Synthesis, NMR Characterization, and Antileukemic Activity of N-Nonanoylpiperazinyl-5α-Androstane-3α,17β-Diol A-Ring Derivatives



Abstract
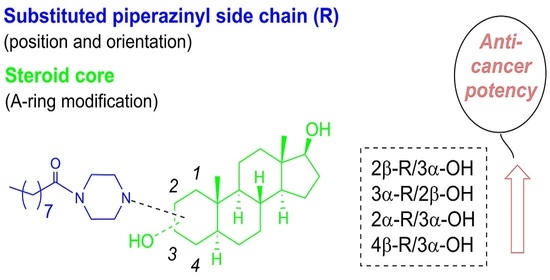
:
1. Introduction
2. Results and Discussion
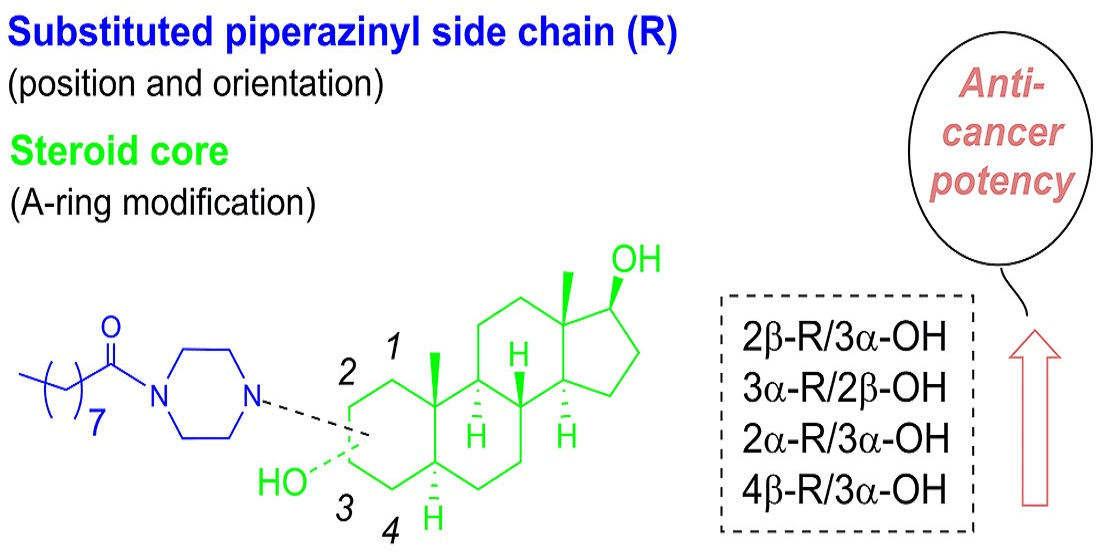
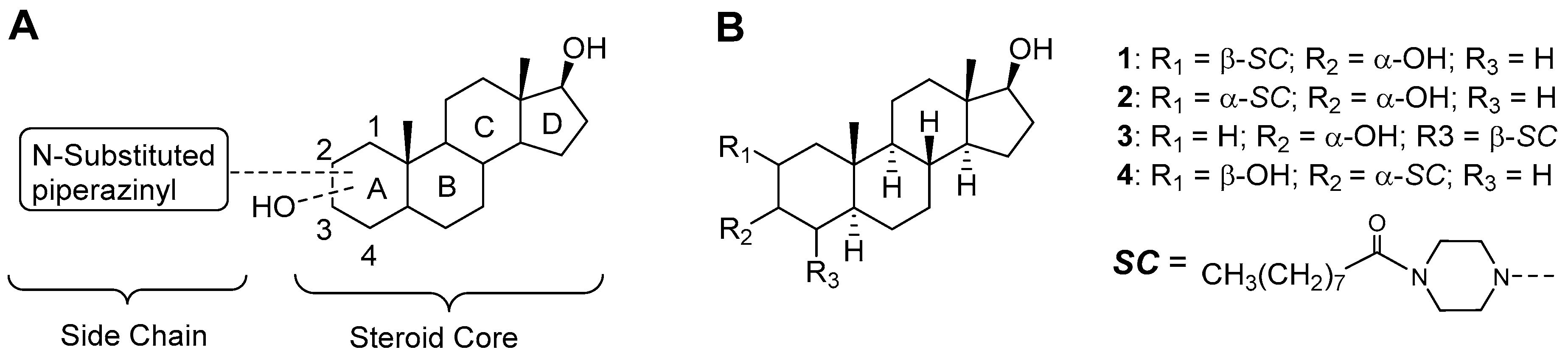
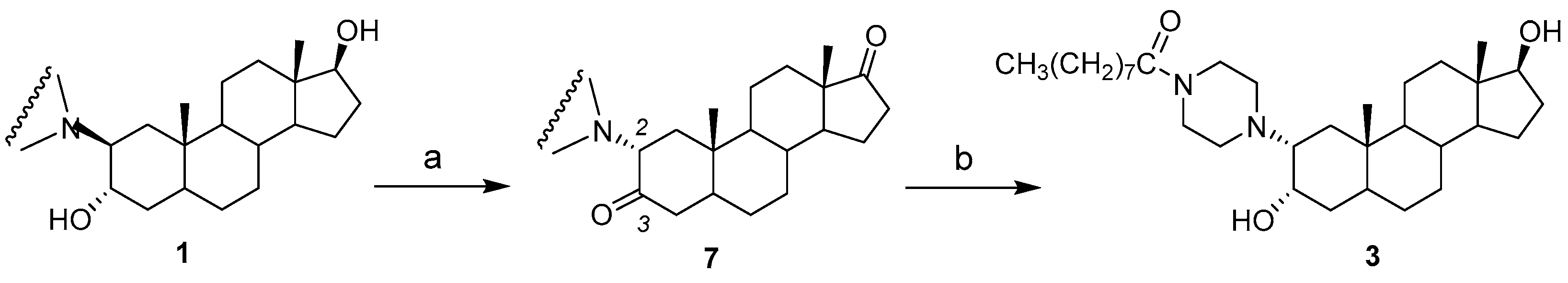
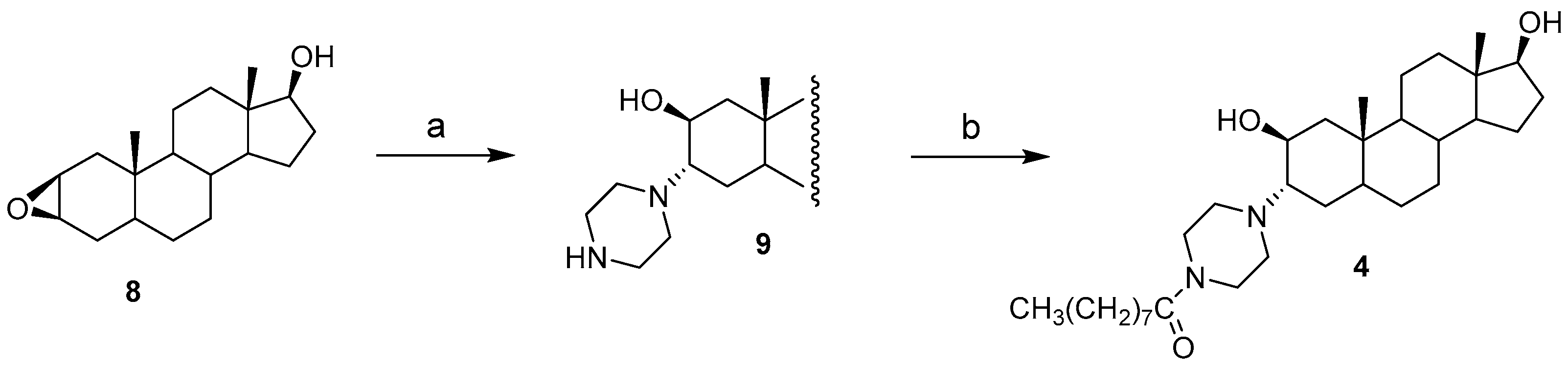
2.1. Chemical Synthesis of Compounds 1–4
2.2. Nuclear Magnetic Resonance (NMR) Characterization
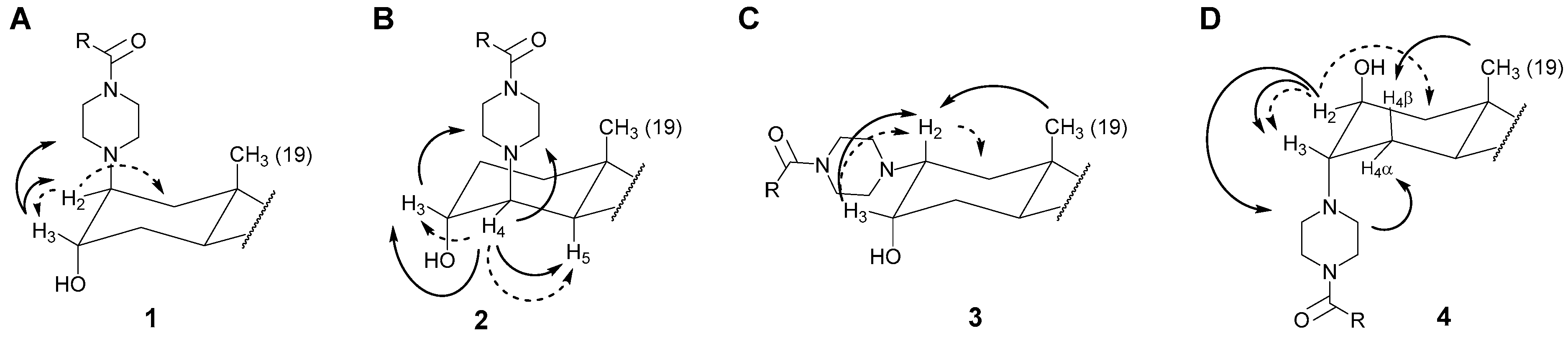
2.2.1. Assignments of Carbons and Protons
2.2.2. Positioning and Orientation of A-Ring Sidechain and OH
2.3. Inhibition of HL-60 Cell Proliferation
3. Materials and Methods
3.1. General
3.2. Synthesis of Compounds 1 and 2
3.3. Synthesis of Compound 3
3.4. Synthesis of Compound 4
3.5. Cell Proliferation Assay
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- He, Q.; Na, X. The effects and mechanisms of a novel 2-aminosteroid on murine WEHI-3B leukemia cells in vitro and in vivo. Leukemia Res. 2001, 25, 455–461. [Google Scholar] [CrossRef]
- Thibeault, D.; Roy, J.; DeRoy, P.; Poirier, D. Chemical synthesis of 2β-amino-5α-androstane-3α,17β-diol N-derivatives and their antiproliferative effect on HL-60 human leukemia cells. Bioorg. Med. Chem. 2008, 16, 5062–5077. [Google Scholar] [CrossRef] [PubMed]
- Roy, J.; Maltais, R.; Jegham, H.; Poirier, D. Libraries of 2β-(N-substituted piperazino)-5α-androstane-3α,17β-diols: Chemical synthesis and cytotoxic effects on human leukemia HL-60 cells and on normal lymphocytes. Mol. Divers. 2011, 15, 317–339. [Google Scholar] [CrossRef] [PubMed]
