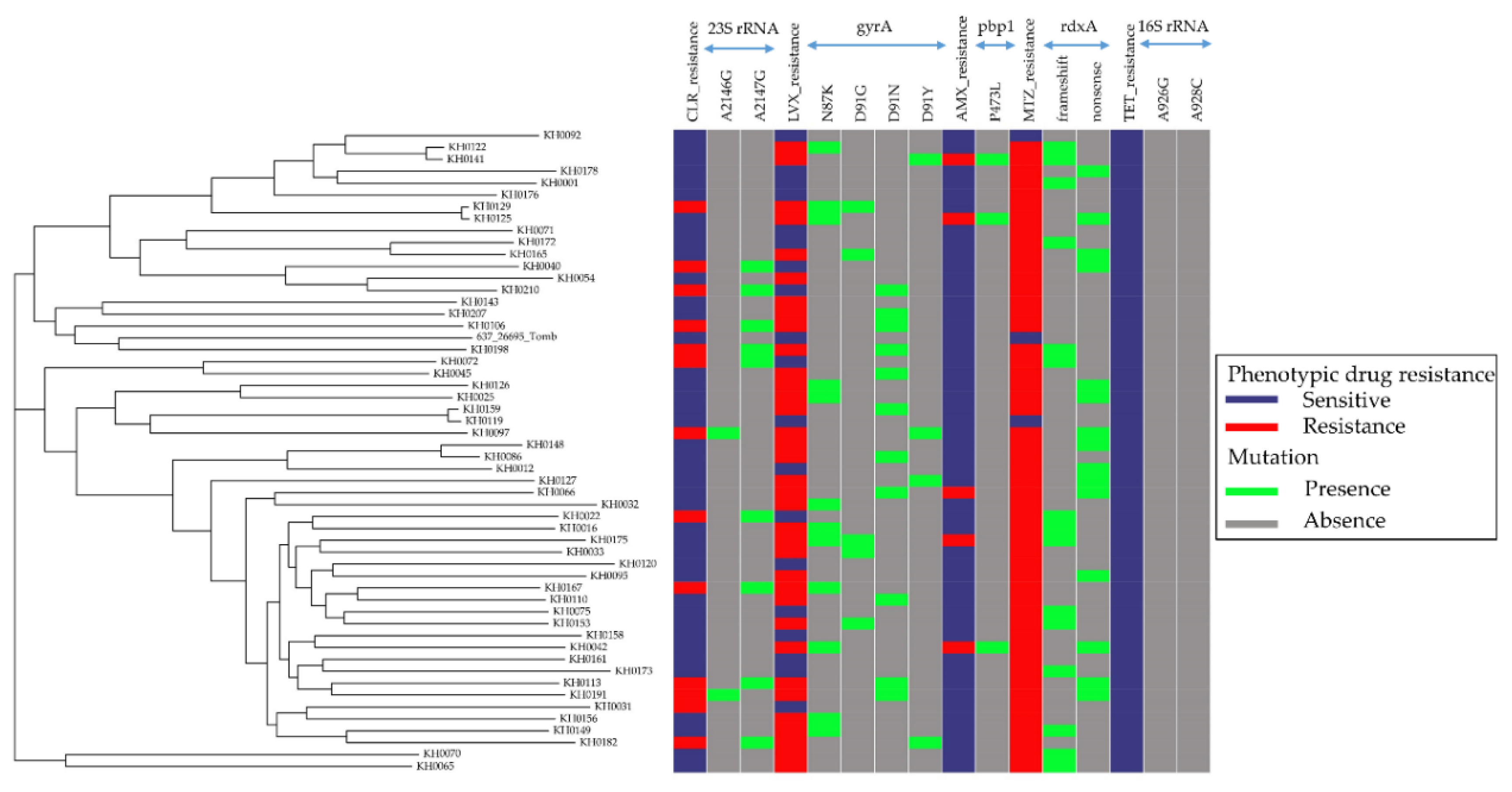
3.2. Relationship between Antibiotic Resistance Patterns and Strain-Relatedness in Cambodian H. pylori Isolates
3.3. Plasmid Detection from Cambodian H. pylori Isolates
3.4. Genetic Determinants of Cambodian H. pylori Antibiotic Resistance
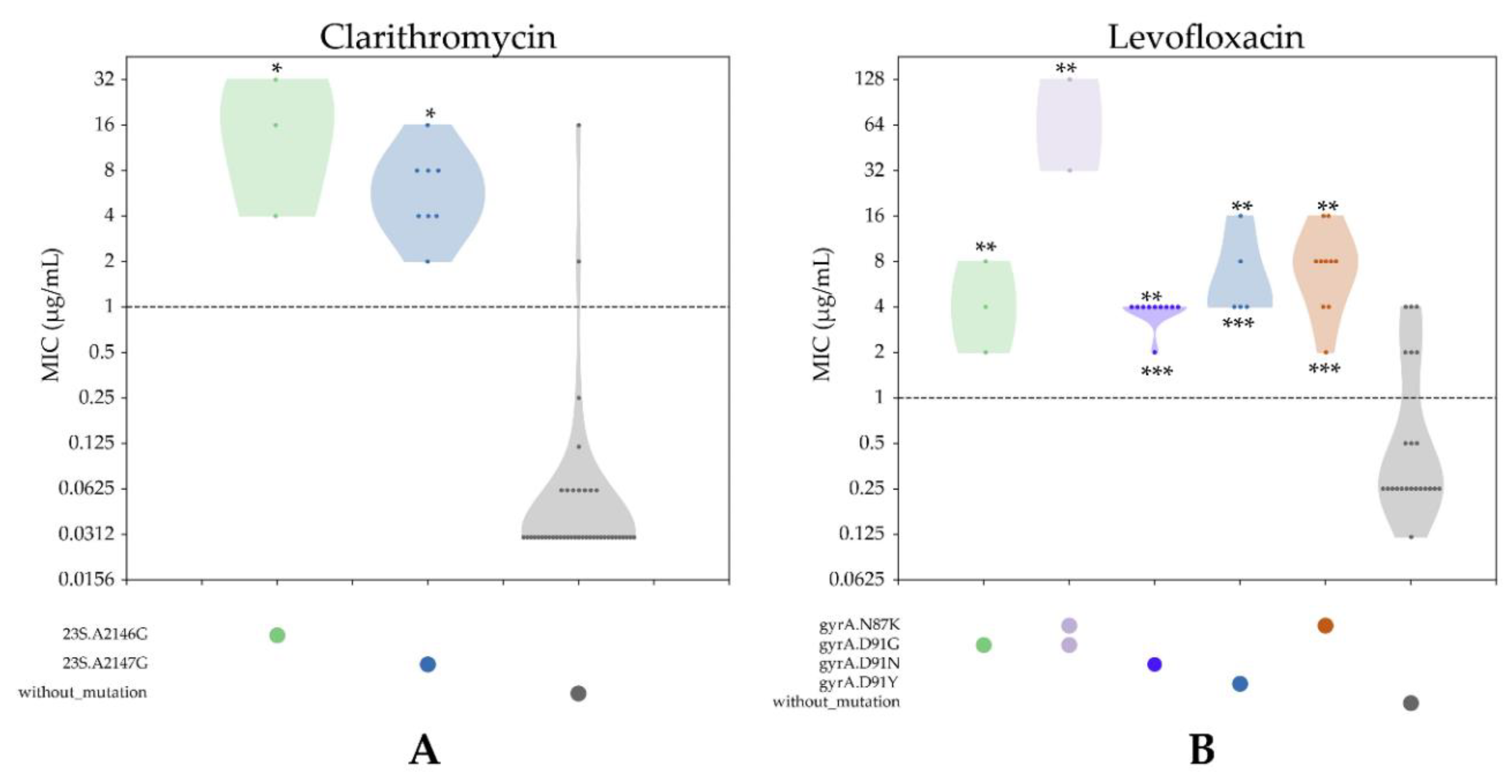
3.4.1. CLR Resistance
3.4.2. LVX Resistance
3.4.3. AMX Resistance
3.4.4. MTZ Resistance
3.4.5. TET Resistance
3.5. Comparison between Antibiotic Susceptibility Genotypes and Phenotypes
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Brogan, D.M.; Mossialos, E. A critical analysis of the review on antimicrobial resistance report and the infectious disease financing facility. Glob. Health 2016, 12, 8. [Google Scholar] [CrossRef] [PubMed]
- Michael, C.A.; Dominey-Howes, D.; Labbate, M. The antimicrobial resistance crisis: Causes, consequences, and management. Front. Public Health 2014, 2, 145. [Google Scholar] [CrossRef] [PubMed]
- Dang, B.N.; Graham, D.Y. Helicobacter pylori infection and antibiotic resistance: A WHO high priority? Nat. Rev. Gastroenterol. Hepatol. 2017, 14, 383–384. [Google Scholar] [CrossRef] [PubMed]
- Tacconelli, E.; Carrara, E.; Savoldi, A.; Harbarth, S.; Mendelson, M.; Monnet, D.L.; Pulcini, C.; Kahlmeter, G.; Kluytmans, J.; Carmeli, Y.; et al. Discovery, research, and development of new antibiotics: The WHO priority list of antibiotic-resistant bacteria and tuberculosis. Lancet Infect. Dis. 2018, 18, 318–327. [Google Scholar] [CrossRef]
- Moss, S.F. The clinical evidence linking Helicobacter pylori to gastric cancer. Cell. Mol. Gastroenterol. Hepatol. 2017, 3, 183–191. [Google Scholar] [CrossRef] [PubMed]
- Malfertheiner, P.; Megraud, F.; O’Morain, C.A.; Atherton, J.; Axon, A.T.; Bazzoli, F.; Gensini, G.F.; Gisbert, J.P.; Graham, D.Y.; Rokkas, T.; et al. Management of Helicobacter pylori infection—The Maastricht IV/Florence Consensus report. Gut 2012, 61, 646–664. [Google Scholar] [CrossRef]
