Strategies to Better Target Fungal Squalene Monooxygenase

Department of Biochemistry, Albert Einstein College of Medicine, Bronx, NY 10461, USA
J. Fungi 2021, 7(1), 49; https://0-doi-org.brum.beds.ac.uk/10.3390/jof7010049
Submission received: 12 December 2020
/
Revised: 4 January 2021
/
Accepted: 5 January 2021
/
Published: 13 January 2021
(This article belongs to the Special Issue The Application of Structural Biology in Antifungal Drug Discovery)
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Fungal pathogens present a challenge in medicine and agriculture. They also harm ecosystems and threaten biodiversity. The allylamine class of antimycotics targets the enzyme squalene monooxygenase. This enzyme occupies a key position in the sterol biosynthesis pathway in eukaryotes, catalyzing the rate-limiting reaction by introducing an oxygen atom to the squalene substrate converting it to 2,3-oxidosqualene. Currently, terbinafine—the most widely used allylamine—is mostly used for treating superficial fungal infections. The ability to better target this enzyme will have significant implications for human health in the treatment of fungal infections. The human orthologue can also be targeted for cholesterol-lowering therapeutics and in cancer therapies. This review will focus on the structural basis for improving the current therapeutics for fungal squalene monooxygenase.
1. Fungi—A Growing Problem
The fungal kingdom is incredibly large and diverse [1] with fungi occupying important roles in our environment, ecosystems, and our daily lives [1]. Amongst diversity, the odds are that some fungal organisms would be pathogenic to animals and plants. Indeed, several hundred fungal species are pathogens. In healthy people, pathogenic fungi can cause superficial infections. However, those with weakened immune systems are at a far greater risk of both superficial and invasive infections. In 2017, it was estimated that fungal infections cost $7.2 billion to the healthcare system in the United States, with $4.5 billion accounting for hospitalizations with the rest covering outpatient visits [2]. The same study showed that candidiasis and aspergillosis accounted for a large proportion of the hospitalization costs, whereas the intended purpose of over half of the outpatient visits was for treatment for dermatophyte infections of the skin, hair, and nails with an estimated total cost of $802 million [2].
In agriculture, major crops such as wheat, rice, maize, potatoes, and soybeans are susceptible to fungal phytopathogens. Millions of tons of crops are destroyed by fungal phytopathogens every year. Fisher et al. estimate that the crops lost to fungi could feed ~8.5% of the world’s population for a year [3]. Crop loss due to fungi is exacerbated by the use of monocultures; one prominent example of this is the Cavendish banana, which is susceptible to the Fusarium fungi. These fungi have been demolishing banana plantations in Asia for a long time and just last year, it was reported to spread to Latin America, threatening the global banana supply and driving up banana prices. Such threats to the global food security have resulted in increasing usage of antifungal pesticides [4]. Heavy use of pesticides comes at a cost—some fungi can develop resistance and, in the case of Aspergillus fumigatus, cross-resistance to medical antifungal drugs has been reported [5]. In addition, there are concerns over the impact of azole pesticides on the endocrine system [6].
There is also an emergence of infectious diseases caused by fungi in natural habitats that devastate plants and animals, damage ecosystems, and threaten biodiversity [3]. In 2006, a new disease caused mass mortalities in a population of bats in North America. The fungal pathogen Geomyces destructans was identified to be responsible for this disease, termed the “white-nose syndrome” as bats had fungi growing on their snouts and wings during hibernation [7]. This disease caused a 70% decline in the affected bat populations [8]. Ecosystems in parts of Central America were reshaped by the loss of about 40% of the amphibian population due to a skin-infecting fungus Batrachochytrium dendrobatidis [9]. This pathogen was originally identified in the 1970s in southern Mexico. It is now found on all continents with amphibian populations [10].
Climate change is one of the drivers impacting the dynamics and distribution of fungal organisms. One recent example of this is the newly emergent drug-resistant pathogen Candida auris, an organism that was not recorded to have infected humans until 2009 [11]. Most fungi grow better at low temperatures, for example, B. dendrobatidis does not grow well above 25 °C. Thus, it easily infects cold-blooded amphibians. Casadevall et al. argue that C. auris was driven by climate change to grow better at higher temperatures and, as a result of crossing this “temperature barrier,” became pathogenic for humans [12]. Given the size of the fungal kingdom and the growing impact of climate change, the probability of such an event occurring again is high.
Fungi pose a threat to human existence, which is likely to get worse in the coming years. Thus, treatment of fungal infections will play an even more crucial role in medicine, the agricultural industry, and biodiversity preservation as time goes on. In the following sections, the antifungal drugs will be discussed with particular focus on the allylamine antifungals and the enzyme they target, squalene monooxygenase (Erg1, SM—an abbreviation for the mammalian homolog). In 2019, the structure of the human homolog of this enzyme was determined by X-ray crystallography. This provided homology modeling opportunities that would allow for the design of better inhibitors for fungal enzymes [13].
2. Blocking Sterol Biosynthesis
To combat fungal infections, the two classes of antifungals that target the mevalonate pathway to block fungal sterol/ergosterol production are the azole drugs (e.g., fluconazole, posaconazole) and the allylamines (e.g., terbinafine and naftifine; Figure 1). Ergosterol depletion changes the composition of the lipid bilayer altering the fluidity of the cell membrane and inhibits fungal growth [14]. This inhibition also allows for the accumulation of toxic ergosterol precursors in the fungal cell. The azoles target the fungal enzyme lanosterol 14 α-demethylase and have been very successful in the clinical and agricultural settings. However, there are still challenges to overcome such as toxicity, antifungal drug resistance, and the emergence of cross-resistance between agricultural and medical azoles [15]. Still, advances are being made, and the latest generation of azoles, the tetrazoles, are currently in clinical trials. VT-1161 is currently in phase III clinical trials for the treatment of recurrent vulvovaginal candidiasis [16,17]. VT-1598 is in phase I trials for the treatment of C. auris infections [18]. This progress with the development of the azole drugs has partially been driven by the need for novel antifungals in the medicine and agriculture and partially by extensive functional and structural characterization of lanosterol 14 α-demethylase from Saccharomyces cerevisiae [19,20], the pathogenic yeast Candida albicans [21], and others covered in a comprehensive review by Monk et al. [15].