- Ayan, D.; Maltais, R.; Hospital, A.; Poirier, D. Chemical synthesis, cytotoxicity, selectivity and bioavailability of 5alpha-androstane-3alpha,17beta-diol derivatives. Bioorg. Med. Chem. 2014, 22, 5847–5859. [Google Scholar] [CrossRef] [PubMed]
- Gupta, A.; Kumar, B.S.; Negi, A.S. Current status on development of steroids as anticancer agents. J. Steroid Biochem. Mol. Biol. 2013, 137, 242–270. [Google Scholar] [CrossRef] [PubMed]
- Jegham, H.; Maltais, R.; Roy, J.; Doillon, C.; Poirier, D. Biological evaluation of a new family of aminosteroids that display a selective toxicity for various malignant cell lines. Anti-Cancer Drugs 2012, 23, 803–814. [Google Scholar] [CrossRef] [PubMed]
- Kenmogne, L.C.; Ayan, D.; Roy, J.; Maltais, R.; Poirier, D. The aminosteroid derivative RM-133 shows in vitro and in vivo antitumor activity in human ovarian and pancreatic cancers. PLoS ONE 2015, 10, e0144890. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maltais, R.; Hospital, A.; Delhomme, A.; Roy, J.; Poirier, D. Chemical synthesis, NMR analysis and evaluation on a cancer xenograft model (HL-60) of the aminosteroid derivative RM-133. Steroids 2014, 82, 68–76. [Google Scholar] [CrossRef] [PubMed]
- Roy, J.; DeRoy, P.; Poirier, D. 2β-(N-substituted piperazino)-5α-androstane-3α,17β-diols: Parallel solid-phase synthesis and antiproliferative activity on human leukemia HL-60 mcells. J. Comb. Chem. 2007, 9, 347–358. [Google Scholar] [CrossRef] [PubMed]
