
Application of Laser-Induced Breakdown Spectroscopy in the Quantitative Analysis of Elements—K, Na, Ca, and Mg in Liquid Solutions


Abstract
:1. Introduction
2. Materials and Methods
2.1. Preparation of Samples
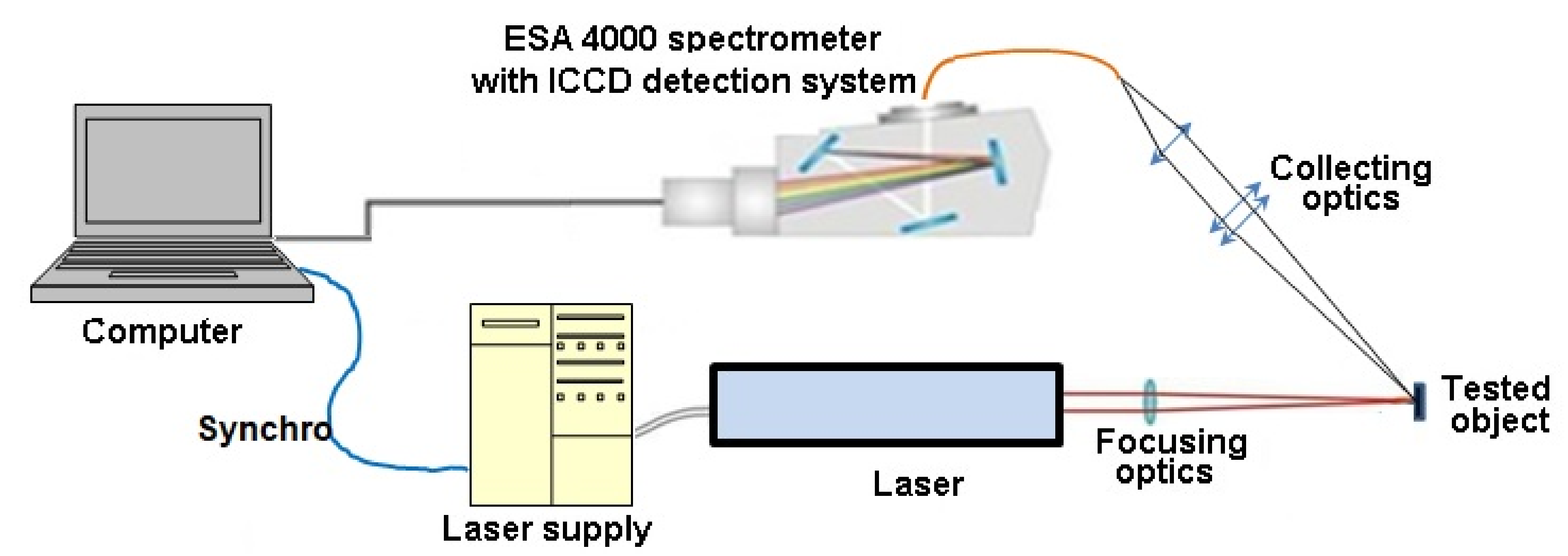
2.2. Experimental Setup
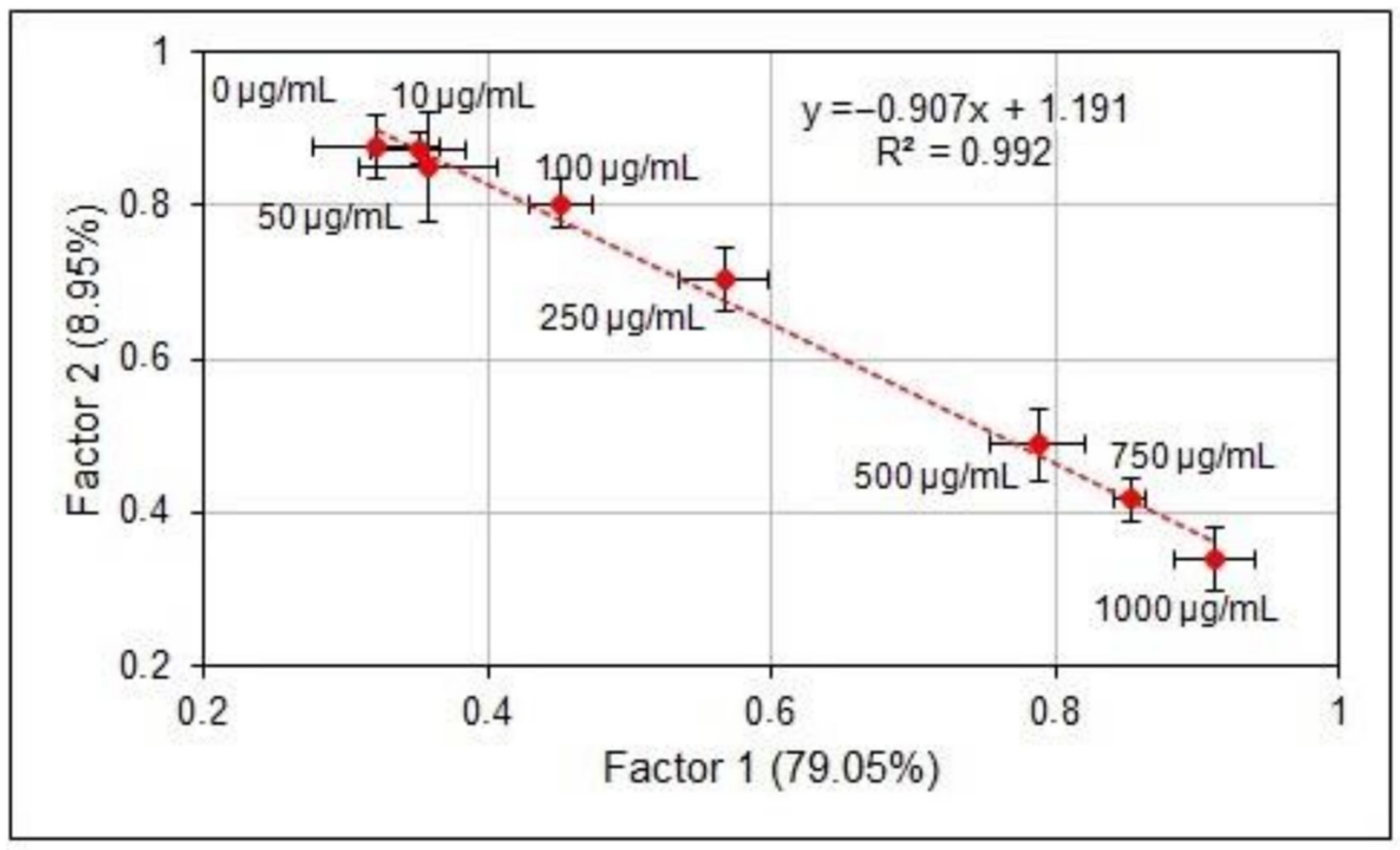
2.3. Statistical Processing
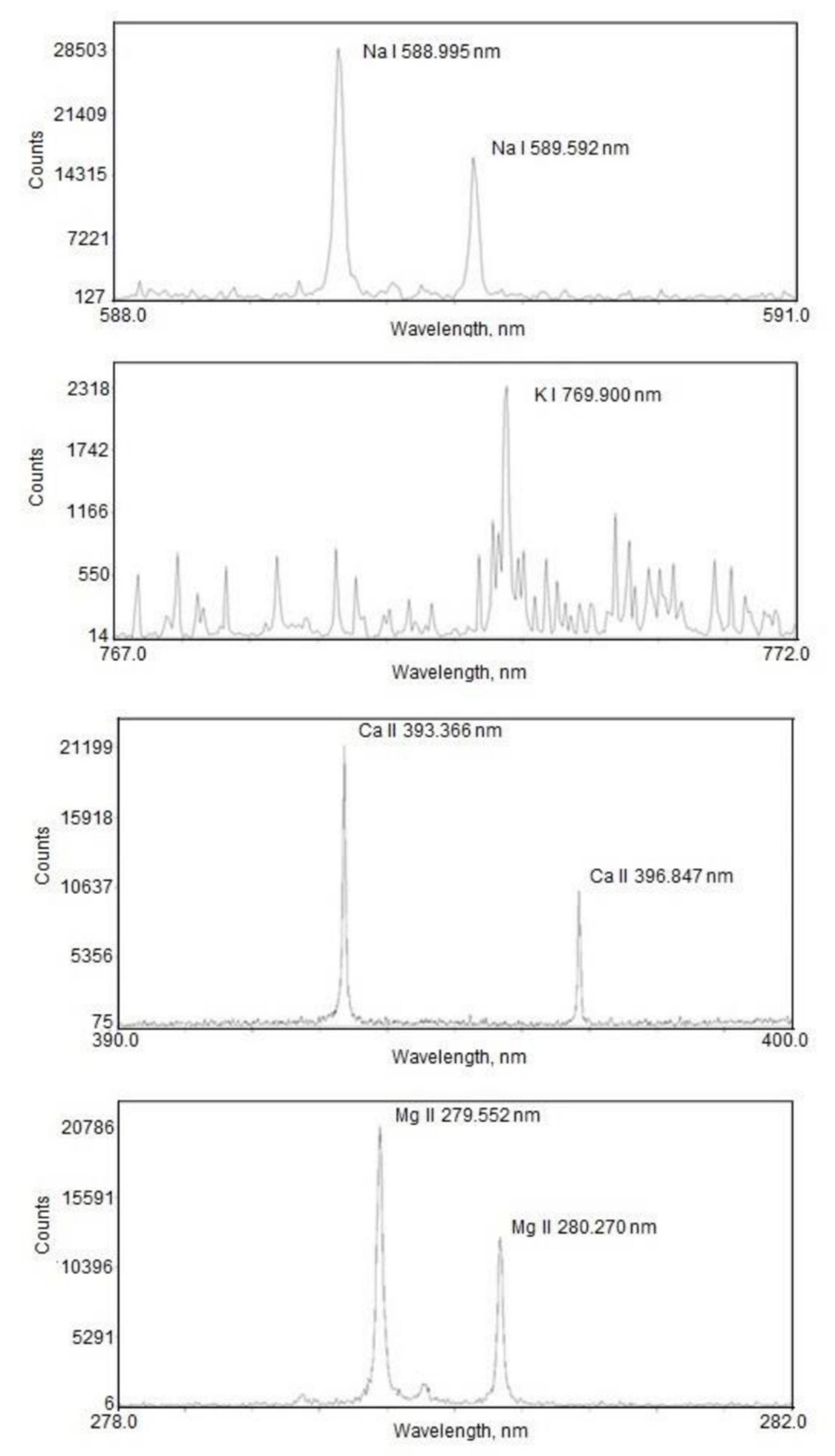
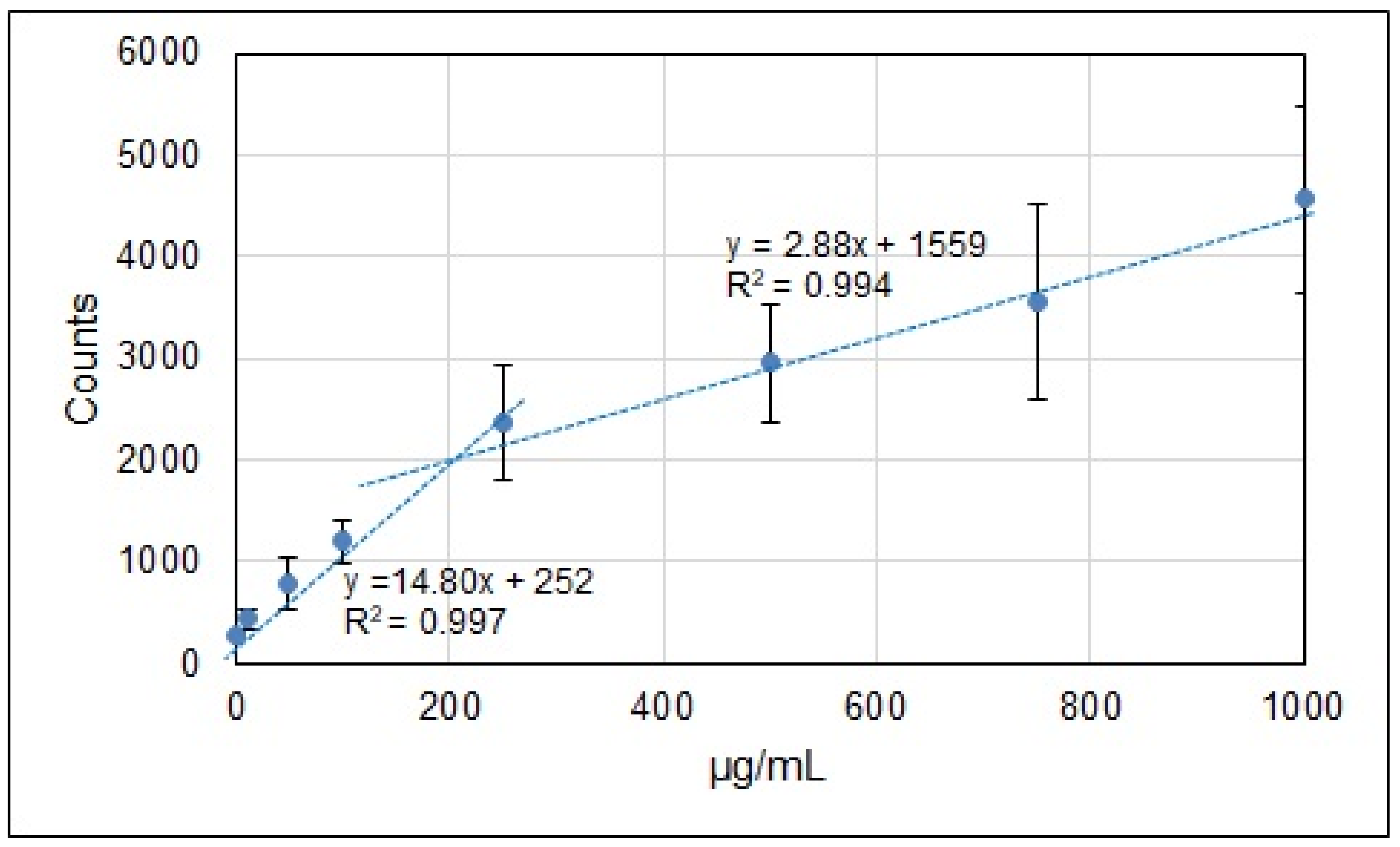
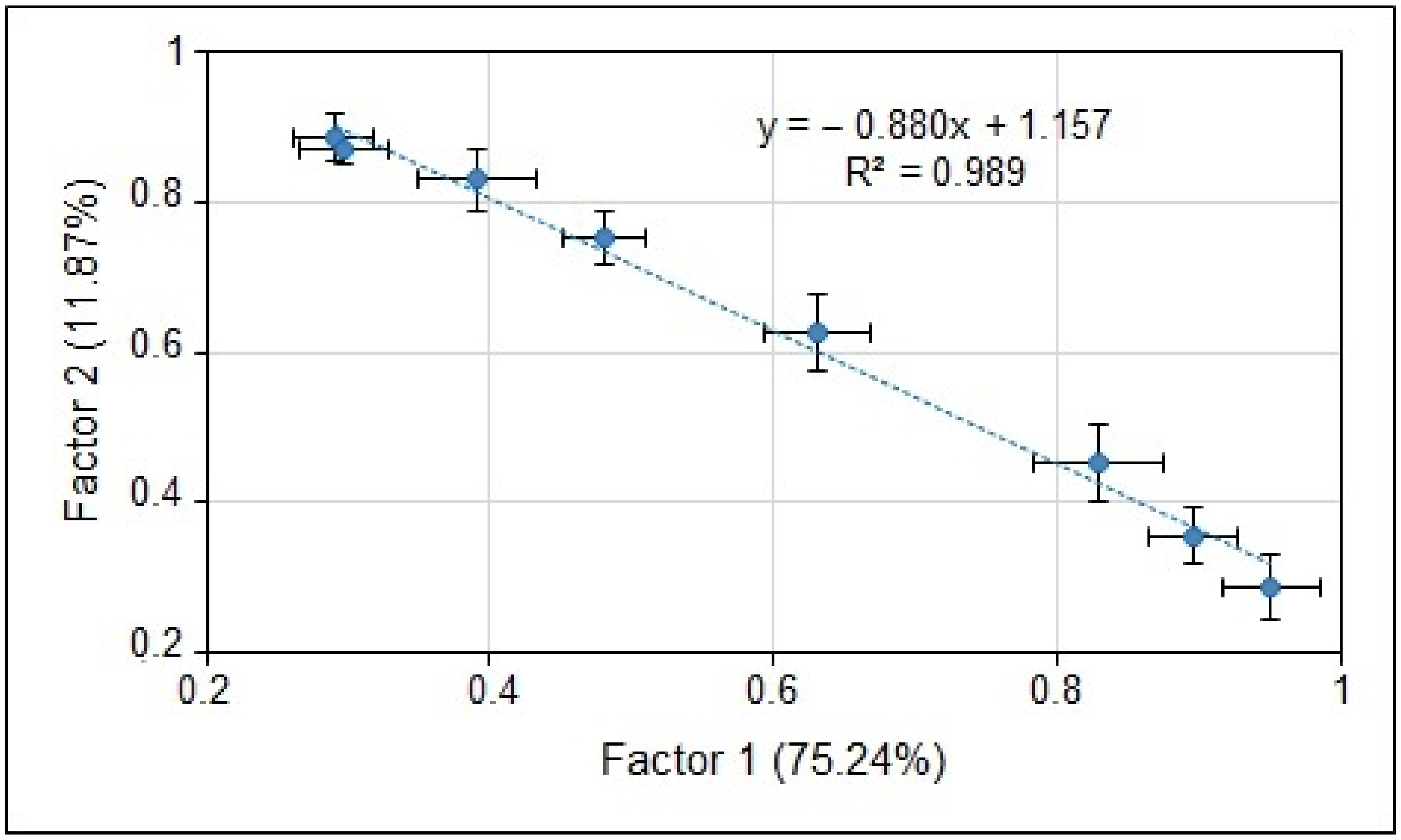
3. Results and Discussion
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lenntech, Composition of Seawater. Available online: https://www.lenntech.com/composition-seawater.htm (accessed on 15 February 2022).
