
Chiral Radical Cation Salts of Me-EDT-TTF and DM-EDT-TTF with Octahedral, Linear and Tetrahedral Monoanions †

 , , , and
, , , and
Abstract
:
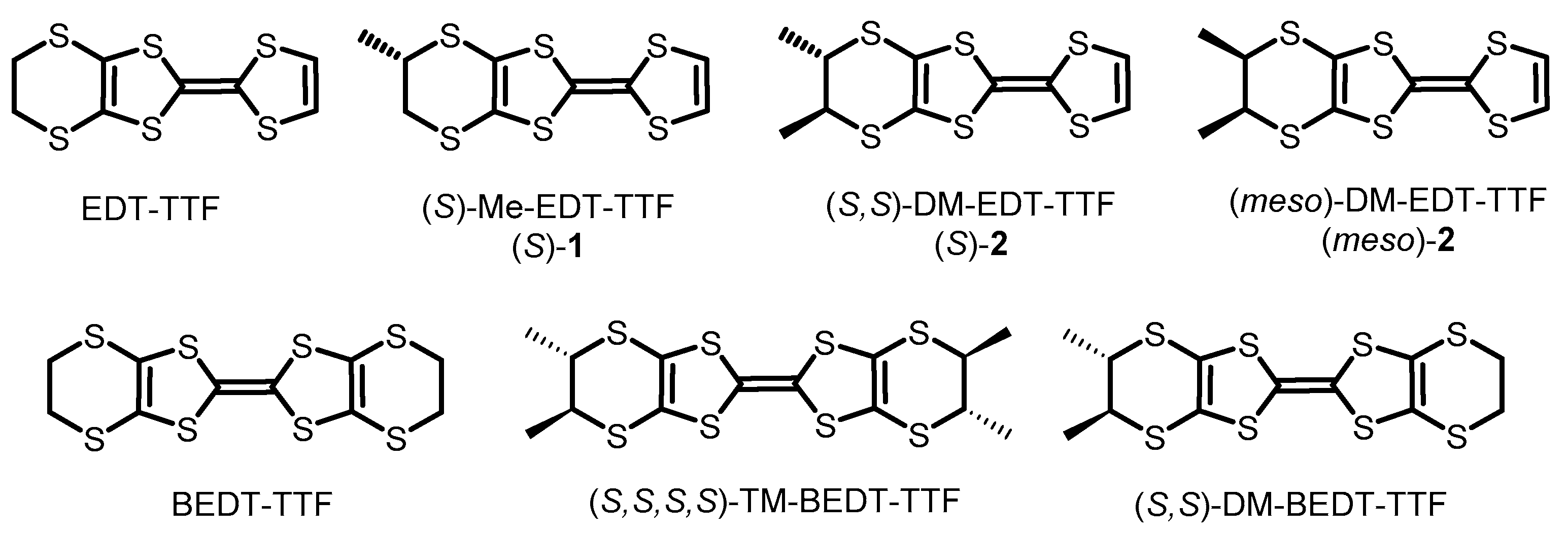
1. Introduction
2. Results and Discussion
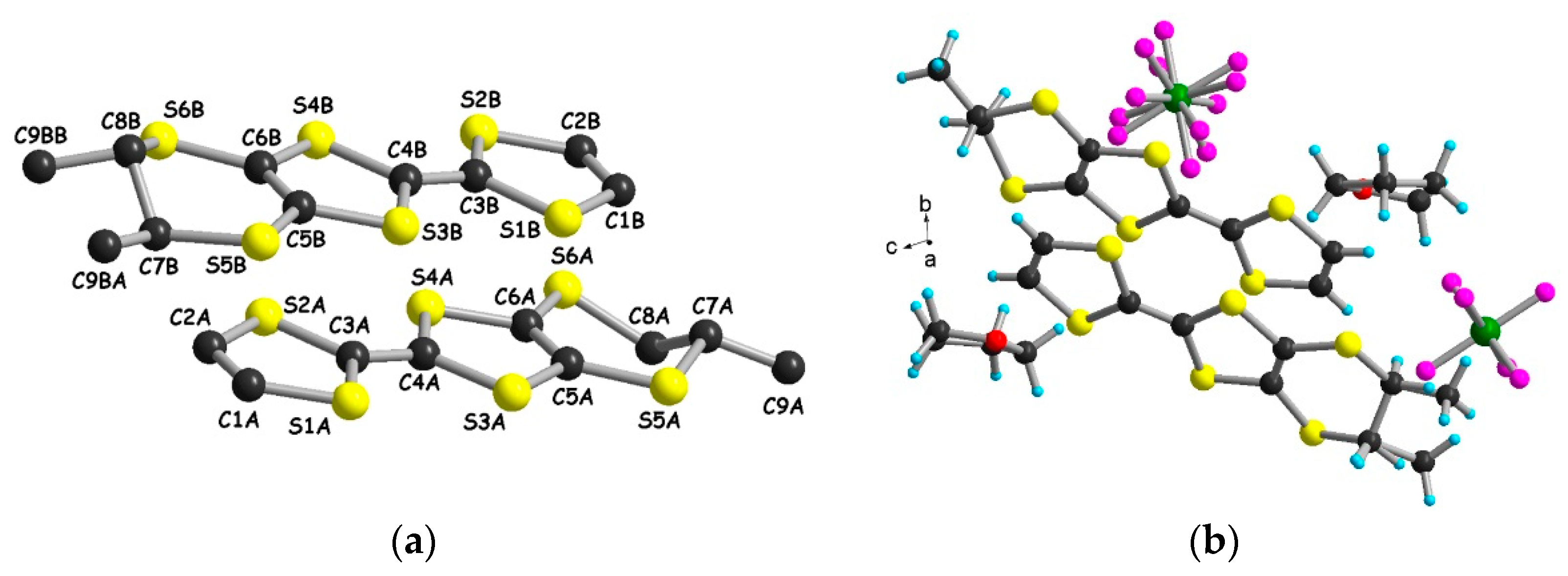
2.1. Radical Cation Salts of Me-EDT-TTF (1) with the AsF6– Anion
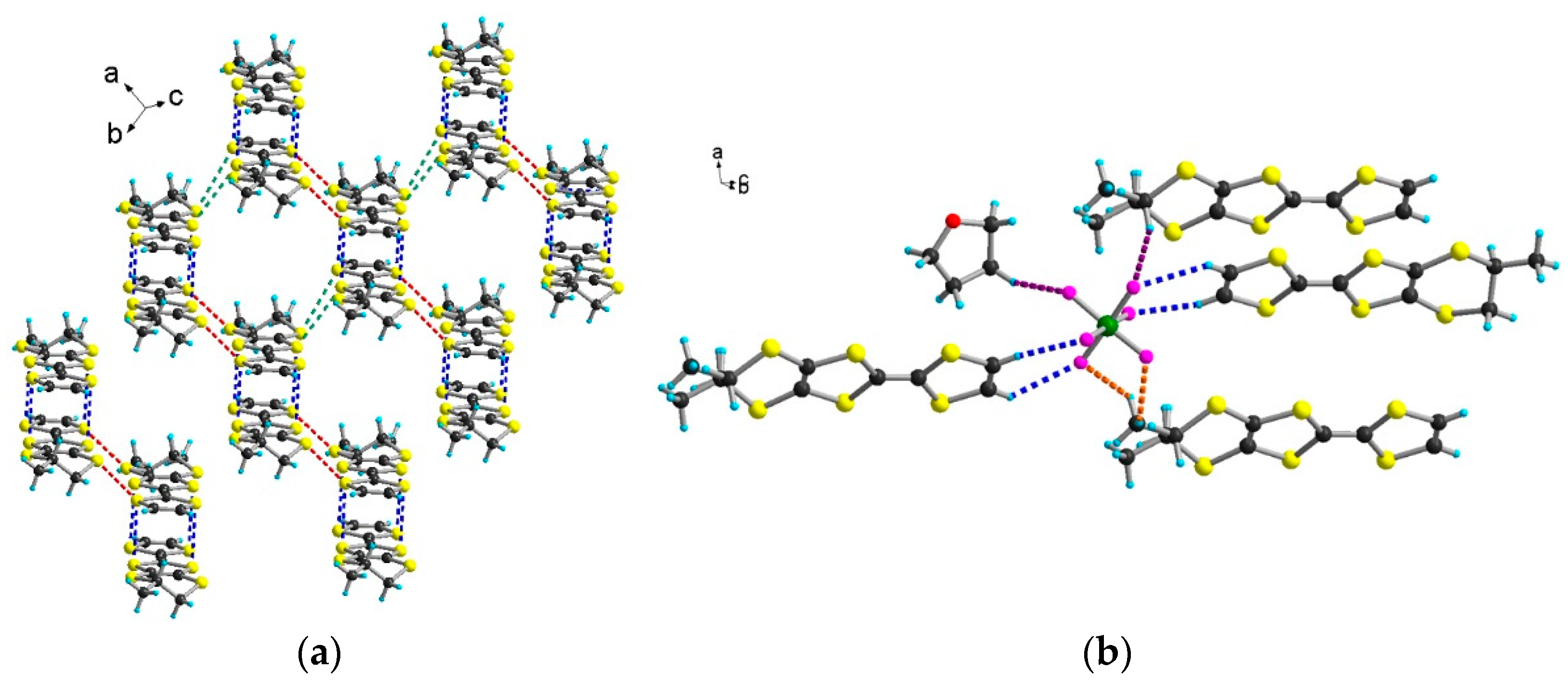
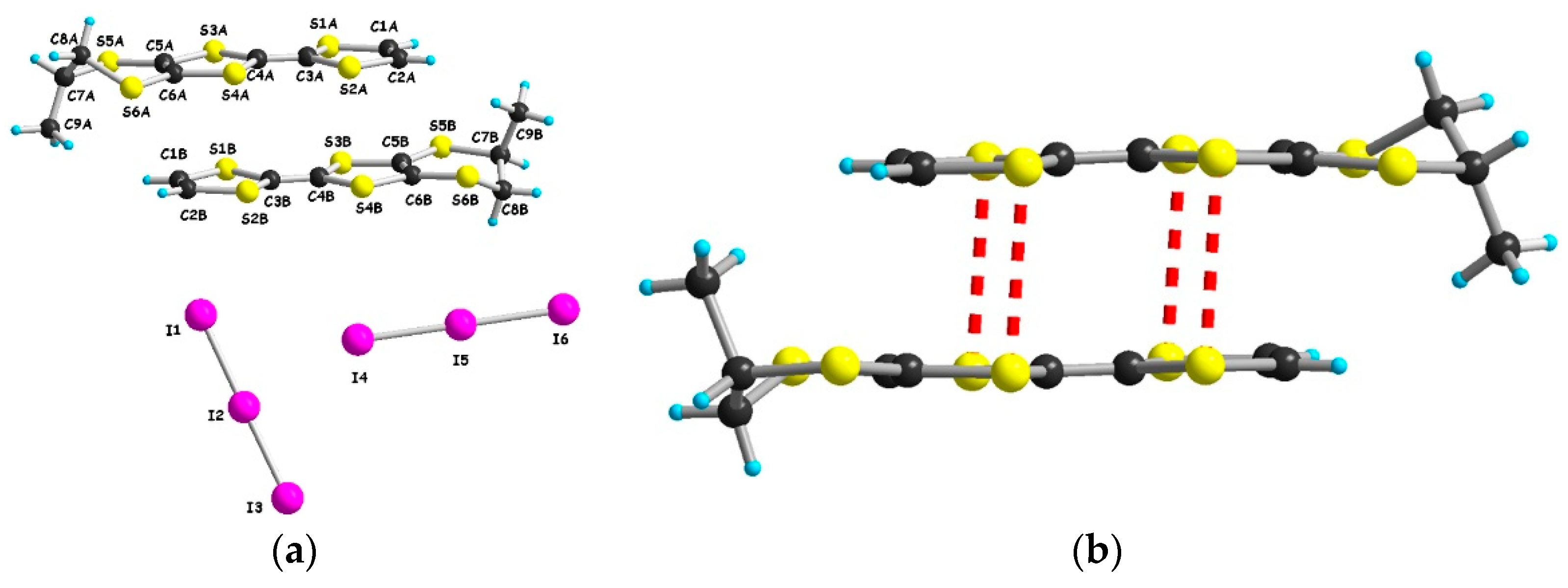
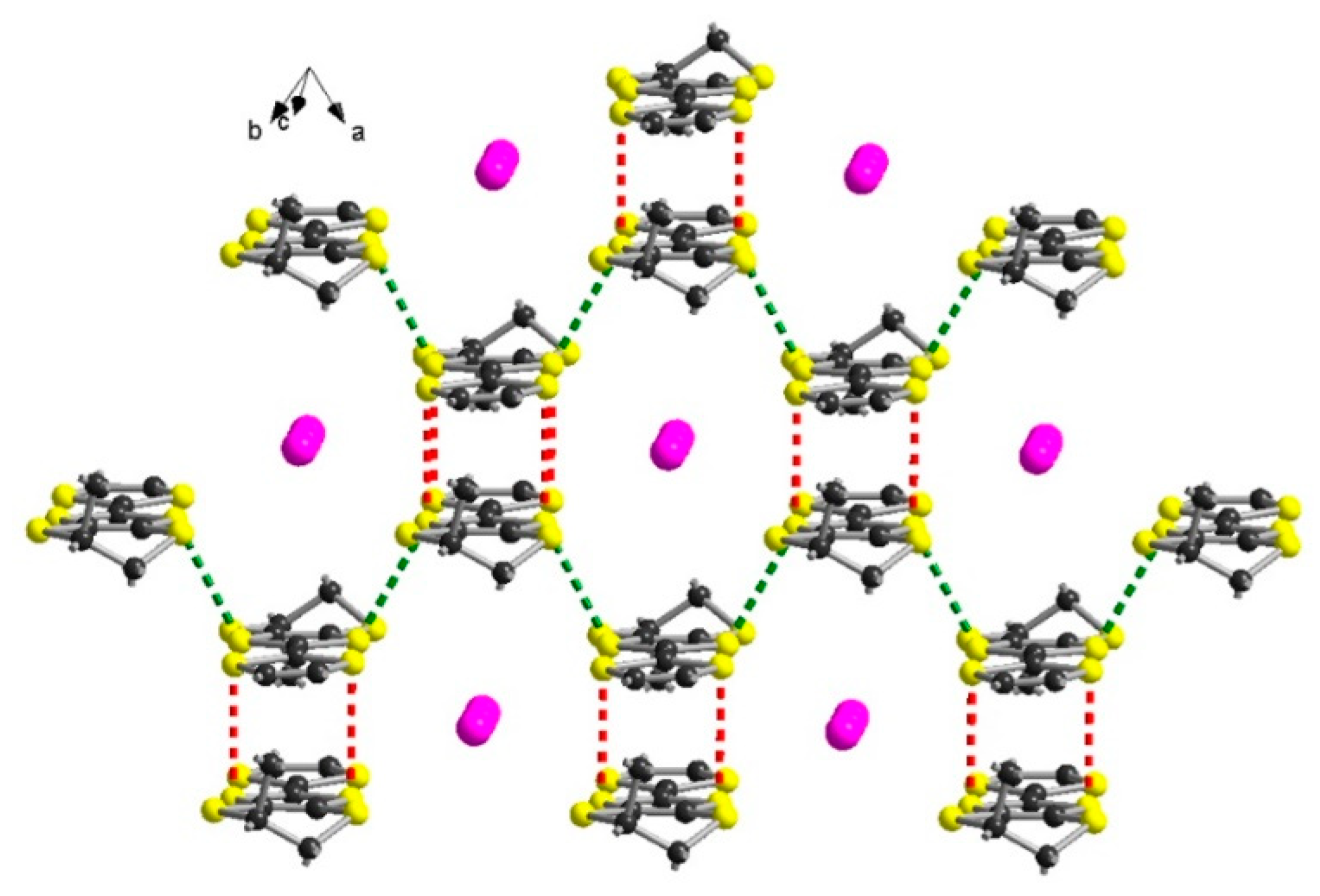
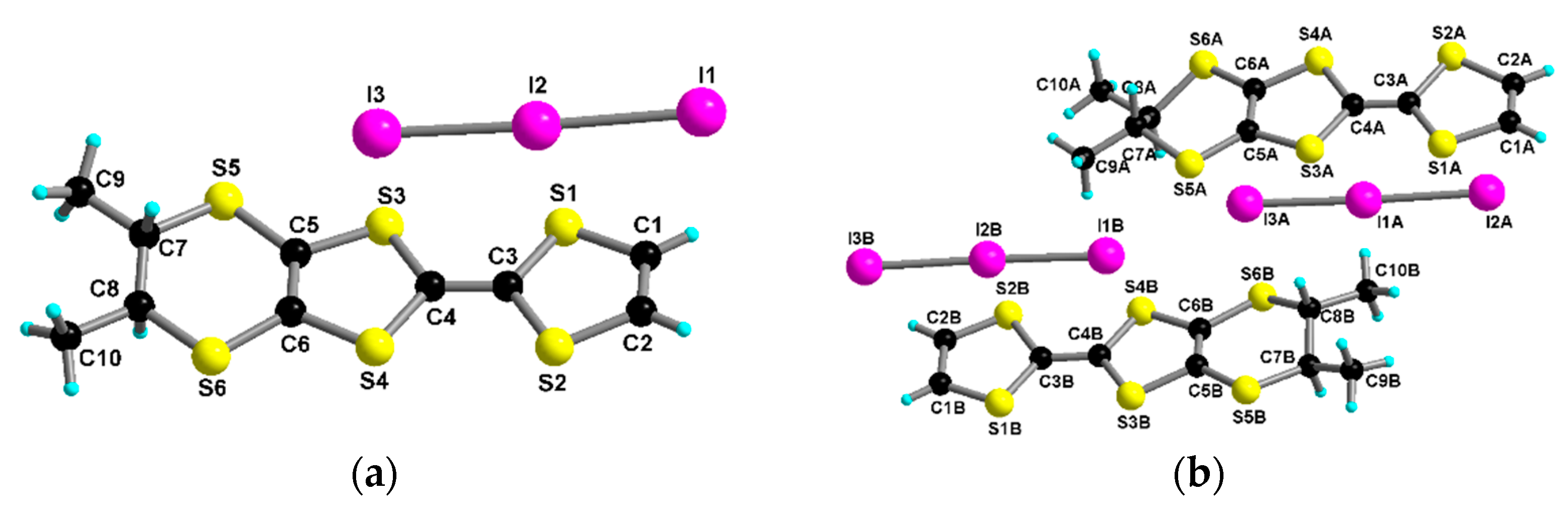
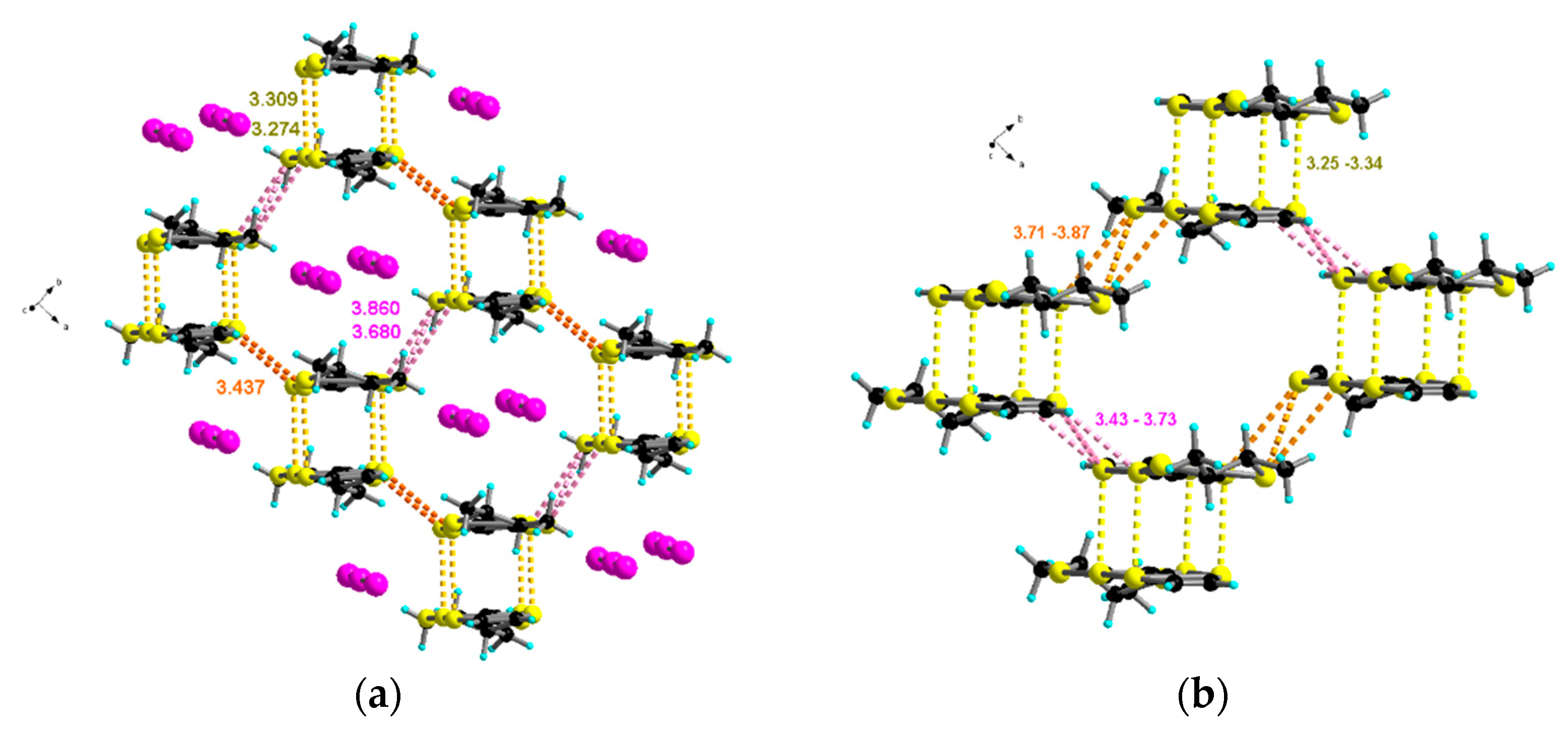
2.2. Radical Cation Salts of Me-EDT-TTF (1) and DM-EDT-TTF (2) with the I3– Anion
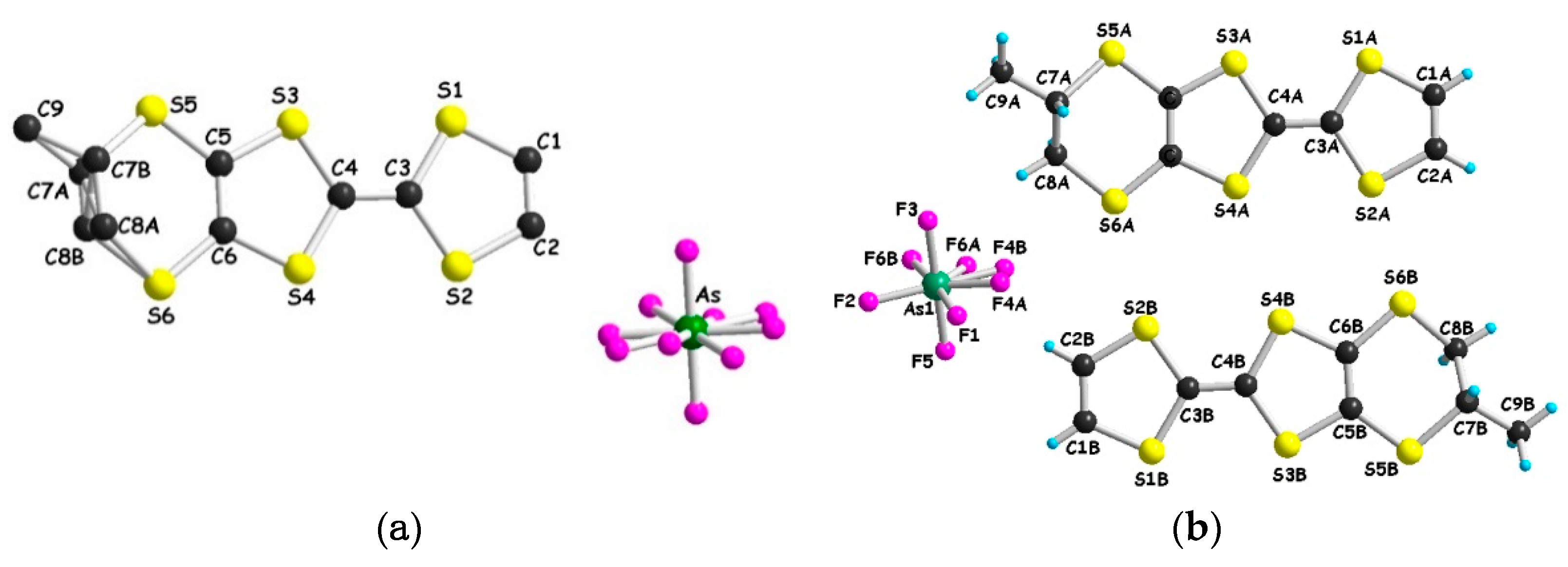
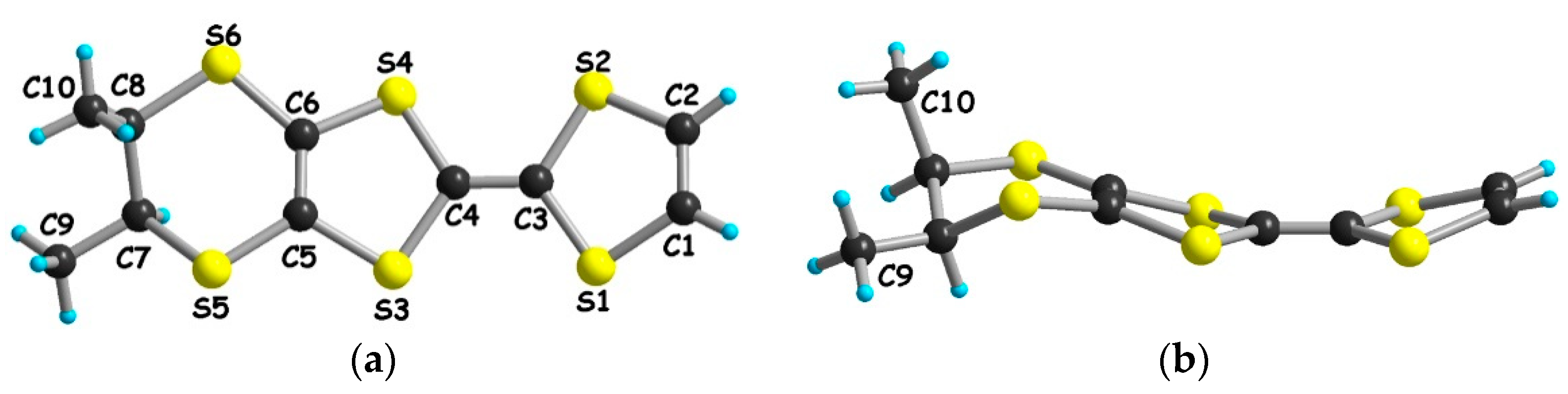
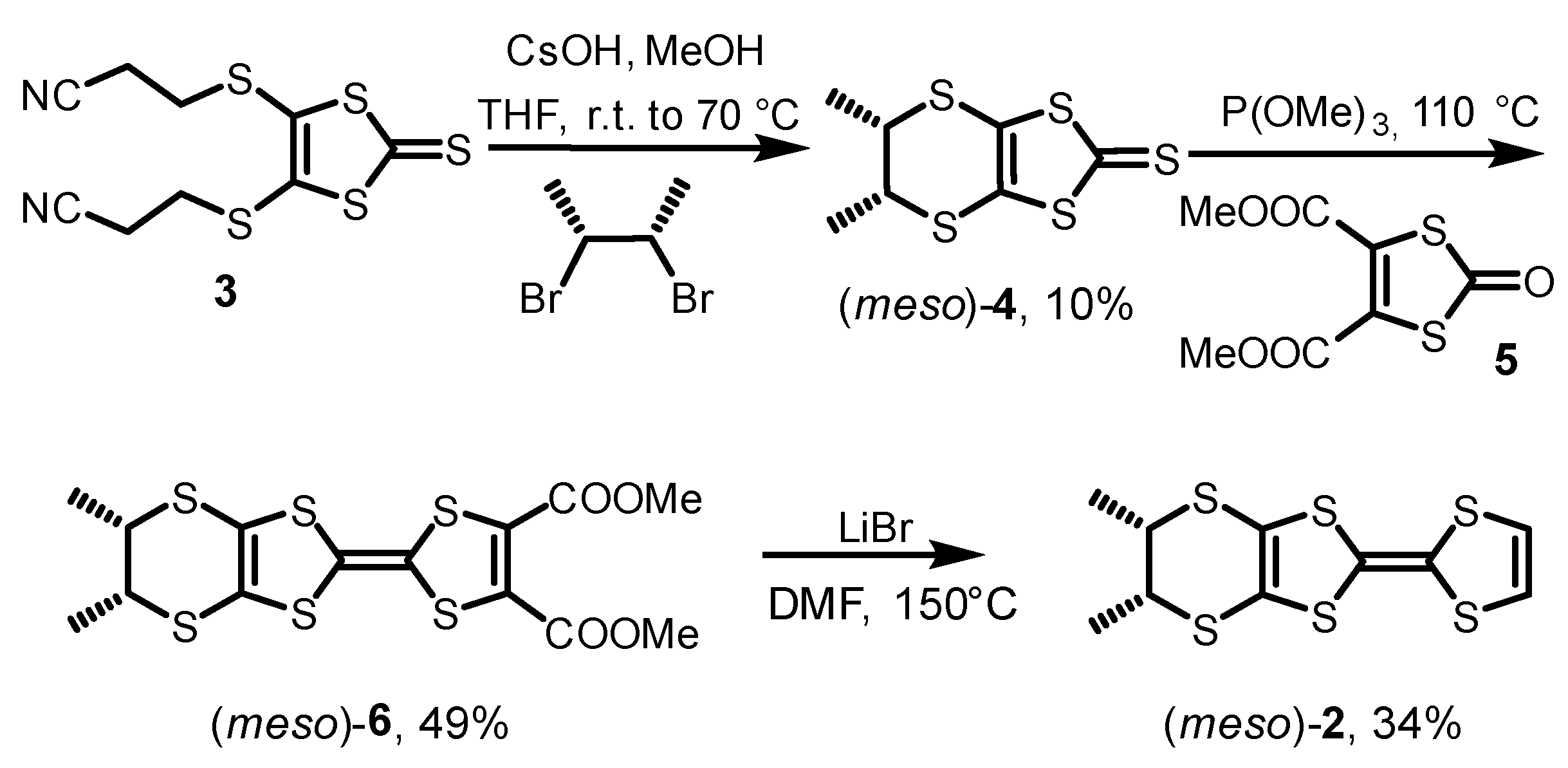
2.3. Synthesis and Structure of (meso)-DM-EDT-TTF
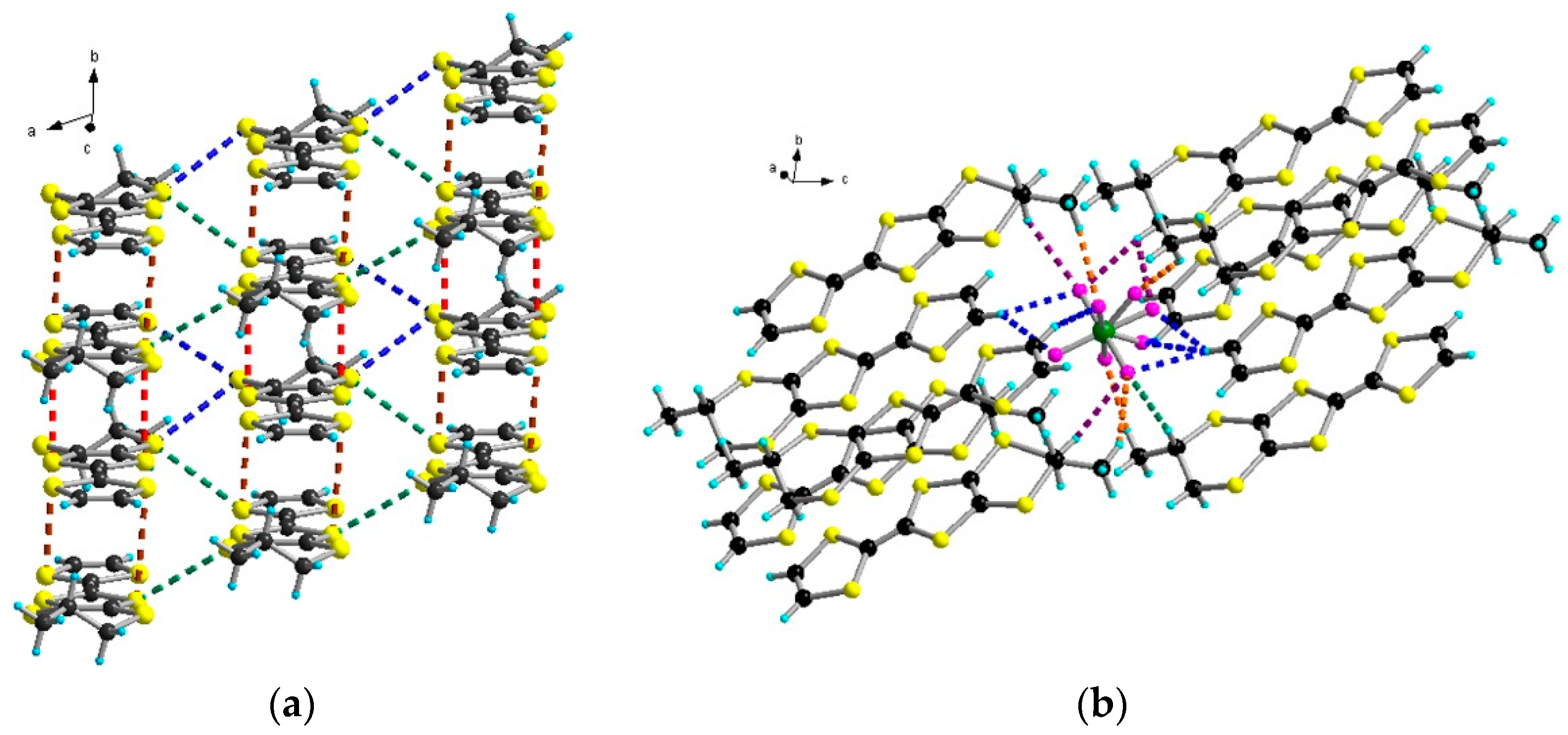
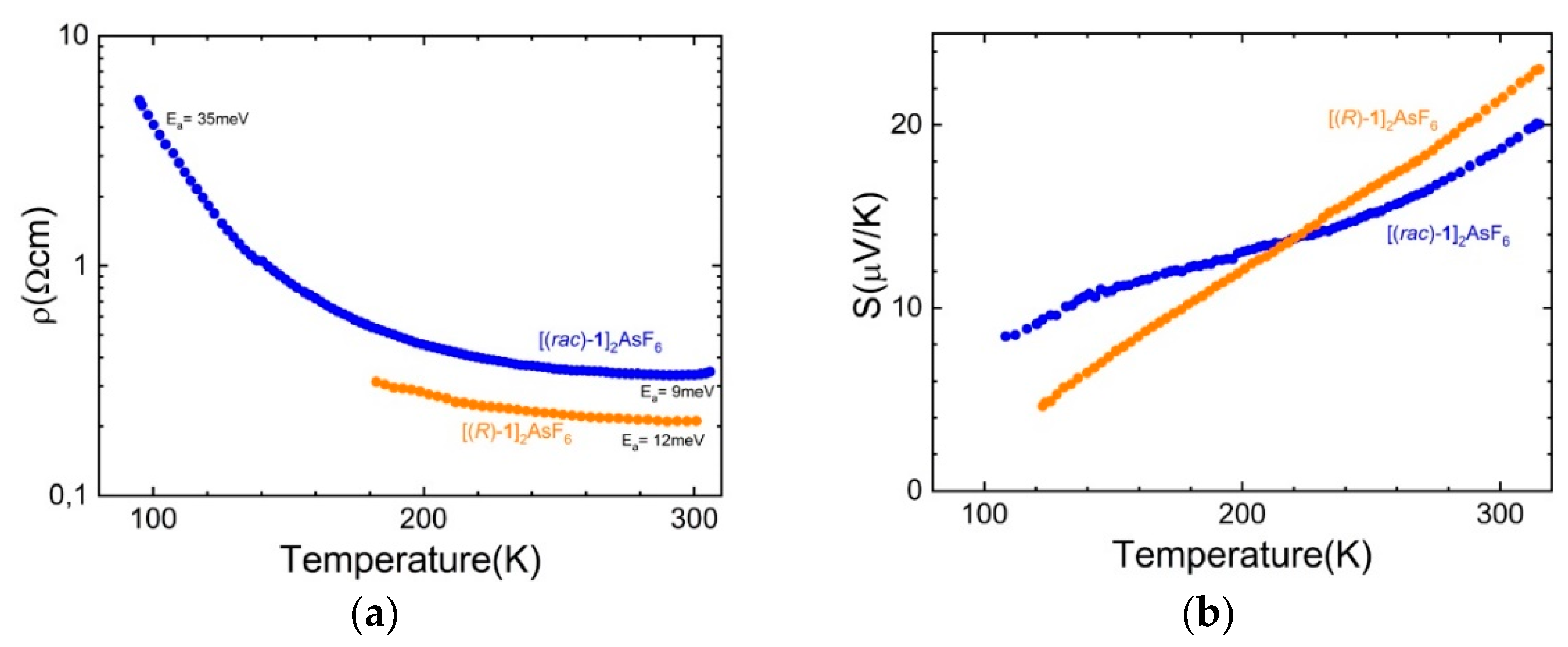
2.4. Radical Cation Salts of (meso)-DM-EDT-TTF
3. Conclusions
4. Materials and Methods
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Acknowledgments
Conflicts of Interest
References
- Avarvari, N.; Wallis, J.D. Strategies Towards Chiral Molecular Conductors. J. Mater. Chem. 2009, 19, 4061–4076. [Google Scholar] [CrossRef] [Green Version]
- Pop, F.; Zigon, N.; Avarvari, N. Main-Group-Based Electro- and Photoactive Chiral Materials. Chem. Rev. 2019, 119, 8435–8478. [Google Scholar] [CrossRef]
- Mroweh, N.; Auban-Senzier, P.; Vanthuyne, N.; Canadell, E.; Avarvari, N. Chiral EDT-TTF precursors with one stereogenic centre: Substituent size modulation of the conducting properties in the (R-EDT-TTF)2PF6 (R = Me or Et) series. J. Mater. Chem. C 2019, 7, 12664–12673. [Google Scholar] [CrossRef] [Green Version]