- Thibeault, D.; Poirier, D. An efficient method for the regioselective aminolysis of 2,3α-steroidal epoxyde. Synlett 2003, 8, 1192–1194. [Google Scholar]
- Barton, D.H.R. The stereochemistry of cyclohexane derivatives. J. Chem. Soc. 1953, 1027–1040. [Google Scholar] [CrossRef]
- Matthews, G.J.; Hassner, A. Organic Reactions in Organic Chemistry; Fried, J., Edwards, J.A., Eds.; Van Nostrand Reinhold Company: New York, NY, USA, 1972; Volume 2, pp. 1–53. [Google Scholar]
- Anderson, A.; Boyd, A.C.; Byford, A.; Campbell, A.C.; Gemmell, D.K.; Hamilton, N.M.; Hill, D.R.; Hill-Venning, C.; Lambert, J.J.; Maidment, M.S.; et al. Anestetic activity of novel water-soluble 2β-morpholinyl steroids and their modulatory effects at GABAA receptors. J. Med. Chem. 1997, 40, 1668–1681. [Google Scholar] [CrossRef]
- He, Q.; Xu, Y.H. Synthesis of 2β-(4’-methyl-1’-piperazino)-3α-hydroxyl-16,17-substituted-5α-androstanes. Acta Pharm Sin. 1992, 27, 101–106. [Google Scholar]
- Lewett, C.L.; Savage, D.S. Amino-steroids. Part III.1 2- and 3-Amino-5α-androstanes. J. Chem. Soc. 1968, 1134–1140. [Google Scholar] [CrossRef]
- Mancuso, A.J.; Huang, S.L.; Swern, D. Oxidation of long-chain and related alcohols to carbonyls by dimethylsulfoxide “activated” by oxalyl chloride. J. Org. Chem. 1978, 43, 2480–2482. [Google Scholar] [CrossRef]