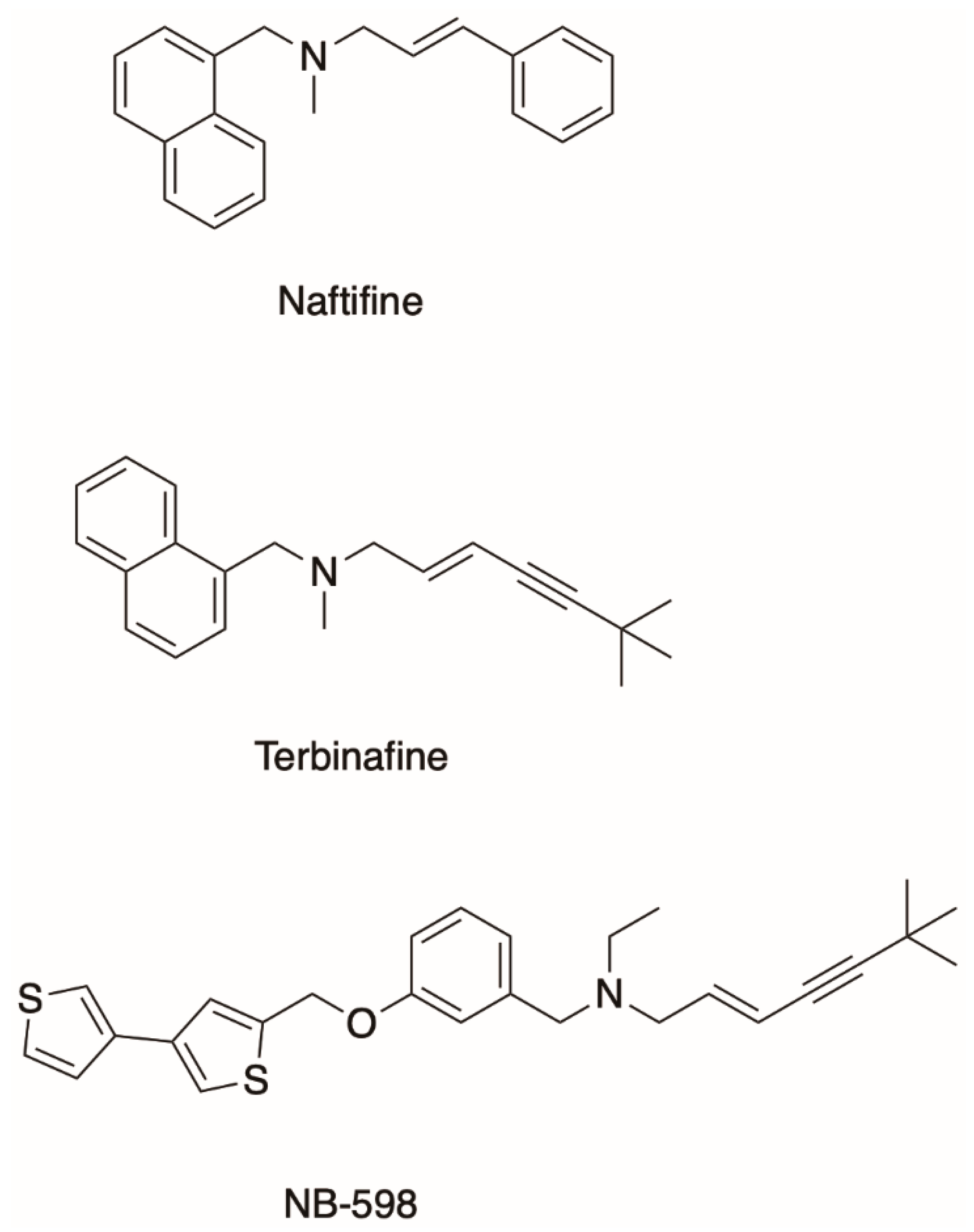
On the other hand, the allylamine class of antifungals targets Erg1, a well-established but underdeveloped drug target [22]. SM is an endoplasmic reticulum-associated enzyme that catalyzes a rate-limiting step of cholesterol biosynthesis in mammals and ergosterol biosynthesis in fungi by converting squalene to 2,3-oxidosqualene (Figure 2). It is classified as a class E monooxygenase as it uses the flavin adenosine dinucleotide (FAD) as a cofactor, to receive electrons from NADPH-cytochrome P450 reductase (P450R) to carry out the epoxidation and the reduction of molecular oxygen to water [23]. There are only a handful of allylamine drugs, including terbinafine, naftifine, and butenafine. Terbinafine was derived from its predecessor naftifine by a substitution of a phenyl ring to a tert-butylacetylene group (Figure 1), improving the activity of the drug several-fold [24,25]. Allylamines have been successfully used in the treatment of superficial fungal infections such as tinea pedis (athlete’s foot), tinea capitis (scalp and hair), and onychomycosis (nail infection). These infections are very common. It is estimated that 20–25% of the global population have superficial mycoses [26]. These are generally caused by filamentous fungi that utilize keratin for growth, such as Trichophyton spp., Microsporum spp., and Epidermophyton spp.
3. Terbinafine
In general, dermatophytes are more susceptible to Erg1 inhibition than other fungi. This is partially due to the lipophilic nature of terbinafine which tends to localize at the sites of infection by dermatophytes such as skin, hair, and nails [25]. These fungi are also less tolerant to the accumulation of squalene [27]. In vitro studies show that terbinafine minimum inhibitory concentration (MIC) is higher for yeasts such as C. albicans and molds such as Aspergillus fumigatus [24,27]. Terbinafine can be effectively used in combination with other antifungals, but in the clinical setting, this must be done in a controlled manner as unmonitored use has the potential to select for multidrug-resistant fungal strains. A combination of the triazoles itraconazole or voriconazole with terbinafine has a synergistic effect against A. fumigatus, Aspergillus flavus, and Aspergillus niger [28]. Synergistic effects were observed in terbinafine/azole drug combinations for Candida glabrata strains with reduced azole susceptibility [29].
Although terbinafine is not used systemically as often as it is used topically, it is a well-tolerated drug. The absorption of the orally administered terbinafine hydrochloride is over 70% and it is mainly localized to the adipose tissue, skin, and nails [30,31]. The elimination of terbinafine from nails and skin takes a long time with it still detected in nails 30 weeks after cessation of treatment [32]. Approximately 70% of terbinafine is excreted by the kidneys into urine and the rest via the feces [31]. Terbinafine is subject to metabolism by the cytochrome P450 liver enzymes and one of the main metabolites found in blood plasma is the N-demethylated derivative of terbinafine [33]. Drug–drug interactions are important to consider in the oral administration of terbinafine as it has been found to inhibit CYP2D6 [34]. This liver enzyme metabolizes about 25% of drugs [35]. However, terbinafine was not found to inhibit CYP3A4, an important enzyme that is responsible for metabolizing ~50% of drugs [36]. Some azoles, on the other hand, can have stronger interactions with liver enzymes, for example, ketoconazole is a potent inhibitor of CYP3A4 [37].
Terbinafine has a much lower binding affinity for the human SM than for Erg1 [27,38]. On the contrary, NB-598 is a very potent inhibitor of the human SM but not of Erg1 (Figure 1) [13]. SM has been proposed as a target for cholesterol-lowering medications with the potential for fewer side effects than the current gold standard for treatment of hypercholesterolemia statins which inhibit HMG-CoA reductase. Fewer side effects would result from targeting the mevalonate pathway at a later step after the formation of isoprenoids required for the biosynthesis of dolichol and prenylated proteins. There is also a growing body of work that links SM to cancer [39]. One of the first cancer types to have links to SM was hepatocellular carcinoma, where SM was found to be overexpressed [40]. More recently, NB-598 was found to halt neuroendocrine cancer cell replication via inhibition of SM, due to a toxic effect of squalene accumulation [41]. In addition to the synthetic SM inhibitors such as NB-598 and compound-4, there are some natural ones like garlic extract compounds and resveratrol found in wine. There are no SM inhibitors currently in clinical use. For a comprehensive review of mammalian SM and its relevance to human health please refer to the review by Chua et al. [42].
The good pharmacokinetic profile of terbinafine makes it attractive for use outside of medicine. For example, the efficacy of terbinafine as a treatment for Batrachochytrium dendrobatidis infections in the amphibians was tested on alpine tree frogs. Unfortunately, terbinafine treatments did not cure the mycoses in these frogs [43]. Efforts into preserving the little brown bat population have been made by determining safe dosages of terbinafine [44]. Another study developed a subcutaneous implant with terbinafine and monitored its release in a saline solution. Although the terbinafine was released over 28 weeks at therapeutic concentrations, this was not repeated in an animal study [45].
4. Squalene Monooxygenase—Fine-Tuning Sterol Homeostasis
The sterol biosynthesis pathway is highly regulated. Several mechanisms are in place to prevent the accumulation of free sterols, which are toxic to the cells. Transcriptional regulation as well as regulation through the rate-limiting enzyme HMG-CoA reductase are key mechanisms for controlling the production of sterols [46,47]. SM plays a role in this process, fine-tuning sterol homeostasis in both fungi and mammals. Accumulation of the end product or one of the intermediates in the pathway results in SM degradation via the endoplasmic reticulum-associated protein degradation (ERAD) pathway.