- Mroweh, N.; Auban-Senzier, P.; Vanthuyne, N.; Lopes, E.B.; Almeida, M.; Canadell, E.; Avarvari, N. Chiral Conducting Me-EDT-TTF and Et-EDT-TTF Based Radical Cation Salts with the Perchlorate Anion. Crystals 2020, 10, 1069. [Google Scholar] [CrossRef]
- Pop, F.; Auban-Senzier, P.; Frąckowiak, A.; Ptaszyński, K.; Olejniczak, I.; Wallis, J.D.; Canadell, E.; Avarvari, N. Chirality Driven Metallic versus Semiconducting Behavior in a Complete Series of Radical Cation Salts Based on Dimethyl-Ethylenedithio-Tetrathiafulvalene (DM-EDT-TTF). J. Am. Chem. Soc. 2013, 135, 17176–17186. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rikken, G.L.J.A.; Fölling, J.; Wyder, P. Electrical Magnetochiral Anisotropy. Phys. Rev. Lett. 2001, 87, 236602. [Google Scholar] [CrossRef] [PubMed]
- Krstić, V.; Roth, S.; Burghard, M.; Kern, K.; Rikken, G.L.J.A. Magneto-Chiral Anisotropy in Charge Transport Through Single-Walled Carbon Nanotubes. J. Chem. Phys. 2002, 117, 11315–11319. [Google Scholar] [CrossRef] [Green Version]
- Pop, F.; Auban-Senzier, P.; Canadell, E.; Rikken, G.L.J.A.; Avarvari, N. Electrical magneto-chiral anisotropy in a bulk chiral molecular conductor. Nat. Commun. 2014, 5, 3757. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pop, F.; Auban-Senzier, P.; Canadell, E.; Avarvari, N. Anion size control of the packing in the metallic versus semiconducting chiral radical cation salts (DM-EDT-TTF)2XF6 (X = P, As, Sb). Chem. Commun. 2016, 52, 12438–12441. [Google Scholar] [CrossRef] [Green Version]