- Banerjee, P.; Prasad, B. Determination of concentration of total sodium and potassium in surface and groundwater using a flame photometer. Appl. Water Sci. 2020, 10, 113. [Google Scholar] [CrossRef]
- Orzepowski, W.; Pulikowski, K. Magnesium, calcium, potassium and sodium content in ground water and surface water in arable lands in the commune of Kąty Wrocławskie. J. Elementol. 2008, 13, 605–614. [Google Scholar]
- Pain, R.W. Body fluid compartments. Anaesth. Intensive Care 1977, 5, 284–294. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pawelski, S.; Maj, S. Normy i Kliniczna Interpretacja Badań Diagnostycznych w Medycynie Wewnętrznej, 4th ed.; PZWL: Warszawa, Poland, 1987; pp. 33–110, 341–398. [Google Scholar]
- EFSA. Dietary Reference Values for Nutrients. Summary Report. Available online: https://0-doi-org.brum.beds.ac.uk/10.2903/sp.efsa.2017.e15121 (accessed on 8 December 2017).
- Pehrsson, P.; Patterson, K.; Perry, C. The Mineral Content of US Drinking and Municipal Water, USDA, Agricultural Research Service, Human Nutrition Research Center. Available online: https://www.ars.usda.gov/arsuserfiles/80400525/articles/ndbc32_watermin.pdf (accessed on 14 February 2022).