- Fischbach, L.; Evans, E.L. Meta-analysis: The effect of antibiotic resistance status on the efficacy of triple and quadruple first-line therapies for Helicobacter pylori. Aliment. Pharmacol. Ther. 2007, 26, 343–357. [Google Scholar] [CrossRef] [PubMed]
- Fallone, C.A.; Chiba, N.; van Zanten, S.V.; Fischbach, L.; Gisbert, J.P.; Hunt, R.H.; Jones, N.L.; Render, C.; Leontiadis, G.I.; Moayyedi, P.; et al. The Toronto consensus for the treatment of Helicobacter pylori infection in adults. Gastroenterology 2016, 151, 51–69 e14. [Google Scholar] [CrossRef]
- Malfertheiner, P.; Megraud, F.; O’Morain, C.A.; Gisbert, J.P.; Kuipers, E.J.; Axon, A.T.; Bazzoli, F.; Gasbarrini, A.; Atherton, J.; Graham, D.Y.; et al. Management of Helicobacter pylori infection—The Maastricht V/Florence consensus report. Gut 2017, 66, 6–30. [Google Scholar] [CrossRef]
- Fock, K.M.; Katelaris, P.; Sugano, K.; Ang, T.L.; Hunt, R.; Talley, N.J.; Lam, S.K.; Xiao, S.D.; Tan, H.J.; Wu, C.Y.; et al. Second Asia-Pacific Consensus Guidelines for Helicobacter pylori infection. J. Gastroenterol. Hepatol. 2009, 24, 1587–1600. [Google Scholar] [CrossRef]
- Sugano, K.; Tack, J.; Kuipers, E.J.; Graham, D.Y.; El-Omar, E.M.; Miura, S.; Haruma, K.; Asaka, M.; Uemura, N.; Malfertheiner, P.; et al. Kyoto global consensus report on Helicobacter pylori gastritis. Gut 2015, 64, 1353–1367. [Google Scholar] [CrossRef]
- Savoldi, A.; Carrara, E.; Graham, D.Y.; Conti, M.; Tacconelli, E. Prevalence of antibiotic resistance in Helicobacter pylori: A systematic review and meta-analysis in World Health Organization Regions. Gastroenterology 2018, 155, 1372–1382 e1317. [Google Scholar] [CrossRef]