- Mroweh, N.; Mézière, C.; Allain, M.; Auban-Senzier, P.; Canadell, E.; Avarvari, N. Conservation of structural arrangements and 3:1 stoichiometry in a series of crystalline conductors of TMTTF, TMTSF, BEDT-TTF, and chiral DM-EDT-TTF with the oxo-bis[pentafluorotantalate(V)] dianion. Chem. Sci. 2020, 11, 10078–10091. [Google Scholar] [CrossRef]
- Yagubskii, E.B.; Shchegolev, I.F.; Laukhin, V.N.; Kononovich, P.A.; Kartsovnik, M.V.; Zvarykina, A.V.; Buravov, L.I. Normal-pressure superconductivity in an organic metal (BEDT-TTF)2I3 [bis (ethylene dithiolo) tetrathiofulvalene triiodide]. JETP Lett. 1984, 39, 12–16. [Google Scholar]
- Crabtree, G.W.; Carlson, K.D.; Hall, L.N.; Copps, P.T.; Wang, H.H.; Emge, T.J.; Beno, M.A.; Williams, J.M. Superconductivity at ambient pressure in di[bis(ethylenedithio)tetrathiafulvalene] triiodide, (BEDT-TTF)2I3. Phys. Rev. B 1984, 30, 2958–2960. [Google Scholar] [CrossRef]
- Tokumoto, M.; Murata, K.; Bando, H.; Anzai, H.; Saito, G.; Kajimura, K.; Ishiguro, T. Ambient-pressure superconductivity at 8 K in the organic conductor β-(BEDT-TTF)2I3. Solid State Commun. 1985, 54, 1031–1034. [Google Scholar] [CrossRef]
- Wallis, J.D.; Karrer, A.; Dunitz, J.D. Chiral metals? A chiral substrate for organic conductors and superconductors. Helv. Chim. Acta 1986, 69, 69–70. [Google Scholar] [CrossRef]