In fungi, the Erg1 degradation was found to be regulated by lanosterol accumulation. S. cerevisiae cells treated with fluconazole, an inhibitor of lanosterol 14 α-demethylase, resulted in the accumulation of lanosterol and rapid degradation of Erg1 [48]. Really interesting new gene (RING) finger-type ubiquitin ligase Dao10 (E3 enzyme) targets Erg1 for proteasomal degradation [48]. Dao10 was reported to ubiquitinate the K311 residue of Erg1, this residue is located in the catalytic domain of the enzyme. It is unclear if there are other ubiquitination sites for Doa10 in Erg1. However, there is precedent to suggest that there may be ubiquitination sites other than lysines for Doa10p [49,50]. Doa10 requires the ubiquitin-conjugating enzymes (E2 enzymes) Ubc6p and Ubc7, with Ubc6 attaching the initial ubiquitin molecule to Doa10 substrates and Ubc7 carrying out polyubiquitination. Ubc6 was found to attach ubiquitin not only to lysine residues, but also to amino acids with hydroxyl groups (serine and threonine) thereby expanding ubiquitination sites for Doa10 [49,50].
The mammalian SM is targeted for degradation by ERAD, but unlike the fungal enzymes, it was shown to be sensitive to the accumulation of cholesterol and not lanosterol. SM has an N-terminal extension of 100 residues termed the N100 degron, which is absent in fungi. A 12-amino acid (Q62-L73) amphipathic helix was identified in this N100 degron domain, which is hypothesized to unfold and flip out of the lipid bilayer [51]. This exposes the loop for ubiquitination by the mammalian equivalent of Doa10, membrane-associated RING-CH-type finger 6 (MARCH6), an ER-localized E3 ubiquitin ligase [52]. Interestingly, four serine residues were found to be ubiquitinated by MARCH6, thus providing more evidence that Doa10 may ubiquitinate other residues in addition to the lysine residues in Erg1 [51].
5. Squalene Monooxygenase Expression and Purification
It has been over 20 years since C. albicans Erg1 was expressed in S. cerevisiae by Ryder et al. [53]. Similarly, human SM was also expressed in Escherichia coli and affinity-purified in 1999 [54]. The first experimentally determined crystal structure was released almost 20 years later [13]. This paucity of structural information is largely attributed to the fact that membrane proteins are notoriously difficult to purify and crystalize. This is reflected in the number of membrane protein structures deposited in the protein data bank (PDB) compared to the soluble proteins. Currently, the total number of entries in the PDB exceeds 170,000, and there are about 4300 entries for membrane proteins and of these only about 1200 are unique structures (mpstruc, https://blanco.biomol.uci.edu/mpstruc/). Often, the transmembrane domains of the membrane protein get truncated for ease of expression and purification. The choice of the detergent for solubilization of membrane proteins from the lipid bilayer in a monodisperse manner presents a challenge. Originally, SM was solubilized from rat liver microsomes using Triton-X100, which also acted instead of the supernatant protein factor needed to carry out squalene epoxidation [55]. It was subsequently discovered that supernatant protein factor contains a Sec14-like lipid-binding domain which can bind both squalene and 2,3-oxidosqualene. This was confirmed in X-ray crystal structures and this supernatant protein is now thought to ensure substrate flow to SM [56]. The fungal enzyme, on the contrary, is weakly stimulated by the soluble fraction and is inhibited by Triton-X100 [25,57].
Efforts have been made to hyperexpress full-length ScErg1, CaErg1, and human SM in the laboratory of Dr Brian Monk (Sagatova et al., unpublished). These constructs were successfully expressed in a well-established S. cerevisiae expression system designed for overexpression of membrane proteins in a hypersensitive host [58,59]. The successful hyperexpression was determined using Western blot in addition to the MIC susceptibility assays to terbinafine. However, membrane solubilization using a detergent that allows for the protein to be successfully purified and subsequently crystallized proved challenging. A non-ionic detergent n-octyl-β-d-glucoside solubilized ScErg1 as seen by size exclusion chromatography. Unfortunately, subsequent crystallization attempts were not successful.
For the current X-ray crystal structure of human SM, N-terminally truncated enzyme (118–574) was used for the crystallographic work, purified from E. coli and solubilized using the detergent 3-[(3-cholamidopropyl)dimethylammonio]-1-propanesulfonate (CHAPS) which had been determined to have the most stabilizing effect on SM using a thermal shift assay [13]. The authors confirmed the functionality of the truncated enzyme using a liquid chromatography–mass spectrometry method that they developed to directly detect the product 2,3-oxidosqualene. The conventional assay of squalene monooxygenase activity involves incubation of total cell extracts with a radioactively labeled substrate [11,25,57]. The LC–MS method is superior to the previously used method of thin-layer chromatography in terms of time, throughput, and accuracy. The LC–MS assay can be further utilized in developing the next-generation inhibitors of SM.
In addition to detergent screening, to obtain protein crystals of the fungal enzyme, one might consider the following strategies. The Erg1s of the yeasts S. cerevisiae and C. albicans have a longer loop between β-strands 6 and 7 (residues 109–139, S. cerevisiae numbering). This motif is much shorter in the human enzyme (residues 210–220), the dermatophytes, and the mold A. fumigatus. This short loop in the human enzyme facilitates crystal packing, i.e., the loops interact between unit cells. This extended loop in ScErg1 and CaErg1 may have presented a hindrance in crystal packing, thus truncating this region in ScErg1 or CaErg1 may prove successful (Figure 3). Elucidating the Erg1 crystal structure of a dermatophyte, such as Trichophyton rubrum, could be possible without such a truncation, as it does not have the elongated loop and has a relatively short N-terminus, which could be reduced by 32 residues to resemble the human SM structure.