- Mahachai, V.; Vilaichone, R.K.; Pittayanon, R.; Rojborwonwitaya, J.; Leelakusolvong, S.; Maneerattanaporn, M.; Chotivitayatarakorn, P.; Treeprasertsuk, S.; Kositchaiwat, C.; Pisespongsa, P.; et al. Helicobacter pylori management in ASEAN: The Bangkok consensus report. J. Gastroenterol. Hepatol. 2018, 33, 37–56. [Google Scholar] [CrossRef] [PubMed]
- Kuo, Y.T.; Liou, J.M.; El-Omar, E.M.; Wu, J.Y.; Leow, A.H.R.; Goh, K.L.; Das, R.; Lu, H.; Lin, J.T.; Tu, Y.K.; et al. Primary antibiotic resistance in Helicobacter pylori in the Asia-Pacific region: A systematic review and meta-analysis. Lancet Gastroenterol. Hepatol. 2017, 2, 707–715. [Google Scholar] [CrossRef]
- Nishizawa, T.; Suzuki, H. Mechanisms of Helicobacter pylori antibiotic resistance and molecular testing. Front. Mol. Biosci. 2014, 1, 19. [Google Scholar] [CrossRef] [PubMed]
- Anjum, M.F.; Zankari, E.; Hasman, H. Molecular methods for detection of antimicrobial resistance. Microbiol. Spectr. 2017, 5. [Google Scholar] [CrossRef] [PubMed]
- Jiang, F.; Wu, Z.; Zheng, Y.; Frana, T.S.; Sahin, O.; Zhang, Q.; Li, G. Genotypes and antimicrobial susceptibility profiles of hemolytic Escherichia coli from diarrheic piglets. Foodborne Pathog. Dis. 2019, 16, 94–103. [Google Scholar] [CrossRef] [PubMed]
- Ellington, M.J.; Ekelund, O.; Aarestrup, F.M.; Canton, R.; Doumith, M.; Giske, C.; Grundman, H.; Hasman, H.; Holden, M.T.G.; Hopkins, K.L.; et al. The role of whole genome sequencing in antimicrobial susceptibility testing of bacteria: Report from the EUCAST Subcommittee. Clin. Microbiol. Infect. 2017, 23, 2–22. [Google Scholar] [CrossRef] [PubMed]
- Pankhurst, L.J.; Del Ojo Elias, C.; Votintseva, A.A.; Walker, T.M.; Cole, K.; Davies, J.; Fermont, J.M.; Gascoyne-Binzi, D.M.; Kohl, T.A.; Kong, C.; et al. Rapid, comprehensive, and affordable mycobacterial diagnosis with whole-genome sequencing: A prospective study. Lancet Respir. Med. 2016, 4, 49–58. [Google Scholar] [CrossRef]