- Pop, F.; Laroussi, S.; Cauchy, T.; Gómez-García, C.J.; Wallis, J.D.; Avarvari, N. Tetramethyl-Bis(ethylenedithio)-Tetrathiafulvalene (TM-BEDT-TTF) Revisited: Crystal Structures, Chiroptical Properties, Theoretical Calculations, and a Complete Series of Conducting Radical Cation Salts. Chirality 2013, 25, 466–474. [Google Scholar] [CrossRef] [PubMed]
- Karrer, A.; Wallis, J.D.; Dunitz, J.D.; Hilti, B.; Mayer, C.W.; Bürkle, M.; Pfeiffer, J. Structures and Electrical Properties of Some New Organic Conductors Derived from the Donor Molecule TMET (S,S,S,S-Bis(dimethylethylenedithio) tetrathiafulvalene). Helv. Chim. Acta 1987, 70, 942–953. [Google Scholar] [CrossRef]
- Galán-Mascarós, J.R.; Coronado, E.; Goddard, P.A.; Singleton, J.; Coldea, A.I.; Wallis, J.D.; Coles, S.J.; Alberola, A. A Chiral Ferromagnetic Molecular Metal. J. Am. Chem. Soc. 2010, 132, 9271–9273. [Google Scholar] [CrossRef] [PubMed]
- Pop, F.; Mézière, C.; Allain, M.; Auban-Senzier, P.; Tajima, N.; Hirobe, D.; Yamamoto, H.M.; Canadell, E.; Avarvari, N. Unusual stoichiometry, band structure and band filling in conducting enantiopure radical cation salts of TM-BEDT-TTF showing helical packing of the donors. J. Mater. Chem. C 2021, 9. [Google Scholar] [CrossRef]
- Matsumiya, S.; Izuoka, A.; Sugawara, T.; Taruishi, T.; Kawada, Y. Effect of Methyl Substitution on Conformation and Molecular Arrangement of BEDT-TTF Derivatives in the Crystalline Environment. Bull. Chem. Soc. Jpn. 1993, 66, 513–522. [Google Scholar] [CrossRef]
- Mroweh, N.; Mézière, C.; Pop, F.; Auban-Senzier, P.; Alemany, P.; Canadell, E.; Avarvari, N. In Search of Chiral Molecular Superconductors: κ-[(S,S)-DM-BEDT-TTF]2ClO4 Revisited. Adv. Mater. 2020, 32, 2002811. [Google Scholar] [CrossRef]