- Menéndez-Velázquez, A.; Núñez-Álvarez, C.; Del Olmo-Aguado, S.; Merayo-Lloves, J.; Fernández-Vega, A.; Osborne, N.N. Comparison of the Mineral Content of Tap Water and Bottled Waters. J. Gen. Intern. Med. 2001, 16, 168–175. [Google Scholar] [CrossRef]
- Olivier, J.; Symington, E.A.; Jonker, C.Z.; Rampedi, I.T.; Van Eeden, T.S. Comparison of the mineral composition of leaves and infusions of traditional and herbal teas. S. Afr. J. Sci. 2012, 108, 1–7. [Google Scholar] [CrossRef]
- Janda, K.; Jakubczyk, K.; Baranowska-Bosiacka, I.; Kapczuk, P.; Kochman, J.; Rębacz-Maron, E.; Gutowska, I. Mineral composition and antioxidant potential of coffee beverages depending on the brewing method. Foods 2020, 9, 121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taylor, A.; Day, M.P.; Hill, S.; Marshall, J.; Patriarca, M.; White, M. Atomic spectrometry update. Clinical and biological materials, foods and beverages. J. Anal. At. Spectrom. 2013, 28, 425–459. [Google Scholar] [CrossRef] [Green Version]
- PerkinElmer, Atomic Spectroscopy—A Guide to Selecting the Appropriate Technique and System. Available online: https://www.perkinelmer.com/lab-solutions/resources/docs/BROWorldLeaderAAICPMSICPMS.pdf (accessed on 14 February 2022).