- Gupta, S.K.; Padmanabhan, B.R.; Diene, S.M.; Lopez-Rojas, R.; Kempf, M.; Landraud, L.; Rolain, J.M. ARG-ANNOT, a new bioinformatic tool to discover antibiotic resistance genes in bacterial genomes. Antimicrob. Agents Chemother. 2014, 58, 212–220. [Google Scholar] [CrossRef] [PubMed]
- McArthur, A.G.; Waglechner, N.; Nizam, F.; Yan, A.; Azad, M.A.; Baylay, A.J.; Bhullar, K.; Canova, M.J.; De Pascale, G.; Ejim, L.; et al. The comprehensive antibiotic resistance database. Antimicrob. Agents Chemother. 2013, 57, 3348–3357. [Google Scholar] [CrossRef] [PubMed]
- Lakin, S.M.; Dean, C.; Noyes, N.R.; Dettenwanger, A.; Ross, A.S.; Doster, E.; Rovira, P.; Abdo, Z.; Jones, K.L.; Ruiz, J.; et al. MEGARes: An antimicrobial resistance database for high throughput sequencing. Nucleic Acids Res. 2017, 45, D574–D580. [Google Scholar] [CrossRef] [PubMed]
- Zankari, E.; Hasman, H.; Cosentino, S.; Vestergaard, M.; Rasmussen, S.; Lund, O.; Aarestrup, F.M.; Larsen, M.V. Identification of acquired antimicrobial resistance genes. J. Antimicrob. Chemother. 2012, 67, 2640–2644. [Google Scholar] [CrossRef] [PubMed]
- Yamaoka, Y.; Kodama, T.; Kita, M.; Imanishi, J.; Kashima, K.; Graham, D.Y. Relationship of vacA genotypes of Helicobacter pylori to cagA status, cytotoxin production, and clinical outcome. Helicobacter 1998, 3, 241–253. [Google Scholar] [CrossRef]
- National Committee for Clinical Laboratory Standards. Performance Standards for Antimicrobial Susceptibility Testing; Approved Standard M7-A5 and Informational Supplement M100-S10; NCCLS: Wayne, PA, USA, 2000. [Google Scholar]
- Roosaare, M.; Puustusmaa, M.; Mols, M.; Vaher, M.; Remm, M. PlasmidSeeker: Identification of known plasmids from bacterial whole genome sequencing reads. PeerJ 2018, 6, e4588. [Google Scholar] [CrossRef] [PubMed]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed]