- Matsumiya, S.; Izuoka, A.; Sugawara, T.; Taruishi, T.; Kawada, Y.; Tokumoto, M. Crystal Structure and Conductivity of Chiral Radical Ion Salts (Me2ET)2X. Bull. Chem. Soc. Jpn. 1993, 66, 1949–1954. [Google Scholar] [CrossRef]
- Kimura, S.; Maejima, T.; Suzuki, H.; Chiba, R.; Mori, H.; Kawamoto, T.; Mori, T.; Moriyama, H.; Nishio, Y.; Kajita, K. A new organic superconductor β-(meso-DMBEDT-TTF)2PF6. Chem. Commun. 2004, 21, 2454–2455. [Google Scholar] [CrossRef] [PubMed]
- Kimura, S.; Suzuki, H.; Maejima, T.; Mori, H.; Yamaura, J.-I.; Kakiuchi, T.; Sawa, H.; Moriyama, H. Checkerboard-Type Charge-Ordered State of a Pressure-Induced Superconductor, β-(meso-DMBEDT-TTF)2PF6. J. Am. Chem. Soc. 2006, 128, 1456–1457. [Google Scholar] [CrossRef] [PubMed]
- Hountas, A.; Terzis, A.; Papavassiliou, G.C.; Hilti, B.; Pfeiffer, J. Structures of the Conducting Salts of Ethylenedithiotetrathiafulvalene (EDTTTF) and Methylenedithiotetrathiafulvalene (MDTTTF): (EDTTTF)I3 and (MDTTTF)I3. Acta Cryst. C 1990, C46, 220–223. [Google Scholar] [CrossRef] [Green Version]
- Cauchy, T.; Pop, F.; Cuny, J.; Avarvari, N. Conformational Study and Chiroptical Properties of Chiral Dimethyl-Ethylenedithio-Tetrathiafulvalene (DM-EDT-TTF). Chimia 2018, 72, 389–393. [Google Scholar] [CrossRef]
- Abhervé, A.; Mroweh, N.; Cauchy, T.; Pop, F.; Cui, H.; Kato, R.; Vanthuyne, N.; Alemany, P.; Canadell, E.; Avarvari, N. Conducting chiral nickel(II) bis(dithiolene) complexes: Structural and electron transport modulation with the charge and the number of stereogenic centres. J. Mater. Chem. C 2021, 9, 4119–4140. [Google Scholar] [CrossRef]