- Miziolek, A.W.; Palleschi, V.; Schechter, I. Laser-Induced Breakdown Spectroscopy. In Fundamentals and Applications; Cambridge University Press: Cambridge, UK, 2006; pp. 1–170. [Google Scholar]
- Jzi, S.C.; Motto-Ros, V.; Trichard, F.; Markushin, Y.; Melikechi, N.; De Giacomo, A. Sample treatment and preparation for laser-induced breakdown spectroscopy. Spectrochim. Acta Part B At. Spectrosc. 2016, 115, 52–63. [Google Scholar]
- Kaiser, J.; Novotný, K.; Martind, M.Z.; Hrdlička, A.; Malina, R.; Hartl, M.; Adam, V.; Kizek, R. Trace elemental analysis by laser-induced breakdown spectroscopy-Biological applications. Surf. Sci. Rep. 2012, 67, 233–243. [Google Scholar] [CrossRef]
- Singh, V.K.; Rai, A.K. Prospects for laser-induced breakdown spectroscopy for biomedical applications: A review. Lasers Med. Sci. 2011, 26, 673–687. [Google Scholar] [CrossRef] [PubMed]
- Peng, J.; Liu, F.; Zhou, F.; Song, K.; Zhang, C.; Ye, L.; He, Y. Challenging applications for multi-element analysis by laser-induced breakdown spectroscopy in agriculture (A review). TrAC 2016, 85, 260–272. [Google Scholar] [CrossRef]
- Santos, D., Jr.; Nunes, L.C.; De Carvalho, G.G.A.; Da Silva Gomes, M.; De Souza, P.F.; De Oliveira Leme, F.; Cofani dos Santos, L.G.; Krug, F.J. Laser-induced breakdown spectroscopy for analysis of plant materials: A review. Spectrochim. Acta Part B At. Spectrosc. 2012, 71–72, 3–13. [Google Scholar] [CrossRef]
- Lin, Q.; Wang, S.; Duan, Y.; Tuchin, V.V. Ex vivo three-dimensional elemental imaging of mouse brain tissue block by laser induced breakdown spectroscopy. J. Biophotonics 2021, 14, e202000479. [Google Scholar] [CrossRef] [PubMed]
- Wei, Z.; Lin, Q.; Lazareva, E.N.; Dyachenko, P.A.; Yang, J.; Duan, Y.; Tuchin, V.V. Optical clearing of laser-induced tissue plasma. Laser Phys. Lett. 2021, 18, 085603. [Google Scholar] [CrossRef]
- Skrzeczanowski, W.; Długaszek, M. Al and Si quantitative analysis in aqueous solutions by LIBS method. Talanta 2021, 225, 121916. [Google Scholar] [CrossRef] [PubMed]
- Panek, T. Statistical Methods in Multivariate Comparative Analysis; SGH Warsaw School of Economics: Warsaw, Poland, 2009; pp. 175–225. (In Polish) [Google Scholar]
- Pořízka, P.; Klus, J.; Kepes, E.; Prochazka, D.; Hahn, D.; Kaiser, J. On the utilization of principal component analysis in laser-induced breakdown spectroscopy data analysis, a review. Spectrochim. Acta Part B At. Spectrosc. 2018, 148, 65–82. [Google Scholar] [CrossRef]
- Golik, S.S.; Bukin, O.A.; Il’in, A.A.; Sokolova, E.B.; Kolesnikov, A.V.; Babiy, M.Y.; Kul’chin, Y.N.; Gal’chenko, A.A. Determination of detection limits for elements in water by femtosecond laser-induced breakdown spectroscopy. J. Appl. Spectrosc. 2012, 79, 471–476. [Google Scholar] [CrossRef]