- Bankevich, A.; Nurk, S.; Antipov, D.; Gurevich, A.A.; Dvorkin, M.; Kulikov, A.S.; Lesin, V.M.; Nikolenko, S.I.; Pham, S.; Prjibelski, A.D.; et al. SPAdes: A new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 2012, 19, 455–477. [Google Scholar] [CrossRef] [PubMed]
- Gurevich, A.; Saveliev, V.; Vyahhi, N.; Tesler, G. QUAST: Quality assessment tool for genome assemblies. Bioinformatics 2013, 29, 1072–1075. [Google Scholar] [CrossRef] [PubMed]
- Seemann, T. Snippy: Fast Bacterial Variant Calling from NGS Reads. 2015. Available online: https://github.com/tseemann/snippy (accessed on 1 January 2018).
- Croucher, N.J.; Page, A.J.; Connor, T.R.; Delaney, A.J.; Keane, J.A.; Bentley, S.D.; Parkhill, J.; Harris, S.R. Rapid phylogenetic analysis of large samples of recombinant bacterial whole genome sequences using Gubbins. Nucleic Acids Res. 2015, 43, e15. [Google Scholar] [CrossRef]
- Stamatakis, A. RAxML version 8: A tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 2014, 30, 1312–1313. [Google Scholar] [CrossRef]
- Ogino, S.; Gulley, M.L.; den Dunnen, J.T.; Wilson, R.B.; The Association for Molecular Pathology Training; Education Committee Association for Molecular Pathology. Standard mutation nomenclature in molecular diagnostics: Practical and educational challenges. J. Mol. Diagn. JMD 2007, 9, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Hadfield, J.; Croucher, N.J.; Goater, R.J.; Abudahab, K.; Aanensen, D.M.; Harris, S.R. Phandango: An interactive viewer for bacterial population genomics. Bioinformatics 2017, 34, 292–293. [Google Scholar] [CrossRef] [PubMed]
- Signorell, A. DescTools: Tools for Descriptive Statistics. R Package Version 0.99. Available online: https://www.r-pkg.org/pkg/DescTools (accessed on 14 March 2019).