- Cadot, C.; Laplante, Y.; Kamal, F.; Luu-The, V.; Poirier, D. C6-(N,N-butyl-methyl-heptanamide) derivatives of estrone and estradiol as inhibitors of type 1 17β-hydroxysteroid dehydrogenase: Chemical synthesis and biological evaluation. Bioorg. Med. Chem. 2007, 15, 714–726. [Google Scholar] [CrossRef] [PubMed]
- Blunt, J.W.; Stothers, J.B. 13C N.m.r. spectra of steroids—A survey and commentary. J. Magn. Reson. 1977, 9, 439–464. [Google Scholar] [CrossRef]
- Poirier, D.; Maltais, R. NMR-assisted structure elucidation of an anticancer steroid-β-enaminone derivative. Magnetochemistry 2017, 3, 37. [Google Scholar] [CrossRef] [Green Version]
- Tchédam-Ngatcha, B.; Trottier, M.C.; Poirier, D. 13C Nuclear magnetic resonance spectroscopy data of a variety of androsterone and epi-androsterone derivatives substituted at position 3beta or/and 3alpha. Curr. Top. Steroid Res. 2011, 8, 35–46. [Google Scholar]
- Fielding, L. 1H and 13C NMR studies of some steroidal neuromuscular blocking drugs: Solution conformations and dynamics. Magn. Reson. Chem. 1998, 36, 387–397. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}

| C and H Assignment | 1 (1H) | 1 (13C) | 2 (1H) | 2 (13C) | 3 (1H) | 3 (13C) | 4 (1H) | 4 (13C) |
|---|---|---|---|---|---|---|---|---|
| 2β-Chain- | -3α-OH | 4β-Chain- | -3α-OH | 2α-Chain- | -3α-OH | 3α-Chain- | -2β-OH | |
| CH2-1 | 1.40/1.82 | 34.5 | 1.40/1.52 | 34.0 | 1.30/1.73 | 36.9 | 1.54/1.68 | 42.4 |
| CH-2 or CH2-2 | 2.40 | 66.5 | 1.90 | 26.9 | 2.24 | 63.0 | 4.09 | 68.5 |
| CH-3 | 4.10 | 66.6 | 4.10 | 66.6 | 4.15 | 66.8 | 2.25 | 65.8 |
| CH-4 or CH2-4 | 1.35 | 34.2 | 2.34 | 70.5 | 1.55 | 36.5 | 1.42/1.82 | 26.6 |