- Atzori, M.; Pop, F.; Auban-Senzier, P.; Clérac, R.; Canadell, E.; Mercuri, M.L.; Avarvari, N. Complete Series of Chiral Paramagnetic Molecular Conductors Based on Tetramethyl-bis(ethylenedithio)-tetrathiafulvalene (TM-BEDT-TTF) and Chloranilate-Bridged Heterobimetallic Honeycomb Layers. Inorg. Chem. 2015, 54, 3643–3653. [Google Scholar] [CrossRef]
- Coronado, E.; Galán-Mascarós, J.R.; Gómez-García, C.; Laukhin, V. Coexistence of ferromagnetism and metallic conductivity in a molecule-based layered compound. Nature 2000, 408, 447–449. [Google Scholar] [CrossRef] [PubMed]
- Rashid, S.; Turner, S.S.; Day, P.; Howard, J.A.K.; Guionneau, P.; McInnes, E.J.L.; Mabbs, F.E.; Clark, R.J.H.; Firth, S.; Biggs, T. New Superconducting Charge-Transfer Salts (BEDT-TTF)4[A·M(C2O4)3]·C6H5NO2(A = H3O or NH4, M = Cr or Fe, BEDT-TTF = Bis(Ethylenedithio)Tetrathiafulvalene). J. Mater. Chem. 2001, 11, 2095–2101. [Google Scholar] [CrossRef]
- Uji, S.; Shinagawa, H.; Terashima, T.; Yakabe, T.; Terai, Y.; Tokumoto, M.; Kobayashi, A.; Tanaka, H.; Kobayashi, H. Magnetic-Field-Induced Superconductivity in a Two-Dimensional Organic Conductor. Nature. 2001, 410, 908–910. [Google Scholar] [CrossRef] [PubMed]
- Kurmoo, M.; Graham, A.W.; Day, P.; Coles, S.J.; Hursthouse, M.B.; Caulfield, J.L.; Singleton, J.; Pratt, F.L.; Hayes, W.; Ducasse, L.; et al. Superconducting and Semiconducting Magnetic Charge Transfer Salts: (BEDT-TTF)4AFe(C2O4)3∙C6H5CN (A = H2O, K, NH4). J. Am. Chem. Soc. 1995, 117, 12209–12217. [Google Scholar] [CrossRef]