- Versalovic, J.; Shortridge, D.; Kibler, K.; Griffy, M.V.; Beyer, J.; Flamm, R.K.; Tanaka, S.K.; Graham, D.Y.; Go, M.F. Mutations in 23S rRNA are associated with clarithromycin resistance in Helicobacter pylori. Antimicrob. Agents Chemother. 1996, 40, 477–480. [Google Scholar] [CrossRef] [PubMed]
- Kwon, Y.H.; Kim, J.Y.; Kim, N.; Park, J.H.; Nam, R.H.; Lee, S.M.; Kim, J.W.; Kim, J.M.; Park, J.Y.; Lee, D.H. Specific mutations of penicillin-binding protein 1A in 77 clinically acquired amoxicillin-resistant Helicobacter pylori strains in comparison with 77 amoxicillin-susceptible strains. Helicobacter 2017, 22. [Google Scholar] [CrossRef] [PubMed]
- Paul, R.; Postius, S.; Melchers, K.; Schafer, K.P. Mutations of the Helicobacter pylori genes rdxA and pbp1 cause resistance against metronidazole and amoxicillin. Antimicrob. Agents Chemother. 2001, 45, 962–965. [Google Scholar] [CrossRef] [PubMed]
- Jenks, P.J.; Ferrero, R.L.; Labigne, A. The role of the rdxA gene in the evolution of metronidazole resistance in Helicobacter pylori. J. Antimicrob. Chemother. 1999, 43, 753–758. [Google Scholar] [CrossRef] [PubMed]
- Gerrits, M.M.; de Zoete, M.R.; Arents, N.L.; Kuipers, E.J.; Kusters, J.G. 16S rRNA mutation-mediated tetracycline resistance in Helicobacter pylori. Antimicrob. Agents Chemother. 2002, 46, 2996–3000. [Google Scholar] [CrossRef] [PubMed]
- Ang, T.L.; Fock, K.M.; Ang, D.; Kwek, A.B.; Teo, E.K.; Dhamodaran, S. The changing profile of Helicobacter pylori antibiotic resistance in Singapore: A 15-year study. Helicobacter 2016, 21, 261–265. [Google Scholar] [CrossRef] [PubMed]
- Vilaichone, R.K.; Quach, D.T.; Yamaoka, Y.; Sugano, K.; Mahachai, V. Prevalence and pattern of antibiotic resistant strains of Helicobacter pylori infection in ASEAN. Asian Pac. J. Cancer Prev. APJCP 2018, 19, 1411–1413. [Google Scholar] [CrossRef]
- De Francesco, V.; Zullo, A.; Fiorini, G.; Saracino, I.M.; Pavoni, M.; Vaira, D. Role of MIC levels of resistance to clarithromycin and metronidazole in Helicobacter pylori eradication. J. Antimicrob. Chemother. 2019, 74, 772–774. [Google Scholar] [CrossRef] [PubMed]
- Om, C.; Daily, F.; Vlieghe, E.; McLaughlin, J.C.; McLaws, M.L. Pervasive antibiotic misuse in the Cambodian community: Antibiotic-seeking behaviour with unrestricted access. Antimicrob. Resist. Infect. Control 2017, 6, 30. [Google Scholar] [CrossRef] [PubMed]
- Om, C.; Daily, F.; Vlieghe, E.; McLaughlin, J.C.; McLaws, M.L. “If it’s a broad spectrum, it can shoot better”: Inappropriate antibiotic prescribing in Cambodia. Antimicrob. Resist. Infect. Control 2016, 5, 58. [Google Scholar] [CrossRef] [PubMed]
- Munita, J.M.; Arias, C.A. Mechanisms of antibiotic resistance. Microbiol. Spectr. 2016, 4. [Google Scholar] [CrossRef]