6. Squalene Monooxygenase Structural Information
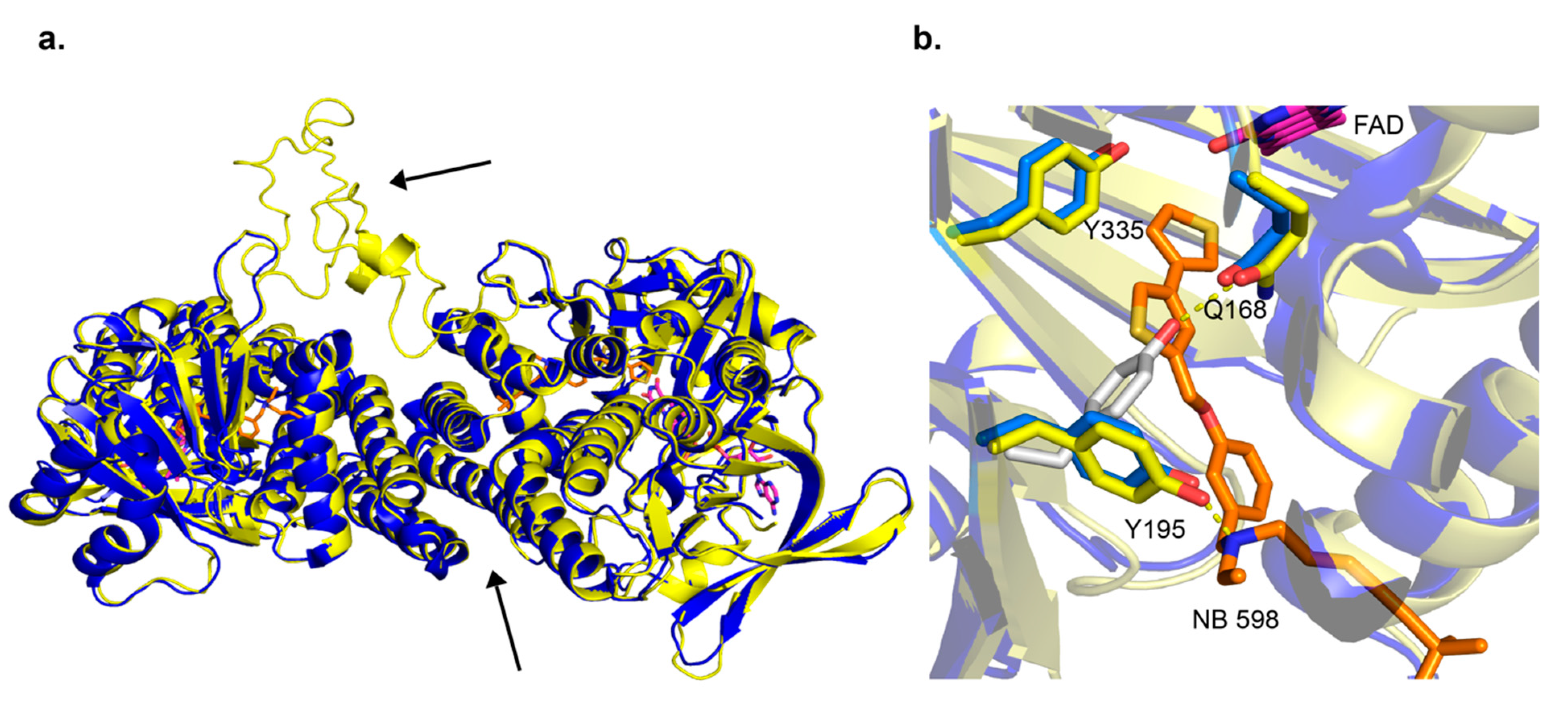
There had been no published structures for SM until last year when the human SM structure was published by Padyana et al. [13]. Three X-ray crystal structures were deposited into the PDB, one in complex with NB-598 (PDB ID 6C6P), one in complex with compound-4 (PDB ID 6C6N), and an unliganded structure (PDB ID 6C6R). The structures show a homodimer in the asymmetric unit, with C-terminal α-helices 11 and 12, residues 516–537 and 545–567, respectively, forming the interacting interface. Those C-terminal α-helices are putative membrane-associated domains, so it makes sense that they will form stabilizing hydrophobic interactions (Figure 3a). As shown by Padyana et al. in thermal stability assays, the SM (118–574) was more stable than the SM (118–488) by 4 °C. The FAD-binding domain was identified to have the GR2 Rossmann fold. The inhibitor-binding site was identified with both NB-598 and compound-4 binding in an extended conformation into a relatively hydrophobic binding pocket consistent with the isoprenoid substrate squalene. The clinically relevant ligand terbinafine was also modeled into the unliganded structure of SM. The authors identified interactions of the binding site and terbinafine and mapped some of the resistance mutations which occur in Erg1 [13] (discussed in later sections). This provides useful information as there is no X-ray crystal structure available for the fungal enzymes to date. The human SM structure is good for homology modeling and other computational techniques, as the substrate-binding site is relatively conserved among human SM, and the Erg1s of the dermatophyte fungi T. rubrum and Trichophyton interdigitale, and yeasts like C. albicans and S. cerevisiae (Figure 4).
A detailed in silico homology model was determined in 2011 (S. cerevisiae Erg1) using a crystal structure of p-hydroxybenzoate hydroxylase from Pseudomonas fluorescens (PDB ID 1PBE) and an aromatic hydrolase from E. coli (PDB ID 2QA1) [63]. Both these enzymes have FAD-binding domains but low sequence homology to the fungal enzyme. The ScErg1 model identified the FAD and the substrate-binding domains, including the characteristic GR2 Rossmann fold [64]. The authors used a 3D-Jury approach to get the overall 3D fold and Modeler was used for the missing loops and sidechains. Molecular dynamics simulations were done to model terbinafine binding to Erg1. In addition, they used a mutagenesis study to verify their findings [65]. Mutagenesis studies of the S. cerevisiae enzyme showed that G27S, G30S mutations reduced the activity of the enzyme in vitro. The authors hypothesized that this was due to the proximity of these residues to the FAD cofactor. These residues are conserved in the human and the dermatophyte T. rubrum enzymes (Figure 4). The SM structure by Padyana et al. shows that the SM-equivalent glycine residues are in 3–4 Å proximity to the FAD molecule [13]. Several residues important for terbinafine binding were identified in the in silico model. Among them was Y90, which showed that its hydroxyl group projected to make a 2.4 Å hydrogen bond with the amine group of terbinafine. In the structural model of SM, Padyana et al. found that the equivalent residue Y195 makes a hydrogen bond with the tertiary amine of NB-598 and compound-4, the only hydrogen bond between the enzyme and those inhibitors (Figure 3b) [13]. In silico model identified this important interaction of ScErg1 with terbinafine in the lipophilic binding cavity, which illustrates the power of such predictive models. Furthermore, the progress of computational techniques has been immense since that time and protein structure predictions are getting better and better. As an example, DeepMind’s artificial intelligence system called AlphaFold outperformed its competitors in predicting protein structures and achieved results that are very close to the experimentally determined structures in the Critical Assessment of Structure Prediction in 2020 [66].
Another interesting interaction found in the binding site of ligand-free SM (PDB ID 6C6R) is the change of conformation of Y195 to form a hydrogen bond with the amine of Q168. Both residues are conserved in the fungal enzyme (Figure 3b). This is a very important interaction, which is redirected when an inhibitor is bound. It is worth noting that a tertiary amine is present in all SM and Erg1 inhibitors. The importance of this interaction and the Y195 residue is highlighted by an ~90% loss of activity by SM when this tyrosine is mutated to phenylalanine [13]. Another important tyrosine is located close to the flavin moiety of FAD (PDB ID 6C6P). Y335 forms a hydrogen bond network with the N5 of flavin via a water molecule. This is also conserved in fungi, and if the side chain of it is rotated about 30 degrees, it can make a hydrogen bond to Y195. It is therefore tempting to hypothesize this residue network is involved in proton-coupled electron transfer to the FAD. However, it is not yet know how the binding of squalene affects the conformation of the residues in the binding cavity as there is no crystal structure with the substrate.
7. Terbinafine Resistance Mutations
There are several ways that fungi can become tolerant and less susceptible to antifungals such as their stress response mechanisms, drug efflux via efflux pumps, gene duplication, and mutations in the drug target gene [67]. In dermatophytes, the main mechanisms of resistance to terbinafine are through point mutations in Erg1. Some of the first mutations found in clinical isolates harbored L393F and F397L mutations in T. rubrum Erg1 [68,69]. The potency of the F397L mutation was demonstrated when the S. cerevisiae strain was transformed with C. albicans Erg1 with and without the equivalent mutation increasing the MIC90 more than 16-fold [69]. It is worth pointing out that when resistant clinical isolate strains are compared with the susceptible ones, the changes in MIC may not only be attributable to a particular mutation as other resistance/tolerance mechanisms may be in play.
These same mutations L393F and F397L were subsequently found in clinical isolates of T. interdigitale [70]. The F397 position of the residues changes to a less bulky side chain like leucine, isoleucine, and valine, and the L393 is mutated to either a bulky phenylalanine or a polar serine [71]. Mutagenesis studies carried out on ScErg1 to find strains less susceptible to terbinafine have found an equivalent residue mutation to F397L (F402L; Figure 4) as well several additional ones [65]. The structurally equivalent residues of SM are found in the inhibitor-binding pocket [13]. Since there is only one polar interaction between the binding cavity and the inhibitor, most of the mutations change the size of the hydrophobic side chains, thereby altering hydrophobic interactions.