- Martin, L.; Lopez, J.R.; Akutsu, H.; Nakazawa, Y.; Imajo, S. Bulk Kosterlitz−Thouless Type Molecular Superconductor β″-(BEDTTTF)2[(H2O)(NH4)2Cr(C2O4)3]·18-crown-6. Inorg. Chem. 2017, 56, 14045–14052. [Google Scholar] [CrossRef] [Green Version]
- Chaikin, P.M.; Kwak, J.F. Apparatus for thermopower measurements on organic conductors. Rev. Sci. Instrum. 1975, 46, 218–220. [Google Scholar] [CrossRef]
- Almeida, M.; Alcácer, L.; Oostra, S. Anisotropy of thermopower in N-methyl-N-ethylmorpholinium bistetracyanoquinodimethane, MEM(TCNQ)2, in the region of the high-temperature phase transitions. Phys. Rev. B 1984, 30, 2839–2844. [Google Scholar] [CrossRef]
- Lopes, E.B. INETI-Sacavém; Internal Report; INETI Press: Sacavém, Portugal, 1991. [Google Scholar]
- Huebener, R.P. Thermoelectric Power of Lattice Vacancies in Gold. Phys. Rev. 1964, 135, A1281–A1291. [Google Scholar] [CrossRef]
















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Bond lengths (Å) | ||||
|---|---|---|---|---|
| [(S)-1]2AsF6 | [(rac)-1]AsF6 | |||
| C3A–C4A | 1.367(9) | C3–C4 | 1.363(5) | |
| S1A–C3A | 1.714(7) | S1–C3 | 1.739(4) | |
| A | S2A–C3A | 1.759(7) | S2–C3 | 1.739(3) |
| S3A–C4A | 1.744(7) | S3–C4 | 1.740(3) | |
| S4A–C4A | 1.736(7) | S4–C4 | 1.733(3) | |
| C3B–C4B | 1.374(10) | |||
| S1B–C3B | 1.737(7) | |||
| B | S2B–C3B | 1.753(7) | ||
| S3B–C4B | 1.733(7) | |||
| S4B–C4B | 1.726(7) | |||
| Bond Lengths (Å) | ||||
|---|---|---|---|---|