| CH-5 | 1.66 | 40.6 | 1.77 | 45.7 | 1.60 | 40.0 | 1.48 | 41.1 |
| CH2-6 | 1.30 | 29.0 | 1.40 | 29.2 | 1.30 | 28.9 | 1.28 | 29.2 |
| CH2-7 | 0.92/1.71 | 32.7 | 0.91/1.80 | 34.0 | 0.95/1.88 | 32.8 | 0.92/1.71 | 32.8 |
| CH-8 | 1.42 | 36.6 | 1.40 | 37.0 | 1.45 | 36.6 | 1.46 | 36.5 |
| CH-9 | 0.75 | 56.9 | 0.73 | 57.4 | 0.82 | 56.3 | 0.72 | 57.1 |
| C-10 | - | 37.3 | - | 37.2 | - | 37.7 | - | 37.1 |
| CH2-11 | 1.38/1.62 | 21.8 | 1.33/1.58 | 21.1 | 1.38/1.70 | 21.6 | 1.33/1.60 | 21.7 |
| CH2-12 | 1.05/1.84 | 38.1 | 1.04/1.84 | 38.1 | 1.10/1.85 | 38.0 | 1.05/1.84 | 38.1 |
| C-13 | - | 44.2 | - | 44.0 | - | 44.1 | - | 44.2 |
| CH-14 | 0.95 | 52.4 | 0.93 | 52.4 | 1.00 | 52.4 | 0.96 | 52.4 |
| CH2-15 | 1.25/1.60 | 24.3 | 1.27/1.62 | 24.3 | 1.30/1.62 | 24.3 | 1.26/1.60 | 24.3 |
| CH2-16 | 1.46/1.98 | 30.6 | 1.45/1.98 | 30.6 | 1.50/2.00 | 30.6 | 1.47/1.98 | 30.6 |
| CH-17 | 3.57 | 82.5 | 3.57 | 82.5 | 3.57 | 82.5 | 3.57 | 82.5 |
| CH3-18 | 0.74 | 11.7 | 0.74 | 11.6 | 0.75 | 11.7 | 0.74 | 11.7 |
| CH3-19 | 1.04 | 14.6 | 1.11 | 14.5 | 0.86 | 12.9 | 1.05 | 15.6 |
| CH2-2′/CH2-6′ | 2.53/2.63 | 51.4/51.9 | 2.63/2.68 | 53.8/54.4 | 2.65/2.70 | 50.9/51.3 | 2.50/2.56 | 51.4/51.9 |
| CH2-3′/CH2-5′ | 3.57 | 43.1/47.2 | 3.50/3.57 | 43.6/47.7 | 3.58 | 43.0/47.1 | 3.57 | 43.1/47.2 |
| C-1″ | - | 174.1 | - | 174.1 | - | 174.1 | - | 174.0 |
| CH2-2″ | 2.40 | 34.0 | 2.38 | 34.0 | 2.40 | 34.0 | 2.40 | 34.0 |
| CH2-3″ | 1.61 | 26.6 | 1.61 | 26.6 | 1.62 | 26.6 | 1.61 | 26.6 |