- Gerrits, M.M.; van Vliet, A.H.; Kuipers, E.J.; Kusters, J.G. Helicobacter pylori and antimicrobial resistance: Molecular mechanisms and clinical implications. Lancet Infect. Dis. 2006, 6, 699–709. [Google Scholar] [CrossRef]
- Matta, A.J.; Zambrano, D.C.; Pazos, A.J. Punctual mutations in 23S rRNA gene of clarithromycin-resistant Helicobacter pylori in Colombian populations. World J. Gastroenterol. 2018, 24, 1531–1539. [Google Scholar] [CrossRef] [PubMed]
- Melnyk, A.H.; Wong, A.; Kassen, R. The fitness costs of antibiotic resistance mutations. Evol. Appl. 2015, 8, 273–283. [Google Scholar] [CrossRef] [PubMed]
- Cambau, E.; Allerheiligen, V.; Coulon, C.; Corbel, C.; Lascols, C.; Deforges, L.; Soussy, C.J.; Delchier, J.C.; Megraud, F. Evaluation of a new test, genotype HelicoDR, for molecular detection of antibiotic resistance in Helicobacter pylori. J. Clin. Microbiol. 2009, 47, 3600–3607. [Google Scholar] [CrossRef] [PubMed]
- Taylor, D.E.; Ge, Z.; Purych, D.; Lo, T.; Hiratsuka, K. Cloning and sequence analysis of two copies of a 23S rRNA gene from Helicobacter pylori and association of clarithromycin resistance with 23S rRNA mutations. Antimicrob. Agents Chemother. 1997, 41, 2621–2628. [Google Scholar] [CrossRef]
- Pernodet, J.L.; Boccard, F.; Alegre, M.T.; Blondelet-Rouault, M.H.; Guerineau, M. Resistance to macrolides, lincosamides and streptogramin type B antibiotics due to a mutation in an rRNA operon of Streptomyces ambofaciens. EMBO J. 1988, 7, 277–282. [Google Scholar] [CrossRef]
- Kageyama, C.; Sato, M.; Sakae, H.; Obayashi, Y.; Kawahara, Y.; Mima, T.; Matsushita, O.; Yokota, K.; Mizuno, M.; Okada, H. Increase in antibiotic resistant Helicobacter pylori in a University Hospital in Japan. Infect. Drug Resist. 2019, 12, 597–602. [Google Scholar] [CrossRef]
- Gerrits, M.M.; Godoy, A.P.; Kuipers, E.J.; Ribeiro, M.L.; Stoof, J.; Mendonca, S.; van Vliet, A.H.; Pedrazzoli, J., Jr.; Kusters, J.G. Multiple mutations in or adjacent to the conserved penicillin-binding protein motifs of the penicillin-binding protein 1A confer amoxicillin resistance to Helicobacter pylori. Helicobacter 2006, 11, 181–187. [Google Scholar] [CrossRef] [PubMed]
- Chua, E.G.; Debowski, A.W.; Webberley, K.M.; Peters, F.; Lamichhane, B.; Loke, M.F.; Vadivelu, J.; Tay, C.Y.; Marshall, B.J.; Wise, M.J. Analysis of core protein clusters identifies candidate variable sites conferring metronidazole resistance in Helicobacter pylori. Gastroenterol. Rep. 2019, 7, 42–49. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, M.; Shibayama, K.; Yahara, K. A genome-wide association study identifies a horizontally transferred bacterial surface adhesin gene associated with antimicrobial resistant strains. Sci. Rep. 2016, 6, 37811. [Google Scholar] [CrossRef] [PubMed] [Green Version]