Terbinafine only partially inhibits human SM. Recently, Padyana et al. reported an IC50 of 7.7 µM and 65% inhibition at 100 µM [13]. Terbinafine modeled into SM places the naphthalene ring close to residues I197 and L324, which overlaps with the smaller benzene ring of NB-598. In contrast, in dermatophytes, those residues are valines. The authors hypothesized that the smaller residues accommodate the presence of the bulky naphthalene moiety of terbinafine better than the bigger leucine and isoleucine residues. Interestingly, the frequently mutated residues F397 and L393 are located adjacently to those valines in the binding pocket, highlighting the importance of hydrophobic interactions in drug binding. F415I/V, H440Y, and Q408L mutations have been identified. These are located closer to the trimethyl end of the terbinafine molecule [71,72] when modelled into SM. However, these mutations do not confer reduced susceptibility to terbinafine to the same extent as L393F and F397L. Double mutants H440Y F484Y and I121M V237I have been reported and were classified as “low-level” resistance mutations [73]. The T. rubrum strain with the double mutations resulted in an MIC of 0.125 μg/mL in comparison with T. rubrum with the “high-level” L393F mutation (MIC > 8 μg/mL) and the susceptible strain (0.016 μg/mL).
8. Concluding Remarks
Considerable progress has been made concerning understanding squalene monooxygenase, including the identification of important differences between the fungal and the mammalian enzymes, their structural characterization, and the links of the human enzyme to cancer.
Overcoming resistance mutations in dermatophytes and improving the efficacy of allylamines in yeasts such as C. albicans and A. fumigatus will have significant benefits for medicine and possibly in agriculture. Elucidating the structure of the fungal enzyme will shed more light on how to better target Erg1 and develop better antifungals. In the meantime, the human SM crystal structure provides a good avenue for finding better ways to purify and crystalize the fungal enzyme and for providing homology models to aid in developing novel antifungals. Hydrophobic interactions play an important role in drug binding to SM. However, there are polar residues in the active site that can be targeted such as Q168 (SM numbering), a residue conserved in fungi. Increasing the water solubility of allylamines will allow for better distribution throughout the body and not mainly in the skin and adipose tissue. This may increase the efficacy of drugs in treating disseminated infections. The same may also be true for targeting SM. NB-598 is a potent inhibitor but its toxicity has prevented clinical trials. In dogs, administering NB-598 has resulted in a reduction of cholesterol, triacylglycerol, and LDL-cholesterol [74]. Unfortunately, dermatitis-like toxicity effects were observed. This effect was thought to be due to the accumulation of squalene in the skin. In a recent study, the effects of NB-598 were investigated in cynomolgus monkeys and repeated in dogs [75]. In addition to dermatitis, gastrointestinal toxicity was observed in monkeys to the point where the animals had to be euthanized after 3 or 4 days of dosing [75]. It was confirmed that squalene accumulation in the skin is the result of the co-localization of NB-598. Much more work needs to be done to develop next-generation inhibitors including consideration of more appropriate methods of drug delivery.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
All the data used here was publicly available in the form of published articles, crystal structures added to the PDB, and homology model deposited into the SWISS Prot database.
Conflicts of Interest
The author declares no conflict of interest.
References
- Taylor, D.L.; Hollingsworth, T.N.; McFarland, J.W.; Lennon, N.J.; Nusbaum, C.; Ruess, R.W. A first comprehensive census of fungi in soil reveals both hyperdiversity and fine-scale niche partitioning. Ecol. Monogr. 2014, 84, 3–20. [Google Scholar] [CrossRef]
- Benedict, K.; Jackson, B.R.; Chiller, T.; Beer, K.D. Estimation of direct healthcare costs of fungal diseases in the United States. Clin. Infect. Dis. 2019, 68, 1791–1797. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fisher, M.C.; Henk, D.A.; Briggs, C.J.; Brownstein, J.S.; Madoff, L.C.; McCraw, S.L.; Gurr, S.J. Emerging fungal threats to animal, plant and ecosystem health. Nature 2012, 484, 186–194. [Google Scholar] [CrossRef] [PubMed]
- Brauer, V.S.; Rezende, C.P.; Pessoni, A.M.; de Paula, R.G.; Rangappa, K.S.; Nayaka, S.C.; Gupta, V.K.; Almeida, F. Antifungal agents in agriculture: Friends and foes of Public Health. Biomolecules 2019, 9, 521. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Snelders, E.; Van Der Lee, H.A.L.; Kuijpers, J.; Rijs, A.J.M.M.; Varga, J.; Samson, R.A.; Mellado, E.; Donders, A.R.T.; Melchers, W.J.G.; Verweij, P.E. Emergence of Azole Resistance in Aspergillus fumigatus and Spread of a Single Resistance Mechanism. PLoS Med. 2008, 5, e219. [Google Scholar] [CrossRef]
- Egbuta, C.; Lo, J.; Ghosh, D. Mechanism of Inhibition of Estrogen Biosynthesis by Azole Fungicides. Endocrinology 2014, 155, 4622–4628. [Google Scholar] [CrossRef]
- Lorch, J.M.; Meteyer, C.U.; Behr, M.J.; Boyles, J.G.; Cryan, P.M.; Hicks, A.C.; Ballmann, A.E.; Coleman, J.T.H.; Redell, D.N.; Reeder, D.M.; et al. Experimental infection of bats with Geomyces destructans causes white-nose syndrome. Nat. Cell Biol. 2011, 480, 376–378. [Google Scholar] [CrossRef]
- Frick, W.F.; Pollock, J.F.; Hicks, A.C.; Langwig, K.E.; Reynolds, D.S.; Turner, G.G.; Butchkoski, C.M.; Kunz, T.H. An emerging disease causes regional population collapse of a common North American bat species. Science 2010, 329, 679–682. [Google Scholar] [CrossRef] [Green Version]
- Crawford, A.J.; Lips, K.R.; Bermingham, E. Epidemic disease decimates amphibian abundance, species diversity, and evolutionary history in the highlands of central Panama. Proc. Natl. Acad. Sci. USA 2010, 107, 13777–13782. [Google Scholar] [CrossRef] [Green Version]
- Fisher, M.C.; Garner, T.W.; Walker, S.F. Global emergence of Batrachochytrium dendrobatidis and amphibian chytridiomycosis in space, time, and host. Annu. Rev. Microbiol. 2009, 63, 291–310. [Google Scholar] [CrossRef] [Green Version]
- Satoh, K.; Makimura, K.; Hasumi, Y.; Nishiyama, Y.; Uchida, K.; Yamaguchi, H. Candida auris sp. nov., a novel ascomycetous yeast isolated from the external ear canal of an inpatient in a Japanese hospital. Microbiol. Immunol. 2009, 53, 41–44. [Google Scholar] [CrossRef] [PubMed]
- Casadevall, A.; Kontoyiannis, D.P.; Robert, V. On the emergence of Candida auris: Climate change, azoles, swamps, and birds. mBio 2019, 10, e01397-19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Padyana, A.K.; Gross, S.; Jin, L.; Cianchetta, G.; Narayanaswamy, R.; Wang, F.; Wang, R.; Fang, C.; Lv, X.; Biller, S.A.; et al. Structure and inhibition mechanism of the catalytic domain of human squalene epoxidase. Nat. Commun. 2019, 10, 97. [Google Scholar]