| CH2-4″ | 1.34 | 30.3 | 1.34 | 30.3 | 1.36 | 30.3 | 1.35 | 30.3 |
| CH2-5″ | 1.34 | 30.5 | 1.34 | 30.5 | 1.36 | 30.5 | 1.35 | 30.5 |
| CH2-6″ | 1.34 | 30.5 | 1.34 | 30.4 | 1.36 | 30.4 | 1.35 | 30.4 |
| CH2-7″ | 1.34 | 33.0 | 1.34 | 33.0 | 1.36 | 33.0 | 1.33 | 33.0 |
| CH2-8″ | 1.34 | 23.7 | 1.34 | 23.7 | 1.35 | 23.7 | 1.33 | 23.7 |
| CH2-9″ | 0.92 | 14.4 | 0.92 | 14.4 | 0.92 | 14.4 | 0.92 | 14.5 |
| Compound | 1 | 2 | 3 | 4 |
|---|---|---|---|---|
| Structure |  |  |  |  |
| Substitution | 2β-R (axial) 3α-OH (axial) | 4β-R (axial) 3α-OH (axial) | 2α-R (equatorial) 3α-OH (axial) | 3α-R (axial) 2β-OH (axial) |
| AA (%)a | 82 | 1 | 42 | 51 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Poirier, D.; Raad, I.; Roy, J.; Maltais, R. Synthesis, NMR Characterization, and Antileukemic Activity of N-Nonanoylpiperazinyl-5α-Androstane-3α,17β-Diol A-Ring Derivatives. Magnetochemistry 2021, 7, 3. https://0-doi-org.brum.beds.ac.uk/10.3390/magnetochemistry7010003
Poirier D, Raad I, Roy J, Maltais R. Synthesis, NMR Characterization, and Antileukemic Activity of N-Nonanoylpiperazinyl-5α-Androstane-3α,17β-Diol A-Ring Derivatives. Magnetochemistry. 2021; 7(1):3. https://0-doi-org.brum.beds.ac.uk/10.3390/magnetochemistry7010003
Chicago/Turabian StylePoirier, Donald, Imad Raad, Jenny Roy, and René Maltais. 2021. "Synthesis, NMR Characterization, and Antileukemic Activity of N-Nonanoylpiperazinyl-5α-Androstane-3α,17β-Diol A-Ring Derivatives" Magnetochemistry 7, no. 1: 3. https://0-doi-org.brum.beds.ac.uk/10.3390/magnetochemistry7010003