{kind=link}
{kind=link}
{kind=link}
| Resistance Pattern | Number of Strains | Percentage % (95%CI) |
|---|---|---|
| Susceptible to all | 2 | 3.6 (0.4–12.5) |
| All resistance | 53 | 96.4 (87.5–99.6) |
| CLR | 14 | 25.5 (14.7–39) |
| LVX | 37 | 67.3 (53.3–79.3) |
| AMX | 5 | 9.1 (3–20) |
| MTZ | 53 | 96.4 (87.5–99.6) |
| TET | 0 | 0 (0–6.5) |
| Mono Resistance | 12 | 21.8 (11.8–35) |
| CLR only | 0 | 0 (0–6.5) |
| LVX only | 0 | 0 (0–6.5) |
| AMX only | 0 | 0 (0–6.5) |
| MTZ only | 12 | 21.8 (11.8–35) |
| TET only | 0 | 0 (0–6.5) |
| Multiple resistance | 42 | 76.4 (62.9–86.8) |
| LVX + MTZ | 22 | 40 (27–54.1) |
| CLR + MTZ | 4 | 7.3 (2–17.6) |
| AMX + LVX + MTZ | 5 | 9.1 (3–20) |
| CLR + LVX + MTZ | 10 | 18.2 (9.1–30.9) |
| Antibiotic | Susceptible Phenotype | Resistant Phenotype | Agreement | Kappa Coefficiency | p-Value | ||
|---|---|---|---|---|---|---|---|
| Resistant Genotype | Susceptible Genotype | Resistant Genotype | Susceptible Genotype | ||||
| AMX | 0 | 48 | 3 | 2 | 96.20% | 0.73 (95% CI, 0.47–0.99) | 3.29 × 10−8 |
| MTZ | 0 | 2 | 30 | 21 | 60.40% | 0.1 (95% CI, 0.02–0.21) | 0.1 |
| LVX | 0 | 17 | 29 | 7 | 86.80% | 0.73 (95% CI, 0.47–0.99) | 3.81 × 10−8 |
| CLR | 0 | 40 | 11 | 2 | 96.20% | 0.89 (95% CI, 0.62–1.00) | 6.35 × 10−11 |
| TET | 0 | 53 | 0 | 0 | - | - | - |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tuan, V.P.; Narith, D.; Tshibangu-Kabamba, E.; Dung, H.D.Q.; Viet, P.T.; Sokomoth, S.; Binh, T.T.; Sokhem, S.; Tri, T.D.; Ngov, S.; et al. A Next-Generation Sequencing-Based Approach to Identify Genetic Determinants of Antibiotic Resistance in Cambodian Helicobacter pylori Clinical Isolates. J. Clin. Med. 2019, 8, 858. https://0-doi-org.brum.beds.ac.uk/10.3390/jcm8060858
Tuan VP, Narith D, Tshibangu-Kabamba E, Dung HDQ, Viet PT, Sokomoth S, Binh TT, Sokhem S, Tri TD, Ngov S, et al. A Next-Generation Sequencing-Based Approach to Identify Genetic Determinants of Antibiotic Resistance in Cambodian Helicobacter pylori Clinical Isolates. Journal of Clinical Medicine. 2019; 8(6):858. https://0-doi-org.brum.beds.ac.uk/10.3390/jcm8060858
Chicago/Turabian StyleTuan, Vo Phuoc, Dou Narith, Evariste Tshibangu-Kabamba, Ho Dang Quy Dung, Pham Thanh Viet, Sin Sokomoth, Tran Thanh Binh, Sok Sokhem, Tran Dinh Tri, Seng Ngov, and et al. 2019. "A Next-Generation Sequencing-Based Approach to Identify Genetic Determinants of Antibiotic Resistance in Cambodian Helicobacter pylori Clinical Isolates" Journal of Clinical Medicine 8, no. 6: 858. https://0-doi-org.brum.beds.ac.uk/10.3390/jcm8060858