- Rodriguez, R.J.; Taylor, F.R.; Parks, L.W. A requirement for ergosterol to permit growth of yeast sterol auxotrophs on cholestanol. Biochem. Biophys. Res. Commun. 1982, 106, 435–441. [Google Scholar] [CrossRef]
- Monk, B.C.; Sagatova, A.A.; Hosseini, P.; Ruma, Y.N.; Wilson, R.K.; Keniya, M.V. Fungal lanosterol 14α-demethylase: A target for next-generation antifungal design. Biochim. Biophys. Acta (BBA) Proteins Proteom. 2020, 1868, 140206. [Google Scholar] [CrossRef]
- Warrilow, A.G.S.; Hull, C.M.; Parker, J.E.; Garvey, E.P.; Hoekstra, W.J.; Moore, W.R.; Schotzinger, R.J.; Kelly, D.E.; Kelly, S. The clinical candidate VT-1161 is a highly potent inhibitor of Candida albicans CYP51 but fails to bind the human enzyme. Antimicrob. Agents Chemother. 2014, 58, 7121–7127. [Google Scholar] [CrossRef] [Green Version]
- Sobel, J.; Brand, S.; Degenhardt, T.; Person, K.; Nyirjesy, P.; Schotzinger, R.; Tavakkol, A. Results from a phase 2, randomized, double-blind, placebo-controlled, dose ranging study to evaluate the efficacy and safety of VT 1161 oral tablets in the treatment of patients with recurrent vulvovaginal candidiasis. Am. J. Obstet. Gynecol. 2017, 217, 715. [Google Scholar] [CrossRef]
- Wiederhold, N.P.; Lockhart, S.R.; Najvar, L.K.; Berkow, E.L.; Jaramillo, R.; Olivo, M.; Garvey, E.P.; Yates, C.M.; Schotzinger, R.J.; Catanoet, G.; et al. The fungal Cyp51-specific inhibitor VT-1598 demonstrates in vitro and in vivo activity against Candida auris. Antimicrob. Agents Chemother. 2019, 63. [Google Scholar] [CrossRef] [Green Version]
- Monk, B.C.; Tomasiak, T.M.; Keniya, M.V.; Huschmann, F.U.; Tyndall, J.D.A.; Iii, J.D.O.; Cannon, R.D.; McDonald, J.G.; Rodriguez, A.; Finer-Moore, J.S.; et al. Architecture of a single membrane spanning cytochrome P450 suggests constraints that orient the catalytic domain relative to a bilayer. Proc. Natl. Acad. Sci. USA 2014, 111, 3865–3870. [Google Scholar] [CrossRef] [Green Version]
- Sagatova, A.A.; Keniya, M.V.; Wilson, R.K.; Sabherwal, M.; Tyndall, J.D.A.; Monk, B.C. Triazole resistance mediated by mutations of a conserved active site tyrosine in fungal lanosterol 14α-demethylase. Sci. Rep. 2016, 6, 26213. [Google Scholar] [CrossRef] [Green Version]
- Hargrove, T.Y.; Friggeri, L.; Wawrzak, Z.; Qi, A.; Hoekstra, W.J.; Schotzinger, R.J.; York, J.D.; Guengerich, F.P.; Lepesheva, G.I. Structural analyses of Candida albicans sterol 14alpha-demethylase complexed with azole drugs address the molecular basis of azole-mediated inhibition of fungal sterol biosynthesis. J. Biol. Chem. 2017, 292, 6728–6743. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ryder, N. The mechanism of action of terbinafine. Clin. Exp. Dermatol. 1989, 14, 98–100. [Google Scholar] [CrossRef] [PubMed]
- Mascotti, M.L.; Ayub, M.J.; Furnham, N.; Thornton, J.M.; Laskowski, R.A. Chopping and Changing: The Evolution of the Flavin-dependent Monooxygenases. J. Mol. Biol. 2016, 428, 3131–3146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fernández-Torres, B.; Carrillo, A.J.; Martín, E.; Del Palacio, A.; Moore, M.K.; Valverde, A.; Serrano, M.; Guarro, J. In vitro activities of 10 antifungal drugs against 508 dermatophyte strains. Antimicrob. Agents Chemother. 2001, 45, 2524–2528. [Google Scholar]
- Favre, B.; Ryder, N.S. Characterization of squalene epoxidase activity from the dermatophyte Trichophyton rubrum and its inhibition by terbinafine and other antimycotic agents. Antimicrob. Agents Chemother. 1996, 40, 443–447. [Google Scholar] [CrossRef] [Green Version]
- Havlickova, B.; Czaika, V.A.; Friedrich, M. Epidemiological trends in skin mycoses worldwide. Mycoses 2008, 51, 2–15. [Google Scholar] [CrossRef]
- Ryder, N. Terbinafine: Mode of action and properties of the squalene epoxidase inhibition. Br. J. Dermatol. 1992, 126, 2–7. [Google Scholar] [CrossRef]
- Ryder, N.S.; Leitner, I. Synergistic interaction of terbinafine with triazoles or amphotericin B against Aspergillus species. Med. Mycol. 2001, 39, 91–95. [Google Scholar] [CrossRef] [Green Version]
- Perea, S.; Gonzalez, G.; Fothergill, A.W.; Sutton, D.A.; Rinaldi, M.G. In vitro activities of terbinafine in combination with fluconazole, itraconazole, voriconazole, and posaconazole against clinical isolates of Candida glabrata with decreased susceptibility to azoles. J. Clin. Microbiol. 2002, 40, 1831–1833. [Google Scholar] [CrossRef] [Green Version]
- Faergemann, J.; Zehender, H.; Jones, T.; Maibach, I. Terbinafine levels in serum, stratum corneum, dermis-epidermis (without stratum corneum), hair, sebum and eccrine sweat. Acta Derm. Venereol. 1991, 71, 322–326. [Google Scholar]
- Jensen, J.C. Clinical pharmacokinetics of terbinafine (Lamisil). Clin. Exp. Dermatol. 1989, 14, 110–113. [Google Scholar] [CrossRef] [PubMed]
- Darkes, M.J.; Scott, L.J.; Goa, K.L. Terbinafine: A review of its use in onychomycosis in adults. Am. J. Clin. Dermatol. 2003, 4, 39–65. [Google Scholar] [CrossRef] [PubMed]
- Kovarik, J.; Kirkesseli, S.; Humbert, H.; Grass, P.; Kutz, K. Dose-proportional pharmacokinetics of terbinafine and its N-demethylated metabolite in healthy volunteers. Br. J. Dermatol. 1992, 126, 8–13. [Google Scholar] [CrossRef] [PubMed]
- Abdel-Rahman, S.M.; Gotschall, R.R.; Kauffman, R.E.; Leeder, J.S.; Kearns, G.L. Investigation of terbinafine as a CYP2D6 inhibitor in vivo. Clin. Pharmacol. Ther. 1999, 65, 465–472. [Google Scholar] [CrossRef]
- He, Z.-X.; Chen, X.-W.; Zhou, Z.-W.; Zhou, S.-F. Impact of physiological, pathological and environmental factors on the expression and activity of human cytochrome P450 2D6 and implications in precision medicine. Drug Metab. Rev. 2015, 47, 470–519. [Google Scholar] [CrossRef] [PubMed]
- Vickers, A.E.; Sinclair, J.R.; Zollinger, M.; Heitz, F.; Glänzel, U.; Johanson, L.; Fischer, V. Multiple cytochrome P-450s involved in the metabolism of terbinafine suggest a limited potential for drug-drug interactions. Drug Metab. Dispos. 1999, 27, 1029–1038. [Google Scholar]
- Ekroos, M.; Sjögren, T. Structural basis for ligand promiscuity in cytochrome P450 3A4. Proc. Natl. Acad. Sci. USA 2006, 103, 13682–13687. [Google Scholar] [CrossRef] [Green Version]
- Ryder, N.S.; Dupont, M.C. Inhibition of squalene epoxidase by allylamine antimycotic compounds. A comparative study of the fungal and mammalian enzymes. Biochem. J. 1985, 230, 765–770. [Google Scholar] [CrossRef] [Green Version]
- Cirmena, G.; Franceschelli, P.; Isnaldi, E.; Ferrando, L.; De Mariano, M.; Ballestrero, A.; Zoppoli, G. Squalene epoxidase as a promising metabolic target in cancer treatment. Cancer Lett. 2018, 425, 13–20. [Google Scholar] [CrossRef]