- Huang, J.-S.; Ke, C.-B.; Huang, L.-S.; Lin, K.-C. The correlation between ion production and emission intensity in the laser-induced breakdown spectroscopy of liquid droplets. Spectrochim. Acta Part B At. Spectrosc. 2002, 57, 35–48. [Google Scholar] [CrossRef]
- Zhu, D.; Wu, L.; Wang, B.; Chen, J.; Lu, J.; Ni, X. Determination of Ca and Mg in aqueous solution by laser-induced breakdown spectroscopy using absorbent paper substrates. Appl. Opt. 2011, 50, 5695–5699. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Units | K | Na | Ca | Mg | Refs. | |
|---|---|---|---|---|---|---|
| Sea water | mg/L | 10556 | 380 | 400 | 1262 | [1] |
| Surface water | mg/L | 16–63 | 7–24 | 58–178 * | 58–178 * | [2,3] * |
| Groundwater | mg/L | 6–27 | 18–110 | 162–280 | 25–74 | [3] |
| Intracellular fluids | mmol/L | 156 | 10 | [4] | ||
| Interstitial fluids | mmol/L | 3.8 | 144.0 | [4] | ||
| Serum | mEq/L | 4 | 142 | 5 | 2 | [5] |
| Red cells | mEq/L | 85.78–107.95 | 3.98–19.67 | 0.61–1.97 | 1.6–6.70 | [5] |
| 24h urine excretion | mEq/L | 25–100 | 120–220 | 100–250 | 4–16 | [5] |
| Units | K | Na | Ca | Mg | Refs. | |
|---|---|---|---|---|---|---|
| Tap water | mg/100 g | nd 1–20.4 | 0.1–39.1 | nd 1–10 | nd 1–6 | [7] |
| Low-mineralized waters | mg/L | 1–56 | 4–15 | 1–110 | [8] | |
| High-mineralized waters | mg/L | 900–1419 | 5–176 | 4–60 | [8] | |
| Black tea infusion | mg/250 mL | 42.96 | 0.23 | 0.58 | 2.10 | [9] |
| Coffee infusion | mg/L | 887.37–1547.70 | 24.74–27.81 | 16.34–25.71 | 77.15–116.30 | [10] |
| K | Na | Ca | Mg | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| CRM | LIBS | Δ, % | CRM | LIBS | Δ, % | CRM | LIBS | Δ, % | CRM | LIBS | Δ, % | |
| SRM 1577b Bovine Liver | 92.8 | 104.6 ± 52.0 | 13 | 22.6 | 28.8 ± 6.6 | 27 | 1.1 | - | - | 5.6 | 5.3 | 5 |
| NCT-TL-1 Tea Leaves | 165.0 | 157.0 ± 51.0 | 5 | 0.2 | - | - | 56.5 | 45.2 | 20 | 21.8 | 19.8 | 9 |
| NCS ZC91002 Human Hair | 0.1 | - | - | 2.8 | 3.4 ± 0.9 | 21 | 10.7 | 11.35 | 6 | 1.0 | 1.7 | 70 |
| LOD (μg/mL) | 2.8 | 3.4 | 3.1 | 3.3 | ||||||||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Skrzeczanowski, W.; Długaszek, M. Application of Laser-Induced Breakdown Spectroscopy in the Quantitative Analysis of Elements—K, Na, Ca, and Mg in Liquid Solutions. Materials 2022, 15, 3736. https://0-doi-org.brum.beds.ac.uk/10.3390/ma15103736
Skrzeczanowski W, Długaszek M. Application of Laser-Induced Breakdown Spectroscopy in the Quantitative Analysis of Elements—K, Na, Ca, and Mg in Liquid Solutions. Materials. 2022; 15(10):3736. https://0-doi-org.brum.beds.ac.uk/10.3390/ma15103736
Chicago/Turabian StyleSkrzeczanowski, Wojciech, and Maria Długaszek. 2022. "Application of Laser-Induced Breakdown Spectroscopy in the Quantitative Analysis of Elements—K, Na, Ca, and Mg in Liquid Solutions" Materials 15, no. 10: 3736. https://0-doi-org.brum.beds.ac.uk/10.3390/ma15103736