| [(S)-1]AsF6·C4H8O | [(R)-1]AsF6·C4H8O | |||
| A | C3A–C4A | 1.396(13) | C3A–C4A | 1.4214(13) |
| S1A–C3A | 1.737(9) | S1A–C3A | 1.734(12) | |
| S2A–C3A | 1.710(10) | S2A–C3A | 1.7068(98) | |
| S3A–C4A | 1.705(9) | S3A–C4A | 1.6963(91) | |
| S4A–C4A | 1.727(9) | S4A–C4A | 1.7142(12) | |
| B | C3B–C4B | 1.389(13) | C3B–C4B | 1.3577(13) |
| S1B–C3B | 1.729(10) | S1B–C3B | 1.7306(89) | |
| S2B–C3B | 1.710(10) | S2B–C3B | 1.7154(12) | |
| S3B–C4B | 1.705(10) | S3B–C4B | 1.7128(12) | |
| S4B–C4B | 1.735(9) | S4B–C4B | 1.7485(88) | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mroweh, N.; Bogdan, A.; Pop, F.; Auban-Senzier, P.; Vanthuyne, N.; Lopes, E.B.; Almeida, M.; Avarvari, N. Chiral Radical Cation Salts of Me-EDT-TTF and DM-EDT-TTF with Octahedral, Linear and Tetrahedral Monoanions. Magnetochemistry 2021, 7, 87. https://0-doi-org.brum.beds.ac.uk/10.3390/magnetochemistry7060087
Mroweh N, Bogdan A, Pop F, Auban-Senzier P, Vanthuyne N, Lopes EB, Almeida M, Avarvari N. Chiral Radical Cation Salts of Me-EDT-TTF and DM-EDT-TTF with Octahedral, Linear and Tetrahedral Monoanions. Magnetochemistry. 2021; 7(6):87. https://0-doi-org.brum.beds.ac.uk/10.3390/magnetochemistry7060087
Chicago/Turabian StyleMroweh, Nabil, Alexandra Bogdan, Flavia Pop, Pascale Auban-Senzier, Nicolas Vanthuyne, Elsa B. Lopes, Manuel Almeida, and Narcis Avarvari. 2021. "Chiral Radical Cation Salts of Me-EDT-TTF and DM-EDT-TTF with Octahedral, Linear and Tetrahedral Monoanions" Magnetochemistry 7, no. 6: 87. https://0-doi-org.brum.beds.ac.uk/10.3390/magnetochemistry7060087