- Sui, Z.; Zhou, J.; Cheng, Z.; Lu, P. Squalene epoxidase (SQLE) promotes the growth and migration of the hepatocellular carcinoma cells. Tumor Biol. 2015, 36, 6173–6179. [Google Scholar] [CrossRef]
- Mahoney, C.E.; Pirman, D.; Chubukov, V.; Sleger, T.; Hayes, S.; Fan, Z.P.; Allen, E.L.; Chen, Y.; Huang, L.; Liu, M.; et al. A chemical biology screen identifies a vulnerability of neuroendocrine cancer cells to SQLE inhibition. Nat. Commun. 2019, 10, 96. [Google Scholar]
- Chua, N.K.; Coates, H.W.; Brown, A.J. Squalene monooxygenase: A journey to the heart of cholesterol synthesis. Prog. Lipid Res. 2020, 79, 101033. [Google Scholar] [CrossRef] [PubMed]
- Roberts, A.A.; Berger, L.; Robertson, S.G.; Webb, R.J.; Kosch, T.A.; McFadden, M.; Skerratt, L.F.; Glass, B.D.; Motti, C.A.; Brannelly, L.A. The efficacy and pharmacokinetics of terbinafine against the frog-killing fungus (Batrachochytrium dendrobatidis). Med. Mycol. 2018, 57, 204–214. [Google Scholar] [CrossRef] [PubMed]
- Court, M.H.; Robbins, A.H.; Whitford, A.M.; Beck, E.V.; Tseng, F.S.; Reeder, D.M. Pharmacokinetics of terbinafine in little brown myotis (Myotis lucifugus) infected with Pseudogymnoascus destructans. Am. J. Veter. Res. 2017, 78, 90–99. [Google Scholar] [CrossRef] [PubMed]
- Souza, M.J.; Cairns, T.; Yarbrogh, J.; Cox, S.K. In vitro investigation of a terbinafine impregnated subcutaneous implant for veterinary use. J. Drug Deliv. 2012, 2012, 436710. [Google Scholar] [CrossRef] [Green Version]
- Jordá, T.; Puig, S. Regulation of Ergosterol Biosynthesis in Saccharomyces cerevisiae. Genes 2020, 11, 795. [Google Scholar] [CrossRef]
- Burg, J.S.; Espenshade, P.J. Regulation of HMG-CoA reductase in mammals and yeast. Prog. Lipid Res. 2011, 50, 403–410. [Google Scholar] [CrossRef] [Green Version]
- Foresti, O.; Ruggiano, A.; Hannibal-Bach, H.K.; Ejsing, C.S.; Carvalho, P. Sterol homeostasis requires regulated degradation of squalene monooxygenase by the ubiquitin ligase Doa10/Teb4. eLife 2013, 2, e00953. [Google Scholar] [CrossRef]
- Boban, M.; Ljungdahl, P.O.; Foisner, R. Atypical ubiquitylation in yeast targets lysine-less Asi2 for proteasomal degradation. J. Biol. Chem. 2014, 290, 2489–2495. [Google Scholar] [CrossRef] [Green Version]
- Weber, A.; Cohen, I.; Popp, O.; Dittmar, G.; Reiss, Y.; Sommer, T.; Ravid, T.; Jarosch, E. Sequential poly-ubiquitylation by specialized conjugating enzymes expands the versatility of a quality control ubiquitin ligase. Mol. Cell 2016, 63, 827–839. [Google Scholar] [CrossRef] [Green Version]
- Chua, N.K.; Howe, V.; Jatana, N.; Thukral, L.; Brown, A.J. A conserved degron containing an amphipathic helix regulates the cholesterol-mediated turnover of human squalene monooxygenase, a rate-limiting enzyme in cholesterol synthesis. J. Biol. Chem. 2017, 292, 19959–19973. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stevenson, J.; Luu, W.; Kristiana, I.; Brown, A.J. Squalene mono-oxygenase, a key enzyme in cholesterol synthesis, is stabilized by unsaturated fatty acids. Biochem. J. 2014, 461, 435–442. [Google Scholar] [CrossRef] [PubMed]
- Favre, B.; Ryder, N.S. Cloning and expression of squalene epoxidase from the pathogenic yeast Candida albicans. Gene 1997, 189, 119–126. [Google Scholar] [CrossRef]
- Laden, B.P.; Tang, Y.; Porter, T.D. Cloning, Heterologous Expression, and Enzymological Characterization of Human Squalene Monooxygenase. Arch. Biochem. Biophys. 2000, 374, 381–388. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ono, T.; Bloch, K. Solubilization and partial characterization of rat liver squalene epoxidase. J. Biol. Chem. 1975, 250, 1571–1579. [Google Scholar] [CrossRef]
- Christen, M.; Marcaida, M.J.; Lamprakis, C.; Aeschimann, W.; Vaithilingam, J.; Schneider, P.; Hilbert, M.; Schneider, G.; Cascella, M.; Stocker, A. Structural insights on cholesterol endosynthesis: Binding of squalene and 2,3-oxidosqualene to supernatant protein factor. J. Struct. Biol. 2015, 190, 261–270. [Google Scholar] [CrossRef]
- Ryder, N.S.; Dupont, M.-C. Properties of a particulate squalene epoxidase from Candida albicans. Biochim. Biophys. Acta 1984, 794, 466–471. [Google Scholar] [CrossRef]
- Lamping, E.; Monk, B.C.; Niimi, K.; Holmes, A.R.; Tsao, S.; Tanabe, K.; Niimi, M.; Uehara, Y.; Cannon, R.D. Characterization of three classes of membrane proteins involved in fungal azole resistance by functional hyperexpression in Saccharomyces cerevisiae. Eukaryot. Cell 2007, 6, 1150–1165. [Google Scholar] [CrossRef] [Green Version]
- Monk, B.C.; Cannon, R.D.; Nakamura, K.; Niimi, M.; Niimi, K.; Holmes, A.R.; Lamping, E.; Harding, D.R.K.; Goffeau, A.; Decottignies, A. Yeast Membrane Protein Expression System and Its Application in Drug Screening. Patent WO2003018817 A1, 20 May 2014. [Google Scholar]
- Bienert, S.; Waterhouse, A.; De Beer, T.A.P.; Tauriello, G.; Studer, G.; Bordoli, L.; Schwede, T. The SWISS-MODEL Repository—New features and functionality. Nucleic Acids Res. 2017, 45, D313–D319. [Google Scholar] [CrossRef] [Green Version]
- Sievers, F.; Wilm, A.; Dineen, D.; Gibson, T.J.; Karplus, K.; Li, W.; Lopez, R.; McWilliam, H.; Remmert, M.; Söding, J.; et al. Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol. Syst. Biol. 2011, 7, 539. [Google Scholar] [CrossRef]
- Robert, X.; Gouet, P. Deciphering key features in protein structures with the new ENDscript server. Nucleic Acids Res. 2014, 42, W320–W324. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nowosielski, M.; Hoffmann, M.; Wyrwicz, L.S.; Stepniak, P.; Plewczynski, D.M.; Lazniewski, M.; Ginalski, K.; Rychlewski, L. Detailed mechanism of squalene epoxidase inhibition by terbinafine. J. Chem. Inf. Model. 2011, 51, 455–462. [Google Scholar] [CrossRef] [PubMed]
- Dym, O.; Eisenberg, D. Sequence-structure analysis of FAD-containing proteins. Protein Sci. 2001, 10, 1712–1728. [Google Scholar] [CrossRef] [PubMed]
- Leber, R.; Fuchsbichler, S.; Klobučníková, V.; Schweighofer, N.; Pitters, E.; Wohlfarter, K.; Lederer, M.; Landl, K.; Ruckenstuhl, C.; Hapala, I.; et al. Molecular mechanism of terbinafine resistance in Saccharomyces cerevisiae. Antimicrob. Agents Chemother. 2003, 47, 3890–3900. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Callaway, E. ‘It will change everything’: DeepMind’s AI makes gigantic leap in solving protein structures. Nature 2020, 588, 203–204. [Google Scholar] [CrossRef]
- Martinez-Rossi, N.M.; Bitencourt, T.A.; Peres, N.T.A.; Lang, E.A.S.; Gomes, E.V.; Quaresemin, N.R.; Martins, M.P.; Lopes, L.; Rossi, A. Dermatophyte Resistance to Antifungal Drugs: Mechanisms and Prospectus. Front. Microbiol. 2018, 9, 1108. [Google Scholar] [CrossRef] [Green Version]
- Osborne, C.S.; Leitner, I.; Favre, B.; Ryder, N.S. Amino acid substitution in Trichophyton rubrum squalene epoxidase sssociated with resistance to terbinafine. Antimicrob. Agents Chemother. 2005, 49, 2840–2844. [Google Scholar] [CrossRef] [Green Version]
- Osborne, C.S.; Leitner, I.; Hofbauer, B.; Fielding, C.A.; Favre, B.; Ryder, N.S. Biological, biochemical, and molecular characterization of a new clinical Trichophyton rubrum isolate resistant to terbinafine. Antimicrob. Agents Chemother. 2006, 50, 2234–2236. [Google Scholar] [CrossRef] [Green Version]
- Singh, A.; Masih, A.; Khurana, A.; Singh, P.K.; Gupta, M.; Hagen, F.; Meis, J.F.; Chowdhary, A. High terbinafine resistance in Trichophyton interdigitale isolates in Delhi, India harbouring mutations in the squalene epoxidase gene. Mycoses 2018, 61, 477–484. [Google Scholar] [CrossRef]
- Yamada, T.; Maeda, M.; Alshahni, M.M.; Tanaka, R.; Yaguchi, T.; Bontems, O.; Salamin, K.; Fratti, M.; Monod, M. Terbinafine resistance of Trichophyton clinical isolates caused by specific point mutations in the squalene epoxidase gene. Antimicrob. Agents Chemother. 2017, 61, e00115-17. [Google Scholar] [CrossRef] [Green Version]
- Hsieh, A.; Quenan, S.; Riat, A.; Toutous-Trellu, L.; Fontao, L. A new mutation in the SQLE gene of Trichophyton mentagrophytes associated to terbinafine resistance in a couple with disseminated tinea corporis. J. Med. Mycol. 2019, 29, 352–355. [Google Scholar] [CrossRef] [PubMed]
- Saunte, D.; Hare, R.K.; Jørgensen, K.M.; Jørgensen, R.; Deleuran, M.; Zachariae, C.O.; Thomsen, S.F.; Bjørnskov-Halkier, L.; Kofoed, K.; Arendrup, M.C. Emerging terbinafine resistance in trichophyton: Clinical characteristics, squalene epoxidase gene mutations, and a reliable EUCAST method for detection. Antimicrob. Agents Chemother. 2019, 63, e01126-19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Horie, M.; Tsuchiya, Y.; Hayashi, M.; Iida, Y.; Iwasawa, Y.; Nagata, Y.; Sawasaki, Y.; Fukuzumi, H.; Kitani, K.; Kamei, T. NB-598: A potent competitive inhibitor of squalene epoxidase. J. Biol. Chem. 1990, 265, 18075–18078. [Google Scholar] [CrossRef]
- Nagaraja, R.; Olaharski, A.; Narayanaswamy, R.; Mahoney, C.; Pirman, D.; Gross, S.; Roddy, T.P.; Popovici-Muller, J.; Smolen, G.A.; Silverman, L. Preclinical toxicology profile of squalene epoxidase inhibitors. Toxicol. Appl. Pharmacol. 2020, 401, 115103. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Allylamines terbinafine and naftifine and the human squalene monooxygenase inhibitor NB-598.
Figure 1.
Allylamines terbinafine and naftifine and the human squalene monooxygenase inhibitor NB-598.

Figure 2.
Ergosterol biosynthesis pathway. The simple arrow indicates one catalytic step from substrate to product and the dotted arrow represents presence of several additional catalytic steps.
Figure 2.
Ergosterol biosynthesis pathway. The simple arrow indicates one catalytic step from substrate to product and the dotted arrow represents presence of several additional catalytic steps.

Figure 3.
Overlay of human SM X-ray crystal structure and the S. cerevisiae Erg1 homology model. The homology model was downloaded from SWISS-MODEL with PDB ID 6C6N used as a template [60]. The ScErg1 is in yellow and SM (PDB ID 6C6P) is in blue. FAD and NB-598 are represented with sticks with carbons colored magenta and orange, respectively. (a) Two molecules were found in the asymmetric unit and the interface of C-terminal α-helices is indicated with an arrow. The extended loop of ScErg1 is shown with an arrow. (b) The residues Q168, Y195, and Y335 (Q63, Y90, and Y261, S. cerevisiae numbering) are shown as sticks. In gray is the Y195 residue position in the absence of the inhibitor (PDB ID 6C6R). The hydrogen bonds are shown as yellow dashed lines.
Figure 3.
Overlay of human SM X-ray crystal structure and the S. cerevisiae Erg1 homology model. The homology model was downloaded from SWISS-MODEL with PDB ID 6C6N used as a template [60]. The ScErg1 is in yellow and SM (PDB ID 6C6P) is in blue. FAD and NB-598 are represented with sticks with carbons colored magenta and orange, respectively. (a) Two molecules were found in the asymmetric unit and the interface of C-terminal α-helices is indicated with an arrow. The extended loop of ScErg1 is shown with an arrow. (b) The residues Q168, Y195, and Y335 (Q63, Y90, and Y261, S. cerevisiae numbering) are shown as sticks. In gray is the Y195 residue position in the absence of the inhibitor (PDB ID 6C6R). The hydrogen bonds are shown as yellow dashed lines.

Figure 4.
Sequence alignment of the human SM and fungal Erg1s. Sequence alignment was done using Clustal Omega [61] and the figure was generated using the ESPript 3.0 [62] online server using PDB ID 6C6P for secondary structure designation. The sequences were obtained in Uniprot H. sapiens (accession number Q145334), S. cerevisiae strain S288c (accession number P32476), A. fumigatus Z5 (accession number A0A0J5PRX5), T. interdigitale strain MR 816 (accession number A0A059JE48), T. rubrum (accession number Q4JEY0), C. albicans strain SC5314 (accession number Q92206). Conserved residues are highlighted in red and similar residues are shown in the red font. Mutations which confer reduced terbinafine susceptibility in dermatophytes T. rubrum and T. interdigitale are designated with triangles. The secondary structure is annotated as α for α-helices (curved line), β for β-strands (arrow), and η for 310-helices, the dots on the top of the sequences appear every ten residues.
Figure 4.
Sequence alignment of the human SM and fungal Erg1s. Sequence alignment was done using Clustal Omega [61] and the figure was generated using the ESPript 3.0 [62] online server using PDB ID 6C6P for secondary structure designation. The sequences were obtained in Uniprot H. sapiens (accession number Q145334), S. cerevisiae strain S288c (accession number P32476), A. fumigatus Z5 (accession number A0A0J5PRX5), T. interdigitale strain MR 816 (accession number A0A059JE48), T. rubrum (accession number Q4JEY0), C. albicans strain SC5314 (accession number Q92206). Conserved residues are highlighted in red and similar residues are shown in the red font. Mutations which confer reduced terbinafine susceptibility in dermatophytes T. rubrum and T. interdigitale are designated with triangles. The secondary structure is annotated as α for α-helices (curved line), β for β-strands (arrow), and η for 310-helices, the dots on the top of the sequences appear every ten residues.

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Sagatova, A.A. Strategies to Better Target Fungal Squalene Monooxygenase. J. Fungi 2021, 7, 49. https://0-doi-org.brum.beds.ac.uk/10.3390/jof7010049
AMA Style
Sagatova AA. Strategies to Better Target Fungal Squalene Monooxygenase. Journal of Fungi. 2021; 7(1):49. https://0-doi-org.brum.beds.ac.uk/10.3390/jof7010049
Chicago/Turabian StyleSagatova, Alia A. 2021. "Strategies to Better Target Fungal Squalene Monooxygenase" Journal of Fungi 7, no. 1: 49. https://0-doi-org.brum.beds.ac.uk/10.3390/jof7010049
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.